


Abstract
Multicellular organismal development is controlled by a complex network of transcription factors, promoters and enhancers. Although reliable computational and experimental methods exist for enhancer detection, prediction of their target genes remains a major challenge. On the basis of available literature and ChIP-seq and ChIP-chip data for enhanceosome factor p300 and the transcriptional regulator Gli3, we found that genomic proximity and conserved synteny predict target genes with a relatively low recall of 12–27% within 2 Mb intervals centered at the enhancers. Here, we show that functional similarities between enhancer binding proteins and their transcriptional targets and proximity in the protein–protein interactome improve prediction of target genes. We used all four features to train random forest classifiers that predict target genes with a recall of 58% in 2 Mb intervals that may contain dozens of genes, representing a better than two-fold improvement over the performance of prediction based on single features alone. Genome-wide ChIP data is still relatively poorly understood, and it remains difficult to assign biological significance to binding events. Our study represents a first step in integrating various genomic features in order to elucidate the genomic network of long-range regulatory interactions.
INTRODUCTION
Decoding the regulatory program that controls metazoan development is a major barrier to the understanding of multicellular complexity in higher organisms. A substantial fraction of this program is likely to be encoded in gene deserts which harbor highly conserved non-coding elements (HCNEs), located up to several hundred kilobases away from the nearest gene (1,2). Many of these intergenic and intronic regions represent evolutionarily conserved enhancers and silencers, which we will refer to as ‘enhancers’ in the following. Enhancers coordinate tissue and developmental stage-specific expression of their target genes by inducing changes in chromatin conformation in order to bring distant regulatory elements into spatial proximity of the transcription start sites (TSS) of their target genes. Extensive experimental and computational work has been carried out on the detection of enhancer regions (3,4).
The advent of high‐throughput chromatin immunoprecipitation assays (ChIP-chip and ChIP-seq) has made genome-wide in vivo mapping of protein–DNA interactions possible. In agreement with the observation that evolutionarily conserved regulatory elements are located primarily in intergenic regions, <10% of transcription factors have >50% of their binding sites within 2.5 kb of a transcription start site (5). Recently, Visel et al. (6) employed ChIP-seq to identify several thousand genomic loci in mouse embryonic tissues which were bound by the enhancer-associated p300 protein. p300 is a transcriptional coactivator (7) that is recruited by other DNA binding proteins in a tissue and cell-type specific manner to form an enhanceosome complex with regulatory activity (8). About 87% of the p300 bound loci regions showed tissue-specific enhancer activity (6).
Global correlations with expression data (6) and strong biases for HCNEs to occur in the vicinity of transcription factors and developmental genes (2) support the assumption that enhancers regulate nearby genes. To date, computational and experimental approaches for enhancer detection have employed proximity-based cutoffs on genomic distance or nearest gene assignments to associate putative enhancer regions to their target genes (4,9–11). Although the genes located closest to the enhancers are reasonable candidates for the target genes, this is not a general rule. For instance, a Pax6 enhancer is located in an intron of a neighboring gene (12,13). Interactions between enhancers and their target genes can span large genomic distances. For instance, an enhancer of the sonic hedgehog (Shh) gene is located 1 Mb upstream of the Shh gene (14). For these reasons, enhancer targets cannot be reliably predicted by simple computational rules based on genomic proximity.
Besides genomic distance, conserved synteny is the only feature that has been considered to possess predictive power for enhancer-target gene interactions (15,16). Conserved synteny generally describes a relative order of two or more genomic loci that is conserved in more than one species (Supplementary Figure S1). This might reflect a certain pattern of co-evolution between regulatory region and target gene. The Vista enhancer browser (17) allows manual investigation of flanking genes, and some genome browsers like SynBrowse include information about conserved synteny (18), but no automated approaches exist that specifically predict the target genes of a number of predicted or known enhancers (19). Consequently, existing approaches for enhancer detection (4,20,21) remain incomplete and fail to integrate important developmental target genes into larger regulatory modules and networks that control multicellular organismal development.
One impediment to progress in this area is the paucity of experimental enhancer-target gene interaction data. Commonly used in vivo assays for enhancer activity that use co-injection of enhancer and minimal promoter reporter genes (2,6,22) provide evidence about the tissue specificity of the enhancer but do not indicate which genes are targets of the enhancer. On the other hand, chromosome conformation capture (3C) assays (23,24) test for physical interactions between enhancer and promoter regions, and thus can be used to identify enhancer target genes. However, no large-scale data set of enhancer-specific chromatin interactions is available with which to assess the quality of prediction methods.
To our knowledge, there has been no previous large-scale computational analysis of the prediction of enhancer targets. Ahituv et al. (15) mapped conserved blocks of synteny (CBSs) that were homologous among human/mouse/chicken or human/mouse/frog genomes and identified ∼2000 CBSs > 200 kb for each comparison. They postulated that such CBSs were enriched for long‐range regulatory interactions between enhancers and target genes because the prevalence and distribution of chromosomal aberrations leading to position effects showed a clear bias not only for mapping onto CBS but also for longer CBS size. Using a similar definition based on alignments between human and zebrafish genomes, Akalin et al. identified a set of genomic regulatory blocks (GRBs) located within conserved human/zebrafish-syntenic regions and predicted a set of 269 target genes of within the GRBs (25). The authors postulated that HCNEs within the GRBs are enhancers and that transcription factor genes within the GRBs are their targets, but did not develop a method for predicting target genes on a genome-wide basis or of predicting target genes of a specific enhancer protein. In this work, we present a method to predict the target genes of potential enhancers identified as bound DNA sequences in ChIP-seq and ChIP-chip experiments. We evaluated our method using published data for p300 and Gli3 in embryonic mouse tissues (6,26). Our method uses an integrative approach based on random forest analysis of a combination of genomic proximity, conserved synteny as well as distance in protein–protein interaction (PPI) networks, and Gene Ontology (GO) similarities between regulator and putative target gene. Our algorithm showed a substantially better accurracy than predictions based on any single feature in isolation.
MATERIALS AND METHODS
Genome data and alignments
We downloaded pairwise net alignments generated by blastz (27) for mouse (Mus musculus, mm9) against opossum (Monodelphis domestica, monDom4), chicken (Gallus gallus, galGal3), frog (Xenopus tropicalis, xenTro2), zebrafish (Danio rerio, danRer5) and fugu (Takifugu rubripes, fr2). We initially used data from human and dog in our analysis; however including these data sets did not improve the results (Supplementary Figure S2), and therefore these two genomes were not used for further analysis. In addition, mouse RefSeq annotations for 22 468 genes were downloaded from the UCSC Genome Browser (28). The phylogenetic distances between these species are shown in Supplementary Figure S3, whereby the branch lengths reflect the average number of subsitutions per site as calculated from genome-wide blastz alignments (29).
p300 ChIP-seq data
Visel et al. (6) used chromatin immunoprecipitation with the enhancer-associated protein p300 followed by massively parallel sequencing to map the in vivo binding sites of p300 in mouse embryonic forebrain, midbrain and limb tissue. We downloaded p300 ChIP-seq peaks and lists of upregulated genes that were identified by comparing forebrain and limb expression with E11.5 whole embryo gene expression as measured on Affymetrix GeneChip MouseGenome 430 2.0 arrays. The limb data (30) are based on E11.5 proximal hindlimb expression (GEO series GSE10516, samples GSM264689, GSM264690 and GSM264691). The ChIP-seq also includes P300 bound sites from midbrain, but we did not use this data because no set of midbrain upregulated genes were defined by Visel et al. We focused on upregulated genes since Visel et al. (6) only observed ChIP-seq peak enrichments in the vicinity of genes that are significantly upregulated in the corresponding tissue, indicating that p300 acts as a coactivator rather than as a repressor. In total 2453 ChIP-seq peaks were obtained for embryonic mouse forebrain tissue and 2105 for limb. Additionally, 1062 and 748 significantly upregulated probe sets were obtained that correspond to 555 upregulated genes with RefSeq IDs for forebrain and 347 for limb (6). Affymetrix gene expression microarrays are not able to reliably distinguish between different transcripts of genes. Therefore, one representative transcript was chosen for each gene according to whether a transcript was in the set of differentially expressed probesets, or failing that, arbitrarily as the leftmost transcript on the Watson strand of the chromosome. This reduced the number of RefSeq IDs to 19 569.
Gli3 ChIP-chip data
We downloaded Supplementary Data sets 1 and 2 from Vokes et al. (26). These data sets contain 5274 Gli binding regions and 753 responsive genes that were identified using pairwise and multiple sample comparison of expression levels (Affymetrix Mouse Exon 1.0 ST arrays) for overexpressed and mutated Gli3 versus wildtype and anterior versus posterior forelimbs (26).
Genomic distances between enhancer and target gene
For each gene in a genomic window centered at the enhancer, we calculated the genomic distance between enhancer and target gene as the minimal distance between the endpoints of the enhancer region and the TSS of the candidate target genes. For the genomic distance‐based predictions, the gene with the minimal distance was predicted to be the target gene.
Calculation of conserved synteny score (CSS)
We defined for each enhancer e a genomic interval in the reference species r by selecting all genes for which the genomic distance between enhancer and TSS of the gene g is less than a maximal distance threshold dr(e,g) < Θ (Supplementary Figure S1). For each gene g in this region, we define a conserved synteny score (CSS) by testing in other species s = 1, … ,k whether the distance ds(e,g) between aligned regions of enhancer and TSS is smaller than the threshold Θ. The CSS is then calculated as the sum of phylogenetic distances ϕ(r,s) (Supplementary Figure S3) between the reference r and species s, where ds(e,g) < Θ.
| (1) |
δs(e,g) was also taken to be zero if an orthologous gene and enhancer could not be identified in the other species. Since some genomes in our analysis are not finished and annotations are incomplete, we identified orthologous genes on the basis of the presence of aligned sequences around the promoter region as defined by the TSS ±1 kb. This assumes that the enhancer specifically interacts with the promoters of their target genes. This is supported by our recent finding that the occurrence of intergenic HCNEs correlates with conservation in promoter regions of nearby genes (31), which we interpret as evidence for similar evolutionary constraints acting on the enhancers as well as the promoter regions.
For the enhancer sequence, orthologous sequences were identified on the basis of aligned sequence in the other species. We note that rearrangements that disrupt collinearity are not penalized by our scoring scheme because according to our assumption, enhancers can retain their function even after chromosomal rearrangements that invert genes or change their order.
Gene Ontology similarity definition
We calculated for each GO term (t) in the ontology an information content value (IC) defined as 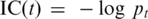 , where pt is the number of genes annotated by GO term t divided by the total number of annotated genes. The similarity between two terms can be calculated as the IC of their most informative common ancestor (MICA) (32). This can be used to calculate the similarity (sim) between one set of terms, to another set of terms, each of which belongs to a particular gene (gi, gj):
, where pt is the number of genes annotated by GO term t divided by the total number of annotated genes. The similarity between two terms can be calculated as the IC of their most informative common ancestor (MICA) (32). This can be used to calculate the similarity (sim) between one set of terms, to another set of terms, each of which belongs to a particular gene (gi, gj):
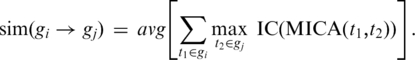 |
(2) |
Note, that 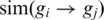 is not necessarily equal to
is not necessarily equal to 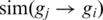 . As previously described (33), we defined the similarity between two genes as the symmetric version of the formula above by calculating:
. As previously described (33), we defined the similarity between two genes as the symmetric version of the formula above by calculating:
| (3) |
Distance computation for PPI networks
In order to define the similarity between two genes, we created a network based on the data of the STRING 8.2 database (34), physical and functional interactions. The network consists of 138 156 interactions including 194 direct interactions between p300 and other proteins. In a previous study, we have shown that global network similarity measures are better suited for defining functionally associated groups of genes (35). We constructed a mouse-specific adjacency matrix, which was transformed into a column-normalized adjacency matrix (A). The random walk starts at a certain node corresponding to a gene gi at time point t0 and randomly visits adjacent nodes. The random walk distance pt + 1(gi,gj) is defined as the probability of the random walker being at node gj at time point t + 1 given that the walker started at gi. For a vector of starting probabilities p0, the state probabilities pt + 1 can be computed iteratively:
| (4) |
whereby r denotes the restart probability (r = 0.7). For 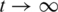 , the state probabilities converge to a stationary distribution p∞ that can be written as:
, the state probabilities converge to a stationary distribution p∞ that can be written as:
| (5) |
The matrix I denotes the identity matrix and the starting probabilities p0 were set to 1 for gi and 0 for all other genes. For two genes gi and gj, we define a symmetric PPI distance score by taking the average of the probabilities to encounter gj when starting at gi and vice versa.
Binary and discriminative random forest classifier
We first developed a binary random forest classifier (36) for the problem of deciding whether a single gene is an enhancer target based on the four features: genomic distance to an enhancer, CSS, PPI distance and GO similarity (`binary RF'). The classifier learns to predict the class from the four features and to output the ratio of trees that voted for this class. In case of missing values, we assigned the median GO similarity or PPI distance value between p300 and all other genes for the respective feature. We used an implementation of Breiman's algorithm that uses random selection of features at each node to determine a split (37) (R package ‘randomForest', version 4.5-34) and to train a random forest of 1000 randomly generated decision trees. The final prediction was made by selecting among all genes in the interval the one with the highest probability (i.e. the highest number of trees voting for it).
The binary RF can yield only a yes/no decision as to whether a gene is an enhancer target or not, and is not designed to rank all the candidate genes in the interval. We therefore implemented a second classifier (`discriminative RF') that evaluates each gene pair gi and gj in the interval using feature values as well as pairwise rankings and then decides among the following outcomes:
gi is the target gene
gj is the target gene
neither gi nor gj is the target
This RF takes 12 input features, corresponding to eight feature values for both genes (genomic distance, CSS, GO similarity, and PPI similarity for gi and for gj) and four features that assign gi either to ‘winner’ (W) or ‘looser’ (L) or ‘tied’ (equal, E) in comparison with the respective feature of gj. Since GO and PPI annotations are incomplete, we added two labels ‘W?’ and ‘L?’ to model the uncertainty that is associated with gene pairs for which one value is missing (`NA'). Then, for each of the four comparisons between genes gi and gj, a feature f is assigned to gi:
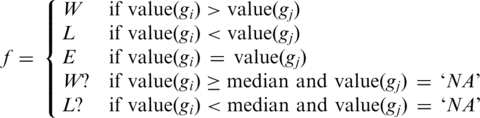 |
A random forest of 1000 trees was trained using these 12 features. The output consisted of the probabilities for the three classes and the class with the majority vote. The final prediction was made by summing over all probabilities for target gene assignments in pairwise comparisons for all pairs in the interval and reporting the gene with the highest sum as the target gene. A schematic overview of both classifiers is shown in Supplementary Figure S4.
Statistical analysis
For evaluation of various values of the maximal distance parameter Θ on the forebrain and limb data, we used only the p300 enhancers with distance <Θ to a differentially upregulated gene. Depending on the distance parameter Θ, it may be that multiple differentially upregulated genes are located in a given genomic interval. In such cases, we counted the prediction as a ‘correct prediction’ if at least one of the upregulated genes was unambiguously predicted as a target gene by any of the prediction methods.
We calculated the precision of a method as 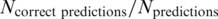 and recall as
and recall as 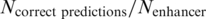 . Precision indicates the probability that a prediction is correct and recall denotes the ratio of enhancers for which a correct prediction could be made. Precision and recall values are highly similar for most methods, only differing in cases where multiple genes are assigned the same maximal score by a method. These cases were counted as ‘no prediction’ in the precision and recall calculations.
. Precision indicates the probability that a prediction is correct and recall denotes the ratio of enhancers for which a correct prediction could be made. Precision and recall values are highly similar for most methods, only differing in cases where multiple genes are assigned the same maximal score by a method. These cases were counted as ‘no prediction’ in the precision and recall calculations.
For the training of the random forest classifiers, we split the enhancer sets into 80% training samples and calculated the precision and recall values on the remaining 20% validation samples. This was done 10 times, the values in Figure 4 represent the means of the different iterations. For both models, we subsampled the training set so that each possible outcome occurred an equal amount of times. The predictions in Supplementary Data S1 contain leave-one-out predictions for the Θ = 1000 kb data and predictions for p300 enhancers that are >1 Mb away of an upregulated gene. For these enhancers, random forest classiers were trained on the complete data set for Θ = 1000 kb.
Figure 4.

Evaluation of random forest classifier predictions on p300 ChIP-seq data for (A) limb, (B) forebrain and (C) 2430 Gli3 bound regions identified from ChIP-chip experiments (26). Average precision-recall values for predictions based on distance, and two random forest classifiers are shown. For the random forest models, the data were split into 80% training and 20% validation sets. The results shown are mean values after 10 repeated evaluations and standard errors. Combination of genomic, functional and protein interactome data allows correct target gene identification in 56–61% of cases for genomic intervals of 2 Mb.
RESULTS
Conserved synteny predictions of enhancer targets have low recall
Previously identified candidate enhancer regions have been shown to be enriched in the vicinity of transcription factors and developmental genes (2,38) and to maintain conserved synteny (15,16). However, it is not clear to what degree conserved synteny or genomic proximity can be used to predict target genes. We therefore initially compared the performance of predictions based on genomic proximity (i.e. the nearest gene is taken to be the target of an enhancer), conserved synteny and randomly choosing one of the genes in a window around the enhancer. The CSS was calculated on the basis of a conserved association between the enhancer and the promoter regions of putative target genes in related genomes (Supplementary Figure S1). We also evaluated ortholog predictions based on protein sequence similarity, but this approach showed slightly lower precision and recall values than using the genomic alignments of promoter sequences (Supplementary Figure S2). The conserved syntenies were weighted by the evolutionary distances between mouse and the target species. For each of the three methods, we evaluated the quality of predictions by calculating precision and recall for various maximal distance thresholds Θ that define a genomic window centered around the enhancer region. Several studies have outlined that HCNEs and thus putative enhancers span genomic regions of several hundreds of kilobases around their target genes (2,38). We therefore assessed the performance of predictions for 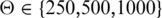 kb. We chose an arbitrary maximal cutoff of Θ = 1000 kb because the great majority of experimentally validated enhancer/target gene pairs are separated by less than this amount (c.f. Figure 1A).
kb. We chose an arbitrary maximal cutoff of Θ = 1000 kb because the great majority of experimentally validated enhancer/target gene pairs are separated by less than this amount (c.f. Figure 1A).
Figure 1.

(A) Distance distribution between enhancers and target genes for 31 regulatory interactions from the literature. (B–D) Evaluation of precision (B), recall (C) and average precision and recall (D) for predictions based on conserved synteny, genomic distance or random predictions of a gene in the genomic interval defined by a maximal distance threshold 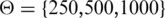 kb around the enhancer. (E) Distance distribution between p300 enhancers and putative target genes (6). (F–H) Evaluation of precision (F), recall (G) and average precision and recall (H) on different sets of p300 enhancers. Although literature data and ChIP-seq data show substantially different distributions of enhancer target gene distances, conserved synteny shows the highest precision values for both data sets.
kb around the enhancer. (E) Distance distribution between p300 enhancers and putative target genes (6). (F–H) Evaluation of precision (F), recall (G) and average precision and recall (H) on different sets of p300 enhancers. Although literature data and ChIP-seq data show substantially different distributions of enhancer target gene distances, conserved synteny shows the highest precision values for both data sets.
At present, there is no database of enhancer targets, and information in the literature is sparse. We therefore compiled a set of 31 known enhancer-target gene interactions from the literature in order to estimate the quality of predictions that are based on genomic distance or conseved synteny. We included all interactions from either human or mouse that were identified either by observations of phenotypes due to genomic rearrangements (14), similar activation and expression pattern of enhancer and target gene (39) and 3C experiments (24). We assumed that enhancer activities are conserved in human and mouse and mapped human enhancers to the homologous sequences using blastz alignments (27) (See Supplementary Table S1 for a list of the 31 experimentally validated enhancer-target gene interactions). Synteny-based predictions showed a precision ∼90% in contrast to ∼61% for genomic distance (Figure 1 A–D). However, recall values for synteny-based predictions only reach a level of 69% for Θ = 250 kb, 62% for Θ = 500 kb and only 37% for Θ = 1000 kb, probably because it is not usually possible to assign an enhancer unambiguously to a single target gene on the basis of synteny alone. In all comparisons, predictions based on CSS and genomic distance perform substantially better than random.
Binding by the transcriptional coactivator, p300 is thought to be a marker for enhancer activity. For instance, in one series of lacZ reporter gene assays in transgenic mice, 75 of 86 (87%) p300 ChIP-seq peaks showed enhancer activity (6).
In the following, we will refer to the p300 ChIP-seq peaks as `p300 enhancers'. It should be noted that a p300 ChIP-seq peak does not necessarily represent a biologically relevant enhancer, which is a limitation of our approach. For our evaluation, we extracted 1862 enhancers from a set of about 4500 p300 enhancers identified by Visel et al. (6) under the assumption that upregulated genes located in the same genomic region as p300 enhancers represent the target genes. Although the target genes of p300 enhancers are likely to also be differentially expressed, we note that this assumption may not be correct in all cases because the upregulation can be due to secondary effects. As with the gold-standard targets from the literature, we compared predictions for various maximal distance thresholds 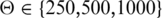 kb that define a genomic window centered around the p300 enhancer, and assessed the performance of synteny-based and genomic proximity-based predictions relative to random guessing. Figure 1E–H shows precision and recall values for the predictions based on each of the two features and random guessing for the merged p300 enhancers from limb and forebrain. In agreement with our observations on the known target gene interactions, conserved synteny alone exhibits a higher precision compared with the use of distance alone. However, conserved synteny could only unambiguously assign a minority of enhancers to their target genes leading to a recall of <20% for Θ = 1000 kb.
kb that define a genomic window centered around the p300 enhancer, and assessed the performance of synteny-based and genomic proximity-based predictions relative to random guessing. Figure 1E–H shows precision and recall values for the predictions based on each of the two features and random guessing for the merged p300 enhancers from limb and forebrain. In agreement with our observations on the known target gene interactions, conserved synteny alone exhibits a higher precision compared with the use of distance alone. However, conserved synteny could only unambiguously assign a minority of enhancers to their target genes leading to a recall of <20% for Θ = 1000 kb.
The difference in the quality of predictions for the two sets is likely to be related at least partially to the different distribution of distances between enhancer and target gene (Figure 2A and E). The results do suggest that using conserved synteny or genomic distance alone is not able to generate accurate target gene predictions for the p300 enhancers.
Figure 2.

We hypothesize that the enhancer binding protein and its target genes show a tendency for shared functions such as `transcription factor activity' and are located in the vicinity of one another in the protein–protein interactome. This is illustrated in the example of the potential regulation of Sox9 by p300. (A) PPIs involving p300 and SOX9. p300 and SOX9 directly interact with one another (40), and also share a number of known or predicted intermediary interaction partners in the protein interaction network (34). (B) SOX9 has 21 GO annotations, and p300 has 35 GO annotations. The eight shared annotations are shown. (C) UCSC Genome Browser view on the Sox9 locus. Seven p300 enhancers from mouse limb tissue (6) show the highest degree of conserved synteny with the Sox9 promoter region, however, only the enhancers p300.5 and p300.6 can unambiguously be assigned to the Sox9 promoter. For the remaining enhancers, multiple genes including Sox9 exhibit the same degree of conserved synteny. However, high GO similarity between p300 and Sox9 as well as their proximity in the PPI network suggest that the target gene of these enhancers is Sox9.
GO similiarity and proximity in PPI networks may be used to improve prediction of enhancer target genes
The above‐mentioned results demonstrated that genomic distance and conserved synteny are of limited utility in predicting the target genes of p300 enhancers. Although CSS has reasonably high precision values, it often fails to unambiguously assign an enhancer to a target gene because multiple genes in the interval exhibit the same degree of conserved synteny. This accounts for 20–50% of p300 enhancers and thus represents a major limitation in the use of conserved synteny. For instance, seven p300 enhancers for limb tissues are located in the Sox9 locus and might account for the upregulation of Sox9, observed by Visel et al. (6). An analysis based on genomic proximity alone would not identify Sox9 as the target, and an analysis based on conserved synteny would identify up to five additional genes in the vicinity as potential targets. In some cases, transcription factors regulate genes with which they also physically interact, e.g. Runx2 and Dlx5 (41,42). We therefore hypothesized that p300 and its targets are located more proximal to each other in PPI networks (Supplementary Figure S5) than p300 and non-target genes. We additionally hypothesized that functional similarity between p300 and targets is greater than between p300 and non-targets. p300 has a number of GO annotations related to organ development, regulation of transcription factor activity, response to stimuli including calcium, transcription cofactor activity and others (Supplementary Table S2). GO analysis of the limb and forebrain upregulated genes from Visel et al. (6) revealed that both sets are significantly enriched in terms such as ‘developmental process’ and ‘transcription factor activity’ (Supplementary Table S3). These observations reflect the known role of p300 in development (43,44).
If we take the upregulation of Sox9 in the above‐mentioned experiment as evidence that Sox9 is the target gene of the enhancers, then the observation that Sox9 is a direct protein interaction partner of p300, and that it shares a number of GO annotations with p300 could be used to identify Sox9, and not one of the other five genes showing conserved synteny, as the correct target gene. This observation motivates our approach (Figure 2).
We therefore tested whether GO similarity and PPI distance can be used to resolve the ambiguity in cases where CSS fails to unambiguously assign an enhancer to a target gene. Figure 3 shows that in case of ties in CSS, target genes show higher GO similarity and are closer to p300 in PPI networks than non-target genes. This observation motivated us to develop an integrative approach that combines all four features in a random forest classifier.
Figure 3.

Since conserved synteny often fails to unambiguously predict a target gene, we tested whether GO similarity and PPI distances may help to resolve cases where multiple genes exhibit equal degrees of conserved synteny. For p300 enhancers from limb and forebrain, we identified all genes in intervals at Θ = 500 kb with highest CSS but that cannot uniquely be assigned to the enhancer. We grouped this set into CSS classes that correspond to evolutionary distances from Supplementary Figure S3 with the exception that the label `fish' indicates 1.72 < CSS ≤ 2.3] and `multiple' corresponds to CSS>2.3. (A and B) GO similarities for target and non-target genes for p300 limb (A) and forebrain (B) enhancers. (C and D) PPI distance for target and non-target genes for p300 limb (C) and forebrain (D) enhancers. Comparison of target genes versus non-target genes within these subsets showed for all subclasses that target genes show a tendency for higher GO similarity and closer distances in PPI networks. *P < 0.05, Wilcoxon test with Benjamini–Hochberg multiple testing correction.
Accurate target gene prediction using random forest classifiers and combination of features
Decision tree induction is a supervised learning method for classifying data. During the learning phase, a tree is constructed iteratively, whereby at each node a test is derived that splits the local training set into two subsets so that the heterogeneity of the resulting subsets is minimized. Typically, the learning phase is stopped as soon as the heterogeneity falls below a certain threshold. Random forests (RFs) are an extension of decision trees to collections of trees that use randomization in the selection of features for splitting the learning sample at each node (37). The final classification is made by taking the majority vote for all trees in the forest (36). Alternatively, classification probabilities can be defined as the ratio of trees voting for a certain class.
We evaluated two random forest approaches (Supplementary Figure S4). The first was a binary RF classifier which separately calculates the probability of each gene of being a target; these probabilities can then be used to rank the k genes in a given interval according to the probability of being a target gene. The second approach involved a discriminative RF classifier which compares all gene pairs in the interval and chooses the gene that was the most frequently predicted target gene in the set of all pairs (see `Materials and Methods' section).
In order to make the methods more easily comparable, we will use the average precision-recall (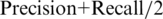 ) as performance measure, analogously to Schweikert et al. (45). Individual precision and recall values for limb and forebrain target gene predictions can be found in Supplementary Tables S4 and S5. Both classifiers were compared with the genomic distance-based method. Figure 4A and B shows the average precision-recall values of the three approaches for
) as performance measure, analogously to Schweikert et al. (45). Individual precision and recall values for limb and forebrain target gene predictions can be found in Supplementary Tables S4 and S5. Both classifiers were compared with the genomic distance-based method. Figure 4A and B shows the average precision-recall values of the three approaches for 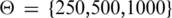 kb. Increasing Θ leads to higher number of genes in an interval. For Θ = 250 kb an average of 8.5 genes is located in the genomic window around the putative p300 from the limb data set. This number increases to 25 genes for Θ = 1000 kb. Supplementary Table S6 shows the average number of genes and differentially expressed genes per interval. Binary and discriminative random forest classifiers show substantially better performance than the distance-based approach. This is true for all comparisons of random forest classifiers versus predictions based on any single feature (Supplementary Tables S4 and S5).
kb. Increasing Θ leads to higher number of genes in an interval. For Θ = 250 kb an average of 8.5 genes is located in the genomic window around the putative p300 from the limb data set. This number increases to 25 genes for Θ = 1000 kb. Supplementary Table S6 shows the average number of genes and differentially expressed genes per interval. Binary and discriminative random forest classifiers show substantially better performance than the distance-based approach. This is true for all comparisons of random forest classifiers versus predictions based on any single feature (Supplementary Tables S4 and S5).
Since cases have been reported where the distance to the target genes exeeds 1 Mb, we also applied the classifier on a putative p300 enhancers that are located up to 2 Mb away of the nearest differentially expressed gene. The average precision-recall value stays almost constant at a level of 58% (Supplementary Tables S5).
We also applied the classifier on 1372 p300 forebrain enhancers and 1324 limb enhancers that are not in proximity of an upregulated gene (Θ = 1000 kb). The predictions include previously reported Bmp7 limb enhancer and a Sox2 enhancer, that is active in rhombencephalon (46,47), suggesting that at least a proportion of the predictions is valid. As expected, GO enrichment analysis shows a similar pattern as the upregulated genes from Visel et al. (Supplementary Tables S3, S8 and S9). It is likely that the enrichment of ‘transcription factor activity’ and ‘developmental process’ is a consequence of using GO similarity in the prediction. However, both predicted target gene sets include significantly enriched terms that are unique to the respective set, such as Notch and Wnt signaling in limb and nervous system development in forebrain (Supplementary Tables S8 and S9). Supplementary Figure S6 shows the distributions of distances between p300 enhancer and predicted target genes. Forebrain enhancer-target gene pairs have a median distance of 388.2 kb with a median of two intervening genes and limb pairs have a distance of 395.3 kb spanning over three genes. A complete list of predictions for the p300 peaks is provided as Supplementary Data S1.
Prediction of Gli3 target genes in a limb ChIP-chip data set
To test whether our method might be applicable to experiments with other enhancers, we analyzed a ChIP-chip data set for Gli3 in mouse limbs (26). Gli3 is a transcription factor that is activated upon Shh signaling which specificies the anterior posterior axis in the developing limb bud and thus regulates the number of digits. Vokes et al. defined a high‐quality set of 5274 Gli3 bound regions of which 2430 are located <1 Mb away of an Shh‐repsonsive gene as identified by differential expression (26). Similar to the p300 data, conserved synteny predicts targets with higher precision but lower recall and the random forest approaches showed substantially better performance than any single feature‐based prediction (Figure 4C and Supplementary Table S7).
DISCUSSION
Current research on long-range regulatory interactions is strongly focused on the computational detection and experimental validation of cis-regulatory elements (4,9). ChIP-seq experiments on the transcriptional coactivator p300 have proven to be a highly reliable method for experimental detection of enhancer regions in various tissues (6,8), but the identified sequences still have to be linked to their transcriptional targets.
Previous studies have postulated that evolutionarily constraints on enhancer-target gene interactions are likely to be responsible for the maintainance the conserved synteny in large genomic intervals (15,16,25). Kikuta et al. and Akalin et al. defined target genes as transcription factors with an HCNE density peak in human-zebrafish conserved-syntenic regions that were termed GRBs (16,25). This is in agreement with the observations that HCNEs are clustered around developmental genes and transcription factors (2,48), but it may not reflect the general pattern of enhancer-target gene interactions since previously defined GRBs (25) do not represent an exhaustive genome-wide collection of genes targeted by long-range regulation. For example, only 579 (12.7%) of ChIP-seq peaks from limb and forebrain overlap with aligned regions between mouse and zebrafish genomes and only 64 (7.7%) of upregulated genes in limb and forebrain overlap with the mouse orthologs of the GRB target genes. Therefore, p300 ChIP-seq data define a more general class of enhancer-target gene interactions that are less conserved and not exclusively restricted to transcription factors.
Two observations prompted us to use proximity in PPI networks and GO functional similarity as features for predicting enhancer targets. Feed forward loops and autoregulatory loops are common in gene regulatory networks (49). p300 binding in genomic regions that display conserved synteny with the Sox9 promoter suggests that it could be involved in the activation of SOX9 transcription (Figure 2). p300 also directly interacts with the SOX9 protein (40) and shares a number of known or predicted intermediary interaction partners in the protein interaction network (34). This suggested the hypothesis that the enhancer binding protein might display a relative proximity to its targets in the protein interaction network. Second, we hypothesized that the regulator would have a higher GO similarity to its targets than to non-target genes. Although GO similarity alone predicts target genes at larger distances (Θ > 500 kb) with comparable recall values as genomic distance (Supplementary Table S4, S5 and S7), it cannot be utilized to predict non-transcription factor targets of very specific functions of p300 targets that are involved in cell adhesion (50) or erythropoiesis (51). The motivation of the random forest approach was therefore to exploit the complementary aspects of the four features. Our results demonstrate that the combination of features dramatically improved the prediction of target genes in genomic intervals of up to 2 Mb centered at the location of a p300 enhancer, with a recall of 58% compared with only 27% for genomic proximity and 12% for conserved synteny (Supplementary Table S5). The analysis of a second data set on Gli3 binding in embryonic mouse limbs displayed a similar advantage for the random forest predictions.
Since available data on enhancer-target gene interactions are extremely limited, we chose to interpret an upregulation of a gene in the vicinity of an enhancer to be the effect of direct regulation. This represents a limitation of our study, as the assumption that genes not found to be differentially expressed are not target genes may be incorrect, for instance because the differential expression may occur at a time point that was not measured. Another limitation is the assumption of our model that enhancers can regulate only one target gene. Enhancers may be active in various tissues (2) and multiple enhancers may coordinate the expression of one target gene (24). Nevertheless, under the assumptions of our study, we have shown that genomic distance, conserved synteny, PPI distance and functional similarity can be combined to dramatically improve predictions of the target genes.
The random forest classifiers that have been trained on limb and forebrain enhancers cannot be directly transferred to enhancer-target gene prediction in other tissues. Using the limited data now available, we have observed that the random forest classifiers are specific not only for the immunoprecipitated factor but also for the tissue (Supplementary Figure S7). However, more data will be needed to evaluate if this reflects variability between experiments or tissue specificity characteristics of regulatory interactions. With this proviso, our methodology can be applied to new ChIP-seq data to prioritize candidate enhancer-target gene interactions for validation experiments, and may also be useful for assessing the most biologically relevant hits identified by high-throughput chromosome conformation capture assays that have been developed to globally map chromatin interactions (23,52,53). ChIP-seq is still a relatively new protocol and contains biases that are poorly understood (54); however, with more experimental data sets becoming publicly available, more detailed analyses can be performed to further evaluate how to combine functional classification of binding events and the association to target genes into an integrative downstream analysis of ChIP-seq experiments.
SUPPLEMENTARY DATA
Supplementary Data are availble at NAR Online.
FUNDING
Bundesministerium für Bildung und Forschung (BMBF, project number 0313911); Deutsche Forschungsgemeinschaft (SFB 760). Funding for open access charge: German Research Foundation (DFG).
Conflict of interest statement. None declared.
REFERENCES
- 1.Ovcharenko I, Loots GG, Nobrega MA, Hardison RC, Miller W, Stubbs L. Evolution and functional classification of vertebrate gene deserts. Genome Res. 2005;15:137–145. doi: 10.1101/gr.3015505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Krys Kelly K, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 4.Pennacchio LM, Loots GG, Nobrega MA, Ovcharenko I. Predicting tissue-specific enhancers in the human genome. Genome Res. 2007;17:201–211. doi: 10.1101/gr.5972507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/P300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol. Cell. 1998;1:277–287. doi: 10.1016/s1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 9.Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, Ovcharenko I. Genome-wide discovery of human heart enhancers. Genome Res. 2010;20:381–392. doi: 10.1101/gr.098657.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warner JB, Philippakis AA, Jaeger SA, He FS, Lin J, Bulyk ML. Systematic identification of mammalian regulatory motifs’ target genes and functions. Nat. Methods. 2008;5:347–353. doi: 10.1038/nmeth.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell. 2010;18:662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleinjan DA, Seawright A, Elgar G, Heyningen V. Characterization of a novel gene adjacent to Pax6, revealing synteny conservation with functional significance. Mamm. Genome. 2002;13:102–107. doi: 10.1007/s00335-001-3058-y. [DOI] [PubMed] [Google Scholar]
- 13.Kleinjan DA, Seawright A, Mella S, Carr CB, Tyas DA, Simpson TI, Mason JO, Price DJ, van Heyningen V. Long-range downstream enhancers are essential for Pax6 expression. Dev. Biol. 2006;299:563–581. doi: 10.1016/j.ydbio.2006.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lettice LA, Heaney SJH, Purdie LA, Li Li, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 15.Ahituv N, Prabhakar S, Poulin F, Rubin EM, Couronne O. Mapping cis-regulatory domains in the human genome using multi-species conservation of synteny. Hum. Mol. Genet. 2005;14:3057–3063. doi: 10.1093/hmg/ddi338. [DOI] [PubMed] [Google Scholar]
- 16.Kikuta H, Laplante M, Navratilova P, Komisarczuk AZ, Engström PG, Fredman D, Akalin A, Caccamo M, Sealy I, Howe K, et al. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007;17:545–555. doi: 10.1101/gr.6086307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visel A, Minovitsky S, Dubchak I, Pennacchio L. VISTA enhancer browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 2007;35(Database issue):D88–D92. doi: 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan X, Stein L, Brendel V. Synbrowse: a synteny browser for comparative sequence analysis. Bioinformatics. 2005;21:3461–3468. doi: 10.1093/bioinformatics/bti555. [DOI] [PubMed] [Google Scholar]
- 19.Farnham PJ. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009;10:605–616. doi: 10.1038/nrg2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frith MC, Li MC, Weng Z. Cluster-buster: Finding dense clusters of motifs in DNA sequences. Nucleic Acids Res. 2003;31:3666–3668. doi: 10.1093/nar/gkg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinha S, Liang Y, Siggia E. Stubb: a program for discovery and analysis of cis-regulatory modules. Nucleic Acids Res. 2006;34(Web Server issue):W555–W559. doi: 10.1093/nar/gkl224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller M, Chang B, Albert S, Fischer N, Tora L, Strähle U. Intronic enhancers control expression of zebrafish sonic hedgehog in floor plate and notochord. Development. 1999;126:2103–2116. doi: 10.1242/dev.126.10.2103. [DOI] [PubMed] [Google Scholar]
- 23.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 24.D'haene B, Attanasio C, Beysen D, Dostie J, Lemire E, Bouchard P, Field M, Jones K, Lorenz B, Menten B, et al. Disease-causing 7.4 kb cis-regulatory deletion disrupting conserved non-coding sequences and their interaction with the FOXL2 promotor: implications for mutation screening. PLoS Genet. 2009;5:e1000522. doi: 10.1371/journal.pgen.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akalin A, Fredman D, Arner E, Dong X, Bryne J, Suzuki H, Daub C, Hayashizaki Y, Lenhard B. Transcriptional features of genomic regulatory blocks. Genome Biol. 2009;10:R38. doi: 10.1186/gb-2009-10-4-r38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vokes SA, Ji H, Wong WH, McMahon AP. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008;22:2651–2663. doi: 10.1101/gad.1693008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W. Human-mouse alignments with blastz. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, et al. The UCSC genome browser database: 2008 update. Nucleic Acids Res. 2008;36(Database issue):D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller W, Rosenbloom K, Hardison RC, Hou M, Taylor J, Raney B, Burhans R, King DC, Baertsch R, Blankenberg D, et al. 28-way vertebrate alignment and conservation track in the UCSC genome browser. Genome Res. 2007;17:1797–1808. doi: 10.1101/gr.6761107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krawchuk D, Kania A. Identification of genes controlled by LMX1B in the developing mouse limb bud. Dev. Dyn. 2008;237:1183–1192. doi: 10.1002/dvdy.21514. [DOI] [PubMed] [Google Scholar]
- 31.Rödelsperger C, Köhler S, Schulz MH, Manke T, Bauer S, Robinson PN. Short ultraconserved promoter regions delineate a class of preferentially expressed alternatively spliced transcripts. Genomics. 2009;94:308–316. doi: 10.1016/j.ygeno.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 32.Resnik P. Proceedings of the 14th International Joint Conference on Artificial Intelligence. Morgan Kaufmann Publishers Inc.; 1995. Using information content to evaluate semantic similarity in a taxonomy; pp. 448–453. [Google Scholar]
- 33.Köhler S, Schulz MH, Krawitz P, Bauer S, Dölken S, Ott CE, Mundlos C, Horn D, Mundlos S, Robinson PN. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 2009;85:457–64. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412–416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Köhler S, Bauer S, Horn S, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 2008;82:949–58. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geurts P, Irrthum A, Wehenkel L. Supervised learning with decision tree-based methods in computational and systems biology. Mol. Biosyst. 2009;5:1593–1605. doi: 10.1039/b907946g. [DOI] [PubMed] [Google Scholar]
- 37.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 38.Sandelin A, Bailey P, Bruce S, Engström PG, Klos JM, Wasserman WW, Ericson J, Lenhard B. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004;5:99. doi: 10.1186/1471-2164-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, Kent WJ, Haussler D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006;441:87–90. doi: 10.1038/nature04696. [DOI] [PubMed] [Google Scholar]
- 40.Furumatsu T, Tsuda M, Yoshida K, Taniguchi N, Ito T, Hashimoto M, Ito T, Asahara H. Sox9 and p300 cooperatively regulate chromatin-mediated transcription. J. Biol. Chem. 2005;280:35203–35208. doi: 10.1074/jbc.M502409200. [DOI] [PubMed] [Google Scholar]
- 41.Roca H, Phimphilai M, Gopalakrishnan R, Xiao G, Franceschi RT. Cooperative interactions between RUNX2 and homeodomain protein-binding sites are critical for the osteoblast-specific expression of the bone sialoprotein gene. J. Biol. Chem. 2005;280:30845–30855. doi: 10.1074/jbc.M503942200. [DOI] [PubMed] [Google Scholar]
- 42.Holleville N, Matéos S, Bontoux M, Bollerot K, Monsoro-Burq A. Dlx5 drives Runx2 expression and osteogenic differentiation in developing cranial suture mesenchyme. Dev. Biol. 2007;304:860–874. doi: 10.1016/j.ydbio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh AK, Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell Physiol. 2007;213:663–671. doi: 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- 44.Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth J, Marino S, Wittwer J, Scheidweiler A, Eckner R. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 2003;22:5175–5185. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schweikert G, Zien A, Zeller G, Behr J, Dieterich C, Ong CS, Philips P, de Bona F, Hartmann L, Bohlen A, et al. mGene: accurate SVM-based gene finding with an application to nematode genomes. Genome Res. 2009;19:2133–2143. doi: 10.1101/gr.090597.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams D, Karolak M, Robertson E, Oxburgh L. Control of kidney, eye and limb expression of Bmp7 by an enhancer element highly conserved between species. Dev. Biol. 2007;311:679–690. doi: 10.1016/j.ydbio.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uchikawa M, Ishida Y, Takemoto T, Kamachi Y, Kondoh H. Functional analysis of chicken Sox2 enhancers highlights an array of diverse regulatory elements that are conserved in mammals. Dev Cell. 2003;4:509–519. doi: 10.1016/s1534-5807(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 48.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 49.Kielbasa SM, Martin Vingron M. Transcriptional autoregulatory loops are highly conserved in vertebrate evolution. PLoS ONE. 2008;3:e3210. doi: 10.1371/journal.pone.0003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim Y, Lee S, Ye S, Lee JW. Epigenetic regulation of integrin-linked kinase expression depending on adhesion of gastric carcinoma cells. Am. J. Physiol Cell Physiol. 2007;292:C857–C866. doi: 10.1152/ajpcell.00169.2006. [DOI] [PubMed] [Google Scholar]
- 51.Engel JD, Tanimoto K. Looping, linking, and chromatin activity: new insights into beta-globin locus regulation. Cell. 2000;100:499–502. doi: 10.1016/s0092-8674(00)80686-8. [DOI] [PubMed] [Google Scholar]
- 52.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Bin Mohamed Y, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.