


Abstract
Early in F plasmid conjugative transfer, the F relaxase, TraI, cleaves one plasmid strand at a site within the origin of transfer called nic. The reaction covalently links TraI Tyr16 to the 5′-ssDNA phosphate. Ultimately, TraI reverses the cleavage reaction to circularize the plasmid strand. The joining reaction requires a ssDNA 3′-hydroxyl; a second cleavage reaction at nic, regenerated by extension from the plasmid cleavage site, may generate this hydroxyl. Here we confirm that TraI is transported to the recipient during transfer. We track the secondary cleavage reaction and provide evidence it occurs in the donor and F ssDNA is transferred to the recipient with a free 3′-hydroxyl. Phe substitutions for four Tyr within the TraI active site implicate only Tyr16 in the two cleavage reactions required for transfer. Therefore, two TraI molecules are required for F plasmid transfer. Analysis of TraI translocation on various linear and circular ssDNA substrates supports the assertion that TraI slowly dissociates from the 3′-end of cleaved F plasmid, likely a characteristic essential for plasmid re-circularization.
INTRODUCTION
During bacterial conjugation, a specific ssDNA plasmid strand, called the T-strand, is transferred from donor to recipient, likely in a mechanism similar to rolling-circle replication (RCR) (1). Rolling-circle replication is one of many replication mechanisms used by bacteriophages, viruses and plasmids [reviewed in (2–4)]. RCR is initiated by Rep class proteins. Rep proteins bind DNA within the origin of replication and cleave one strand of DNA at a specific site called nick, resulting in a covalent attachment between a Rep Tyr and a ssDNA 5′-phosphate. The cleavage reaction is reversible, meaning that Rep can rejoin nicked ssDNA. Rep recruits a helicase to the cleavage site, and a replication complex can assemble at the free 3′-OH, extending from the site using the 3′-OH as a primer. Replication is terminated following extension of the newly synthesized strand beyond the Rep protein nick site, yielding two identical Rep nick sites. A secondary nick site is generated at the replication start site and a tertiary nick site is generated at the replication end. Because Rep proteins likely form dimers or oligomers (5–6), one monomer of a Rep dimer could generate the initial nick while a second monomer could cleave at the secondary nick site. The action of the second monomer would create a free ssDNA end that could be joined by the initial Rep monomer to the ssDNA end covalently attached to it. The result would be circularization of the original plasmid strand and release of the initial Rep monomer. Similarly, the initial Rep monomer then cleaves the tertiary nick site to free a 3′-OH and the second Rep monomer circularizes the newly synthesized plasmid strand. Therefore, a Rep complex must perform three cleavage and two rejoining reactions in order to re-circularize the original and a newly synthesized plasmid. Alternatively, a flip-flop mechanism of re-circularization has been identified in which a Rep protein has two catalytic Tyr within the same Rep active site (7). The required reactions then can be performed by a single Rep protein.
Relaxase proteins involved in bacterial conjugation and Rep proteins share a similar fold (8–10). Relaxases, like Rep proteins, cleave ssDNA through a transesterification that yields a phosphotyrosyl linkage (11). Relaxase TrwC from conjugative plasmid R388 employs two catalytic Tyr to perform cleavage and rejoining reactions similar to reactions of Rep proteins during RCR (12–13). TrwC Y18 links to the 5′-phosphate at nic within the R388 origin of transfer (oriT) during the initiation of plasmid transfer and pilots plasmid ssDNA into the recipient. In the recipient, Y26 probably catalyzes a secondary cleavage reaction on the newly synthesized oriT, generating the 3′-hydroxyl required to circularize the transferred plasmid (13). F TraI, like R388 TrwC, contains two pairs of adjacent Tyrs (Y16, Y17; Y23, Y24). Y16 catalyzes the primary cleavage reaction (8) and in principle Y23 could catalyze a secondary cleavage reaction (14). However, Y23 is not located near the divalent cation required for cleavage in the crystal structure (8). Moreover, there is no obvious ssDNA binding surface close to Y23. Therefore, Y23-catalyzed cleavage would require significant structural rearrangement and/or a different mode of ssDNA recognition and catalysis (8,15). TraI cleavage activity against a supercoiled DNA substrate is substantially more efficient in the presence of proteins that participate in a complex, called the relaxosome, that initiates F plasmid transfer (16–18). In addition to relaxase activities, TraI and TrwC both possess processive ssDNA-dependent helicase activities that unwind DNA by unidirectional translocation on ssDNA in a 5′–3′-direction (19–20).
Both TrwC and TraI belong to the MobF group of relaxases (21) and we wanted to determine whether TrwC from R388 and TraI from F plasmid perform similar reactions during plasmid transfer. We provide evidence that F plasmid TraI employs a different mechanism to transfer F plasmid. Testing TraI variants for their ability to facilitate plasmid transfer, we find that F TraI requires only one catalytically active Tyr (Y16), similar to the RP4 plasmid TraI from the MobP group of relaxases (22). Our second objective was to present a model of TraI activity during bacterial conjugation. We track the secondary cleavage reaction and provide evidence that plasmid ssDNA is transferred in a cleaved form with a free 3′-hydroxyl. We also describe F plasmid TraI translocation on various linear and circular ssDNA substrates and study cleavage and ligation reactions in vitro. Our data indicate that (1) TraI can recognize and slowly dissociate from a particular nucleotide sequence on the 3′-end of linear ssDNA and (2) a hairpin located on the 3′-end of F plasmid oriT catalyzes the ssDNA joining reaction.
MATERIALS AND METHODS
Oligonucleotides, expression constructs, strains and materials
Oligonucleotides used in this study were purchased from Integrated DNA Technologies. Fluorophore-labeled oligonucleotides were HPLC-purified by Integrated DNA Technologies while other oligonucleotides were desalted but not further purified. Sequences of oligonucleotides used in binding and ATPase assays are given in Table 1 and Supplementary Table SI. Sequences of oligonucleotides used for PCR amplification and mutagenesis are available upon request. The construction of TraI expression plasmid pET24a-traI has been described (23). BamHI, AatII, AccI, DraIII, T4 DNA polymerase, T4 DNA ligase and the pACYC177 vector were purchased from New England Biolabs. The pUC18 vector was purchased from Stratagene. Pyruvate kinase, lactate dehydrogenase, phosphoenolpyruvate and nicotinamide adenine dinucleotide in reduced form (NADH) were purchased from Sigma. Escherichia coli strain XK1502 containing F plasmid with the traI gene substituted by tetracycline resistance cassette (XK1502/FΔTraI) was kindly provided by Prof. Beth Traxler (University of Washington, USA) (24).
Table 1.
Sequences of oligonucleotides used in the study
| ssDNA label | ssDNA sequence |
|---|---|
| oriT17 oligonucleotide | TTT GCG TGG GGT GTG GT |
| A oligonucleotide | GGT TCT GTT TCA TGA TGT CAC G |
| B oligonucleotide | GGG CTG GCA AGC CAC GTT TGG TG |
| Oligonucleotide70 | ACT TAA AAC AGC ATT CAT AAG TTA CCT CAA TTT CGG ATA AAT GAA TAA AAT TAC GCC CCG CCC TGC CAC T |
| 5′oriT60-mer | TTTTCATAACACTCTATTTATAAAGAAAAATCAGCAAAAACTTGTT TTTGCGTGGGGTGT |
| -90 -30 5′oriT60-mer | AATATCATAAAGAGAGTAAGAGAAACTAATTTTTCATAACACTCTA TTTATAAAGAAAAA |
| 3′oriT30-mer | GGTGCTTTTGGTGGTGAGAACCACCAACCT |
| 3′oriT60-mer | GGTGCTTTTGGTGGTGAGAACCACCAACCTGTTGAGCCTTTTTGTG GAGTGGGTTAAATT |
| Hairpin | GCAAAAACTTGTTTTTGCGTGGGGTGT |
| No hairpin | GCTTTTTCTTGTTTTTGCGTGGGGTGT |
| Hairpin G144′C | GCAAAAACTTGTTTTTGCGTGGGCTGT |
| No hairpin G144′C | CGTTTTTGTTGTTTTTGCGTGGGCTGT |
| Hairpin R100 | GCAAAAACTTGTTTTTGCGTAGTGTGT |
| No hairpin R100 | CGTTTTTGTTGTTTTTGCGTAGTGTGT |
| Hairpin 3random | GCAAAAACTTGTTTTTGCCACGGGTGT |
| No hairpin 3random | CGTTTTTGTTGTTTTTGCCACGGGTGT |
| F oriT hairpin | GCAAAAACTTGTTTTTGCGTGGGGTGT GGT |
| F oriT no hairpin | GCTTTTTCTTGTTTTTGCGTGGGGTGT GGT |
Underlined bases indicate differences from F oriT hairpin oligonucleotide.
Preparation of circular ssDNA
Circular ssDNA from vector pUC18 was extracted from M13 phage using M13KO7 helper phage (New England Biolabs) and purified as described (25).
TraI protein and TraI protein variants purification and mutagenesis
TraI and TraI relaxase domain (TraI36) were expressed, purified and quantified as described (26,27). Mutagenesis was performed as described (28) using the QuikChange mutagenesis kit (Stratagene) with one modification: LA Taq DNA polymerase (TaKaRa Bio Inc.) was used in place of PfuTurbo DNA polymerase. Mutant plasmids were generated from vector pET24a-traI and engineered substitutions were verified by DNA sequencing.
Construction of oriT inserts in pACYC177
pACYC177 plasmid constructs are illustrated in Figure 1. The oriT region coordinate system used herein is that defined by Finlay et al. (29). A 300-bp F plasmid oriT region from 81 to 380 nt (30) was PCR amplified using the F′ from E. coli K12 ER2738 strain as a template. PCR primers contained AatII and BamHI restriction sites. The amplification product was digested with AatII and BamHI restriction enzymes and ligated into a pACYC177 vector digested with these enzymes. The insert was verified by DNA sequencing. The resulting plasmid was named pACYC177o300. A 35-bp long F plasmid oriT section from nucleotide 133 to 168 was generated using primers 5′-ATGCAAAAACTTGTTTTTGCGTGGGGTGTGGTGCTTTGTT-3′ and 5′-AAAGCACCACACCCCACGCAAAAACAAGTTTTTGC-3′. Primers were annealed by 10 min incubation at 95°C and slow cooling to room temperature. The product was ligated into pACYC177 digested with AccI and DraIII. The insert was verified by DNA sequencing. The resulting plasmid was named pACYC177o35. pACYC177o2ak contains both 300 and 35 bp long oriT regions, cloned in that order using the two steps described above. Sequences of the inserts were verified by DNA sequencing.
Figure 1.

Map of pACYC177 plasmid and variants. (A) pACYC177 plasmid and variants with one F plasmid oriT are illustrated. (B) pACYC177 plasmid variant with two F plasmid oriT regions is shown on the left. The deletant form, one possible product of pACYC177o2ak plasmid transfer, is shown on the right. Kan, kanamycin resistant gene; Amp, ampicilin resistant gene; oriR, origin of replication; oriT300, 300bp long F plasmid oriT region from nucleotide 81 to 380; oriT35, 35bp long F plasmid oriT region from nucleotide 133 to 168; oriT, F plasmid origin of transfer.
Affinity for ssDNA
Affinities of proteins for ssDNA were measured by following fluorescence emission intensity and anisotropy changes of a 3′-carboxytetramethyl-rhodamine-labeled oligonucleotide upon protein binding at 25°C. Data were collected on an AVIV ATF-105 automatic titrating fluorometer. Prior to measurement, fluorescently labeled oligonucleotides were diluted to 4 nM in binding buffer (100 mM NaCl, 20 mM Tris–HCl pH 7.5 and 1 mM EDTA). TraI in binding buffer was titrated into the oligonucleotide dilution. The fluorophore was excited at 520 nm and emission data were collected at 580 nM. Each data point was averaged over 15 s and equilibration time was 75 s. Fluorescence data were fit to a single site binding model (1) using KaleidaGraph (Synergy Software) and equation 1:
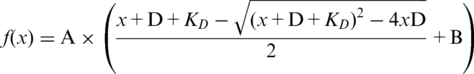 |
(1) |
where D is the concentration of labeled ssDNA, x is concentration of protein and KD is dissociation constant. A is amplitude and A × B is lower baseline.
ATPase activity assay
The ssDNA-dependent ATPase activity of TraI was measured according to Dash et al. (31) using the pyruvate kinase/lactate dehydrogenase coupled enzyme assay. TraI (1 or 20 nM) was mixed with a circular ssDNA or linear ssDNA substrate and incubated for 5 min at 37°C in 1 ml of assay solution [1 mM phosphoenolpyruvate, 0.3 mM NADH, 100 mg/ml pyruvate kinase, 100 mg/ml lactate dehydrogenase, 40 mM Tris–HCl (pH 7.5), 5 mM MgCl2 and 76.4 mM (NH3)2SO4]. ATP (1 mM final) was added to initiate the reaction cascade. The decrease in NADH concentration was monitored by the change in optical density at 340 nm at 37°C for 15 min in a UV-506 spectrophotometer (Shimadzu). The NADH concentration was calculated by using an extinction coefficient of 6220 M−1 cm−1. ATPase rates were determined from the rate of change in absorbance of NADH at 340 nm. The ATPase rate dependency on ssDNA substrate concentration was estimated by fit of the Michelis–Menten equation using KaleidaGraph (Synergy Software).
Oligonucleotide cleavage assay
About 20 nM of 5′-Cy5-labeled oligonucleotide was mixed with 1 µM or 20 nM TraI or TraI relaxase domain in reaction buffer [20 mM MgCl2, 100 mM NaCl, 40 mM Tris–HCl (pH 7.5)]. The reactions were incubated at 37°C for 1 h. Afterward, 1 µM Y16F TraI protein was added to the reactions indicated and all reactions were incubated for an additional hour at 37°C. Reactions were terminated by the addition of ∼2 mg/ml of Proteinase K. Samples were mixed with an equal volume of 100% formamide (v/v), heated at 95°C for 5 min prior to gel loading and run on a 16% polyacrylamide-8.3 M urea gel at 20 V per 1 cm of gel length for 2.5 h. Gels were imaged on a Typhoon 9410 Variable Mode Imager (GE Healthcare) using excitation at 648 nm and emission at 668 nm.
Oligonucleotide joining assay
About 20 nM of 3′-Cy5-labeled oriT 17-mer oligonucleotide and 1 µM TraI protein and TraI relaxase domain were mixed in reaction buffer [20 mM MgCl2, 100 mM NaCl, 40 mM Tris–HCl (pH 7.5)] and incubated at 37°C for 1 h. 10 µM of 27-base unlabeled oligonucleotide with an oriT sequence and a 3′ that ends at nic, the TraI cleavage site, was added to the reaction and incubated at 37°C for an additional hour. The reactions were stopped with the addition of ∼2 mg/ml of Proteinase K. Samples were mixed with an equal volume of 100% formamide (v/v), heated at 95°C for 5 min prior to gel loading and run on a 16% polyacrylamide–8.3M urea gel at 20V per 1 cm of gel for 2.5 h. Gels were imaged on a Typhoon 9410 Variable Mode Imager (GE Healthcare) using excitation at 648 nm and emission at 668 nm.
Conjugative transfer assays
XK1502/FΔTraI (24) was transformed with pET24a-traI and used as the plasmid donor strain. Streptomycin resistant E. coli strain TB1 was used as the recipient. Both donor and recipient cells were grown at 37°C to A600 ∼0.6, pelleted at 4000 × g for 10 min and resuspended in an equivalent volume of phosphate buffered saline (PBS) buffer. About 500 µl of recipients were mixed with 50 µl of donors, incubated at 37°C for 30 or 60 min and vortexed for 30 s to disrupt mating. Transconjugants were selected on LB-Agar plates with 25 µg/ml tetracycline (Tet) and 50 µg/ml streptomycin (Strep) (LB-tet-strep), while donors were selected on LB-Agar plates with 25 µg/ml Tet and 35 µg/ml kanamycin (Kan) (LB-tet-kan). The T7 promoter of the pET24a-traI vector was not induced during the experiment. Transconjugation efficiency was calculated as the number of transconjugants per donor.
To monitor TraI activity in conjugation recipient cells, E. coli XK1502/FΔTraI was transformed with pET24a-traI Y16F/Y17F/Y23F/Y24F or pET24a-traI-Y16F and used as the donor strain. E. coli strain TB1 carrying vector pET24a-traI served as the recipient. Transconjugants were selected on LB-tet-strep plates.
To monitor TraI activity in the conjugation donor, E. coli strain TB1 was chosen as the recipient cell and tetracycline-resistant E. coli K12 strain ER2738 carrying pACYC177o2ak was used as the donor. Transconjugants were selected on LB-Agar plates with 50 µg/ml Strep, 35 µg/ml Kan and/or 50 µg/ml ampicillin (Amp).
A two-step mating was performed as indicated in Figure 2 in LB media instead of PBS. In the first step, E. coli strain XK1502/FΔTraI transformed with pET24a-traI was used as the donor and E. coli strain BL21 carrying pACYC177o300 served as the recipient. Cells were incubated at 37°C for 60 min. Transconjugants were selected on LB-tet-kan-amp plates. In the second step, the mixture of cells from the first step was used as the donor and E. coli strain TB1 served as the recipient. The final mixture of cells was incubated at 37°C for 60 min and transconjugants were selected on LB-strep-kan-amp plates.
Figure 2.

Scheme of two-step mating. Bacteria are represented by rounded rectangles and particular E. coli strains are indicated in bold. Plasmids are indicated together with their antibiotic resistance. In the first step, FΔTraI plasmid is transferred with help of non-transferable plasmid pET24a-traI. In the second step, transferred FΔTraI plasmid and transferred TraI protein facilitate transfer of pACYC177o300 plasmid. Kan, kanamycin; Amp, ampicillin; Tet, tetracycline; Strep, streptomycin.
RESULTS
TraI Y16 is essential for F plasmid conjugation
F TraI performs ssDNA cleavage and joining reactions to initiate and terminate plasmid transfer. Like other members of the MobF group of relaxases, TraI contains two pairs of adjacent Tyr residues, Y16/Y17 and Y23/Y24, located within or near the relaxase active site (32–33). These residues are the best candidates to catalyze cleavage and joining reactions. TraI Y16 is involved in the cleavage of ssDNA at oriT (8,23,28). Gonzalez-Perez et al. (13) provided evidence that the TrwC relaxase from R388 plasmid, a member of the MobF group of relaxases, initially cleaves the oriT with Y18, and employs a second Tyr, Y26, to perform secondary cleavage at nic, resulting in two ssDNA molecules covalently attached to TrwC. The second cleavage reaction generates a free 3′-hydroxyl at nic that is required to circularize the transferred plasmid within the recipient (13). To catalyze the cleavage reactions, TrwC employs two Tyr, Y18 and Y26 (13). We set out to test whether F plasmid TraI performs reactions analogous to TrwC by measuring conjugation efficiencies facilitated by TraI and TraI variants. We used the XK1502 donor cell line carrying an F variant having a deleted TraI gene (FΔTraI) (24). The transfer efficiency of XK1502/FΔTraI is below the detection limit (<1.6 × 10−7) meaning that no transconjugants were observed on the plates. XK1502/FΔTraI was then complemented with plasmids carrying wild-type (wt) traI or traI variants with individual Tyr for Phe substitutions (TraI Y16F, TraI Y17F, TraI Y23F or TraI Y24F). Conjugation efficiencies are given in Table 2. As expected, complementation of FΔTraI by TraI Y16F shows ∼5000-fold reduced plasmid transfer efficiency (28). Complementation of FΔTraI with TraI Y17F, TraI Y23F or TraI Y24F, however, yielded similar high transfer efficiencies that were only slightly reduced (to 30–50%) relative to complementation with wt TraI. These data indicate that TraI Y16 is essential for F plasmid conjugation and suggest that the mechanism of F plasmid transfer differs from the proposed R388 plasmid transfer mechanism, not requiring a second cleavage reaction catalyzed by the same TraI molecule.
Table 2.
Transfer Efficiency of F ΔTraI plasmid complemented with TraI
| Donor and recipient co-incubation time |
||
|---|---|---|
| TraI variant | 30 min | 60 min |
| TraI wt | 0.039 ± 0.007 | 0.12 ± 0.04 |
| TraI Y16F | ND | 2.5 ± 1.6 × 10−5 |
| TraI Y17F | 8.6 ± 2.0 × 10−3 | 0.037 ± 0.004 |
| TraI Y23F | 0.033 ± 0.032 | 0.063 ± 0.050 |
| TraI Y24F | 0.028 ± 0.011 | 0.062 ± 0.009 |
| TraI Y16F/Y17F | <1.7 × 10−5 | 1.5 × 10−5 |
| TraI Y23F/Y24F | 0.016 ± 0.001 | 0.033 ± 0.001 |
| TraI Y16F/Y23F/Y24F | ND | 0.5 ± 0.3 × 10−5 |
| TraI Y17F/Y23F/Y24F | ND | 0.07 ± 0.05 |
| TraI Y16F/Y17F/Y23F/Y24F | ND | 1.3 ± 1.4 × 10−5 |
| TraI from ER2738 | 0.090 ± 0.012 | 0.29 ± 0.07 |
| No TraI | ND | <1.6 × 10−7 |
Transfer efficiencies, calculated as number of transconjugants per donor, are averages of 2–4 independent measurements and are listed with the standard deviation. The E. coli K12 ER2738 strain was used as a control for wt F plasmid transfer efficiency. ND, not determined.
TraI Y16 is the only Tyr catalyzing ssDNA cleavage required for F plasmid transfer
To rule out the possibility that TraI ligation or cleavage can be catalyzed by Y17, Y23 or Y24, we generated TraI double and triple Phe substitutions. Table 2 shows transfer efficiencies of the FΔTraI plasmid complemented with these variants. The TraI Y23F/Y24F double mutant, like the single mutants, facilitates transfer with a slightly reduced (to 28%) efficiency relative to wt TraI. The TraI Y17F/Y23F/Y24F triple mutant also shows slightly reduced transfer efficiency of FΔTraI plasmid (to 60% of wt TraI). In contrast, FΔTraI transfer is reduced ∼5000-fold when complemented with TraI Y16F, TraI Y16F/Y17F, TraI Y16F/Y23F/Y24F or TraI Y16F/Y17F/Y23F/Y24F. The Y16F substitution renders TraI essentially inactive. The presence of detectable FΔTraI transfer for each of the Y16F variants, however, indicates that cleavage and ligation at nic was performed. Additional substitutions at Y17, Y23 or Y24 do not alter the transfer efficiency of a Y16F variant. These results suggest that the activity seen in Y16F variants is not due to a contribution by Y17, Y23 or Y24, but instead probably results from a low level of misincorporation or post-translational modification of Phe for Tyr at position 16 as previously suggested by Pansegrau et al. (34).
TraI is transported from donor to recipient cell
Relaxases of different plasmids are transported in multiple copies into recipient cells, including relaxases MobA of plasmid RSF1010 (35), TrwC of plasmid R388 (36), TraA of plasmid pATC58 (37) and VirD2 of Ti plasmid (38). Cleavage or joining reactions are likely catalyzed by TrwC of R388 within the recipient because an anti-TrwC relaxase domain antibody expressed within the recipient significantly inhibits R388 plasmid transfer (39). Knowledge of the location of TraI during transfer will provide clues about how and where F plasmid DNA is processed during conjugation. Mihajlovic, Zechner and colleagues have referred to unpublished data indicating TraI is transported to the recipient during conjugative transfer of the F-like R1 plasmid (18). To confirm that TraI is transferred to the recipient, we performed a two-step mating as illustrated in Figure 2. In the first step, the donor, tetracycline (Tet) resistant XK1502/FΔTraI complemented with pet24a-traI, transfers the FΔTraI plasmid into a BL21 recipient. No detectable FΔTraI plasmid transfer occurs in absence of the pet24a-traI plasmid (<1.6 × 10−7) (24). The BL21 recipient harbors the pACYC177o300 plasmid that contains a 300 bp F plasmid oriT region and genes conferring kanamycin (Kan) and ampicillin (Amp) resistance. The pACYC177o300 plasmid cannot be transferred without complementation by all essential F transfer genes. Therefore, transfer of pACYC177o300 in the second round is consistent with transfer of FΔTraI plasmid and TraI to the BL21 strain. At the end of the first round, the transfer efficiency of FΔTraI into BL21 is 0.16 ± 0.15 (average ± SD; n = 3). When the cells from the first incubation were combined with the streptomycin resistant TB1 strain, the efficiency of transfer of pACYC177o300 TB1 yielding Strep, Kan and Amp resistant transconjugants is 0.012 ± 0.006, well above the threshold for detection. These transfer efficiency data support the contention that (1) TraI protein is indeed transported from donor into recipient cell and (2) transferred TraI is biologically active and capable of facilitating all reactions required for plasmid transfer.
Cleavage reactions are performed in donor
Assuming that the plasmid transfer mechanism is similar to RCR (1), three cleavage and two joining reactions must take place to complete plasmid transfer (2). The first cleavage reaction takes place in the donor cell prior to donor–recipient contact (17,40) and results in a TraI-ssDNA covalent linkage via Y16. The 3′-end of the cleaved DNA generated by this reaction serves as a primer for DNA polymerase, allowing complementary strand synthesis, thereby generating a complete oriT. Gonzales-Perez and colleagues have suggested that a secondary cleavage reaction of the R388 plasmid at a newly synthesized oriT is performed in the recipient cell by a second catalytically active Tyr of the TrwC relaxase (12–13). We have demonstrated that (1) TraI Y16 is essential for plasmid transfer and (2) Y16 is the only F TraI relaxase Tyr required for F plasmid transfer. Thus our data imply that the first two cleavage reactions must be catalyzed by two different TraI molecules. We have also provided evidence that TraI is transported into the recipient during F plasmid conjugation, likely in multiple copies per recipient cell as shown for relaxases from different plasmids (35–38). The TraI secondary cleavage reaction therefore might be catalyzed by the TraI molecule that was transported into the recipient.
To identify whether the secondary cleavage reaction is catalyzed within the donor or the recipient, we measured conjugation of XK1502/FΔTraI complemented with pet24a-traI Y16F or pet24a-traI Y16F/Y17F/Y23F/Y24F plasmid as donors, and TB1 recipient cells expressing TraI protein. If the secondary cleavage reaction is performed in the recipient, we expect that expression of wt TraI molecules in the recipient will efficiently catalyze the secondary cleavage reaction. As a result, FΔTraI plasmid transfer complemented with pet24a-traI Y16F or pet24a-traI Y16F/Y17F/Y23F/Y24F will likely be discernibly increased. The probability that the donor at a given time has one TraI molecule with a Tyr at position 16 is much higher than the probability that two such TraI molecules are generated. The efficiency of transfer of FΔTraI facilitated by donor-expressed TraI Y16F or TraI Y16F/Y17F/Y23F/Y24F into a TB1 recipient expressing TraI protein is ∼1 × 10−5. These transfer efficiencies are the same as when a TB1 recipient that does not express wt TraI is used. The similar transfer efficiencies support the contention that secondary cleavage is performed in the donor cell. If so, F plasmid is delivered into the recipient as cleaved ssDNA with a free 3′-hydroxyl.
TraI efficiently terminates plasmid transfer at a short oriT
To gain insight into how TraI locates and cleaves a newly synthesized oriT within the donor cell, we modified pACYC177. Two sequences derived from the F plasmid oriT were inserted on either side of the Kan resistance gene (Figure 1). The first 300 bp oriT segment contains elements required for plasmid transfer initiation and termination (41). In contrast, the second 35 bp oriT segment (Figure 1) only contains sequences recognized by TraI and is therefore competent only for plasmid transfer termination. Transfer efficiencies of plasmids containing only one of the two oriT regions are 2.9 (±1.7) × 10−1 and <1.2 × 10−6 for pACYC177o300 and pACYC177o35, respectively. These data confirm that plasmid conjugation cannot initiate from the 35 bp oriT segment while it can initiate from the 300 bp segment. Conjugation with donor strain ER2738 carrying plasmid pACYC177o2ak, which contains both 300 and 35 bp oriT regions, and StrR recipient strain TB1 yields AmpR/StrepR/KanS and AmpR/StrepR/KanR transconjugants. The AmpR/StrepR/KanS transconjugants can result from initiation on the 300 bp oriT region and termination on the 35 bp oriT region, causing transfer of a plasmid conferring AmpR but lacking the kanamycin resistance gene. Comparing AmpR/StrepR/KanS transconjugants and AmpR/StrepR/KanR transconjugants gives the probability of termination on the 35 bp oriT region. Of transconjugants, 80 ± 5% are AmpR/StrepR/KanS, indicating that for these transconjugants, transfer was initiated at the 300 bp oriT segment and terminated at the 35 bp oriT region. Another 20 ± 1% of transconjugants were AmpR/StrepR/KanR, indicating that for these, transfer was initiated at the 300 bp oriT segment and terminated at some point beyond the 35 bp oriT region, most likely at the 300 bp oriT segment. The observed frequencies are in agreement with results from similar experiments using significantly larger oriT fragments (41). These data suggest that TraI can effectively recognize and cleave short oriT ssDNA sequences without accessory proteins, consistent with TraI actively scanning plasmid ssDNA by translocation.
TraI translocates rapidly on ssDNA
TraI is a highly processive ssDNA-dependent helicase that can separate double-stranded DNA at a rate of ∼1100 bp/s with a kinetic step of 6–8 bp (42). TraI can also translocate along ssDNA in a 5′-to-3′ direction powered by ATP hydrolysis. If TraI is transported to the recipient covalently attached to plasmid ssDNA, as would be expected if it is responsible for joining plasmid ends to generate a closed circular form, TraI may translocate along the incoming plasmid ssDNA to locate the 3′-cut site prior to rejoining the ends. We examined the details of TraI translocation on circular and linear ssDNA hoping to gain insight into how TraI might recognize the 3′-end of the transferred ssDNA.
TraI translocation was assessed indirectly by measuring ssDNA-dependent ATP hydrolysis. Under steady state conditions, the apparent rate of TraI translocation will be affected by different steps based upon assay conditions. For example, given the processivity of TraI translocation, use of a circular ssDNA molecule as a substrate should accurately depict the rate of TraI translocation (19). However, when using short linear ssDNA substrates, TraI association or dissociation may be a rate-limiting step. We assessed the ssDNA-dependent ATP hydrolysis activity of TraI using two ssDNA substrates: the 2713-base circular ssDNA from pUC18 (plus strand) and a 70-base ssDNA oligonucleotide (oligo70) from F plasmid (sequence is given in Table 1). The Michaelis constant, K′, which is the ssDNA concentration required to reach half-maximal ATP hydrolysis, was 3.8 ± 0.2 and 0.45 ± 0.04 µg/ml for the linear oligo70 and circular ssDNA, respectively. The significant difference in K′ indicates that TraI requires a much higher concentration of linear than circular ssDNA to reach the same rate of ATP hydrolysis. The difference in K′ values also demonstrates that (i) TraI dissociates much less frequently from circular than from linear ssDNA and (ii) TraI requires a much longer ssDNA to dissociate from a circular ssDNA substrate than a linear substrate. Thus, the difference in K′ when using the two different substrates reflects TraI translocation on ssDNA (19). In contrast, we observed similar maximal rates of ATP hydrolysis (kcat) of 310 ± 20 s−1 and 380 ± 10 s−1 for the linear oligo70 and circular ssDNA substrates, respectively. The similar kcat values suggest that TraI translocation rates are similar on both linear and circular ssDNA. This result is expected because the translocation velocity of TraI should be identical on both linear and circular ssDNA substrates.
TraI translocation on short linear ssDNA is influenced by nucleotide sequence
TraI ATPase activity during translocation is very strongly stimulated by the presence of single-stranded salmon testes DNA (31), whereas Lahue and Matson reported low levels of TraI ATPase activity on a uniform linear ssDNA substrate several hundred bases long (19). Suspecting that the apparent difference in ATPase activity might derive from the sequences of the ssDNA substrates, we tested a variety of linear ssDNA oligonucleotides with lengths ranging from 19 to 87 bases as substrates for TraI ATPase activity (ssDNA translocation) (Supplementary Table SI). Standard conditions for measuring TraI ATPase activity were 20 nM TraI and 1 µg/ml ssDNA. Under these conditions, TraI shows robust ATPase activity with pUC18 circular ssDNA but reduced activity with less favored linear substrates. Therefore, using a single condition, we could obtain information on the Michaelis constant (K′) of the ssDNA substrate, which in turn is strongly influenced by differences in association and dissociation of TraI from the oligonucleotide substrate. All TraI ATPase activity data were compared relative to TraI ATP hydrolysis rate during translocation on oligo70. TraI translocates effectively on seven ssDNA substrates out of 27 tested in our basic conditions (Supplementary Table SI). Oligonucleotide20 is the shortest ssDNA that induced relatively high TraI ATPase activity.
The influence of ssDNA length and secondary structure on TraI ssDNA-dependent ATPase activity appears to be minor. For example, the AA and AAA oligonucleotides are concatamers of the A oligonucleotide and while AA generates a greater TraI ATPase activity than A (Table 3), the observed rate with the AA and AAA oligonucleotides is similar. Supplementary Table SI also lists ssDNA sequences of various lengths and secondary structures along with the TraI ATPase activity observed when they are used as substrates. Oligonucleotide53 does not form any secondary structure according to mfold (43) and yet is a poor substrate for TraI ssDNA-dependent ATPase activity. Oligonucleotide20, oligonucleotide36 and oligo70 also do not form any significant secondary structure but serve as substrates for TraI ssDNA-dependent ATPase activity. Oligonucleotide38, oligonucleotide64, oligonucleotide67, oligonucleotide87 and B oligonucleotide all form stable secondary structures. Oligonucleotide38, oligonucleotide67 and B are poor substrates for the TraI ATPase activity, while oligonucleotide64 and oligonucleotide 87 generate a robust TraI ATPase activity. It therefore appears that while the ssDNA sequence clearly affects TraI ssDNA-dependent ATPase activity, the ability of a sequence to serve as a substrate is not obviously and directly linked to the structure or conformation of the oligonucleotide. We then focused on short sequences that yielded different ATPase activities to try to determine the limits of their effects.
Table 3.
TraI Translocation on ssDNA with Hairpin
| ssDNA | Length | Relative rate |
|---|---|---|
| A oligonucleotide | 22 | 0.24 ± 0.03 |
| B oligonucleotide | 23 | 0.004 ± 0.003 |
| AA oligonucleotide | 44 | 0.68 ± 0.07 |
| AB oligonucleotide | 45 | 0.023 ± 0.010 |
| BA oligonucleotide | 45 | 0.42 ± 0.09 |
| BB oligonucleotide | 46 | ND |
| AAA oligonucleotide | 66 | 0.61 ± 0.04 |
| AAB oligonucleotide | 67 | 0.37a |
| ABB oligonucleotide | 68 | 0.017 ± 0.007 |
| BAB oligonucleotide | 68 | 0.072a |
| BBA oligonucleotide | 68 | 0.24 ± 0.03 |
| BBB oligonucleotide | 69 | 0.009 ± 0.004 |
| Oligo70 | 70 | 1 |
Rates of ATP hydrolysis, relative to that elicited by 70-base Oligo70 oligonucleotide, were measured at 20 nM TraI and 1 µg/ml of ssDNA. Data are averages of two to six independent measurements. Error was calculated as standard deviation error. Sequences of A and B oligonucleotides are given in Table 1.
aSingle measurement.
We chose the A and B oligonucleotide substrates and synthesized oligonucleotides that contained multiple units of these sequences in different combinations (Table 3). The A oligonucleotide is an excellent substrate for TraI ssDNA-dependent ATPase activity, while the B oligonucleotide forms a secondary structure according to mfold (43) and is a poor substrate for the TraI ATPase activity. We found that the AA, BA, AA, AAA, BBA and AAB oligonucleotide combinations stimulate strong TraI ATPase activity whereas AB, ABB, BAB and BBB oligonucleotides do not stimulate significant TraI ATPase activity. These results are not obviously linked to the affinity of TraI for the ssDNA. TraI binds A, AA, AB and BA oligonucleotides with similar affinities (data not shown). We therefore conclude that the B oligonucleotide sequence can impair TraI ssDNA-dependent ATPase activity, but exerts this influence only when it is located on the 3′-end of the ssDNA. The exception is the AAB oligonucleotide, but it is possible that an effect by the B sequence may be masked by the presence of an excellent substrate at the 5′-end. These data indicate that the sequence of ssDNA at the 3′-end is important. We speculate that the particular nucleotide sequence located on the 3′-end of ssDNA can reduce the TraI dissociation rate, as suggested previously by Lahue and Matson (19), thereby limiting TraI ATPase activity.
TraI can recognize 3′-end of TraI nicked F plasmid
During ssDNA strand transfer, TraI, like other relaxases (35–38), is transported from donor cell to recipient. In the recipient, TraI, still covalently linked to the ssDNA, could translocate along the incoming ssDNA, locate the 3′-end, and terminate plasmid transfer by reattaching 3′- and 5′-plasmid ends in a reversal of the cleavage reaction. We tested whether F oriT sequences affect TraI helicase processivity in a manner that could assist these possible activities in the recipient. We synthesized two 60-base oligonucleotides, one including 60 bases 5′ of nic (5′oriT60-mer) and the second including 60 bases 3′ of nic (3′oriT60-mer). The 3′oriT60-mer represents the sequence that might be initially encountered by a TraI molecule loading onto and translocating along the cut strand, while 5′oriT60-mer represents the sequence TraI would reach immediately before joining the two plasmid ends. We found that the 3′oriT60-mer stimulates very strong TraI ATPase activity (Table 4). In contrast, 5′oriT60-mer weakly stimulates TraI ATPase activity. These data indicate that TraI can recognize the 3′-end of F plasmid cleaved at nic site and suggest that TraI responds to the 3′-end with slowed translocation or slow dissociation.
Table 4.
TraI Translocation on ssDNA near oriT
| ssDNA | Length | Relative rate |
|---|---|---|
| oriT17-A oligonucleotide | 39 | 0.44 ± 0.07 |
| oriT17-B oligonucleotide | 40 | 0.009a |
| oriT17-AB oligonucleotide | 62 | 0.22 ± 0.02 |
| 5′oriT60-mer | 60 | 0.006 ± 0.002 |
| -90 -30 5′oriT60-mer | 60 | 0.12 ± 0.04 |
| 3′oriT30-mer | 30 | 0.16 ± 0.06 |
| 3′oriT60-mer | 60 | 0.66 ± 0.09 |
| Oligo70 | 70 | 1 |
Rates of ATP hydrolysis, relative to that elicited by the 70-base Oligo70 oligonucleotide, were measured at 20 nM TraI and 1 µg/ml of ssDNA. Data are averages of two to six independent measurements. Error was calculated as standard deviation error. Sequences of A and B oligonucleotides are given in Table 1.
aSingle measurement.
We wondered whether the weak ATPase activity seen with the 5′oriT60-mer resulted from an interaction of the TraI relaxase domain with its specific binding site. The relaxase and helicase-associated ssDNA binding sites exhibit a strong apparent negative cooperativity, with binding to one site inhibiting binding to the second (27). To test whether the presence of a TraI relaxase binding site on an oligonucleotide affects TraI ATPase activity, we added the oriT17 sequence to the 5′-end of oligonucleotides A (oriT17-A), B (oriT17-B) and AB (oriT17-AB). The high relative ATP hydrolysis activity with the oriT17-A oligonucleotide indicates that the presence of the relaxase binding site does not significantly impair ATP hydrolysis (Table 4). The oriT17-B oligonucleotide very poorly induced ATP hydrolysis, while the oriT17-AB oligonucleotide induces TraI ATPase activity similar to AAB oligonucleotide (Tables 3 and 4). Therefore, adding the 17-base TraI relaxase binding site sequence had no obvious negative impact on ATP hydrolysis, and actually significantly improved catalysis for the longest oligonucleotide tested (oriT17-AB).
TraI ligation reaction is favored with hairpin substrate
The F TraI relaxase domain transesterification reaction is not efficient in vitro (Figure 3) and may require accessory proteins in vivo. Indeed, higher in vitro yields of the TraI transesterification reaction have been observed in the presence of TraM and TraD (16,18). For F (26) and other systems (13), high concentrations of relaxase relative to substrate are required to cleave the majority of the substrate in vitro. We wondered whether the excess of relaxase in these assays was serving as a sink for the product, yielding greater apparent relaxase activity. During the cleavage reaction, the relaxase binds the oligonucleotide substrate, cleaves at nic forming a covalent linkage to the ssDNA on the 3′-side of nic. The relaxase may then reverse the reaction to generate the intact substrate, or release the oligonucleotide section 5′ to nic, the cleavage product. We reasoned that in the presence of excess relaxase, released cleavage product would be bound and trapped, reducing the rate of the reverse reaction and giving the appearance of a more efficient cleavage reaction. To test this possibility, we examined the ability of TraI and the TraI relaxase domain at low (20 nM) and high (1 µM) concentrations to cleave oligonucleotide substrates. Results for the TraI relaxase domain are shown, but the results for TraI were identical.
Figure 3.

The TraI transesterification reaction is not efficient. About 20 nM or 1 µM of TraI relaxase domain and 20 nM of 30-base F oriT hairpin (top gel) or F oriT no hairpin oligonucleotide (bottom gel) 5′-labeled with a Cy5 fluorophore were incubated in reaction buffer for 1 h at 37°C. About 1 µM of TraI Y16F was added where indicated and the incubation was continued for an additional 1 h. Reactions were terminated with proteinase K. Cleaved ssDNA is 3 nt shorter and therefore migrates farther. Oligonucleotide sequences are given in Table 1 and Supplementary Table SII.
As noted previously, the TraI relaxase domain generates significantly more cleavage product at high relative to low concentration (Figure 3, upper). Also, a substrate containing a 5′-hairpin, such as an oligonucleotide containing the F oriT sequence extending at least 25 bases 5′ of nic, is a considerably worse substrate than a similar oligonucleotide lacking the hairpin (Figure 3, lower). We then repeated these experiments, but followed the initial incubation with the addition of 1 µM TraI Y16F and further 1 h incubation. The Phe for Tyr substitution in the Y16F TraI impairs ssDNA cleavage [(23,28), Figure 3] even though the protein binds with wild-type affinity. If the enhanced cleavage observed with high TraI concentrations were the result of the excess protein acting as a sink for the cleavage product, the presence of the Y16F TraI variant should also shift the equilibrium. However, addition of Y16F does not appear to affect the concentration of reaction products. These results indicate that the excess TraI relaxase domain is not serving as a product sink. In addition, these results suggest that interactions between relaxases do not generate more reaction product, at least not interactions involving a catalytically inactive relaxase.
As shown in Figure 3, an oligonucleotide with a 5′-hairpin is a relatively poor cleavage substrate. The hairpin, however, also increases the affinity of the TraI relaxase domain for the oligonucleotide (44). We wondered whether the hairpin might assist TraI in the joining reaction that would terminate the transfer reaction. We first incubated the TraI relaxase domain with a 3′-Cy5-labeled oriT17 oligonucleotide. After cleavage and release of the segment 5′ to nic, the TraI relaxase domain was left linked to a short 3′-Cy5-labeled GGT oligonucleotide. We then added an excess of one of several oligonucleotides that ended at the nic site. Successful oligonucleotide joining was visualized on a 16% polyacrylamide-8.3 M urea gel as a band of increased size (Figure 4). Despite its inability to efficiently cleave the hairpin-containing oligonucleotide, TraI relaxase domain joined oriT oligonucleotides with or without hairpins with similar efficiencies (Figure 4, left). More surprising are the results with the hairpin sequence variants (Figure 4, right). We tested TraI relaxase domain for its ability to join oligonucleotides when the 5′-segment contains 1 (G144′C) or 2 (R100) base substitutions that significantly reduce its affinity for TraI and impair cleavage by TraI relaxase domain (44,45). For one (G144′C) the substitution is for a base involved in a three-way base interaction that appears essential for orienting the scissile phosphate for cleavage (46). Oligonucleotides containing these sequences were readily joined provided that the oligonucleotide also had a 5′-hairpin. The same sequences, when in an oligonucleotide that lacked a hairpin, were poor substrates for joining. While these results might be explained by the significantly increased affinity of TraI relaxase domain for oligonucleotides containing a 5′-hairpin, TraI may exhibit different specificities for oligonucleotides with and without hairpins (44).
Figure 4.

The TraI joining reaction is catalyzed by a 5′-hairpin. About 1 µM of TraI relaxase domain was incubated with 20 nM of 3′-Cy5-labeled F oriT17 oligonucleotide in reaction buffer for 1 h at 37°C. About 10 µM of oligonucleotides Hairpin, No hairpin (F oriT) (left gel) or Hairpin G144′C, No hairpin G144′C (G144′C), Hairpin R100, No hairpin R100 (R100), Hairpin 3random or No hairpin 3random (3 random) (right gel) was added and incubated for an additional 1 h. Reactions were terminated with proteinase K. ‘No’ indicates No hairpin and ‘H’ indicates Hairpin. The Figure is composed of two gels separated by oligonucleotide markers (oligo markers) with ssDNA lengths as indicated. Samples on the left gel were not treated with formamide prior to loading, thus hairpin structures were maintained, causing the different mobilities of the Hairpin and No hairpin oligonucleotides.
DISCUSSION
During bacterial conjugation, plasmids are transported from donor to recipient as ssDNA (1). TraI and related relaxases pilot ssDNA into the recipient (47) in addition to performing relaxase and helicase reactions. Both relaxase and helicase activities of TraI are essential for F transfer. Conjugative transfer of a 200-kb plasmid takes 3.5–4 min to complete (48). This means that plasmid DNA is transferred at the rapid rate of ∼1 kb per second. The implication is that TraI and other transfer proteins must work in concert with donor and recipient DNA polymerases synthesizing the complementary plasmid strand. The work presented here focuses on individual steps of F transfer catalyzed by the TraI relaxase, and the data provide additional details about the process of conjugation and the role of TraI in F plasmid transfer (Figure 5).
Figure 5.

Model of the role of TraI in F plasmid DNA transfer. Two scenarios are presented based on presence (a) or absence (b) of F plasmid complementary strand synthesis within donor cell during plasmid transfer. Individual steps of the reactions are described in the text (I–VI). The donor cell is situated on the left side and recipient cell on the right side. The dashed black line represents the cell wall. The solid black line represents the original F plasmid. Complementary strand synthesis is illustrated by a red line (donor cell) and a green line (recipient cell). The black cylinder represents the secretion pore complex and blue ovals are TraI molecules.
By substituting Phe for single and multiple Tyr residues (Y16, Y17, Y23 and Y24) within F TraI, we demonstrated that only Y16 is essential for F plasmid transfer [Table 2, (28)]. TraI thus differs from R388 TrwC by requiring only a single Tyr. The MobP group of relaxases, the prototype of which is RP4 TraI (21), also employs a single Tyr to facilitate plasmid transfer (22). It has been suggested that RP4 TraI pilots plasmid transfer in a mechanism similar to RCR (1). During RCR, three cleavage and two ligation reactions must be performed to replicate plasmid. If F TraI uses a similar mechanism, at a minimum two F TraI molecules, with a single catalytically active Tyr (Y16), are required to transfer F.
Despite the effect of the Y16F substitution, all variants containing the Y16F substitution still facilitate F transfer at detectable levels when complementing the FΔTraI plasmid. Pansegrau et al. suggested that a small population of proteins might be functional due to misincorporation of Tyr for Phe during translation (34). Carboxylase catalyzed post-translational modification of Phe could have a similar effect. Noirot-Gros and Ehrlich showed that a glutamate residue of the RecA protein catalyzes the cleavage reaction of the newly synthesized oriT of the pC194 plasmid (49). It is therefore possible that a functional TraI might also be also generated due to miscorporation of Glu for Phe. The frequency of in vivo amino acid misincorporation during protein synthesis has been estimated at 1 per 103–104 amino acids incorporated (50–52). Assuming that (1) F plasmid TraI expression is ∼300 TraI molecules per cell, and (2) the probability of misincorporation and/or post-translational modification of Phe16 for Tyr16 in TraI Y16F is 1 × 10−4, then the probability that a cell carries one and two functional TraI molecules is 0.029 and 0.00044, respectively. This probability corresponds to one and two functional TraI molecules per every 33 and 2273 cells, respectively. If one functional TraI molecule within the cell were required to complete the transfer of FΔTraI plasmid complemented with TraI Y16F, we would expect a 33-fold reduction in transfer efficiency. Similarly, if two functional TraI molecules per cell were required for transfer, we would expect FΔTraI complemented with TraI Y16F would show a 2273-fold reduction in plasmid transfer efficiency. FΔTraI plasmid transfer is reduced ∼5000-fold when complemented with the Y16F TraI variant (Table 2). These calculations are consistent with more than one functional TraI molecule being required to complete F plasmid transfer.
We provide evidence that (i) TraI is transported from donor to recipient together with plasmid DNA and (ii) TraI catalyzes the second cleavage reaction in the donor. These two results imply that two separate TraI molecules are catalyzing cleavage reactions. The first TraI molecule catalyzes ssDNA cleavage during initiation and is then transported into the recipient. The second TraI molecule locates and cleaves a newly synthesized oriT in the donor. To understand how the secondary cleavage of the newly synthesized oriT is facilitated within the donor, we cloned oriT regions on both sides of a KanR gene within a plasmid (Figure 1). Our data indicate that plasmid transfer readily terminates at a 35 bp oriT sequence at which it cannot initiate. This sequence contains only the TraI binding site and is double stranded prior to plasmid transfer, both of which prevent initiation from this site (in absence of bound relaxosome proteins, TraI can only bind and cleave its site in single-stranded form). Successful termination at the 35 bp oriT implies that the site occurs as ssDNA to allow for TraI cleavage and joining.
Earlier work demonstrated that complementary strand synthesis of plasmid DNA by DNA Polymerase III (Pol III) is not required in the donor cell during bacterial conjugation (53,54). This suggests that when Pol III is inactive, linear plasmid ssDNA with a free 3′-hydroxyl is delivered to the recipient without a second cleavage reaction. In this model, plasmid transfer would require one cleavage reaction in the donor cell and one circularization reaction in the recipient cell, both by the same relaxase molecule. Our data are consistent with this model. Assuming that 300 TraI molecules are expressed in the donor, and the probability of misincorporation and/or post-translational modification of TraI Y16F that replaces Phe16 with Tyr is between 1 × 10−5 and 1 × 10−6, then the probability that a donor cell produces one fully functional TraI is between 0.0030 and 0.0003. This corresponds to one functional TraI per every 333 to 3333 cells, in agreement with the 5000-fold reduction of FΔTraI plasmid transfer complemented with TraI Y16F. However, there is evidence for complementary strand synthesis of plasmid DNA in the donor cell. Gao et al. (41) show that plasmids recovered from recipient cells can be concatemers of the original plasmid. Both Gao et al. (41) and we show that TraI cleavage probability at the following oriT is ∼ 80%, suggesting that concatemers can be transferred during bacterial conjugation. Altogether, the conjugation system is equipped to deal with complementary strand synthesis in the donor. How likely complementary strand synthesis occurs in the donor during bacterial conjugation remains to be determined.
Rates of TraI ATP hydrolysis measured at one basic condition using oligonucleotides of different length and sequence show a strong dependence on the sequence of the oligonucleotide (Supplementary Table SI). It was shown previously that TraI slowly dissociates from linearized circular ssDNA M13mp7 (19). We observe a strong dependence of the TraI ATP hydrolysis rate on the nucleotide sequence at the 3′-end of linear ssDNA (Table 3). Because the maximal TraI hydrolysis rate is similar for ‘infinitely’ long circular and for linear ssDNA, according to the Michalis-Menten equation the observed changes in hydrolysis rate at our basic condition (Supplementary Table SI) likely indicate changes in association and/or dissociation rates of TraI with linear ssDNA. The presence of the B nucleotide sequence (sequence given in Table 1) at the 3′-end of an oligonucleotide reduces the rate of TraI ATP hydrolysis (Tables 3 and 4). In contrast, placing the same sequence at the 5′-end either increases or does not change the TraI ATP hydrolysis rate. Furthermore, multiple repeats of this sequence within circular ssDNA have no effect on TraI ATP hydrolysis (data not shown). Taken together, these results indicate that the 3′-end of a linear piece of ssDNA has a significant impact on TraI translocation, probably through effects on dissociation. Recognition of the 3′-end of F plasmid by a TraI molecule attached to the 5′-end is likely a crucial step of conjugation termination because the cleaved plasmid ends must be joined together within the recipient.
In the in vitro cleavage assay, the TraI cleavage product is in equilibrium with the intact oligonucleotide substrate (13,21). We previously noted that an oriT substrate containing a 5′-hairpin (F oriT hairpin) was cleaved with poor efficiency. The results given here (Figure 4) demonstrate that TraI relatively efficiently catalyzes a joining reaction involving an oligonucleotide with a hairpin. Thus the more accurate view may be that in the presence of the hairpin, the equilibrium of the TraI cleavage reaction is shifted more toward joining rather than cleavage. The hairpin is not required for TraI binding nor for cleavage at oriT (14,28), but the presence of a hairpin can improve binding of some otherwise poorly bound sequences [G144′C; (44)]. Lucas et al. (55) recently demonstrated that binding and cleavage are two distinguishable steps of relaxase TrwC DNA processing. They concluded that the inverted repeat within the R388 oriT is required to position ssDNA in the TrwC active site for cleavage (55). It is possible that the F oriT hairpin serves a similar purpose. We prefer a different model, though, with the presence of the hairpin altering the interaction between ssDNA and TraI and shifting the equilibrium toward the joined state and away from the cleaved state, thus favoring termination. Even so, we have evidence that the hairpin is not indispensable for transfer. Changes in the hairpin sequence, including disruption, have only a minor influence on plasmid transfer efficiency under the conditions used (44). Additional work is needed to understand the role of the hairpin, both in the joining reaction and in transfer.
In summary, the data presented here support the following model for ssDNA delivery by TraI during bacterial conjugation (Figure 5). During initiation, TraI is recruited to the oriT region by proteins forming the relaxosome (21,56,57) and cleaves ssDNA at nic within the relaxosome. TraI is maintained covalently attached to the 5′-end of the ssDNA prior to plasmid transfer (58). Upon mating pair formation, the TraI molecule covalently attached to ssDNA is uptaken by the type IV secretion system, which exposes the T-strand and allows a second TraI molecule to load onto the T-strand to unwind plasmid DNA (Figure 5, II). During this step, several other TraI molecules are also transported by the type IV secretion system. The model then diverges, depending on whether there is synthesis of a complementary strand in the donor. In the donor, the 3′-end of the cleaved plasmid serves as the primer for Pol III, which initiates complementary strand synthesis (Figure 5, IIIa). Alternatively, Pol III is not loaded (Figure 5, IIIb). In the recipient, the TraI molecule attached to the 5′-end of the T-strand translocates on ssDNA plasmid toward the 3′-end and complementary strand synthesis is initiated (Figure 5, IV). In the donor, TraI translocates towards the 3′-end of the T-strand (Figure 5, IVb) or towards the newly synthesized oriT region (Figure 5, IVa) where TraI catalyzes a second cleavage and covalently attaches to the 5′-end of newly synthesized ssDNA (Figure 5, Va). The T-strand is transported in a cleaved form with free 3′-end (Figure 5, V). In the final step, TraI recognizes the 3′-end and reattaches the T-strand ends in the recipient (Figure 5, V). Following the RCR mechanism, a TraI molecule attached to the 5′-end of newly synthesized ssDNA might act similar to Rep protein and circularize newly synthesized plasmid ssDNA (Figure 5, VIa).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health grant 61017 (to J.F.S.). Funding for open access charge: National Institutes of Health (grant GM61017).
Conflict of interest statement. None declared.
NOTE ADDED IN PROOF
Zechner and colleagues recently demonstrated that FΔTraI is transported to the recipient during transfer (Lang et al., 2010, Mol. Micro., DOI: 10.1111/j.1365-2958.2010.07423.x).
ACKNOWLEDGEMENTS
We thank Beth Traxler for providing XK1502/FΔTraI strain. We also thank members of Schildbach lab for helpful discussions.
REFERENCES
- 1.Lanka E, Wilkins BM. DNA Processing Reactions in Bacterial Conjugation. Annu. Rev. Biochem. 1995;64:141–169. doi: 10.1146/annurev.bi.64.070195.001041. [DOI] [PubMed] [Google Scholar]
- 2.Khan SA. Plasmid rolling-circle replication: highlights of two decades of research. Plasmid. 2005;53:126–136. doi: 10.1016/j.plasmid.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Khan SA. Plasmid rolling-circle replication: recent developments. Mol. Microbiol. 2000;37:477–484. doi: 10.1046/j.1365-2958.2000.02001.x. [DOI] [PubMed] [Google Scholar]
- 4.del Solar G, Giraldo R, Ruiz-Echevarria MJ, Espinosa M, Diaz-Orejas R. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 1998;62:434–464. doi: 10.1128/mmbr.62.2.434-464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boer DR, Ruiz-Maso JA, Lopez-Blanco JR, Blanco AG, Vives-Llacer M, Chacon P, Uson I, Gomis-Ruth FX, Espinosa M, Llorca O, et al. Plasmid replication initiator RepB forms a hexamer reminiscent of ring helicases and has mobile nuclease domains. EMBO J. 2009;28:1666–1678. doi: 10.1038/emboj.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang TL, Kramer MG, Ansari RA, Khan SA. Role of individual monomers of a dimeric initiator protein in the initiation and termination of plasmid rolling circle replication. J. Biol. Chem. 2000;275:13529–13534. doi: 10.1074/jbc.275.18.13529. [DOI] [PubMed] [Google Scholar]
- 7.van Mansfeld AD, van Teeffelen HA, Baas PD, Jansz HS. Two juxtaposed tyrosyl-OH groups participate in phi X174 gene A protein catalysed cleavage and ligation of DNA. Nucleic Acids Res. 1986;14:4229–4238. doi: 10.1093/nar/14.10.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta S, Larkin C, Schildbach JF. Structural insights into single-stranded DNA binding and cleavage by F factor TraI. Structure. 2003;11:1369–1379. doi: 10.1016/j.str.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Monzingo AF, Ozburn A, Xia S, Meyer RJ, Robertus JD. The structure of the minimal relaxase domain of MobA at 2.1 A resolution. J. Mol. Biol. 2007;366:165–178. doi: 10.1016/j.jmb.2006.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guasch A, Lucas M, Moncalian G, Cabezas M, Perez-Luque R, Gomis-Ruth FX, de la Cruz F, Coll M. Recognition and processing of the origin of transfer DNA by conjugative relaxase TrwC. Nat. Struct. Biol. 2003;10:1002–1010. doi: 10.1038/nsb1017. [DOI] [PubMed] [Google Scholar]
- 11.Pansegrau W, Schroder W, Lanka E. Relaxase (TraI) of IncP alpha plasmid RP4 catalyzes a site-specific cleaving-joining reaction of single-stranded DNA. Proc. Natl Acad. Sci. USA. 1993;90:2925–2929. doi: 10.1073/pnas.90.7.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grandoso G, Avila P, Cayon A, Hernando MA, Llosa M, de la Cruz F. Two active-site tyrosyl residues of protein TrwC act sequentially at the origin of transfer during plasmid R388 conjugation. J. Mol. Biol. 2000;295:1163–1172. doi: 10.1006/jmbi.1999.3425. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Perez B, Lucas M, Cooke LA, Vyle JS, de la Cruz F, Moncalian G. Analysis of DNA processing reactions in bacterial conjugation by using suicide oligonucleotides. EMBO J. 2007;26:3847–3857. doi: 10.1038/sj.emboj.7601806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larkin C, Datta S, Harley MJ, Anderson BJ, Ebie A, Hargreaves V, Schildbach JF. Inter- and intramolecular determinants of the specificity of single-stranded DNA binding and cleavage by the F factor relaxase. Structure. 2005;13:1533–1544. doi: 10.1016/j.str.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 15.Lujan SA, Guogas LM, Ragonese H, Matson SW, Redinbo MR. Disrupting antibiotic resistance propagation by inhibiting the conjugative DNA relaxase. Proc. Natl Acad. Sci. USA. 2007;104:12282–12287. doi: 10.1073/pnas.0702760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ragonese H, Haisch D, Villareal E, Choi JH, Matson SW. The F plasmid-encoded TraM protein stimulates relaxosome-mediated cleavage at oriT through an interaction with TraI. Mol. Microbiol. 2007;63:1173–1184. doi: 10.1111/j.1365-2958.2006.05576.x. [DOI] [PubMed] [Google Scholar]
- 17.Sut MV, Mihajlovic S, Lang S, Gruber CJ, Zechner EL. Protein and DNA effectors control the TraI conjugative helicase of plasmid R1. J. Bacteriol. 2009;191:6888–6899. doi: 10.1128/JB.00920-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mihajlovic S, Lang S, Sut MV, Strohmaier H, Gruber CJ, Koraimann G, Cabezon E, Moncalian G, de la Cruz F, Zechner EL. Plasmid r1 conjugative DNA processing is regulated at the coupling protein interface. J. Bacteriol. 2009;191:6877–6887. doi: 10.1128/JB.00918-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahue EE, Matson SW. Escherichia coli DNA helicase I catalyzes a unidirectional and highly processive unwinding reaction. J. Biol. Chem. 1988;263:3208–3215. [PubMed] [Google Scholar]
- 20.Grandoso G, Llosa M, Zabala JC, de la Cruz F. Purification and biochemical characterization of TrwC, the helicase involved in plasmid R388 conjugal DNA transfer. Eur. J. Biochem. 1994;226:403–412. doi: 10.1111/j.1432-1033.1994.tb20065.x. [DOI] [PubMed] [Google Scholar]
- 21.de la Cruz F, Frost LS, Meyer RJ, Zechner EL. Conjugative DNA metabolism in Gram-negative bacteria. FEMS Microbiol. Rev. 2010;34:18–40. doi: 10.1111/j.1574-6976.2009.00195.x. [DOI] [PubMed] [Google Scholar]
- 22.Pansegrau W, Lanka E. Mechanisms of initiation and termination reactions in conjugative DNA processing. Independence of tight substrate binding and catalytic activity of relaxase (TraI) of IncPalpha plasmid RP4. J. Biol. Chem. 1996;271:13068–13076. doi: 10.1074/jbc.271.22.13068. [DOI] [PubMed] [Google Scholar]
- 23.Street LM, Harley MJ, Stern JC, Larkin C, Williams SL, Miller DL, Dohm JA, Rodgers ME, Schildbach JF. Subdomain organization and catalytic residues of the F factor TraI relaxase domain. Biochim. Biophys. Acta. 2003;1646:86–99. doi: 10.1016/s1570-9639(02)00553-8. [DOI] [PubMed] [Google Scholar]
- 24.Haft RJF, Palacios G, Nguyen T, Mally M, Gachelet EG, Zechner EL, Traxler B. General mutagenesis of F plasmid TraI reveals its role in conjugative regulation. J. Bacteriol. 2006;188:6346–6353. doi: 10.1128/JB.00462-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch EF, Maniatis F. Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 26.Stern JC, Schildbach JF. DNA recognition by F Factor TraI36: highly sequence-specific binding of single-stranded DNA. Biochemistry. 2001;40:11586–11595. doi: 10.1021/bi010877q. [DOI] [PubMed] [Google Scholar]
- 27.Dostal L, Schildbach JF. Single-stranded DNA binding by F TraI relaxase and helicase domains is coordinately regulated. J. Bacteriol. 2010;192:3620–3628. doi: 10.1128/JB.00154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkin C, Haft RJ, Harley MJ, Traxler B, Schildbach JF. Roles of active site residues and the HUH motif of the F plasmid TraI relaxase. J. Biol. Chem. 2007;282:33707–33713. doi: 10.1074/jbc.M703210200. [DOI] [PubMed] [Google Scholar]
- 29.Finlay BB, Frost LS, Paranchych W. Nucleotide sequence of the tra YALE region from IncFV plasmid pED208. J. Bacteriol. 1986;168:990–998. doi: 10.1128/jb.168.2.990-998.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frost LS, Ippen-Ihler K, Skurray RA. Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol. Rev. 1994;58:162–210. doi: 10.1128/mr.58.2.162-210.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dash PK, Traxler BA, Panicker MM, Hackney DD, Minkley EG., Jr Biochemical characterization of Escherichia coli DNA helicase I. Mol. Microbiol. 1992;6:1163–1172. doi: 10.1111/j.1365-2958.1992.tb01555.x. [DOI] [PubMed] [Google Scholar]
- 32.Datta S, Larkin C, Schildbach JF. Structural insights into single-stranded DNA binding and cleavage by F factor TraI. Structure. 2003;11:1369–1379. doi: 10.1016/j.str.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Garcillan-Barcia MP, Francia MV, de la Cruz F. The diversity of conjugative relaxases and its application in plasmid classification. FEMS Microbiol. Rev. 2009;33:657–687. doi: 10.1111/j.1574-6976.2009.00168.x. [DOI] [PubMed] [Google Scholar]
- 34.Pansegrau W, Schroder W, Lanka E. Concerted action of three distinct domains in the DNA cleaving-joining reaction catalyzed by relaxase (TraI) of conjugative plasmid RP4. J. Biol. Chem. 1994;269:2782–2789. [PubMed] [Google Scholar]
- 35.Luo ZQ, Isberg RR. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl Acad. Sci. USA. 2004;101:841–846. doi: 10.1073/pnas.0304916101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Draper O, Cesar CE, Machon C, de la Cruz F, Llosa M. Site-specific recombinase and integrase activities of a conjugative relaxase in recipient cells. Proc. Natl Acad. Sci. USA. 2005;102:16385–16390. doi: 10.1073/pnas.0506081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulein R, Guye P, Rhomberg TA, Schmid MC, Schroder G, Vergunst AC, Carena I, Dehio C. A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae into human cells. Proc. Natl Acad. Sci. USA. 2005;102:856–861. doi: 10.1073/pnas.0406796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vergunst AC, van Lier MC, den Dulk-Ras A, Stuve TA, Ouwehand A, Hooykaas PJ. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc. Natl Acad. Sci. USA. 2005;102:832–837. doi: 10.1073/pnas.0406241102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcillan-Barcia MP, Jurado P, Gonzalez-Perez B, Moncalian G, Fernandez LA, de la Cruz F. Conjugative transfer can be inhibited by blocking relaxase activity within recipient cells with intrabodies. Mol. Microbiol. 2007;63:404–416. doi: 10.1111/j.1365-2958.2006.05523.x. [DOI] [PubMed] [Google Scholar]
- 40.Kline BC, Helinski DR. F 1 sex factor of Escherichia coli. Size and purification in the form of a strand-specific relaxation complex of supercoiled deoxyribonucleic acid and protein. Biochemistry. 1971;10:4975–4980. doi: 10.1021/bi00802a022. [DOI] [PubMed] [Google Scholar]
- 41.Gao Q, Luo Y, Deonier RC. Initiation and termination of DNA transfer at F plasmid oriT. Mol. Microbiol. 1994;11:449–458. doi: 10.1111/j.1365-2958.1994.tb00326.x. [DOI] [PubMed] [Google Scholar]
- 42.Sikora B, Eoff RL, Matson SW, Raney KD. DNA unwinding by Escherichia coli DNA helicase I (TraI) provides evidence for a processive monomeric molecular motor. J. Biol. Chem. 2006;281:36110–36116. doi: 10.1074/jbc.M604412200. [DOI] [PubMed] [Google Scholar]
- 43.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams SL, Schildbach JF. Examination of an inverted repeat within the F factor origin of transfer: context dependence of F TraI relaxase DNA specificity. Nucleic Acids Res. 2006;34:426–435. doi: 10.1093/nar/gkj444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harley MJ, Schildbach JF. Swapping single-stranded DNA sequence specificities of relaxases from conjugative plasmids F and R100. Proc. Natl Acad. Sci. USA. 2003;100:11243–11248. doi: 10.1073/pnas.2035001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hekman K, Guja K, Larkin C, Schildbach JF. An intrastrand three-DNA-base interaction is a key specificity determinant of F transfer initiation and of F TraI relaxase DNA recognition and cleavage. Nucleic Acids Res. 2008;36:4565–4572. doi: 10.1093/nar/gkn422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alvarez-Martinez CE, Christie PJ. Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 2009;73:775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrup L, Smidt L, Andersen K, Boe L. Kinetics of conjugative transfer: a study of the plasmid pXO16 from Bacillus thuringiensis subsp. israelensis. Plasmid. 1998;40:30–43. doi: 10.1006/plas.1998.1346. [DOI] [PubMed] [Google Scholar]
- 49.Noirot-Gros MF, Ehrlich SD. Change of a catalytic reaction carried out by a DNA replication protein. Science. 1996;274:777–780. doi: 10.1126/science.274.5288.777. [DOI] [PubMed] [Google Scholar]
- 50.Edelmann P, Gallant J. Mistranslation in E. coli. Cell. 1977;10:131–137. doi: 10.1016/0092-8674(77)90147-7. [DOI] [PubMed] [Google Scholar]
- 51.Bouadloun F, Donner D, Kurland CG. Codon-specific missense errors in vivo. EMBO J. 1983;2:1351–1356. doi: 10.1002/j.1460-2075.1983.tb01591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell. 2004;13:191–200. doi: 10.1016/s1097-2765(04)00005-x. [DOI] [PubMed] [Google Scholar]
- 53.Kingsman A, Willetts N. The requirements for conjugal DNA synthesis in the donor strain during flac transfer. J. Mol. Biol. 1978;122:287–300. doi: 10.1016/0022-2836(78)90191-2. [DOI] [PubMed] [Google Scholar]
- 54.Sarathy PV, Siddiqi O. DNA synthesis during bacterial conjugation. II. Is DNA replication in the Hfr obligatory for chromosome transfer? J. Mol. Biol. 1973;78:443–451. doi: 10.1016/0022-2836(73)90467-1. [DOI] [PubMed] [Google Scholar]
- 55.Lucas M, Gonzalez-Perez B, Cabezas M, Moncalian G, Rivas G, de la Cruz F. Relaxase DNA binding and cleavage are two distinguishable steps in conjugative DNA processing that involve different sequence elements of the nic site. J. Biol. Chem. 2010;285:8918–26. doi: 10.1074/jbc.M109.057539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pansegrau W, Balzer D, Kruft V, Lurz R, Lanka E. In vitro assembly of relaxosomes at the transfer origin of plasmid RP4. Proc. Natl Acad. Sci. USA. 1990;87:6555–6559. doi: 10.1073/pnas.87.17.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Howard MT, Nelson WC, Matson SW. Stepwise assembly of a relaxosome at the F plasmid origin of transfer. J. Biol. Chem. 1995;270:28381–28386. [PubMed] [Google Scholar]
- 58.Zechner EL, Pruger H, Grohmann E, Espinosa M, Hogenauer G. Specific cleavage of chromosomal and plasmid DNA strands in gram- positive and gram-negative bacteria can be detected with nucleotide resolution. Proc. Natl Acad. Sci. USA. 1997;94:7435–7440. doi: 10.1073/pnas.94.14.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.