

Abstract
Increasingly large-scale expression compendia for different species are becoming available. By exploiting the modularity of the coexpression network, these compendia can be used to identify biological processes for which the expression behavior is conserved over different species. However, comparing module networks across species is not trivial. The definition of a biologically meaningful module is not a fixed one and changing the distance threshold that defines the degree of coexpression gives rise to different modules. As a result when comparing modules across species, many different partially overlapping conserved module pairs across species exist and deciding which pair is most relevant is hard. Therefore, we developed a method referred to as conserved modules across organisms (COMODO) that uses an objective selection criterium to identify conserved expression modules between two species. The method uses as input microarray data and a gene homology map and provides as output pairs of conserved modules and searches for the pair of modules for which the number of sharing homologs is statistically most significant relative to the size of the linked modules. To demonstrate its principle, we applied COMODO to study coexpression conservation between the two well-studied bacteria Escherichia coli and Bacillus subtilis. COMODO is available at: http://homes.esat.kuleuven.be/∼kmarchal/Supplementary_Information_Zarrineh_2010/comodo/index.html.
INTRODUCTION
The availability of large-scale expression compendia in combination with gene sequence conservation makes it possible to compare expression networks across organisms, in order to study their evolution or to identify functional counterparts in different species as homologs with ‘conserved expression behavior’ (1–3). Besides custom made data sets that measure exactly the same experimental conditions in the different analyzed species (4), also large heterogeneous compendia based on collecting publicly available expression data sets confer a useful resource for cross-species analysis of coexpression (5,6). In contrast to the custom made homogeneous data sets, such heterogeneous expression compendia do not allow for a direct comparison of the expression patterns between orthologs in the different data sets, but instead rely on the search for ‘conserved expression behavior’. With conserved expression behavior, we refer to the conservation of a mutual relation between genes across species (such as the conservation of the mutual correlation between the expression profiles of a pair of genes across species). This conserved behavior is usually derived by defining coexpression modules (i.e. genes sets that behave similarly in all or a subset of the conditions), inferred by either biclustering (searching for coexpressed gene sets) (6–9) or by the analysis of a coexpression network (a network constructed from the data where the nodes refer to the genes and the weighted edges to the degree of coexpression between the connected nodes) (5,10,11). These conserved modules are then compared across the species. Methods differ in the way they perform this module comparison. A first set of approaches starts from a reference species in which an initial set of modules is built (6–8,10). The corresponding homologous modules are then identified in the target species by using gene homology. The approaches allow determining if the expression of a group of coexpressed genes in the reference organism is fully, partially, or not at all conserved at the level of coexpression in the target organism. To make an exhaustive comparison of all conserved modules between both species, each species has once to be used as a reference and once as a target. These approaches are most often applied using one-to-one gene homology relations (4,7). A second set of approaches obviates the need of reference species: in the multi-species coexpression network proposed by Stuart et al. (5), nodes correspond to genes that are conserved across the studied species (one-to-one map) and edges indicate significant pairwise coexpression levels between those genes in the different species. A clustering approach is used to identify conserved modules in this multi-species coexpression network. Alternatively, coclustering strategies exploit homology and coexpression information to identify in both species simultaneously coexpression modules. Depending on the implementation results focus on modules containing only homologous genes that link up related modules (11) or on finding mixed modules containing both homologous linker genes together with other genes that are coexpressed with those linker genes in a species specific way (9).
The difficulty with most previous methods is that they rely on the choice of a particular coexpression threshold or clustering parameter that determines the final module sizes (e.g. minimal degree of coexpression within a cluster or a minimal correlation coefficient to define subsets of coexpresssed genes in a coexpression network, the number of clusters, etc.). However, choosing such parameter is not trivial as the definition of a relevant biological module is not a fixed one: different parameters can result in equally valid modules differing from each other in number of genes and/or conditions. Moreover, the relation between the degree of coexpression and a particular parameter or threshold usually is data set-dependent (noise level, number of arrays tested, etc.) (12). As it is hard to decide in advance on the most optimal coexpression threshold or parameter to define modules in each of the species-specific compendia and to decide upon the threshold or parameter combination that would allow for a proper cross-species comparison of modules, we developed a cross-species coclustering approach referred to as conserved modules across organisms (COMODO) that exploits homology relations to determine the most optimal ‘conserved coexpression modules’ between two species. COMODO can take as input both one-to-one and many-to-many homology relations. The way we exploit the homology relations makes COMODO mainly suitable to search for processes with conserved coexpression behavior. Modules in a conserved pair are composed of homologous genes that share a mutual coexpression in each of the species, together with additional genes for which the coexpression with the homologous linker genes was found to be species-specific. We applied COMODO to search for conserved modules in two evolutionary distant prokaryotic model organisms: Escherichia coli and Bacillus subtilis. For those prokaryotic organisms we found conserved coexpression modules with a considerably larger fraction of genes than the number of conserved transcriptional units previously reported based on comparative genome analysis (13,14) and that cover a wider range of biological processes with conserved coexpression behavior than previously detected (15). Our results also showed how distantly related bacteria support the coexpression behavior of similar elementary processes with a completely different regulatory program.
MATERIALS AND METHODS
COMODO coclustering procedure
An overview of COMODO is given in Figure 1 while in Figure 2 the detailed steps of the coclustering procedure are displayed.
Figure 1.

Detection of evolutionary conserved expression modules. (A) Input data constitute of expression compendia of two distinct organisms (here E. coli and B. subtilis) (left panel) as well as a homology map between genes of the respective species (here derived from COG) (right panel). In the right panel, nodes correspond to genes and edges indicate the homology relations. (B) The left panel schematically illustrates the concept of module trees. Conceptually all potential modules (indicated by rectangles) in each of the species can be represented as nested chains of partially overlapping modules that can theoretically be obtained by gradually decreasing the threshold that determines the degree of coexpression within a module. Consecutive branches of the module trees give a view of all possible module sizes that originate from seed modules (modules indicated by a star correspond to modules obtained with the most stringent threshold). The chains of nested modules are captured by the symmetric gene–gene threshold matrices in each of the species (right panel). Our cross-species coclustering procedure starts from tightly coexpressed seed modules (indicated by stars) and uses a bottom up approach to traverse these chains of nested modules in both species simultaneously to identify from all possible matching pairs the best matching one (here indicated by the modules connected by a gray line, best is defined based on the Chi-square test statistic). (C) Resulting matching module pairs are referred to as evolutionary conserved module pairs and consist of a core and a variable part.
Figure 2.

Cross-species coclustering procedure. Displays the overall strategy of the coclustering approach: first ‘module seeds’ are selected from the gene–gene threshold matrices in the respective organisms. Module seeds linked by a sufficient number of homologous gene pairs are then gradually extended by traversing the space of possible cluster threshold combinations represented on the gene–gene threshold matrices of the respective species until optimality is reached. (A) Module seed selection step: the left panel represents a zoom in on the gene–gene threshold matrices of, respectively, the first and second organisms. Values on the first subdiagonal of the gene-gene threshold matrix (indicated with white rectangles) are used to select the seed modules. The right panel displays the coexpression values corresponding to this first subdiagonal of the gene–gene threshold submatrices of, respectively, organisms 1 and 2. Groups of genes that are mutually more coexpressed than with any other genes on the first subdiagonal are selected as seeds (gray areas in the plot). To prevent that we would obtain many very small seed modules we set in the gene–gene threshold matrix all values larger than a prespecified maximal coexpression stringency value equal to this value. (B) Extension of seed modules step: module seeds linked by a sufficient number of homologous gene pairs are gradually extended by traversing the space of possible cluster threshold combinations represented on the gene–gene threshold matrices in the respective organisms until optimality is reached. As it is computationally heavy to compare all possible threshold pairs, a combination of a greedy and brute force search was used to find the optimal module pair. This combination of a greedy and brute force search is represented as a dimensional grid of different threshold pairs, each with their corresponding chi-square values. The arrows indicate how the search space was traversed to find an optimal threshold pair. The search starts from the most stringent threshold pair [seed modules (top left)]. Greedy (larger black arrows) and brute force (smaller red arrows) searches are called consecutively to evaluate different thresholds pairs in an efficient way. Plot of consecutive Chi-square values obtained along the search (i.e. for the different evaluated threshold pairs). (C) Optimization criterium: a Pearson’s chi-square test was used to assess the statistical significance of a module pairs i.e. to assess to what extent the number of linking and non-linking gene pairs between two modules differ from what is expected by chance.
Gene–gene threshold matrix
Conceptually all theoretically potential modules in each of the species can be represented as nested chains of partially overlapping modules that were obtained by gradually decreasing the threshold of the distance measure used by the clustering or distance approach (Figure 1). Biologically each chain of nested modules corresponds to the hierarchical organization of a certain cellular processes (e.g. ranging from the production of an essential specific amino acid to a general response on a diauxic shift) (16). Different chains can share genes as the same genes can be involved in more processes. We used a symmetric gene–gene threshold matrix to concisely represent such chains of nested modules (Figure 1). Each axis of this matrix corresponds to the genes of one organism. The order of the genes in the x- and y-axis of the matrix is determined by their assignment to modules under the most stringent tested threshold i.e. genes that are coexpressed at the most stringent tested threshold will be grouped. The values in the ith row and jth column of the gene–gene threshold matrix represent the most stringent threshold at which, respectively, genes i and j appear together in at least in one of the detected modules. For the results shown in the main text the pairwise similarity between the genes was based on the Pearson correlation over all conditions in the compendium. The gene–gene threshold matrix in this case contains for each cell a discretized pairwise correlation value and the gene order on the x- and y-axis of the gene–gene threshold matrix equals the order of the genes at the leaves of a hierarchical clustering applied on the non-discretized gene–gene correlation matrix. The number of bins used for the discretization depends on the parameter step size (see also below). We also built a gene–gene threshold matrix by using the gene thresholds defined by the iterative signature algorithm (ISA) to assign its genes to modules (16) (see Supplementary Text S1 for B. subtilis and E. coli data sets). In the latter case, the gene–gene threshold matrix consists of a compact representation of the overlapping clusters (module tree) that can be obtained using ISA with different threshold combinations.
Selection of seed modules
To select the seed modules, we used the values on the first subdiagonal of the gene–gene threshold matrix (the first subdiagonal contains the values directly under those of the main diagonal of the gene–gene threshold matrix). To identify seeds we selected on this first subdiagonal groups of genes that were locally found to be more coexpressed with each other than with their neighboring genes on the first subdiagonal (Figure 2A). For those genes the value on the first subdiagonal corresponds to the most stringent coexpression threshold at which they can be found together. To prevent that we would obtain many very small seed modules, containing two genes only, in the gene–gene threshold matrix all values larger than a prespecified maximal coexpression stringency value were set equal to this value. This guarantees a minimal number of genes to be present in the seed modules. We could show that within a certain range our coclustering procedure is quite robust against the choice of this prespecified maximal coexpression stringency value (Supplementary Text S1).
Extension of seed modules
COMODO uses a bottom up approach to build its conserved module pairs. It starts from the seed modules in each of the species of interest. Module seeds linked by a sufficient number of homologous gene pairs are gradually extended by traversing the space of possible cluster threshold combinations as represented on the gene–gene threshold matrices in the respective species until optimality is reached (see below for the chi-square optimization criterium). As it is computationally heavy to pairwisely compare all cluster threshold combinations between the two organisms we developed a dedicated search methodology. The search space of all possible combinations of thresholds can be represented in a two dimensional grid as shown in Figure 2B. Moving down the grid corresponds to gradually lowering the thresholds pairs. At each move the optimization criterium is evaluated. The parameter ‘Step’ indicates the size by which the threshold is lowered at each move (in our experiments this was set to 0.05). To move along the grid we applied a combination of a greedy and brute force search. The methodology starts with the thresholds that define the seeds module pairs. By applying a greedy search gradually one or both of the thresholds in a combination are lowered until a local optimum is reached, i.e. further lowering the thresholds does not further improve the optimization criteria. To prevent the methodology from getting trapped in a local optimum, it searches further down in the grid in brute force manner until the stop criteria is reached (see below) to make sure no other threshold pair exists that is more optimal. If a better threshold pair than the current local optimum is found, the whole greedy search procedure is restarted from this more optimal threshold pair.
Two stop criteria are used: first, both thresholds should be larger than a preset value (in our example based on the Pearson correlation coefficient, both thresholds should at least be 0.1). Second, the minimal fraction of homologous versus non-homologous genes in the gene sets obtained by a given threshold pair should be higher than a preset number (in our study it was set to 0.1).
To tune the methodology for bacterial applications, we introduced the following refinement procedure: genes that belong to the same operon tend to show a higher degree of coexpression with each other than with other genes. To prevent our methodology of getting biased towards finding module pairs that are composed of evolutionary conserved operons (these might always get the highest chi-square value), we allowed for all module pairs of which one of the composing modules contains less than five genes the following additional threshold relaxations: the threshold of the group that contains less than five genes was relaxed until more genes were included. In such case, both the initially detected module pair and the module pair obtained after threshold relaxation were retained for further analysis.
The method can be applied on any chains of nested modules for which the relation between the modules is hierarchical, meaning that the module(s) obtained with the more stringent thresholds should be subsets of the ones obtained with a more relaxed threshold. Modules obtained with a more stringent threshold can never contain genes that were not detected at a more relaxed threshold.
Chi-square test statistic as optimization criterium
The definition of the best matching module pair is bound by the number of homologs that is shared by the selected modules in each of the species and corresponds to the pair for which the number of sharing homologs is statistically most significant relative to the size of the linked modules (Figure 2C). We used a Pearson’s chi-square test to assess the statistical significance of a module pairs i.e. to assess to what extent the number of linking and non-linking gene pairs between two modules differ from what is expected by chance. To formulate the Pearson’s chi-square test, consider N1 genes in the genome of the first organism and N2 genes in the genome of the second organism, and M linking homologous gene pairs derived from the COG database. If we pick two genes randomly, one from each organism, the probability that a homologous gene pair has been chosen is equal to 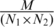 . Therefore, the probability that these genes are not homologous is
. Therefore, the probability that these genes are not homologous is 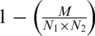 .
.
Given a pair of modules (one for each organism) containing respectively g1 genes from the first organism and g2 genes from the second one (where g1 and g2 << N1 and N2, respectively), the expected number of homologous gene pairs that would appear assuming that the two modules are randomly selected modules can be estimated by:
The expected number of non-homologous gene pairs appearing between them can be estimated by:
We use the Pearson’s chi-square test to assess whether the number of homologous and non-homologous gene pairs in an observed module pair is significantly different from the expected one. A chi-square test with one degree of freedom is as follow:
 |
where O and E stands for observed and expected values, respectively. Note that as the P-value might get very close to zero, we use an optimization criterium that maximizes the actual chi-square values instead of minimizing the corresponding P-values.
Filter procedure
We selected from the raw output the most interesting module pairs for further analysis: we only retained the most significant module pairs (using a minimal threshold on the chi-square value). To remove redundancy we kept in case of overlapping module pairs (different module pairs that share 75% of homologous linker genes) the one with higher chi-square value.
We included the following additional criteria for our specific application: modules of size smaller that six should be linked up to their counterpart modules in the other organism with at least two homologous linker genes, this to avoid small spuriously linked modules. In addition, we required that the number of linker genes comprises at least 20% of the total number of genes in each of the modules to prevent unbalanced growth of one module compared to its counterpart module as the latter modules were very often found not to be biologically meaningful.
Application of the methodology to the E. coli and B. subtilis data sets
Using the Pearson correlation over all conditions as a distance measure and a prespecified maximal coexpression stringency value of 0.7 for seed identification, we obtained conserved module pairs covering 1687 E. coli genes and 2129 B. subtilis genes. After filtering (using a chi-square threshold of 470) and removing overlapping module pairs (see above), we retained 445 E. coli genes and 481 B. subtilis genes being found in 82 non-redundant module pairs. The final 82 conserved module pairs were ordered according to their overlap in gene number in each of the organisms. Modules that shared >30% of their genes were assigned to the same biological process as they were enriched in the same GO categories and pathways. To assess the false discovery rate (FDR) of our results we randomized the expression values in the original compendia and searched for conserved module pairs using the same procedure as described above (process was repeated 50 times, expression data was randomized by reassigning the gene labels to the expression profiles).
Condition selection for module visualization
For visualization purposes heat maps only display the conditions for which the coexpression behavior was most obvious. Relevant conditions were selected by dividing per condition the mean value of the expression levels in the module by the variance (coefficient of variation). If this coefficient of variation exceeds a predefined threshold (one in our case), the corresponding condition is visualized.
Microarray compendia
The microarray compendium of E. coli was obtained from Lemmens et al. (17) and the one of B. subtilis from Fadda et al. (18). They contained, respectively, 870 conditions for E. coli and 231 for B. subtilis.
Homology map and sequence similarity
A total of 5459 homologous gene pairs between E. coli and B. subtilis were annotated based on the COG database (19). This many-to-many COG map was used throughout the paper unless specified otherwise. Orthologous gene pairs between E. coli and B. subtilis were when needed identified by the reciprocal smallest distance approach (20).
Essential genes
Essential genes in B. subtilis and E. coli were downloaded from DEG, a database of essential genes (21,22). This database contains 271 essential genes in B. subtilis, resulting from a single gene deletion experiment (23). For E. coli 620 genes were originally determined to be essential based on genetic footprinting (24) and 303 genes were later identified by single gene deletions (25). As 205 genes were found in common between those two E. coli lists, we obtained in total 712 essential genes for E. coli. Based on the homology relation derived from the COG database, we found 209 homologous pairs of essential genes comprising 191 B. subtilis and 195 E. coli essential genes. From these 195 E. coli genes with homologs in B. subtilis 164 were originally identified by the single gene deletion experiment mentioned above (25).
Enrichment analysis of Gene Ontology terms, metabolic pathways, protein complexes and regulatory data
Gene Ontology (GO) terms, metabolic pathways and protein complexes of E. coli were downloaded from EcoCyc (26). GO terms for B. subtilis were downloaded from the Comprehensive Microbial Resource (CMR) (27). Metabolic pathways and protein complexes of B. subtilis were obtained from BioCyc (28). Transcriptional interactions were downloaded from RegulonDB (29) and DBTBS (30) for E. coli and B. subtilis, respectively. Enrichment analysis was done based on the hypergeometric distribution corrected for multiple testing by the FDR (31).
Operon information
Operon structure was derived from RegulonDB (29) and DBTBS (30) for, respectively, E. coli and B. subtilis. As DBTBS only describes experimentally validated B. subtilis operons, we used for gene sets not covered by DBTBS the following databases with operon predictions: OpeRons (DOOR, http://csbl1.bmb.uga.edu/OperonDB/) (32) and http://www.microbesonline.org/operons/OperonList.html (33). Operon predictions were retained: (i) if databases agree with each other in predicting the same operon structure (this was the most frequent situation), (ii) if they were only predicted by one database, (iii) in the few cases where two databases predicted a different operon structure, we used the structure that was more compatible with our expression results or with the structure of the counterpart operon in E. coli. To identify conserved operons (or homologous operons) between E. coli and B. subtilis we used the following definition: we started from the list of E. coli operons as this was the best annotated. We identified as an operon conserved between E. coli and B. subtilis any annotated operon in B. subtilis for which at least two genes showed homology, based on COG database information. This analysis was also repeated using only strict homology links obtained by the reciprocal smallest distance approach (20) to approximate a definition of ‘orthologous’ operons.
RESULTS
COMODO: a method to identify cross-species expression conservation
As we focused on searching processes across species with evolutionary conserved coexpression behavior, we defined the optimal size of the modules in each of the species as the one that maximizes the fraction of homologous genes that links up both modules in an evolutionary conserved module pair. An overview of the analysis flow is given in Figure 1. To avoid (bi)clustering the data sets using a fixed parameter setting that determines the cluster size in each of the species separately, we relied on a bottom up coclustering approach to build the modules. COMODO is initialized with coexpressed seeds or seed modules obtained in each of the species. These seeds are gradually expanded in each of the species until a pair of modules is obtained for which the number of shared homologs is statistically optimal relative to the size of the linked modules. The optimization criterium is based on a chi-square statistic (‘Material and Methods’ section). Our coclustering procedure, that extends the seed modules until optimality is reached, is based on greedy and brute force procedure described in ‘Materials and Methods’ section.
Eventually pairs of evolutionary conserved modules are obtained, each containing a core and a variable part (Figure 1C). The core part consists of the homologous genes that link up both coexpression modules and for which the mutual coexpression behavior is conserved. The variable part contains the additional genes that belong to the composing modules of a given pair in either one of the organisms. These are the genes that either do not have a homologous counterpart in the other organism or that acquired a coexpression behavior similar to that of the core part in only one of two species (34). Because a module in one species can be linked to more counterparts in the other species (Figure 3), COMODO can be used to study both conservation, but also divergence in expression which makes it optimally suited to be used with a many-to-many homology map.
Figure 3.

Overview of evolutionary conserved modules between E. coli and B. subtilis. A total of 82 evolutionary conserved module pairs of which the matching modules (connected by solid lines) were linked through a statistically significant set of homologs between E. coli and B. subtilis are shown. Node sizes are proportional to the number of coexpressed genes in the modules (indicated in parenthesis) and module ids correspond to those used in Supplementary Table S1. Modules showing an overlap of 30–75% of their genes within each species were connected by dashed lines. Modules that show an overlap of at least 75% in their gene content were merged. Modules to which a similar functional category was assigned were grouped (as indicated by the different panels. Panels with the same color are involved in a similar general process e.g. metabolism).
Identifying evolutionary conserved modules between E. coli and B. subtilis
We applied our methodology to study the degree to which coexpression modules have been conserved between two bacterial model organisms: E. coli and B. subtilis. For both species we used cross-platform microarray compendia covering a wide-range of experimental conditions (‘Materials and Methods’ section). Many-to-many homology relations amongst the genes of the two species were defined based on COG (19). Applying our method resulted in the identification of 82 conserved module pairs in E. coli and B. subtilis that were linked through a statistically significant set of homologous genes. These linked groups are called matching module pairs and they represent processes for which the coexpression is at least partially conserved over the wide evolutionary distance that separates E. coli from B. subtilis. Figure 3 gives an overview of these matching, evolutionary conserved module pairs.
To estimate the potential number of false positives among our detected conserved module pairs, we applied COMODO to a randomized data set from which we did not expect to find any meaningful results (‘Materials and Methods’ section). The FDR estimated as the mean number of significantly detected matching module pairs in random expression compendia was 2.24. In general the chi-square statistic values obtained in the randomized data sets were well below the ones observed for the true data set (t-test, P < 0.05), implying that the size of the core to the variable part is much larger in modules obtained from the true data set than in those obtained from the random data set (Supplementary Text S1 and Figure S1).
In those 82 conserved module pairs, on average 60% of the genes constitute the core part and 40% the variable part. Of those genes in the variable part, 33% did not have a homologous counterpart in the organism of comparison. The other 67% found in the variable part with a homologous counterpart in the other organism could correspond to species-specific members of the regulon represented by the core part. A gene assigned to the core part of one module can also be found in the variable part of another module as the same gene can belong to different regulons that do not completely coincide between species. For instance, EM40-BM40 contains in its core part the orthologous operon nrdEFIH known to be regulated both in E. coli and B. subtilis by NrdR (35,36). In EM59-BM59, containing a Fur-dependent conserved core the same nrdEFIH is in the variable part of the E. coli module. This confirms previous knowledge on nrdEFIH being Fur-dependent in E. coli, but not in B. subtilis (at least not yet observed) (35).
By using a stringent filtering procedure and only maintaining matching module pairs for which the core part was relatively larger compared to the variable part, we focused on the processes for which coexpression behavior was conserved between E. coli and B. subtilis. The number of genes in the evolutionary conserved modules varies largely and ranges between 2 and 100, with a large overrepresentation of small modules (e.g. 28 module pairs containing 2–5 genes only in both matched modules). Smaller modules usually correspond to single operons, subunits of a protein complex or constitute parts of larger biosynthetic pathways. As the size of the conserved modules increases, the modules cover larger pathways. A complete description of the modules can be found in the Supplementary Table S1.
In total 30 of our 82 conserved modules are linked by a single homologous operon: 14 of those by ‘an orthologous operon’ (according to the definition of operon orthology described in ‘Materials and Methods’ section) and 16 by a homologous, but functionally related operon. As by definition genes within an operon (as an estimate of a transcription unit) will be coexpressed, these matching modules, although correctly identified by our method do not contribute more information on the functional relation between the matching operons than the one derived from sequence analysis. Therefore, extrapolation of the operon function between E. coli and B. subtilis should be treated with care for those modules.
Assessing the conservation of coexpression within homologous operons
As the operon structure is an important mechanism to guarantee the conservation of coexpression behavior between genes within a species (13,14,37), we wanted to assess as a validation of our methodology to what extent homologous operons will be found in the core parts of our conserved modules. When using the COG based definition of homologous operons, we could retrieve 289 pairs of E. coli and B. subtilis operons that share at least two homologous genes (‘Materials and Methods’ section). Based on sequence homology several E. coli operons were mapped to at least two different operons in B. subtilis that mutually do not share any gene (this was also observed when the comparison was performed the opposite way around i.e. when B. subtilis operons were mapped to those of E. coli). Of these 289 E. coli operons with a homologous counterpart in B. subtilis, 91 were found as linkers between conserved modules (i.e. 31% recovery rate), resulting in a total of 135 links between conserved modules as some operons can occur in more modules. Of those 135 linking operons, in 61 cases all the genes of the linking operons were found in the core part, in 33 cases one of the operon genes of the linking operons was missing and in 41 cases at least two genes of the linking operons were missing from the core part. Although in some cases lacking some of the operon genes in the core part might point towards differentiation in regulation, for instance, by means of intra-operonic promoters, it seemed that in many cases it was the last operon gene that was no longer found to be coexpressed with the rest of the operon genes in the core parts of the linking modules. This observation can be explained by the increased degradation of the mRNA at the 3′-end of the transcript (38). When using a more stringent definition of homolgous operons (‘Materials and Methods’ section), the fraction of E. coli operons with a counterpart in B. subtilis that was found in the core part (meaning that their genes were found as linker genes in conserved modules) was much higher (50 of the 100 linking operons, i.e. 50% recovery rate). This higher recovery rate might be partially due to the fact that this more strict mapping as an estimate of orthologous operons results in linking operons that mutually share more genes than with the previously used COG-based mapping (as an estimate of homologous operons). When more genes are shared between the linking operons, the chance to find a module pair that meets our selection criteria (sharing at least two coexpressed linker genes) can be met more easily. On the other hand, it definitely also reflects that many of the operons that can be linked through a COG mapping are not each others functional counterparts.
We also found that a considerable part of the orthologous operons could not be retrieved in conserved coexpression modules as their composing genes were not found to be coexpressed in E. coli or B. subtilis, probably due to the still incomplete sampling of conditions in the used expression compendia (this was the case for 50 operon pairs defined using the more stringent definition and for 198 of the conserved operon pairs defined with the less stringent definition). So the 50% recovery rate of orthologous operons (as well as the 31% recovery rate for the homologous operons) in our module cores stems from the incompleteness of the used expression compendia rather than from a bias in the methodology.
Optimized coexpression threshold is module-dependent
Maximizing the statistical significance of the number of linking homologs in the core of a conserved module pair relative to the module sizes in each of the respective species allows us to select in each of the species the modules that best match the conserved processes reflected by the core. Depending on the type of biological process that is conserved in the core the optimal correlation thresholds for the modules in each of the individual species can differ considerably. This is illustrated in Figure 4 where the selected correlation coefficient differs largely between the modules of the different conserved pairs.
Figure 4.

Degree of correlation within a coexpression module versus the number of genes it contains. Number of genes: refers to the total number of genes in the module (adding up genes in core and variable parts). A total of 82 evolutionary conserved modules between E. coli (circles) and B. subtilis (squares) are plotted. In each case the color used to represent a module corresponds to the color scheme in Figure 3 to denote the functional class (or group of related functional classes) a module was assigned to.
Globally the correlation thresholds for the E. coli modules were lower than those of the corresponding B. subtilis modules, most probably because the E. coli compendium is larger and contains more conditions than the one of B. subtilis. When investigating per organism the relation between the used correlation threshold and the number of genes in a selected module, we observed that it is not only the number of genes within a module that determines the selected correlation threshold, but that there is also a clear influence of the type of process the module reflects (Figure 4). House-keeping processes such as ribosomal metabolism and translation (EM34_35_36-BM34_35_36) were found with very strict thresholds despite containing a relatively high number of genes, while for more specialized processes such as e.g. iron acquisition (EM59-BM52_53_59) and motility and flagella synthesis (EM32-BM32) the opposite was observed (Figure 4). This can be related to the number of compendium conditions in which genes are expected to be coexpressed. When using a distance measure that by default considers all conditions (such as Pearson correlation), genes that tend to be active under all conditions (e.g. house-keeping genes) will be found coexpressed with a more stringent correlation threshold than genes that are only coexpressed under a subset of the sampled conditions (e.g. those that belong to the more specialized modules). This observation underlines the need for a module- and data set-dependent determination of the coexpression threshold or clustering parameter that determine the final module sizes during the coclustering of heterogeneous expression compendia.
Comparison with SCSC, a probabilistic coclustering approach
We compared the performance of COMODO with the recently developed coclustering approach SCSC of which the implementation is publicly available (9). Results of this analysis are displayed in Supplementary Text S1. We observed that the intrinsically different way in which the coclustering is performed by, respectively, SCSC and COMODO affects the characteristics of the detected matching module pairs. SCSC partitions the data in each species in a predefined number of modules. This results in sets of loosely connected modules of which the sizes and coexpression level largely depend on the used data set prefiltering and the predefined cluster number. In addition, there is no guarantee that the homologous genes that were added to the modules are amongst the most tightly coexpressed genes in a module. This, in combination with the fact that the identified modules should only be loosely connected by homologs to be identified as a matching pair complicates distinguishing true matching module pairs from spurious matching ones when using SCSC in combination with a many-to-many homology map.
COMODO in contrast chooses the number of genes in the modules of either species to maximize the enrichment of linking homologs relative to the number of variable genes. This criterium results in adapting the size of the modules to the specificities of the conserved processes: as a result COMODO can cover a wide range of module sizes without compromising the quality of the modules (reflected by a good coexpression level). In addition homologous linker genes are by definition as tightly coexpressed as the rest of the genes in a module. This, together with a selection of the most significantly matching module pairs based on the chi-square statistic facilitates prioritizing the most significant matching module pairs. However, because of this bottom up strategy COMODO might unlike SCSC underestimate in the individual species the true sizes of the pathways represented by the cores.
Evolutionary conserved processes and essential genes
Figure 3 gives an overview of the evolutionary conserved modules ordered within a species according to their overlap in genes. Partially overlapping modules (indicated by dashed lines) were assigned to the same functional category. Biological processes involved in carbohydrate metabolism, amino acid metabolism, energy metabolism, nucleotide metabolism, lipid metabolism, translation and ribosomal metabolism, motility and flagella synthesis, DNA repair, cell shape and division, protein folding (heat shock), DNA-binding (cold shock), signal transduction, cofactors and vitamins, and iron acquisition all contain genes for which the mutual coexpression behavior was found to be conserved between E. coli and B. subtilis. As most of these processes with a conserved coexpression behavior are primary processes, we wondered to what extent they contained essential genes, defined as the minimal gene sets required to sustain a living cell. Essential genes are believed to be widespread and highly conserved during evolution (23,24). Previous studies identified a total of 712 essential genes in E. coli and 271 genes in B. subtilis (21,22). Of those, 209 were found to have a counterpart in both species (as homologous gene pairs). Forty-eight (23%) of these essential homologous gene pairs were found as core genes in our conserved modules. The majority of them (37 pairs) belonged to the large conserved module pair involved in translation and ribosomal metabolism. Another 17% of the homologous essential gene pairs appeared in conserved modules that were linked by a smaller number of conserved core genes than the minimum that was required in our selection (i.e. in the module pairs that were linked more weakly by homologous genes pairs and did not pass our stringent selection criteria). For the remainder of the essential genes that were not found in any module, we found that they exhibited a lower degree of coexpression with other genes in the genome than was observed on average (indicating that most likely they are not coexpressed with any other gene in our data set).
In addition, some auxiliary processes not generally considered as essential (23) exhibit a highly conserved coexpression behavior between E. coli and B. subtilis. Remarkably is the group involved in flagella synthesis and motility (EM32–BM32) which recapitulated 68% of the previously characterized motility genes of E. coli and 78% of the genes known to be related to motility in B. subtilis (39). The majority of the genes known to be involved in flagella synthesis with a homologous counterpart in both E. coli and B. subtilis were found in the core part [50 homologous links including 34 linked genes in the core part out of 54 total module genes in E. coli (63%) and 36 linked genes in the core part out of 48 total module genes in B. subtilis (75%)]. The variable part then mainly consisted of genes occurring in one of the two species only [14 out of 20 in E. coli (70%) and 15 out of 22 in B. subtilis (68%)].
Another large group is the one involved in iron acquisition (EM59-BM52_53_59) which contains 70% of the E. coli and 65% of the B. subtilis Fur targets identified by Ollinger et al. (40). Unlike motility and flagella synthesis case, here most of the known Fur targets of E. coli and B. subtilis were not found in the core part. The core part only consists of 26 homologous links [13 out of 52 total module genes in E. coli (25%) and 18 out of 32 total module genes in B. subtilis (56%)] which is a relatively small fraction compared to the large variable parts. The variable part of the E. coli module contained in this case 28 out of the 39 genes (72%) without homologous counterpart in B. subtilis and the variable part of B. subtilis had 7 out of the 14 genes (50%) without counterpart in E. coli. This indicates that the Fur regulon largely changed during evolution to adapt to the specific needs of each organism.
Regulation of evolutionary conserved modules
For all conserved module pairs depicted in Figure 3, the coexpression behavior of their genes has largely been conserved during evolution. This does, however, not necessarily mean that also the regulatory mechanism that is responsible for this coexpression behavior is conserved. To study their regulatory mechanisms, we listed all modules with conserved coexpression behavior and assigned to each module the corresponding transcription and sigma factors by calculating the modules’ enrichment in genes for a given transcription or sigma factor, according to RegulonDB or DBTBS (see Supplementary Table S1). We used the reciprocal smallest distance approach (RSD) (41) to identify the best matching transcription and sigma factors pairs between E. coli and B. subtilis. We then determined whether modules with a conserved coexpression behavior were regulated by matching transcription and sigma factors in both organisms. By doing so, we were able to divide the evolutionary conserved module pairs into three main groups according to the sequence similarity of the transcription and/or sigma factors that were assigned to each of them.
The first group comprises conserved module pairs regulated by reciprocally best matching transcription or sigma factor pairs. To this group belonged 14 of the 82 conserved modules pairs regulated by the pairs NrdR/NrdR (EM39_40-BM39_40), Fur/Fur (EM51_52_53_55_57_58_59-BM51_52_53_55_57_58_59), LexA/LexA (EM67-BM67), BirA/BirA (EM21-BM21) and ArgR/AhrC (EM9_10_79-BM9_10_79) (where the notation corresponds to the E. coli/B. subtilis gene). Each of these best matching transcription factors pairs have previously been identified as functionally conserved counterparts between E. coli and B. subtilis [with Fur/Fur, LexA/LexA and ArgR/AhrC being direct orthologs and BirA/BirA being a best matching xenolog pair as pinpointed by Price et al. (42)]. Moreover, the best matching transcription factors pairs identified by Price et al. (42) as non-functional counterparts were never found to regulate our conserved modules, further confirming the power of using coexpression in inferring functionality. Also in this group we found the conserved modules regulated by two orthologous sigma factor pairs: FliA/SigD (EM32-BM32) and RpoN/SigL (EM79-BM79).
A second group of conserved module pairs appeared to be regulated by transcription or sigma factors showing a homologous link, as predicted by COG, but not being best reciprocal matches. In this group we found four transcription factor pairs: ArcA/ResD (EM24-BM24), FruR/CcpA (EM25-BM25), GalR/CcpA (EM61-BM61) and Gals/CcpA (EM61-BM61) that could be assigned to three conserved module pairs. For the couple ArcA and ResD it is indeed known that they both are sensing aerobic versus anaerobic conditions (15). They both belong to large gene families for which the evolutionary history is hard to resolve and thus inferring functionality from merely sequence homology can be misleading (43). Just like FruR, GalR and GalS in E. coli, CcpA in B. subtilis is still involved in the regulation of carbon sources, but evolved towards a more global function than its homologous counterparts in E. coli. Indeed CcpA is known to be the non-homologous functional counterpart of Crp (15,44). Regarding the sigma factors regulating the modules in this group we observed the pairs: RpoD/SigA (EM1_18_32_39_43_81_82-BM1_18_32_39_43_81_82), RpoH/SigA (EM44_45-BM44_45) and RpoS/SigA (EM62_63-BM62_63). According to the COG homology definition, the house-keeping sigma factor SigA of B. subtilis (45) has three homologs in E. coli, namely RpoD, RpoH and RpoS. These multiple sigma factor copies have resulted in a subfunctionalization in E. coli of the global role executed by the sigma factor SigA in B. subtilis (45,46). This is clearly visible from our results where we found different combinations of respectively RpoD, RpoH and RpoS being responsible for the regulation of at least 12 E. coli modules that were paired with an equal number of B. subtilis modules regulated by SigA.
In the third group of conserved module pairs we found those cases where the assigned transcription regulators do not show any significant sequence similarity with each other, but they appear to regulate genes with similar function in both organisms. For 65 of the 82 conserved module pairs, at least one of the assigned transcription factors was different between E. coli and B. subtilis (summarized in the Supplementary Table S1). For example, the master regulators FlhC and FlhD responsible for regulation of motility and flagella synthesis in E. coli do not have a homologous counterpart in B. subtilis, while the coexpression behavior of their cognate modules is conserved (EM32-BM32). We can thus assume that a non-homologous functional counterpart, such as recently proposed SwrA takes over the mechanism of regulation in B. subtilis (47–49). Indeed SwrA is known to regulate SigD in B. subtilis as FlhC and FlhD do in E. coli (50).
Additional striking examples are the pairs of conserved modules in E. coli and B. subtilis regulated, respectively, by PurR/PurR (EM16_17_18-BM16_17_18), TreR/TreR (EM48-BM48), CysB/YwfK (EM2-BM2), MalT/AbrB (EM47-BM47). A complete list of such non-homologous transcription factors that regulate paired coexpression modules in E. coli and B. subtilis can be found in Supplementary Table S1. PurR is known in both E. coli (51) and B. subtilis (52) to respond to purine excess by repressing genes of the inositol monophosphate (IMP) to adenine monophasphate (AMP) conversion pathway. TreR on the other hand controls the expression of the trehalose utilization operon in both species and its activity is known to be dependent on the cAMP gene activation protein (CAP) in both E. coli and B. subtilis (53). Both pairs of similarly named transcription factors PurR/PurR and TreR/TreR constitute well documented cases of parallel evolution: despite being each others functional counterparts in both E. coli and B. subtilis and being responsible for the regulation of an almost conserved regulon, the proteins in each pair do not exhibit any significant sequence homology, nor any similarity in their molecular mode of action (53–55).
In contrast to these well-documented cases no studies exists that focus on the direct functional comparison of the pairs CysB/YwfK and MalT/AbrB in respectively E. coli and B. subtilis. The functional relation between CysB/YwfK was supported by the fact that both regulators belong to the same LysR-type of activators and they do show a low level of sequence homology (28% of sequence homology) (56). Also, the regulator pair was assigned to conserved modules involved in cysteine biosynthesis, a role which is well documented for CysB and YwfK. Both regulators are also related to sulfate transport (56,57). Phenotypes of E. coli cysB mutants were found to be very similar to those of B. subtilis ywfK mutants (56). The conserved modules regulated by the MalT/AbrB pair were found to be in involved in maltose metabolism. In E. coli MalT is known to regulate seven operons of the maltose regulon (58) that are subjected to catabolite repression (59). For AbrB the direct role on maltose regulation is not reported. Instead AbrB is known to be a dual regulator that regulates a plethora of genes during starvation-induced processes such as those involved in sporulation, production of antibiotics and degradative enzymes (60). The fact that AbrB has been found to modulate the cAMP-CAP system by competing with catabolite repressor proteins during growth on carbon sources that induce partial catabolite repression (61) points towards a possible functional link between MalT and AbrB.
Differentiation in expression by divergence of regulation
We can also find modules containing sets of genes, coexpressed in one species that got split up in different coexpression modules in the second species (Figure 5). We identified such gene sets as follows: a single module in the first organism should be linked to two different modules in the second organism of which the respective core parts do not share >30% of their genes. Such cases might point towards a condition-dependent differentiation in regulation that is observed in one species, but not in the other. Such differentiation in regulation seems to occur, for instance, for heat shock genes (EM44_45–BM44_45), most of which are chaperones and proteases known to protect cells against damage induced by protein unfolding. These genes were found to be coexpressed in E. coli as was also previously observed (62). In B. subtilis the corresponding genes, all known to be regulated by HrcA are split up in two different modules (63). This observation indicates that HrcA induces a difference in expression behavior, depending on the type of transcription factors it is combined with. A potential interacting partner of HrcA could be CtsR, the transcription factor known to regulate the gene clpE (63) that belongs to one of the two evolutionary conserved modules in B. subtilis. Note that HrcA seems not to have a homologous counterpart in E. coli.
Figure 5.

Differentiation in expression. The E. coli module EM44_45 (left panel) is covered by two different modules BM44 and BM45 in B. subtilis (right panel). Genes that belong to the same module are displayed in a gray box and homology relations are denoted by gray edges; numbers on the edges indicate Smith–Waterman alignment scores (z-values). Shaded areas in the right heatmap correspond to conditions where both B. subtilis modules do not overlap.
Expression behavior of linker genes
The fact that the identification of evolutionary conserved module pairs was based on a many-to-many homology map allowed us to study the complex evolutionary history of several of the linker genes (Supplementary Table S2).
At first we focused on linker genes that all showed a mutual homology. We found several of those linker genes modules, being connected by several one-to-many or many-to-many relations: e.g. paired modules that contained at least one gene in E. coli with multiple homologous counterparts in B. subtilis each of which was found in a different conserved module or the opposite way around. Those genes for which we found a divergence in mutual coexpression behavior between the homologous genes within one species could be an indication of their functional divergence as it is known that multiple copies of a particular gene in one species, resulting from horizontal gene transfer or duplication events tend to disappear unless they evolve into non-redundant copies by acquiring novel functions (neo-, subfunctionalization) (64). We found in total 19 cases of potential neo- and/or subfunctionalization (Supplementary Table S2). For instance, the duplicated genes in E. coli with ribonucleotide reductase activity (Figure 6): each gene of the duplicated pairs nrdA/nrdE and nrdE/nrdF belongs to a different module (respectively, EM39-BM39 and EM40-BM40), while the homologous counterparts of these genes in B. subtilis (being nrdE and nrdF) belong to one single coexpression module. Although we found NrdR as the responsible regulator for both sets of paralogous ribonucleotide reductases genes in E. coli, genes within a duplicated pair exhibit a clear difference in expression behavior. Moreover, all three coexpressed genes of the B. subtilis conserved module (nrdEF-ymaA) were reported as essential genes (23), while their most closely related homologs (the E. coli nrdEF genes) were not (21,22), but instead essentiality in E. coli was taken over by the less related homologs (nrdAB), reflecting a clear case of sub/neofunctionalization. Another example of complex transcriptional evolution of homologous gene families relates to the family involved in oligopeptide and dipeptide ABC transport. Supplementary Text S1 and Figure S2 shows how in both organisms homologous genes are coexpressed in different conserved modules. A large fraction of homology links (indicated by blue, green and red lines) occur between members of the DppBCDF system in E. coli with members of the Opp and App transport system in B. subtilis. In each case, a gene in E. coli is linked to two or more genes in B. subtilis covering more than one coexpression module. For example, the E. coli gene dppD (EM26_27) is linked to, respectively, B. subtilis oppD (BM26_28) and dppD, appD (BM27_29_30). In E. coli dppBCDF genes form a dipeptide inner membrane ATP-binding cassette transporter involved in the uptake of heme iron (65). In B. subtilis both the oligopeptide transport system Opp (66) and the AppA system (67) are involved in competence development and sporulation with the App system being able to substitute the Opp system. Although both systems being functionally related in B. subtilis, they exhibit clear differences in their expression behavior pointing towards at least some further specialization (Supplementary Text S1 and Figure S2).
Figure 6.

Expression divergence of duplicated genes in E. coli. Expression behavior of genes in modules EM39 (above the line in the heatmap) and EM40 (below the line in the heatmap) in E. coli (left panel). Shaded areas correspond to conditions not shared between modules. Homologous genes to the B. subtilis nrdEF operon (module BM39_40) were found in two different coexpression modules in E. coli (modules EM39 and EM40). Each module is surrounded by a gray box and homology relations are denoted by gray lines (right panel). Numbers over the lines represent Smith–Waterman alignment scores (z-values).
For another set of homologous linker genes (16 cases) we found the multiple copies of the gene family within one species in the same module, indicating that their expression behavior was retained as a result of either recent multiplication events that did not yet result in further functional divergence, or the need of multiple gene copies for dosage effect.
In addition to these linker genes that all belonged to the same COG, we also found few examples (five cases) where genes not exhibiting any mutual homology in one organism (not belonging to the same COG) were linked to the same gene in the other organism, implying that here two protein domains occurring in one organism in separate genes got fused in the other organism into a single gene. One case for which the fusion was also supported by the literature was the linking gene set purL/purL and purL/purQ (68). The most interesting cases were those where the genes containing the separate or unfused domains belonged to different coexpression modules (frwB/manP and frwC/manP) as this indicates that there is a functional constraint to keep these genes unfused so that they can be differentially expressed.
DISCUSSION
COMODO is a method for cross-species coclustering. It relies on the use of large-scale coexpression compendia for each of the species to be compared. By using a bottom up approach and by exploiting homology relations to identify the optimal size and degree of coexpression in each of the modules that constitute a conserved module pair, COMODO allows identifying in each of the species the modules that best reflect the processes that are conserved in the core. The strength of COMODO relates to its ability of automatically prioritizing best matching module pairs that can cover a large range of different coexpression levels and module sizes. This feature allows the methodology to adapt to closely or evolutionary distant organisms and to identify both processes that are fully or partially conserved across evolution. Moreover, because COMODO can be used in combination with a many-to-many homology map, it is suitable to study functional relations between linker genes that mutually exhibit complex homology relations.
Applying COMODO to large-scale expression compendia allowed comprehensively mapping the processes with conserved coexpression behavior in the divergent bacterial model organisms E. coli and B. subtilis. In contrast to previous studies Price et al. (33) and van Noort et al. (69), COMODO does not use any prior information on previously documented regulon structure or regulatory information and can thus map in an unbiased way modules with conserved coexpression between both species. Because COMODO adapts its module sizes in each species to maximize the relative number of linking homologs, it will not only identify conserved operons for which obviously the conserved coexpression signal is most pronounced, but it will also detect if they exist conserved modules comprising multiple operons.
As it was previously shown, that inferring true orthology is complicated by duplications and horizontal gene transfer (42), we combined the COG many-to-many map with our expression compendia to infer the most likely functional counterparts between E. coli and B. subtilis. Of the 5459 COG links between E. coli and B. subtilis, 355 were found in conserved module pairs. Of those 355 COG links that could be mapped to conserved module pairs, 149 represented reciprocal best hits. Those probably correspond to true functional counterparts. The other 206 most often were links of large gene families that got sub- or neofunctionalized. This figure also indicates that COG largely overestimates the number of true functional relations, although we cannot completely rule out that some of the functional links were not covered due to a lack of certain conditions in the expression data sets.
In general, we found that most of the conserved modules were involved in elementary cellular processes needed to support bacterial cell duplication and inheritance of the genetic information, cell division and the provision of energy (23). The cores of these modules contained regulon members that were indeed shown by comparative studies to occur over a wide range of bacterial species (44,70). Modules involved in transcription, translation and central carbon metabolism contained genes that were previously shown to be differential expressed during the global response to glucose in both B. subtilis and E. coli (15). Despite covering mainly elementary processes our conserved modules contained relatively few essential genes. This, together with the fact that the conserved modules covering elementary processes were rather small (restricted to a single or to maximally a few transcription units, except for those involved in ribosomal metabolism and translation) confirms the previous suggestion that essential processes seem not to be primarily coordinated by the modulation of gene expression (23).
In addition to these smaller modules, we also found larger conserved module pairs that were mainly involved in iron acquisition (Fur regulon) and flagella synthesis. While both processes are fairly conserved at the level of their gene content, mainly the process involved in iron acquisition has underwent major changes in regulon membership in either species.
The mechanism by which genes were transcriptionally coregulated seemed to be much less conserved than their coexpression behavior itself: while the coexpression behavior of complete orthologous regulons was maintained over evolution, the transcription factors responsible for their regulation were only conserved in few cases as was also observed by Price et al. (42). However, in most cases the ortholog of a particular transcription factor known to be responsible for the coexpression behavior in one species did not exist in the other species, suggesting that the role of the disappeared transcription factor must have been taken over by an alternative, yet unknown but non-homologous transcription factor. Furthermore we observed that the variable part in E. coli or B. subtilis of the conserved modules largely consisted of genes specific for one organism, but not occurring in the other one, indicating that bacteria are also flexible in adding new members to an existing regulon (42,44,70). These observations suggest that despite the extreme potential of network rewiring, prokaryotes are extremely robust in preserving the coexpression behavior of some elementary pathways. Probably the operon structure contributes largely to this robustness against rewiring by maintaining a minimal level of coexpression (17).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
KULeuven Research Council [GOA AMBioRICS, GOA/08/011, CoE EF/05/007, SymBioSys, CoE NATAR IOK-C1895-PF/10/010, CREA/08/023, ZKB8933/CREA/08/023/ BOF]; Flemish Interuniversity Council-University Development Cooperation [VLIR-UOS]; Agency for Innovation by Science and Technology [SBO-BioFrame]; Interuniversity Attraction Poles [P6/25-BioMaGNet]; Research Foundation—Flanders [IOK-B9725-G.0329.09]; Human Frontier Science Program [RGY0079/2007C]. Funding for open access charge: Agency for Innovation by Science and Technology [SBO-BioFrame].
Conflict of interest statement. None declared.
REFERENCES
- 1.Fierro AC, Vandenbussche F, Engelen K, Van de Peer Y, Marchal K. Meta analysis of gene expression data within and across species. Curr. Genom. 2008;9:525–534. doi: 10.2174/138920208786847935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tirosh I, Bilu Y, Barkai N. Comparative biology: beyond sequence analysis. Curr. Opin. Biotechnol. 2007;18:371–377. doi: 10.1016/j.copbio.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 3.Lu Y, Huggins P, Bar-Joseph Z. Cross species analysis of microarray expression data. Bioinformatics. 2009;25:1476–1483. doi: 10.1093/bioinformatics/btp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lelandais G, Tanty V, Geneix C, Etchebest C, Jacq C, Devaux F. Genome adaptation to chemical stress: clues from comparative transcriptomics in Saccharomyces cerevisiae and Candida glabrata. Genome Biol. 2008;9:R164. doi: 10.1186/gb-2008-9-11-r164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 6.Bergmann S, Ihmels J, Barkai N. Similarities and differences in genome-wide expression data of six organisms. PLOS Biol. 2004;2:85–93. doi: 10.1371/journal.pbio.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ihmels J, Bergmann S, Berman J, Barkai N. Comparative gene expression analysis by a differential clustering approach: application to the Candida albicans transcription program. PLOS Genet. 2005;1:380–393. doi: 10.1371/journal.pgen.0010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Y, He X, Zhong S. Cross-species microarray analysis with the OSCAR system suggests an INSR -> Pax6 -> NQO1 neuro-protective pathway in aging and Alzheimer's disease. Nucleic Acids Res. 2007;35:W105–W114. doi: 10.1093/nar/gkm408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai J, Xie D, Fan Z, Chipperfield H, Marden J, Wong WH, Zhong S. Modeling co-expression across species for complex traits: insights to the difference of human and mouse embryonic stem cells. PLoS Comput. Biol. 2010;6:e1000707. doi: 10.1371/journal.pcbi.1000707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc. Natl Acad. Sci. USA. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lefebvre C, Aude JC, Glemet E, Neri C. Balancing protein similarity and gene co-expression reveals new links between genetic conservation and developmental diversity in invertebrates. Bioinformatics. 2005;21:1550–1558. doi: 10.1093/bioinformatics/bti215. [DOI] [PubMed] [Google Scholar]
- 12.Van den Bulcke T, Lemmens K, Van de Peer Y, Marchal K. Inferring transcriptional networks by mining ‘Omics’ data. Curr. Bioinformatics. 2006;1:301–313. [Google Scholar]
- 13.Snel B, van Noort V, Huynen MA. Gene co-regulation is highly conserved in the evolution of eukaryotes and prokaryotes. Nucleic Acids Res. 2004;32:4725–4731. doi: 10.1093/nar/gkh815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okuda S, Kawashima S, Kobayashi K, Ogasawara N, Kanehisa M, Goto S. Characterization of relationships between transcriptional units and operon structures in Bacillus subtilis and Escherichia coli. BMC Genomics. 2007;8:48. doi: 10.1186/1471-2164-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vazquez CD, Freyre-Gonzalez JA, Gosset G, Loza JA, Gutierrez-Rios RM. Identification of network topological units coordinating the global expression response to glucose in Bacillus subtilis and its comparison to Escherichia coli. BMC Microbiol. 2009;9:176. doi: 10.1186/1471-2180-9-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergmann S, Ihmels J, Barkai N. Iterative signature algorithm for the analysis of large-scale gene expression data. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 2003;67:031902. doi: 10.1103/PhysRevE.67.031902. [DOI] [PubMed] [Google Scholar]
- 17.Lemmens K, De Bie T, Dhollander T, De Keersmaecker SC, Thijs IM, Schoofs G, De Weerdt A, De Moor B, Vanderleyden J, Collado-Vides J, et al. DISTILLER: a data integration framework to reveal condition dependency of complex regulons in Escherichia coli. Genome Biol. 2009;10:R27. doi: 10.1186/gb-2009-10-3-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fadda A, Fierro AC, Lemmens K, Monsieurs P, Engelen K, Marchal K. Inferring the transcriptional network of Bacillus subtilis. Mol. Biosystems. 2009;5:1840–1852. doi: 10.1039/b907310h. [DOI] [PubMed] [Google Scholar]
- 19.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 20.Wall DP, Deluca T. Ortholog detection using the reciprocal smallest distance algorithm. Mol. Syst. Biol. 2007;396:95–110. doi: 10.1007/978-1-59745-515-2_7. [DOI] [PubMed] [Google Scholar]
- 21.Zhang R, Ou HY, Zhang CT. DEG: a database of essential genes. Nucleic Acids Res. 2004;32:D271–D272. doi: 10.1093/nar/gkh024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009;37:D455–D458. doi: 10.1093/nar/gkn858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, et al. Essential Bacillus subtilis genes. Proc. Natl Acad. Sci. USA. 2003;100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerdes SY, Scholle MD, Campbell JW, Balazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, et al. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J. Bacteriol. 2003;185:5673–5684. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Systems Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keseler IM, Bonavides-Martinez C, Collado-Vides J, Gama-Castro S, Gunsalus RP, Johnson DA, Krummenacker M, Nolan LM, Paley S, Paulsen IT, et al. EcoCyc: A comprehensive view of Escherichia coli biology. Nucleic Acids Res. 2009;37:D464–D470. doi: 10.1093/nar/gkn751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson JD, Umayam LA, Dickinson T, Hickey EK, White O. The comprehensive microbial resource. Nucleic Acids Res. 2001;29:123–125. doi: 10.1093/nar/29.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karp PD, Ouzounis CA, Moore-Kochlacs C, Goldovsky L, Kaipa P, Ahren D, Tsoka S, Darzentas N, Kunin V, Lopez-Bigas N. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005;33:6083–6089. doi: 10.1093/nar/gki892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gama-Castro S, Jimenez-Jacinto V, Peralta-Gil M, Santos-Zavaleta A, Penaloza-Spinola MI, Contreras-Moreira B, Segura-Salazar J, Muniz-Rascado L, Martinez-Flores I, Salgado H, et al. RegulonDB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucleic Acids Res. 2008;36:D120–D124. doi: 10.1093/nar/gkm994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sierro N, Makita Y, de Hoon M, Nakai K. DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res. 2008;36:D93–D96. doi: 10.1093/nar/gkm910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Royal Statist. Soc. B-Methodol. 1995;57:289–300. [Google Scholar]
- 32.Mao FL, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 2009;37:D459–D463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Price MN, Huang KH, Alm EJ, Arkin AP. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 2005;33:880–892. doi: 10.1093/nar/gki232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez JC, Groisman EA. Evolution of transcriptional regulatory circuits in bacteria. Cell. 2009;138:233–244. doi: 10.1016/j.cell.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartig E, Hartmann A, Schatzle M, Albertini AM, Jahn D. The Bacillus subtilis nrdEF genes, encoding a class Ib ribonucleotide reductase, are essential for aerobic and anaerobic growth. Appl. Env. Microbiol. 2006;72:5260–5265. doi: 10.1128/AEM.00599-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torrents E, Grinberg I, Gorovitz-Harris B, Lundstrom H, Borovok I, Aharonowitz Y, Sjoberg BM, Cohen G. NrdR controls differential expression of the Escherichia coli ribonucleotide reductase genes. J. Bacteriol. 2007;189:5012–5021. doi: 10.1128/JB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okuda S, Kawashima S, Goto S, Kanehisa M. Conservation of gene co-regulation between two prokaryotes: Bacillus subtilis and Escherichia coli. Genome informatics. Int. Conf. Genome Inform. 2005;16:116–124. [PubMed] [Google Scholar]
- 38.Grunberg-Manago M. Messenger RNA stability and its role in control of gene expression in bacteria and phages. Ann. Rev. Genet. 1999;33:193–227. doi: 10.1146/annurev.genet.33.1.193. [DOI] [PubMed] [Google Scholar]
- 39.Rajagopala SV, Titz B, Goll J, Parrish JR, Wohlbold K, McKevitt MT, Palzkill T, Mori H, Finley RL, Jr, Uetz P. The protein network of bacterial motility. Mol. Syst. Biol. 2007;3:128. doi: 10.1038/msb4100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ollinger J, Song KB, Antelmann H, Hecker M, Helmann JD. Role of the Fur regulon in iron transport in Bacillus subtilis. J. Bacteriol. 2006;188:3664–3673. doi: 10.1128/JB.188.10.3664-3673.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wall DP, Fraser HB, Hirsh AE. Detecting putative orthologs. Bioinformatics. 2003;19:1710–1711. doi: 10.1093/bioinformatics/btg213. [DOI] [PubMed] [Google Scholar]
- 42.Price MN, Dehal PS, Arkin AP. Orthologous transcription factors in bacteria have different functions and regulate different genes. PLOS Comput. Biol. 2007;3:1739–1750. doi: 10.1371/journal.pcbi.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun G, Sharkova E, Chesnut R, Birkey S, Duggan MF, Sorokin A, Pujic P, Ehrlich SD, Hulett FM. Regulators of aerobic and anaerobic respiration in Bacillus subtilis. J. Bacteriol. 1996;178:1374–1385. doi: 10.1128/jb.178.5.1374-1385.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Babu MM, Teichmann SA, Aravind L. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J. Mol. Biol. 2006;358:614–633. doi: 10.1016/j.jmb.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 45.Paget MSB, Helmann JD. Protein family review - the sigma(70) family of sigma factors. Genome Biol. 2003;4:203. doi: 10.1186/gb-2003-4-1-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wade JT, Roa DC, Grainger DC, Hurd D, Busby SJW, Struhl K, Nudler E. Extensive functional overlap between sigma factors in Escherichia coli. Nature Struct. Mol. Biol. 2006;13:806–814. doi: 10.1038/nsmb1130. [DOI] [PubMed] [Google Scholar]
- 47.Calvio C, Celandroni F, Ghelardi E, Amati G, Salvetti S, Ceciliani F, Galizzi A, Senesi S. Swarming differentiation and swimming motility in Bacillus subtilis are controlled by SwrA, a newly identified dicistronic operon. J. Bacteriol. 2005;187:5356–5366. doi: 10.1128/JB.187.15.5356-5366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calvio C, Osera C, Amati G, Galizzi A. Autoregulation of swrAA and motility in Bacillus subtilis. J. Bacteriol. 2008;190:5720–5728. doi: 10.1128/JB.00455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith TG, Hoover TR. Deciphering bacterial flagellar gene regulatory networks in the genomic era. Adv. Appl. Microbiol. 2009;67:257–295. doi: 10.1016/S0065-2164(08)01008-3. [DOI] [PubMed] [Google Scholar]
- 50.Hamze K, Julkowska D, Autret S, Hinc K, Nagorska K, Sekowska A, Holland IB, Seror SJ. Identification of genes required for different stages of dendritic swarming in Bacillus subtilis, with a novel role for phrC. Microbiology. 2009;155:398–412. doi: 10.1099/mic.0.021477-0. [DOI] [PubMed] [Google Scholar]
- 51.Rolfes RJ, Zalkin H. Escherichia coli gene PurR encoding a repressor protein for purine nucleotide synthesis - cloning, nucleotide-sequence, and interaction with the PurF operator. J. Biol. Chem. 1988;263:19653–19661. [PubMed] [Google Scholar]
- 52.Weng M, Nagy PL, Zalkin H. Identification of the Bacillus subtilis pur operon repressor. Proc. Natl Acad. Sci. USA. 1995;92:7455–7459. doi: 10.1073/pnas.92.16.7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horlacher R, Boos W. Characterization of TreR, the major regulator of the Escherichia coli trehalose system. J. Biol. Chem. 1997;272:13026–13032. doi: 10.1074/jbc.272.20.13026. [DOI] [PubMed] [Google Scholar]
- 54.Fukami-Kobayashi K, Tateno Y, Nishikawa K. Parallel evolution of ligand specificity between LacI/GalR family repressors and periplasmic sugar-binding proteins. Mol. Biol. Evol. 2003;20:267–277. doi: 10.1093/molbev/msg038. [DOI] [PubMed] [Google Scholar]
- 55.Schock F, Dahl MK. Expression of the tre operon of Bacillus subtilis 168 is regulated by the repressor TreR. J. Bacteriol. 1996;178:4576–4581. doi: 10.1128/jb.178.15.4576-4581.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guillouard I, Auger S, Hullo MF, Chetouani F, Danchin A, Martin-Verstraete I. Identification of Bacillus subtilis CysL, a regulator of the cysJI operon, which encodes sulfite reductase. J. Bacteriol. 2002;184:4681–4689. doi: 10.1128/JB.184.17.4681-4689.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sekowska A, Kung HF, Danchin A. Sulfur metabolism in Escherichia coli and related bacteria: facts and fiction. J. Mol. Microbiol. Biotechnol. 2000;2:145–177. [PubMed] [Google Scholar]
- 58.Danot O, Vidal-Ingigliardi D, Raibaud O. Two amino acid residues from the DNA-binding domain of MalT play a crucial role in transcriptional activation. J. Mol. Biol. 1996;262:1–11. doi: 10.1006/jmbi.1996.0493. [DOI] [PubMed] [Google Scholar]
- 59.Eppler T, Postma P, Schutz A, Volker U, Boos W. Glycerol-3-phosphate-induced catabolite repression in Escherichia coli. J. Bacteriol. 2002;184:3044–3052. doi: 10.1128/JB.184.11.3044-3052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robertson JB, Gocht M, Marahiel MA, Zuber P. AbrB, a regulator of gene expression in Bacillus, interacts with the transcription initiation regions of a sporulation gene and an antibiotic biosynthesis gene. Proc. Natl Acad. Sci. USA. 1989;86:8457–8461. doi: 10.1073/pnas.86.21.8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fisher SH, Strauch MA, Atkinson MR, Wray LV., Jr Modulation of Bacillus subtilis catabolite repression by transition state regulatory protein AbrB. J. Bacteriol. 1994;176:1903–1912. doi: 10.1128/jb.176.7.1903-1912.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rasouly A, Schonbrun M, Shenhar Y, Ron EZ. YbeY, a heat shock protein involved in translation in Escherichia coli. J. Bacteriol. 2009;191:2649–2655. doi: 10.1128/JB.01663-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schumann W. The Bacillus subtilis heat shock stimulon. Cell stress chaperones. 2003;8:207–217. doi: 10.1379/1466-1268(2003)008<0207:tbshss>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rastogi S, Liberles DA. Subfunctionalization of duplicated genes as a transition state to neofunctionalization. BMC Evol. Biol. 2005;5:28. doi: 10.1186/1471-2148-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Letoffe S, Delepelaire P, Wandersman C. The housekeeping dipeptide permease is the Escherichia coli heme transporter and functions with two optional peptide binding proteins. Proc. Natl Acad. Sci. USA. 2006;103:12891–12896. doi: 10.1073/pnas.0605440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perego M, Higgins CF, Pearce SR, Gallagher MP, Hoch JA. The oligopeptide transport-system of Bacillus subtilis plays a role in the initiation of sporulation. Mol. Microbiol. 1991;5:173–185. doi: 10.1111/j.1365-2958.1991.tb01838.x. [DOI] [PubMed] [Google Scholar]
- 67.Koide A, Hoch JA. Identification of a second oligopeptide transport system in Bacillus subtilis and determination of its role in sporulation. Mol. Microbiol. 1994;13:417–426. doi: 10.1111/j.1365-2958.1994.tb00436.x. [DOI] [PubMed] [Google Scholar]
- 68.Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- 69.van Noort V, Snel B, Huynen MA. Predicting gene function by conserved coexpression. Trends Genetics. 2003;19:238–242. doi: 10.1016/S0168-9525(03)00056-8. [DOI] [PubMed] [Google Scholar]
- 70.Lozada-Chavez I, Janga SC, Collado-Vides J. Bacterial regulatory networks are extremely flexible in evolution (vol 34, pg 3434, 2006) Nucleic Acids Res. 2006;34:4654. doi: 10.1093/nar/gkl423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.