

Abstract
We have reported previously that treatment of non-obese diabetic (NOD) mice with the invariant natural killer T (iNK T) cell agonist α-galactosylceramide C26:0 (α-GalCer) or its T helper type 2 (Th2)-biasing derivative α-GalCer C20:2 (C20:2) protects against type 1 diabetes (T1D), with C20:2 yielding greater protection. After an initial response to α-GalCer, iNK T cells become anergic upon restimulation. While such anergic iNK T cells can induce tolerogenic dendritic cells (DCs) that mediate protection from T1D, chronic administration of α-GalCer also results in long-lasting anergy accompanied by significantly reduced iNK T cell frequencies, which raises concerns about its long-term therapeutic use. In this study, our objective was to understand more clearly the roles of anergy and induction of tolerogenic DCs in iNK T cell-mediated protection from T1D and to circumvent potential complications associated with α-GalCer. We demonstrate that NOD iNK T cells activated during multi-dose (MD) treatment in vivo with C20:2 enter into and exit from anergy more rapidly than after activation by α-GalCer. Importantly, this shorter duration of iNK T cells in the anergic state promotes the more rapid induction of tolerogenic DCs and reduced iNK T cell death, and enables C20:2 stimulated iNK T cells to elicit enhanced protection from T1D. Our findings further that suggest C20:2 is a more effective therapeutic drug than α-GalCer for protection from T1D. Moreover, the characteristics of C20:2 provide a basis of selection of next-generation iNK T cell agonists for the prevention of T1D.
Keywords: anergy, apoptosis, cytokines, diabetes, iNK T cells
Introduction
Invariant natural killer T (iNK T) cells are a non-conventional subset of T cells that express a canonical invariant T cell antigen receptor (TCR) α-chain, which together with a limited repertoire of TCR-β chains mediates their recognition of glycolipid antigens presented by CD1d molecules [1–8]. Due to their regulatory role in the immune system, iNK T cell deficiencies have been shown to play a role in various diseases, including type 1 diabetes (T1D) [9–13]. Non-obese diabetic (NOD) mice, which develop T1D spontaneously, exhibit both functional and numerical deficiencies in iNK T cells [10–14]. Thus, the activation and/or expansion of iNK T cells has been considered for therapeutic purposes.
Upon activation with the α-galactosyl-ceramide C26:0 (α-GalCer) glycosphingolipid agonist, iNK T cells synthesize and secrete large amounts of both T helper type 1 (Th1)-like [e.g. interferon (IFN)-γ)] and Th2-like [e.g. interferon (IL-4)] cytokines. The transactivation of several other immune cell types by these cytokines significantly amplifies the ensuing immune response [1–8]. We and others have reported that treatment of NOD mice with multiple doses, but not a single dose, of α-GalCer protects from T1D [9–11]. The therapeutic effect of α-GalCer may be associated with a Th2-like environment dependent on the activity of IL-4, which promotes the recruitment of suppressive or ‘tolerogenic’ dendritic cells (DCs) to the draining pancreatic lymph nodes (PLN) [12]. These tolerogenic DCs may suppress autoreactive T cells responsible for islet β cell death, and also generate Th2 and regulatory T (Treg) cell responses that further aid in protection against T1D [8–11,13–17]. Indeed, we have shown that α-GalCer-induced iNK T cell-mediated protection against T1D is dependent upon Treg activity [18].
After initial activation by α-GalCer, iNK T cells become hyporesponsive to glycolipid restimulation and enter a state of anergy, which may last for at least 4–6 weeks [19]. Upon restimulation, anergic iNK T cells fail to proliferate but continue to produce IL-4 and significantly lower levels of IFN-γ and IL-2 [19]. It has been hypothesized that iNK T cell anergy may be an intrinsic regulatory mechanism designed to avoid chronic cytokine production that leads to an uncontrolled inflammatory response and inflicts host damage [4]. Ligation of programmed death-1 (PD-1) to the programmed death ligand-1 (PD-L1) or PD-L2 regulates the induction and maintenance of iNK T cell anergy [20–22].
The state of anergy may mediate the immunoregulatory effects of iNK T cells and subsequent protection from T1D in NOD mice by influencing the induction of tolerogenic DCs [16]. During the 7 days between the initial activation of iNK T cells and induction of anergy after α-GalCer administration, restimulation of these cells promotes the maturation of DCs with immunogenic and inflammatory properties [16]. Conversely, restimulation of previously anergized iNK T cells with α-GalCer induces non-inflammatory DCs with suppressive capabilities [16]. These findings highlight the importance of iNK T cell anergy in α-GalCer-mediated protection from T1D, and may explain why only multiple doses of glycolipid are therapeutically effective. Whereas restimulation of anergic iNK T cells can improve outcomes for the treatment of inflammatory conditions, such anergic iNK T cells are more susceptible to apoptosis [4]. Indeed, chronic administration of α-GalCer results in long-lasting anergy accompanied by significantly reduced iNK T cell frequencies, which raises concerns about its long-term therapeutic use [4,23,24]
To circumvent these potential complications associated with α-GalCer, several synthetic derivatives of α-GalCer have been generated in attempts to elicit immune responses that protect from various diseases. In the case of T1D, we have reasoned that it is preferable to administer an iNK T cell agonist that not only elicits a strong anti-inflammatory IL-4 response but also reduces the amount of proinflammatory secreted IFN-γ. A promising Th2-biasing glycolipid that elicits such a CD1d-dependent response from iNK T cells is α-galactosylceramide C20:2 (C20:2), an N-acyl variant of α-GalCer consisting of a fatty acyl side chain truncated from C26 to C20 that has two sites of unsaturation at carbons 11 and 14 [25].
Although α-GalCer and C20:2 possess a similar binding affinity for CD1d [26], and upon dose titration stimulate very similar in vitro proliferative responses of iNK T cells [24], we found that C20:2 administration in vivo results in the overall reduced inflammatory cytokine (IFN-γ) secretion, iNK T cell expansion and transactivation of T, B and NK cells [23–25]. Our additional kinetic analyses attributed these differences to a reduced capacity of C20:2 to sustain high iNK T cell activation beyond 6 h compared to α-GalCer when administered in equal doses [24]. Moreover, the administration of C20:2 in multiple doses significantly delayed and reduced the incidence of T1D in NOD mice with increased efficacy compared to α-GalCer and was shown to be less dependent on the activity of Tregs[23,24]. These more favourable characteristics of C20:2 prompted us to characterize further the iNK T cell responses elicited by C20:2 that may explain its ability to protect against T1D more efficiently than α-GalCer.
Due to the recent evidence that iNK T cell anergy may control the recruitment of tolerogenic DCs [16], it was of interest to compare the kinetics of anergy induction and its role in DC recruitment by α-GalCer and C20:2. In this study, we report that relative to α-GalCer, C20:2 administration results in quicker iNK T cell entry into and exit from anergy, which correlates directly with the more rapid kinetics of PD-1 and PD-L1 up-regulation on the surface of these cells. Two significant outcomes resulted from the altered kinetics of iNK T cell anergy induction. First, the faster induction of iNK T cell anergy elicited by C20:2 administration resulted in the more rapid induction of tolerogenic DCs. Secondly, the faster recovery from anergy of C20:2 experienced iNK T cells correlated with their increased survival following multi-dose (MD) treatment due to their reduced susceptibility to apoptosis. Our results not only underscore further the importance of iNK T cell anergy in the modulation of immune responses, but also emphasize the significance of the differential kinetics of entry into and exit from anergy induced by different glycolipid agonists in protection against T1D.
Materials and methods
Mice
All experimental mice were 4–6-week-old females and were maintained in a specific pathogen-free facility in the Animal Care and Veterinary Services at the University of Western Ontario (London, ON, Canada) according to institutional guidelines. NOD mice were bred in the animal care facility at the Robarts Research Institute on site. The incidence of T1D in female NOD mice in our colony is typically 25–30% at 15 weeks of age and > 80% by 30 weeks. All experiments were performed in accordance with the Canadian Council for Animal Care guidelines.
Glycolipids, antibodies and reagents
Synthetic α-GalCer (KRN7000) and its vehicle were obtained from Kirin Pharmaceutical Research Laboratories (Gunma, Japan), solubilized in water and injected intraperitoneally (i.p.) into mice (4 µg/dose). C20:2 was synthesized as described previously [25] and dissolved in phosphate-buffered saline (PBS) (Gibco® Invitrogen, Burlington, ON, Canada) containing 0·02% Tween 20 and 0·1% dimethylsulphoxide (DMSO) and injected i.p. (4 µg/dose). Where indicated, the MD administration protocol consists of 4 µg injections of glycolipid or vehicle given every other day for 3 weeks (11 total injections). Fluorescein isothiocynate (FITC)-conjugated anti-TCR-β (H57-597), anti-CD4 (RM4-5), anti-CD8α (53–6·7), anti-CD11c (N418), anti-Siglec H (eBio440c), anti-CD86 (Gl-1); phycoerythrin (PE)-conjugated anti-PD-1 (CD279, RMP1-30), anti-PD-L1 (CD274, MIH5), anti-IFN-γ (XMG1·2), anti-IL-2 (JES6-5H4), anti-IL-4 (11B11), anti-IL-10 (JES5-16E3), anti-IL-12p40/70 (C15·6), anti-I-Ad (AMS-32·1); PE-Cy7-conjugated anti-CD4 (L3T4); peridinin chlorophyll (PerCP)-conjugated anti-CD3ε (145-2C11), anti-CD4 (RM4-5), anti-CD8α (53–2·1), anti-Siglec H (eBio440c); allophycocyanin (APC)-conjugated anti-CD11c (N418), anti-CD40; APC-Cy7-conjugated anti-CD4 (RM4-5), anti-CD11c (N418); and AlexaFlour-700-conjugated anti-CD8α (53–6·7) were purchased from eBiosciences (San Diego, CA, USA), BD Biosciences (Mississauga, ON, Canada) or Cedarlane Laboratories Limited (Burlington, ON, Canada). Fluorescently labelled α-GalCer-loaded and unloaded CD1d tetramers were kindly provided by the NIH tetramer facility (Atlanta, GA, USA). RPMI-1640 tissue culture medium was supplemented with 10% heat-inactivated fetal bovine serum (FBS), 10 mm 4-(2-hydroxyethyl)-1-piperazine-ethanesulphonic acid (HEPES) buffer, 1 mm sodium pyruvate, 2 mm l-glutamine, 100 units/ml penicillin, 0·1 mg/ml streptomycin and 0·05 mm 2-mercaptoethanol (2-ME) (all purchased from Invitrogen).
Cell isolation and flow cytometry
Spleen and PLN lymphocyte suspensions were prepared [24] and cultured (107 cells/ml) for 3 h in supplemented RPMI-1640 culture medium (see above) containing GolgiStop (1:1000 dilution) (BD Biosciences). Intracellular staining for IL-2, IL-4, IL-10, IL-12 and IFN-γ was performed using a BD Cytofix/Cytoperm buffer set (BD Biosciences) in accordance with the manufacturer's instructions, as described [24]. Flow cytometry was performed using a fluorescence activated cell sorter (FACS)Calibur or FACSCanto II together with CellQuest Pro or FACSDiva software (BD Biosciences), respectively [24]. Analyses were conducted using FlowJo software (Treestar Inc., Ashland, OR, USA). Non-viable cells were excluded by electronic gating for all experiments.
In vitro cultures and enzyme-linked immunosorbent assay (ELISA)
Splenocytes were isolated and cultured (5 × 106/ml) for 72 h in the presence of α-GalCer or C20:2 (100 ng/ml), or vehicle. Sandwich ELISAs were performed for mouse cytokines using paired antibody kits for IL-2, IFN-γ and IL-4 (BD Biosciences) in accordance with the manufacturer's instructions. For proliferation assays, 5 × 105 splenocytes were cultured in the presence of glycolipid (100 ng/ml) or vehicle for 72 h. Cells were pulsed with [3H]-thymidine (1 µCi/well; PerkinElmer, Woodbridge, ON, Canada) for the final 18 h of culture, harvested and read on a 1450 Microbeta counter (PerkinElmer). For DC cultures, NOD mice were treated i.p. with a varying number of doses of glycolipid or vehicle (4 µg/dose) spaced 2 days apart. After 2 days of rest, spleen and PLN suspensions were pooled and the total DC population was isolated magnetically with about 85% purity (assayed by FACS) from each treatment group using a CD11c-positive selection kit (Miltenyi Biotec, Auburn, CA, USA). Concurrently, CD8+ peptide-primed T cells were isolated with about 90% purity (assayed by FACS) using a CD8+ T cell-negative selection kit (Miltenyi Biotec) from NOD mice injected (i.p.) 10 days previously with 100 mg of NOD-relevant V7 peptide (NRP-V7) peptide (KYNKANVFL, kindly provided by Dr B. Singh, University of Western Ontario, London, ON, Canada) emulsified in incomplete Freund's adjuvant (IFA) (Sigma Aldrich, Oakville, ON, Canada). Peptide primed-CD8+ T cells were cultured with CD11c+ DC at a 20:1 ratio in the presence of the NRP-V7 peptide (100 µg/ml) for 72 h for the ELISA (2 × 106 T cells + 1 × 105 DCs) and proliferation (5 × 105 T cells + 1 × 104 DCs) assays, respectively.
Apoptosis assay
NOD mice were treated with glycolipid according to our MD protocol as reported previously [18] and rested for varying times. Mice were reinjected with an additional dose of glycolipid (4 µg, i.p.), rested for 2 h and iNK T cells from the spleen and PLN were isolated from the different treatment groups and stained with a Vybrant® Apoptosis Assay (Alexa Fluor® 488 annexin V/propidium iodide) kit no. 2 (Invitrogen) to quantify iNK T cell death.
Statistical analysis
Statistical significance of cell proliferation and cytokine secretion were determined using two-way analysis of variance (anova) comparisons with Bonferroni post-tests, as described [24]. In all experiments, differences were considered statistically significant when P was less than 0·05.
Results
C20:2-activated iNK T cells induce a less sustained and Th2-biased cytokine response
Relative to α-GalCer, C20:2 contains a fatty acyl side chain shortened from C26 to C20 with the introduction of two unsaturated bonds at carbons 11 and 14 [25]. To investigate the responses elicited by C20:2 in NOD mice, cytokine production was analysed after an initial stimulation in vitro (Fig. 1a) and in vivo (Fig. 1b). Consistent with previous reports [23–25,27], C20:2 treatment stimulated a greater Th2-type response than α-GalCer characterized by increased IL-4 and decreased IFN-γ secretion. At early time-points (2 h), C20:2 induced about a twofold greater IFN-γ response compared to α-GalCer. However, at the time of peak secretion (8 h), this trend was reversed and α-GalCer demonstrated a much more robust IFN-γ response (Fig. 1b). Interestingly, C20:2 administration also resulted in a less sustained IFN-γ response that was barely detectable after 24 h compared to α-GalCer, which elicited a vigorous response throughout the time–course (Fig. 1b). Thus, C20:2 functions as a Th2 biasing glycolipid in NOD mice, and the earlier but less sustained IFN-γ response elicited by C20:2 suggests that it may load more rapidly in CD1d on iNK T cells than α-GalCer.
Fig. 1.
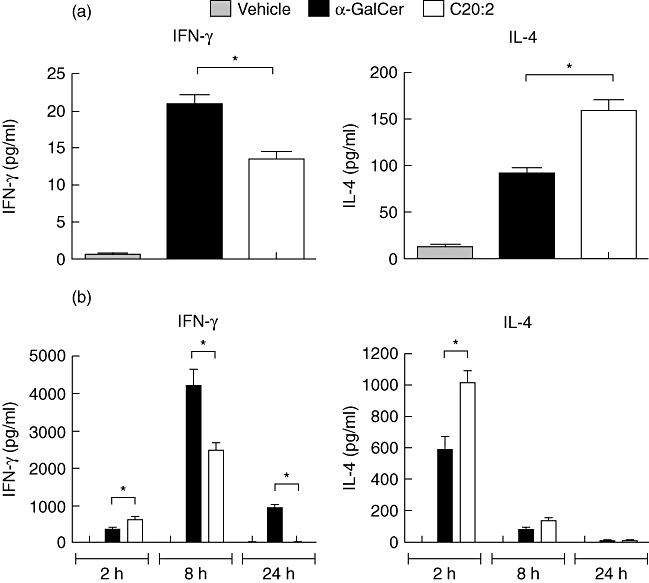
Administration of C20:2 elicits a T helper type 2 (Th2)-biased response. (a) Splenocytes from naive non-obese diabetic (NOD) mice (4–6 weeks old) were stimulated in vitro with α-galactosyl-ceramide C26:0 (α-GalCer), C20:2 or vehicle (100 ng/ml) for 72 h. Culture supernatants were analysed for interferon (IFN)-γ and interleukin (IL)-4 by enzyme-linked immunosorbent assay (ELISA). (b) NOD mice (4–6 weeks old) were injected intraperitoneally (i.p.) with a single dose (4 µg) of glycolipid or vehicle. Serum was collected at 2 h, 8 h or 24 h after injection and ELISAs were performed to measure serum levels of IFN-γ and IL-4. Data are representative of at least three independent experiments yielding similar results and are expressed as mean ± standard deviation (s.d); five mice/treatment group/time-point. *Statistically significant (P < 0·05) difference between compared groups.
iNK T cells enter into and recover from anergy more rapidly after stimulation with C20:2
Due to the essential role of iNK T cell anergy in the recruitment of tolerogenic DCs [16,17], we initially compared the kinetics of anergy induction in NOD iNK T cells elicited by α-GalCer versus C20:2. NOD mice were administered a single dose (4 µg, i.p.) of glycolipid or vehicle and rested for various times (24 h, 48 h, 72 h, 1 week or 1 month). Splenocytes were then cultured and restimulated with the indicated glycolipids (100 ng/ml) for 72 h, and cytokine secretion was analysed by ELISA (Fig. 2a). Consistent with previous results (Fig. 1) [24,25], initial exposure to C20:2 (vehicle/C20:2) elicited a greater Th2 response, characterized by reduced IFN-γ and increased IL-4 compared to α-GalCer (vehicle/α-GalCer). Interestingly, C20:2 also induced a significantly greater IL-2 response. These cytokine levels observed after primary stimulation were then used as a baseline to measure iNK T cell hyporesponsiveness. Both glycolipids induced iNK T cell anergy after treatment with a single dose, as demonstrated by a significant decrease in the levels of IL-2 and IFN-γ in splenocyte cultures analysed after restimulation in vitro. However, relative to α-GalCer, C20:2 yielded a faster induction of and recovery from iNK T cell anergy. This was evident from the more rapid reduction of IFN-γ levels by C20:2 at 48 h as well as the full recovery of IFN-γ and IL-2 responses by 1 month (Fig. 2a). Cultures of iNK T cells from mice that received an α-GalCer injection 1 month prior to restimulation still exhibited significantly lower IFN-γ and IL-2 responses, indicating that these cells remained anergic. The IL-4 responses elicited by α-GalCer and C20:2 were similar and did not vary appreciably during the time–course of activation in vivo, as reported previously for α-GalCer [19].
Fig. 2.
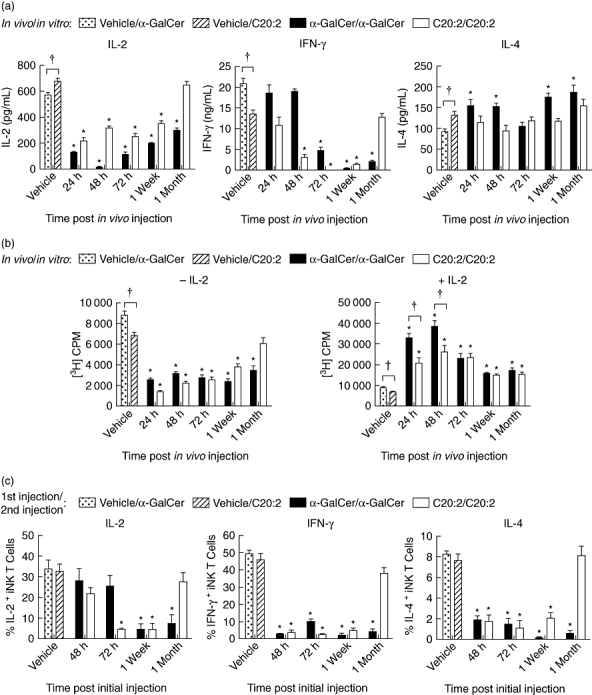
Kinetic analysis of induction of invariant natural killer T (iNK T) cell anergy. Non-obese diabetic (NOD) mice (4–6 weeks old; five mice/treatment group/time-point) were injected intraperitoneally (i.p.) with a single dose (4 µg) of glycolipid or vehicle (Veh). Splenocytes were isolated at various times (24 h, 48 h, 72 h, 1 week, 1 month) and then restimulated in vitro with glycolipid (100 ng/ml) for 72 h (a, b). In (a) supernatants were analysed for interleukin (IL)-2, interferon (IFN)-γ and IL-4 content by enzyme-linked immunosorbent assay (ELISA), and in (b) the proliferative capacity of iNK T cells cultured in the presence or absence of IL-2 (5 ng/ml) was assayed by [3H]-thymidine incorporation. (c) After mice were injected with glycolipid (once with 4 µg, i.p.) or vehicle and rested for various times (48 h, 72 h, 1 week, 1 month), they were reinjected with the same dose of glycolipid. Splenocytes were harvested 2 h after the second injection and then cultured in vitro for 3 h in the presence of a protein transport inhibitor, GolgiStop, to block cytokine secretion. T cell receptor (TCR)-β+tetramer+ iNK T cells were stained for intracellular levels of IL-2, IFN-γ and IL-4. Data are representative of four independent experiments yielding similar results and are expressed as mean ± standard deviation (s.d.). *Significant (P < 0·05) reduction between primary and recall responses (vehicle/α-galactosylceramide C26:0 (α-GalCer) versusα-GalCer/α-GalCer, vehicle/C20:2 versus C20:2/C20:2). †Significant (P < 0·05) difference between primary responses to α-GalCer and C20:2 treatments (vehicle/α-GalCer versus vehicle/C20:2).
C20:2 also restored the proliferative responses of NOD splenocytes to vehicle control levels more rapidly than did α-GalCer, as demonstrated at 1 month post-injection (Fig. 2b, left panel). The addition of exogenous IL-2 (5 ng/ml) to initial cultures stimulated recall responses that were more than twofold greater than their respective primary responses throughout the time–course (Fig. 2b, right panel). These findings support a role for IL-2 in reversing iNK T cell anergy [28], and indicate that the reduced recall responses seen in vitro are a result of iNK T cell anergy.
To determine further whether iNK T cells become anergic in vivo, the levels of intracellular cytokine accumulation in iNK T cells were monitored after glycolipid restimulation. NOD mice were administered a single dose (4 µg, i.p.) of glycolipid or vehicle, rested for various times (48 h, 72 h, 1 week or 1 month), and then reinjected with the same dose of glycolipid. Splenocytes were harvested 2 h later, and iNK T cells were stained for intracellular levels of IL-2, IFN-γ and IL-4 (Fig. S1; see Supporting information at end of paper). In agreement with the in vitro responses, C20:2-treated iNK T cells enter into and recover from anergy more rapidly than those injected with α-GalCer (Fig. 2c). This was evident for levels of intracellular IL-2 detected at 72 h post-injection, and for all three cytokines assayed at 1 month post-injection. In contrast to splenocytes cultured in vitro, which maintain consistent levels of secreted IL-4 upon restimulation (Fig. 2a), iNK T cells activated in vivo exhibited a diminished IL-4 response upon restimulation at 1 month post-injection. However, at 24 h after injection, note that the intracellular levels of IFN-γ and IL-4 were below the limit of detection. This may arise from surface TCR internalization at this early time after activation. Thus, iNK T cells treated with C20:2 enter into and recover from anergy more rapidly than those treated with α-GalCer.
iNK T cells recover faster from anergy after MD treatment with C20:2 than α-GalCer
Having analysed the kinetics of iNK T cell anergy induced by a single dose of glycolipid, we next investigated the effects of MD glycolipid treatment on iNK T cell responsiveness. An MD protocol was examined as it protects NOD mice effectively from T1D, whereas treatment with a single dose of glycolipid offers little protection [24]. NOD mice received MD treatment with glycolipid or vehicle, and then rested for 10 days or 1 month. Splenocytes were restimulated in vitro with glycolipid (100 ng/ml) for 72 h, and were then assayed for their proliferative capacity and cytokine (IL-2, IFN-γ and IL-4) secretion responses. iNK T cells remained anergic at 10 days after MD treatment with either α-GalCer or C20:2, as revealed by the diminished IL-2, IFN-γ and proliferative responses to in vitro restimulation relative to the primary immune response (α-GalCer/α-GalCer versus vehicle/α-GalCer, and C20:2/C20:2 versus vehicle/C20:2) (Fig. 3a). Interestingly, at 1 month post-MD treatment, iNK T cells from mice that received C20:2, but not α-GalCer, were fully recovered from anergy. The IL-2, IFN-γ and proliferative responses of iNK T cells from C20:2-treated mice all returned to levels equivalent to that of the primary response (vehicle/C20:2), while those treated with α-GalCer were still reduced significantly. After restimulation, the levels of secreted IL-4 in culture did not vary appreciably during the time–course of MD treatment with either α-GalCer or C20:2, in agreement with our single-dose findings.
Fig. 3.
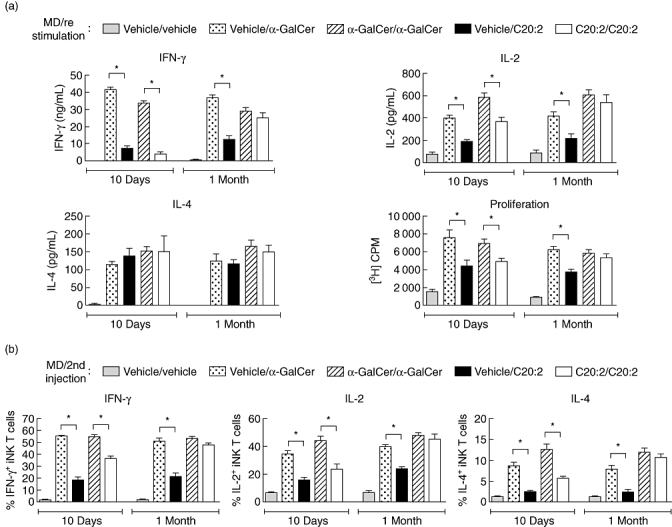
Kinetic analysis of invariant natural killer T (iNK T) cell anergy following treatment with a multi-dose (MD) protocol of glycolipid. Non-obese diabetic (NOD) mice (4–6 weeks old) received MD treatment [4 µg/dose every other day for 3 weeks, intraperitoneally (i.p.)] of glycolipid or vehicle and were then rested for 10 days or 1 month. (a) Splenocytes were isolated after the in vivo treatment and restimulated in vitro with glycolipid (100 ng/ml) for 72 h. The proliferative capacity and levels of cytokine secretion by the splenic iNK T cells were assayed by [3H]-thymidine incorporation and enzyme-linked immunosorbent assay (ELISA), respectively. (b) After 10 days or 1 month of rest, mice were reinjected with an additional dose (4 µg, i.p.) of glycolipid. Splenocytes were harvested 2 h after the final injection, cultured in vitro for 3 h in the presence of a protein transport inhibitor, GolgiStop, and T cell receptor (TCR)-β+tetramer+ iNK T cells were stained for intracellular levels of interleukin (IL)-2, interferon (IFN)-γ and IL-4. Data are representative of three independent experiments yielding similar results and are expressed as mean ± standard deviation (s.d.); five mice/treatment group/time-point. *Significant (P < 0·05) reduction between primary and recall responses (vehicle/α-galactosylceramide C26:0 (α-GalCer) versusα-GalCer/α-GalCer, vehicle/C20:2 versus C20:2/C20:2).
To validate the effect of the MD protocol on cytokine secretion by iNK T cells, intracellular cytokine staining of iNK T cells was performed after in vivo restimulation with glycolipid. NOD mice were treated as above with an MD protocol of glycolipid or vehicle, rested for 10 days or 1 month, and then reinjected with glycolipid (4 µg). Splenocytes were harvested 2 h after the final injection, and iNK T cells were analysed for intracellular cytokines (Fig. S2; see Supporting information at end of paper). Chronic administration of either C20:2 or α-GalCer rendered iNK T cells anergic (Fig. 3b). After 10 days of rest, iNK T cells from both treatment groups still exhibited a decreased IL-2, IFN-γ and IL-4 response upon restimulation. However, after 1 month of rest, iNK T cells treated with C20:2, but not α-GalCer, were fully recovered from anergy (Fig. 3b). Thus, iNK T cells recover from anergy by 1 month after treatment with either a single dose or MD of C20:2, and their recovery time is considerably shorter than that observed for α-GalCer.
C20:2 stimulates a more rapid, but less sustained, up-regulation of PD-1 and PD-L1 expression on iNK T cells
Interaction between the inhibitory signalling molecule PD-1 and its ligand PD-L1 is crucial for the induction of iNK T cell anergy [22,29]. Naive iNK T cells were shown to express low levels of both PD-1 and PD-L1, but not PD-L2, on their surface and to up-regulate the expression of these molecules upon treatment with α-GalCer [22,29]. None the less, these studies were conducted in C57BL/6 mice and did not quantitate the relative levels of up-regulated PD-1, PD-L1 and PD-L2 expression. Moreover, only iNK T cells from the spleen and liver were investigated at the exclusion of iNK T cells in the draining PLN, an important immunological site of T cell activation and regulation during the development of T1D. Therefore, we examined the effects of exposure to α-GalCer and C20:2 on the levels of PD-1 and PD-L1 surface expression on iNK T cells from the spleen and PLN of NOD mice. NOD mice were administered a single dose (4 µg, i.p.) of glycolipid or vehicle and rested for various times (24 h, 48 h, 72 h, 1 week or 1 month) (Fig. S3; see Supporting information at end of paper). Flow cytometric analyses showed that untreated (vehicle) mice expressed low levels of surface PD-1 and PD-L1, but not PD-L2 (not shown), on spleen- and PLN-derived iNK T cells (Fig. 4a). Administration of either α-GalCer or C20:2 up-regulated PD-1 expression on splenic iNK T cells beginning at 24 h and peaking at 48 h or 72 h post-treatment (Fig. 4a, top left panel). However, treatment with α-GalCer led to a significantly greater percentage of PD-1+ iNK T cells during this peak expression time compared to C20:2, demonstrating that it is a stronger iNK T cell agonist. In addition, PD-1 expression levels were still increased significantly 1 month after α-GalCer treatment. In contrast, C20:2 treatment elicited a reduced and less sustained up-regulation of PD-1 on spleen iNK T cells, as noted by the return to steady-state PD-1 expression levels at 1 week post-injection. A similar trend was also observed for the levels of surface expression of PD-L1 in the spleen (Fig. 4a, top right panel).
Fig. 4.
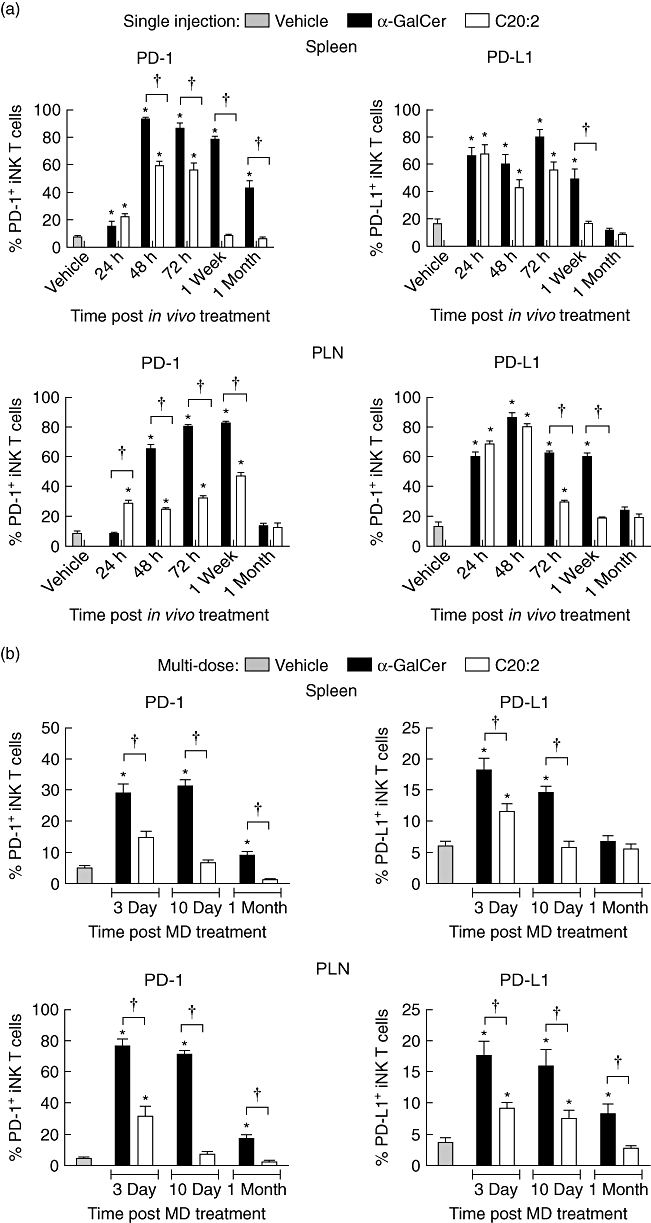
Kinetic analysis of up-regulation of programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1) surface expression on invariant natural killer T (iNK T) cells following glycolipid administration. (a) Non-obese diabetic (NOD) mice (4–6 weeks old) were injected with a single dose [4 µg, intraperitoneally (i.p.)] of glycolipid or vehicle and rested for various times (24 h, 48 h, 72 h, 1 week and 1 month). Spleen and pancreatic lymph node (PLN) lymphocytes were isolated, and the levels of PD-1 and PD-L1 surface expression on T cell receptor (TCR)-β+tetramer+ iNK T cells were analysed by flow cytometry. (b) NOD mice (4–6 weeks old) were treated with a multi-dose (MD) protocol of α-galactosylceramide C26:0 (α-GalCer), C20:2 or vehicle (4 µg/dose every other day for 3 weeks, i.p.) and rested for either 10 days or 1 month. Spleen and PLN lymphocytes were isolated and the levels of PD-1 and PD-L1 surface expression on TCR-β+tetramer+ iNKT cells were analysed by flow cytometry. Data are representative of two independent experiments yielding similar results and are expressed as mean ± standard deviation (s.d.); five mice/treatment group/time-point. *Significant (P < 0·05) difference between the vehicle and glycolipid treatment groups. †Significant (P < 0·05) difference between α-GalCer and C20:2 treatment values.
In the PLN, PD-1 expression on iNK T cells was up-regulated and peaked between 72 h and 1 week after glycolipid exposure (Fig. 4a, bottom left panel), revealing a slight delay compared to the kinetics of PD-1 expression in the spleen (Fig. 4a, top left panel). A significantly greater increase in PD-1 expression occurred during the peak up-regulation stimulated by α-GalCer relative to C20:2. Similar to the kinetics observed in the spleen, PD-L1 surface expression on PLN-derived iNK T cells returned to a steady state level more rapidly after C20:2 treatment (Fig. 4a, bottom right panel). Interestingly, after 24 h, the level of PD-1 expression on iNK T cells in the spleen and PLN following C20:2 administration was higher than those stimulated by α-GalCer (Fig. 4a, left panels). Therefore, a single dose of C20:2 yields a more rapid but less sustained up-regulation of PD-1 and PD-L1 surface expression on splenic and PLN iNK T cells than α-GalCer.
Next, we determined whether this is also the case for MD treatment of NOD mice with C20:2 versusα-GalCer. NOD mice were treated as above with an MD protocol of glycolipid or vehicle, and iNK T cells were analysed for their surface expression of PD-1 and PD-L1 after various rest times (3 days, 10 days or 1 month) (Fig. S4; see Supporting information at end of paper). As expected, the results obtained were similar to those observed for the single-dose injections. C20:2-treated iNK T cells expressed significantly reduced amounts of PD-1 and PD-L1 during peak expression times compared to those treated with α-GalCer, and also down-regulated PD-1 and PD-L1 surface expression to control (vehicle) levels more rapidly (Fig. 4b). The higher and longer-lasting surface expression of PD-1 and PD-L1 on iNK T cells after α-GalCer treatment indicates that these cells remain activated for a longer time than those stimulated by C20:2. Moreover, the faster return of PD-1 and PD-L1 expression to vehicle levels after C20:2 treatment is associated with the more rapid recovery of iNK T cells from anergy.
Restimulation of anergic iNK T cells with glycolipid alters the phenotypic response elicited by CD8α+ mDCs
Upon glycolipid restimulation, anergic iNK T cells can induce the activity of tolerogenic DCs, which are characterized phenotypically by their decreased IL-12 and increased IL-10 secretion responses [16]. Because murine CD8α+ myeloid DCs (mDCs) express significantly higher amounts of CD1d than other DC subsets and are also a major source of the inflammatory cytokine IL-12 in vivo[30–32], we analysed the primary and recall immune responses of CD8α+ mDCs elicited by α-GalCer and C20:2. NOD mice were administered a single dose (4 µg, i.p.) of α-GalCer, C20:2 or vehicle and rested for 1 week to ensure that anergy was induced in both glycolipid treatment groups based on our previous results (Fig. 2). After reinjection of the mice, splenocytes were harvested 6 h later and CD11chighCD8α+ mDCs were stained for intracellular levels of IL-12 and IL-10 (Fig. S5; see Supporting information at end of paper). Upon initial exposure to glycolipid, CD8α+ mDCs responded by synthesizing significantly greater amounts of IL-12 in response to α-GalCer (vehicle/α-GalCer) relative to C20:2 (vehicle/C20:2) (Fig. 5, left panel). The primary IL-10 responses elicited by both glycolipids were equivalent (Fig. 5, right panel), demonstrating further that C20:2 elicits a reduced inflammatory response. The CD8α+ mDC response elicited by anergic iNK T cells differed greatly from those elicited by non-anergic iNK T cells after glycolipid stimulation (α-GalCer/α-GalCer versus vehicle/α-GalCer, C20:2/C20:2 versus vehicle/C20:2). Restimulation of anergic iNK T cells with α-GalCer inhibited the CD8α+ mDC IL-12 response significantly but increased the levels of intracellular IL-10 expressed, albeit this increase was not significant (P > 0·05) relative to the result obtained for T cells from mice treated with a single dose of α-GalCer versus vehicle (compare α-GalCer/α-GalCer versus vehicle/α-GalCer) (Fig. 5). In contrast, restimulation of anergic iNK T cells with C20:2 induced a more pronounced shift in the CD8α+ mDC response. While the relative amount of intracellular IL-12 was decreased to less than that of the vehicle, IL-10 production was increased about twofold (Fig. 5). Thus, an MD treatment with C20:2 induces CD8α+ mDCs that possess a greater anti-inflammatory and possibly ‘tolerogenic’ phenotype than that elicited by α-GalCer.
Fig. 5.
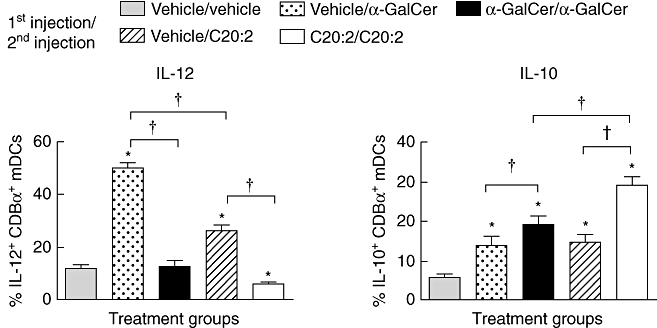
Primary and recall intracellular cytokine responses of CD11chighCD8+ mDCs to α-galactosylceramide C26:0 (α-GalCer) and C20:2. Non-obese diabetic (NOD) mice (4–6 weeks old) were treated with a single dose [4 µg, intraperitoneally (i.p.)] of α-GalCer, C20:2 or vehicle and rested for 1 week to induce invariant natural killer T (iNK T) cell anergy. Mice were then reinjected with the same dose (4 µg, i.p.) of glycolipid. Splenocytes were harvested 6 h after the final injection, cultured in vitro for 3 h in the presence of a protein transport inhibitor, GolgiStop, and CD11chighCD8+ myeloid DCs (mDCs) were stained for intracellular levels of interleukin (IL)-12 and IL-10. Data are representative of two independent experiments yielding similar results; five mice/treatment group. *Significant (P < 0·05) difference between the vehicle/vehicle and glycolipid treatment groups. †Significant (P < 0·05) difference between indicated values.
C20:2 recruits tolerogenic DCs to the spleen and PLN more rapidly than α-GalCer
Protection from T1D by treatment with α-GalCer results in part from the induction of tolerogenic DCs that migrate to the PLN and suppress autoreactive T cells [9,14,16]. Having shown that repeated administration of C20:2 induces CD8α+ mDCs that possess a tolerogenic phenotype, we next determined whether these DCs can transfer antigen-specific tolerance. In addition, due to the reported critical role of anergic iNK T cells in the recruitment of suppressive DCs [16], we investigated whether the faster induction of iNK T cell anergy by C20:2 (Fig. 2) correlates with a more rapid induction of tolerogenic APCs. To mimic our protective MD treatment protocol, NOD mice received one to five i.p. injections of glycolipid (4 µg/dose) spaced 2 days apart. After 2 days of rest, spleen- and PLN-derived CD11c+ DCs were isolated and pooled to assay their capacity to stimulate a CD8+ T cell response from incomplete Freund's adjuvant (IFA) + NRP-V7 (KYNKANVFL) peptide-primed mice. The CD8+ T cells and glycolipid-activated DCs were cultured in a 20 : 1 ratio, respectively, for 72 h in the presence of 100 µg/ml of the cognate peptide NRP-V7 (Fig. 6). Interestingly, the more rapid induction of iNK T cell anergy elicited by C20:2 administration (Fig. 2) correlated with the faster recruitment of tolerogenic DCs to the spleen and PLN induced by C20:2. CD11c+ DCs from vehicle-treated mice strongly activated peptide primed CD8+ T cells upon restimulation with NRP-V7, as evidenced by the robust cell proliferation, IFN-γ and IL-2 responses (Fig. 6). IL-4 secretion was not detected in any of the cultures (data not shown). In agreement with previous findings [16], α-GalCer administered in multiple doses recruited suppressive DCs to the spleen and PLN that elicited reduced CD8+ T cell-proliferative and cytokine (IFN-γ and IL-2) secretion responses. However, this suppressive phenotype was induced only after treatment with five doses of glycolipid. In contrast, C20:2 not only recruited tolerogenic DCs with greater suppressive activity than did α-GalCer, but also these suppressive DCs were induced after only three doses of glycolipid (Fig. 6). Therefore, our MD treatment protocol enables C20:2 to induce iNK T cell anergy and recruit tolerogenic DCs to secondary lymphoid tissues more rapidly than α-GalCer. These data explain partially why C20:2 protects NOD mice from T1D with greater efficacy than does α-GalCer [23,24].
Fig. 6.
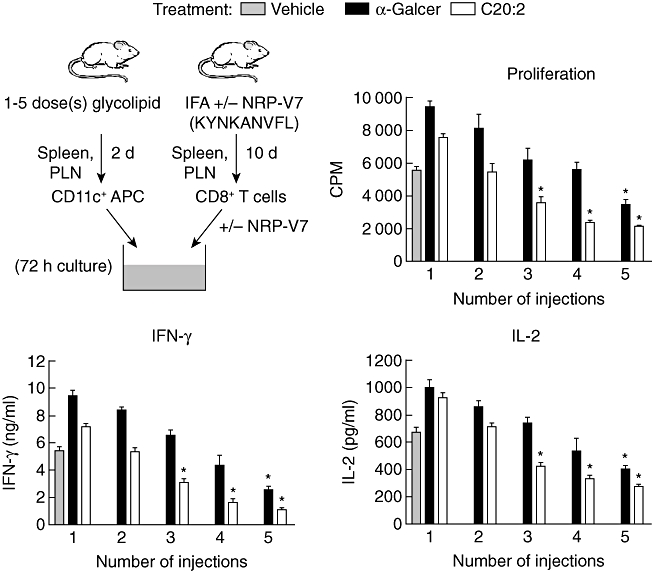
Kinetic analysis of induction of tolerogenic dendritic cells (DCs). Non-obese diabetic (NOD) mice (4–6 weeks old) were treated with a variable number of injections (one to five) of α-galactosylceramide C26:0 (α-GalCer), C20:2 or vehicle [4 µg/dose spaced 2 days apart, intraperitoneally (i.p.)]. After 2 days of rest, spleen and pancreatic lymph node (PLN) suspensions were pooled and CD11c+ DCs from each group were sorted. Concurrently, the NOD-relevant V7 peptide (NRP-V7) peptide emulsified in incomplete Freund's adjuvant (IFA) was administered (100 µg/dose, i.p.) to a separate group of NOD mice to prime their CD8+ T cells. Fluorescence activated cell sorted (FACS) NRP-V7 peptide primed-CD8+ T cells were cultured with CD11c+ DCs in the presence of 100 µg/ml of NRP-V7 for 72 h. T cell proliferation and enzyme-linked immunosorbent assay (ELISA) assays of cytokine [interleukin (IL)-2, interferon (IFN)-γ, IL-4] concentrations in culture supernatants were then performed. Data are representative of two independent experiments yielding similar results and are expressed as mean ± standard deviation (s.d.). *Significant (P < 0·05) reduction compared to vehicle group.
MD administration of C20:2 elicits less iNK T cell apoptosis than does α-GalCer
Although MD treatment with certain glycolipids protect NOD mice from T1D, the iNK T cell frequency in the spleen of these mice can be reduced significantly, due possibly to apoptosis [16,30]. If the therapeutic benefit of such a glycolipid drug were to be tested clinically in humans at risk for T1D, it is essential that the treatment regimen not compromise the patients' immune responses. As iNK T cells mediate many immune responses [33], long-term depletion of this cell subset may have detrimental effects and result in an increased risk of infection. Thus, we investigated whether the faster recovery of iNK T cell anergy after MD administration with C20:2, compared to α-GalCer, results in less apoptosis and increased survival of these iNK T cells.
To determine whether C20:2 administration alters the frequency of iNK T cells in the spleen and PLN, NOD mice were treated by an MD protocol using either α-GalCer, C20:2 or vehicle. After 1 month of rest, the frequencies of iNK T cells in the spleen and PLN measured by flow cytometry showed that both glycolipids reduced the iNK T cell frequency markedly in the spleen but not PLN (Fig. 7a). Whereas the iNK T cell frequency in the spleen of mice treated with C20:2 was still reduced compared to vehicle levels, it was significantly higher than that observed in mice treated with α-GalCer. Note that our result for the iNK T cell frequency in the PLN differs from that reported previously [23] in NOD mice treated with multiple weekly doses of C20:2, in which the frequency of iNK T cells in the PLN was also reduced even at 30 days after the end of treatment. This difference may arise from the various treatment regimens used in the two studies, and be ascribed to the different dose intervals and duration of treatments examined.
Fig. 7.
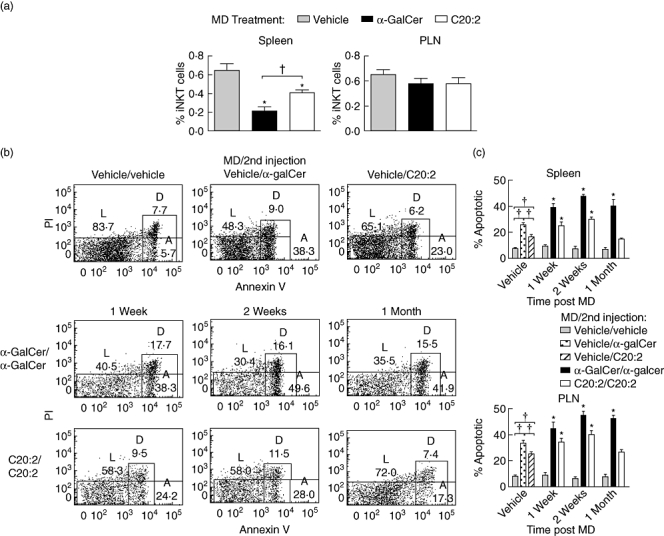
A more rapid recovery from invariant natural killer T (iNK T) cell anergy increases the survival of iNKT cells in the spleen due to their reduced susceptibility to apoptosis. Non-obese diabetic (NOD) mice (4–6 weeks old) were treated with a multi-dose (MD) protocol of α-galactosylceramide C26:0 (α-GalCer), C20:2 or vehicle [4 µg/dose every other day for 3 weeks, intraperitoneally (i.p.)]. (a) Spleen and pancreatic lymph node (PLN) lymphocytes were isolated 1 month post-treatment and the frequencies of T cell receptor (TCR)-β+tetramer+ iNK T cells were analysed by flow cytometry. Data are representative of two independent experiments yielding similar results and are expressed as mean ± standard deviation (s.d.); five mice/treatment group/time-point. *Significant (P < 0·05) difference between the vehicle and glycolipid treatment groups. †Significant (P < 0·05) difference between α-GalCer and C20:2 treatment values. (b) At varying times (1 week, 2 weeks, 1 month) after MD treatment, mice were reinjected (4 µg, i.p.) with an additional dose of glycolipid. Spleen and PLN were isolated 2 h after the final injection, and TCR-β+tetramer+ iNK T cells were analysed for their levels of live (L), apoptotic (A) and dead (D) cells based on annexin V and propidium iodide (PI) staining; five mice/treatment group/time-point. (c) Non-obese (NOD) mice (five mice/treatment group/time-point) were treated (MD protocol) with α-GalCer, C20:2 or vehicle. At different times (1 week, 2 weeks, 1 month) after MD treatment, the mice were reinjected with glycolipid, as described in (b). Spleen and PLN were isolated 2 h after the final injection, and TCR-β+tetramer+ iNK T cells were analysed for their percentage apoptosis based on annexin V and PI staining. The fluorescence activated cell sorter (FACS) plots shown in (b) were quantified, and are presented as histograms. Data are representative of two independent and reproducible experiments. †Significant (P < 0·05) difference between vehicle and primary glycolipid (vehicle/α-GalCer and vehicle/C20:2) responses. *Significant (P < 0·05) difference between primary and recall responses (vehicle/α-GalCer versusα-GalCer/α-GalCer, vehicle/C20:2 versus C20:2/C20:2).
It is possible that the faster recovery of iNK T cell anergy following C20:2 administration may account for a reduced susceptibility to apoptosis, and therefore lead to greater iNK T cell survival. Alternatively, multiple doses of glycolipid may result in an altered chemokine receptor profile on the treated iNK T cells leading to the enhanced migration of cells from the spleen. To address this possibility, iNK T cells from the spleen and PLN were analysed by FACS analyses for their relative susceptibility to apoptosis at various times after an MD treatment with glycolipid (Fig. 7b,c). The FACS plots are shown in Fig. 7b and are quantified in Fig. 7c. In agreement with the more potent immune response elicited by α-GalCer, a single dose of α-GalCer (vehicle MD/α-GalCer) yielded significantly more iNK T cell apoptosis at 2 h after administration compared to C20:2 (vehicle MD/C20:2). At 1 week and 2 weeks after MD treatment, both spleen- and PLN-derived iNK T cells exhibited increased apoptosis compared to their respective single-dose vehicle controls (α-GalCer/α-GalCer versus vehicle/α-GalCer and C20:2/C20:2 versus vehicle/C20:2) (Fig. 7b,c). Interestingly, at 1 month after MD treatment, the level of apoptosis seen in C20:2-treated iNK T cells (C20:2/C20:2) was equivalent to that in vehicle control (vehicle/C20:2)-treated cells, while α-GalCer-treated iNK T cells were still significantly more susceptible to apoptosis (α-GalCer/α-GalCer versus vehicle/α-GalCer). These results correlate closely with those observed for the faster recovery of iNK T cells from anergy following C20:2 administration (Fig. 3), and support the notion that anergic iNK T cells are more prone to apoptosis than non-anergic iNK T cells [16,30]. Moreover, chemokine receptor (CCR7, CXCR5, CCR5 and CXCR3) surface expression levels on iNK T cells were not altered significantly during this time–course of MD treatment with either α-GalCer or C20:2 (our unpublished results), indicating that the reduction in iNK T cell frequency in the spleen may be due to the greater susceptibility of iNK T cells to apoptosis and not to their increased migration to another tissue(s).
Discussion
The dual ability to both activate and suppress immune responses make iNK T cells a promising target for potential drug therapy for various immune diseases. We have shown previously that the potent iNK T cell agonist α-GalCer protects NOD mice against T1D when administered using an MD protocol [10]. While α-GalCer has provided invaluable insight into the mechanisms of iNK T cell activation due to the robust cytokine responses and long-lasting transactivation of other immune cells elicited, it may not be a suitable drug candidate for the treatment of T1D [1–4,8,23,24]. A more promising candidate is the α-GalCer C20:2 N-acyl variant, that protects NOD mice from T1D with a higher efficacy than α-GalCer [23,24]. The overall reduced and predominantly Th2 response (Fig. 1) elicited by C20:2 enables this compound to protect against T1D without a requirement for Treg activity, unlike that observed for α-GalCer [24]. In this study, we characterized further the mechanism of activation of iNK T cells by C20:2 and in particular compared the role of anergy induction by α-GalCer versus C20:2 in protection from T1D. We found that activation by C20:2 triggers a more rapid induction of and recovery from iNK T cell anergy than α-GalCer (Figs 2 and 3). This result was obtained upon stimulation by C20:2 in a single (Fig. 2) and multiple (Fig. 3) dose protocol, and full iNK T cell responsiveness to C20:2 restimulation was detected at 1 month after the initial treatment. The altered kinetics of iNK T cell anergy elicited by C20:2 resulted in two favourable downstream responses. First, the more rapid induction of iNK T cell anergy by C20:2 led to the faster generation of tolerogenic DCs that possess a significantly reduced CD8+ T cell stimulatory capacity (Fig. 6). This is consistent with a role for iNK T cell anergy in the induction of suppressive CD8α+ DCs [16]. Secondly, the faster recovery of iNK T cells from anergy resulted in an increased survival rate of these cells due to their reduced susceptibility to apoptosis after MD treatment with C20:2 versusα-GalCer (Fig. 7). These findings extend the idea that C20:2 is a more beneficial drug for the treatment of T1D than α-GalCer [23,24], and provide support for further investigation of C20:2 as a candidate for clinical trials in humans at risk for T1D.
Treatment of NOD mice with a single dose of C20:2 resulted in a greater Th2 response with increased levels of IL-4 and decreased IFN-γ during their peak time of secretion (Fig. 1b). At 2 h and 24 h post-treatment in vivo, the IFN-γ response stimulated by C20:2 was more rapid, but less sustained, than that induced by α-GalCer. This more rapid C20:2-induced IFN-γ response correlated with a more rapid induction of iNK T cell anergy (Fig. 2). It is possible that a glycolipid containing a shortened fatty acyl side chain (e.g. C20:2) relative to α-GalCer may not completely saturate the A' channel of a CD1d molecule and thus lead to an altered glycolipid presentation and subsequent iNK T cell response [26]. An alternate explanation for these findings relates to the requirements of both glycolipids to initially load onto CD1d molecules. To be loaded onto CD1d, α-GalCer must be internalized and requires the assistance of co-factors such as saposins, GM2 activator proteins and the Niemann–Pick type C1 and C2 proteins [34–38]. Conversely, C20:2 does not require internalization but, rather, complexes preferentially with CD1d molecules on the cell surface [27]. This preferential binding of C20:2 to CD1d on the cell surface activates iNK T cells more rapidly, which is associated with an increased IFN-γ response in the serum at 2 h after C20:2 administration and the more rapid induction of iNK T cell anergy.
As a consequence of the differential CD1d loading requirements of α-GalCer and C20:2, each glycolipid is presented to iNK T cells in a different manner. After internalization of α-GalCer and subsequent loading onto CD1d intracellularly, the α-GalCer/CD1d complex is shuttled to the surface and presented to iNK T cells in the context of a lipid raft, which contains an abundance of co-stimulatory molecules [38–40]. Conversely, the majority (> 80%) of C20:2/CD1d complexes localize on the cell surface outside of lipid rafts [27,41]. Thus, the increased co-stimulation that occurs during α-GalCer-mediated iNK T cell activation may elicit a prolonged stimulation of iNK T cells and lead to a more sustained IFN-γ response. The capacity of anergy to prevent the chronic activation of iNK T cells [42] could explain why C20:2 administration leads to a less sustained activation of iNK T cells (Fig. 1b) [24] and is associated with a more rapid exit of iNK T cells from anergy.
The inhibitory co-receptors PD-1 and PD-L1 are essential for the initiation of anergy [20–22]. PD-L1 is a critical receptor for the induction of tolerogenic DCs and protection against T1D in glycolipid-treated NOD mice [43]. While we found that both α-GalCer and C20:2 up-regulated PD-1 and PD-L1 surface expression on iNK T cells in the spleen and PLN, α-GalCer-stimulated cells displayed significantly higher expression levels and thus appeared to be more highly activated (Fig. 4). Interestingly, the kinetics of PD-1 and PD-L1 expression on iNK T cells elicited by C20:2 correlate directly with the more rapid induction of and recovery from iNK T cell anergy. Administration of C20:2 resulted in a faster up-regulation of PD-1 on iNK T cells in the PLN (Fig. 4a, left panels). As mentioned above, the surface loading capability of C20:2 enables it to complex with CD1d more rapidly than α-GalCer, which is expected to lead to the faster activation of iNK T cells. This is consistent with the more rapid up-regulation of PD-1 surface expression and subsequent induction of anergy observed. In addition, the expression of PD-1 and PD-L1 on C20:2-activated iNK T cells returned to resting levels more rapidly in the spleen and PLN than those treated with α-GalCer by either a single dose or MD dose protocol. Interestingly, this more rapid decrease in PD-1 and PD-L1 expression is compatible with the faster recovery of iNK T cells from anergy after C20:2 administration.
The inhibition of iNK T cell activation and subsequent induction of anergy elicited by PD-1 signalling may stimulate an increase in iNK T cell survival. Upon TCR ligation in conventional T cells, PI3K assists the initiation of the signalling cascade responsible for T cell activation [44–46]. In addition, upon co-stimulation with CD28, PI3K leads to the up-regulation of Bcl-xL, a survival protein that allows activated T cells to become resistant to apoptosis [44–46]. Unlike other inhibitory signalling molecules [e.g. cytotoxic T lymphocyte antigen-4 (CTLA-4), PD-1 signalling does not up-regulate Bcl-xL expression on T cells, suggesting that ligation of PD-1 may render T cells more susceptible to apoptosis [47–49]. This is consistent with reports that anergic iNK T cells are more susceptible to apoptosis [4]. Accordingly, after MD administration of α-GalCer to NOD mice, a significant decrease in the frequency of iNK T cells occurs in the spleen [4]. Conversely, while MD administration of C20:2 decreased the iNK T cell frequency in the spleen, this reduction was significantly less than that observed after α-GalCer treatment (Fig. 7). Hence, the more rapid down-regulation of PD-1 and PD-L1 on iNK T cells combined with the faster recovery from anergy in mice treated with C20:2 relative to those receiving α-GalCer are beneficial, as they lead to less iNK T cell death.
A major reason why certain iNK T cell agonists protect NOD mice from T1D is due probably to the capacity of such activated iNK T cells to modulate the function of CD8α+ DCs [13–15]. After MD treatment of NOD mice with α-GalCer, CD8α+ tolerogenic DCs are recruited to the PLN, where they can inhibit autoreactive T cells, prevent further islet β cell destruction and protect against T1D. Alternatively, this is a mouse strain-specific phenomenon and depending on the genetic origin of iNK T cells and DCs, glycolipid administration can result in the maturation of DCs towards an immunogenic state [43]. Given the high genetic diversity in humans, this will be an important consideration for the application of glycolipid-based therapy in the future.
We have demonstrated that MD treatment of NOD mice with C20:2 induces DCs that are more suppressive in vitro than those exposed to α-GalCer [24]. Analyses of the kinetics of suppressive CD8α+ mDC induction showed that a single dose of C20:2 elicited a robust cytokine response from CD8α+ mDCs, the predominant IL-12-producing DC subset. In comparison to α-GalCer, this single dose of C20:2 induced a significantly reduced intracellular IL-12 response in CD8α+ mDCs, while maintaining equivalent levels of IL-10 (vehicle/α-GalCer versus vehicle/C20:2) (Fig. 5). Thus, C20:2 treated iNK T cells induce a greater Th2-type response owing to their reduced accumulation of intracellular IL-12 and resultant lower IFN-γ secretion by NK cells. Restimulation of anergic iNK T cells by either α-GalCer or C20:2 reduced the level of intracellular IL-12 significantly, a phenotypic trait characteristic of tolerogenic DCs. Interestingly, restimulation with C20:2, but not α-GalCer, also induced a significantly increased IL-10 response by CD8α+ mDCs (vehicle/α-GalCer versusα-GalCer/α-GalCer, vehicle/C20:2 versus C20:2/C20:2) (Fig. 5). This elevated IL-10 response in CD8α+ mDCs from NOD mice treated with C20:2 mediates the more efficient suppression by tolerogenic DCs recruited to the spleen and PLN after MD treatment with C20:2 [24]. Finally, we showed that the more rapid induction of iNK T cell anergy by C20:2 enables C20:2 to recruit tolerogenic DCs more quickly to the spleen and PLN than α-GalCer (Fig. 6). When injections were administered every other day to mimic our MD protocol that protects from T1D, DCs with tolerogenic capabilities were observed following three doses of C20:2 (Fig. 6). Conversely, tolerogenic DCs were observed only after five doses of α-GalCer.
Overall, this study provides novel mechanistic insight and justification for the potential use of the α-GalCer C20:2 N-acyl variant to modulate iNK T cell activation and anergy and afford protection from T1D. Our data show that C20:2 provides several beneficial immune responses that may translate clinically into a more effective and safer therapeutic alternative to α-GalCer. It is possible that a less sustained and Th2-skewed primary immune response together with the faster induction of iNK T cell anergy by C20:2 will reduce the initial side effects of glycolipid administration compared to α-GalCer. In particular, the lower IFN-γ response elicited by C20:2 may limit its capacity to contribute to ongoing chronic inflammatory responses detected typically in T1D patients. In addition, by reducing the time required between the activation and subsequent induction of anergy of iNK T cells from 1 week to 72 h compared to α-GalCer (Fig. 2), C20:2 can recruit tolerogenic DCs more rapidly to the spleen and PLN (Fig. 6). This could diminish the ongoing autoreactive T cell response in T1D patients more rapidly than α-GalCer, thereby reducing the amount of islet β cell destruction. Note also that the faster recovery from iNK T cell anergy induced by repeated C20:2 administration reduced the amount of iNK T cell death compared to that observed for α-GalCer. Due to the expanding role of iNK T cells as regulators of immune responses [5,33,50,51], it is essential that the use of therapeutic drugs that target iNK T cells does not render the host immunocompromised. Our results demonstrate the important role of iNK T cell anergy and how the manipulation of iNK T cells in this state can enhance protection from T1D. It follows that the favourable characteristics of C20:2 make it a prime candidate to consider for future therapeutic use in clinical trials of T1D.
Acknowledgments
We thank Delfina Mazzuca Siroen for her expertise in operating the flow cytometer as well as the staff of the Robarts Barrier Animal Facility for their assistance with the breeding and maintenance of our mice. We also thank all members of our laboratories for their continued support and advice during the preparation of this manuscript. This work was supported by grants from the Juvenile Diabetes Research Foundation International (24-2007-388) and Canadian Institutes of Health Research (MOP 64386) to T. L. Delovitch; Canadian Institutes of Health Research (MOP 86601) to S. M. M. Haeryfar; the Medical Council and the Wellcome Trust (084923/B/08/7) to G. S. Besra; and the National Institutes of Health (RO1 AI45889 and RO1 AI064424) to S. A. Porcelli. S. M. M. Hareyfar is the recipient of a Canada Research Chair in Viral Immunity and Pathogenesis award; R. Tohn was the recipient of a Schulich Graduate Enhancement Scholarship, Department of Microbiology and Immunology Graduate Entrance Fellowship and Ontario Graduate Scholarship; H. Blumenfeld was the recipient of a Schulich Graduate Enhancement Scholarship and Department of Microbiology and Immunology Graduate Entrance Fellowship; S. A. Porcelli was the recipient of an Irma T. Hirschl Cancer Scientist Award; and G. S. Besra was supported by a Personal Research Chair from Mr James Badick, Royal Society Wolfson Research Merit Award and Listar Institute–Jenner Research Fellowship.
Disclosure
Dr Steven A. Porcelli is an inventor of patent applications pertaining to the C20:2 compound and its use in treatment of autoimmunity, and is a consultant for Vaccinex Inc. (Rochester, NY, USA). Dr Terry L. Delovitch is a consultant for Vaccinex Inc. (Rochester, NY, USA). The other authors have no financial conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Kinetic analysis of induction of invariant natural killer T (iNK T) cell anergy.Non-obese diabetic (NOD) mice (4–6 weeks old, five mice/treatment group/time-point) were injected with a single dose (4 µg, intraperitoneally) of glycolipid or vehicle (Veh), rested for various times (48 h, 72 h, 1 week, 1 month), and then reinjected with the same dose of glycolipid. Splenocytes were harvested 2 h later and T cell receptor (TCR)-β+tetramer+ iNK T cells were stained for intracellular levels of interleukin (IL)-2, interferon (IFN)-γ and IL-4 (solid black) in relation to isotype control values (shaded grey).
Fig. S2. Kinetic analysis of invariant natural killer T (iNK T) cell anergy following treatment with a multi-low dose (MD) protocol of glycolipid.Non-obese diabetic (NOD) mice (4–6 weeks old) received MD treatment [4 µg/dose every other day for 3 weeks, intraperitoneally (i.p.)] of glycolipid or vehicle, and after a rest for 10 days or 1 month were reinjected with an additional dose (4 µg, i.p.) of glycolipid. Splenocytes were harvested 2 h after the final injection and T cell receptor (TCR)-β+tetramer+ iNK T cells were stained for intracellular levels of interleukin (IL)-2, interferon (IFN)-γ and IL-4 (solid black) in relation to isotype control values (shaded grey).
Fig. S3. Kinetic analysis of up-regulation of programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1) surface expression on invariant natural killer T (iNK T) cells following single glycolipid administration. Non-obese diabetic (NOD) mice (4–6 weeks old) were injected with a single dose (4 µg, intraperitoneally) of glycolipid or vehicle and then rested for various times (24 h, 48 h, 72 h, 1 week and 1 month). Spleen and pancreatic lymph nodes (PLN) lymphocytes were isolated, and the levels of PD-1 and PD-L1 surface expression (solid black) on T cell receptor (TCR)-β+tetramer+ iNK T cells were analysed by flow cytometry in relation to isotype control values (shaded grey).
Fig. S4. Kinetic analysis of up-regulation of programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1) surface expression on invariant natural killer T (iNK T) cells following treatment with a multi-dose (MD) protocol of glycolipid. Non-obese diabetic (NOD mice) (4–6 weeks old) were treated with a MD protocol of α-galactosylceramide C26:0 (α-GalCer), C20:2 or vehicle (4 µg/dose every other day for 3 weeks, intraperitoneally) and rested for either 3 days, 10 days or 1 month. Spleen and pancreatic lymph nodes (PLN) lymphocytes were isolated and the levels of PD-1 and PD-L1 surface expression (solid black) on TCR-β+tetramer+ iNK T cells were analysed by flow cytometry in relation to isotype control values (shaded grey).
Fig. S5. Primary and recall intracellular cytokine responses of CD11chighCD8+ mDCs to α-galactosylceramide C26:0 (α-GalCer) and C20:2. Non-obese diabetic (NOD) mice (4–6 weeks old) were treated with a single dose [4 µg, intraperitoneally (i.p.)] of α-GalCer, C20:2 or vehicle and rested for 1 week to induce invariant natural killer T (iNK T) cell anergy. Mice were then reinjected with the same dose (4 µg, i.p.) of glycolipid. Splenocytes were harvested 6 h after the final injection and CD11chighCD8+ mDCs were analysed by flow cytometry for their levels of expression of intracellular interleukin (IL-10) and IL-12 (solid black) in relation to isotype control values (shaded grey).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol. 2002;2:557–68. doi: 10.1038/nri854. [DOI] [PubMed] [Google Scholar]
- 2.Taniguchi M, Seino K, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. 2003;4:1164–5. doi: 10.1038/ni1203-1164. [DOI] [PubMed] [Google Scholar]
- 3.Brigl M, Brenner MB. CD1: antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–90. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 4.Van Kaer L. Alpha-galactosylceramide therapy for autoimmune diseases: prospects and obstacles. Nat Rev Immunol. 2005;5:31–42. doi: 10.1038/nri1531. [DOI] [PubMed] [Google Scholar]
- 5.Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007;25:297–36. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 6.Zajonc DM, Kronenberg M. Carbohydrate specificity of the recognition of diverse glycolipids by natural killer T cells. Immunol Rev. 2009;230:188–00. doi: 10.1111/j.1600-065X.2009.00802.x. [DOI] [PubMed] [Google Scholar]
- 7.Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol. 2010;11:197–06. doi: 10.1038/ni.1841. [DOI] [PubMed] [Google Scholar]
- 8.Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 2010;10:501–13. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- 9.Hong S, Wilson MT, Serizawa I, et al. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med. 2001;7:1052–56. doi: 10.1038/nm0901-1052. [DOI] [PubMed] [Google Scholar]
- 10.Sharif S, Arreaza GA, Zucker P, et al. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med. 2001;7:1057–62. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- 11.Wang B, Geng YB, Wang CR. CD1-restricted NK T cells protect nonobese diabetic mice from developing diabetes. J Exp Med. 2001;194:313–20. doi: 10.1084/jem.194.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi QS, Ly D, Zucker P, McGarry M, Delovitch TL. Interleukin-4 but not interleukin-10 protects against spontaneous and recurrent type 1 diabetes by activated CD1d-restricted invariant natural killer T-cells. Diabetes. 2004;53:1303–10. doi: 10.2337/diabetes.53.5.1303. [DOI] [PubMed] [Google Scholar]
- 13.Naumov YN, Bahjat KS, Gausling R, et al. Activation of CD1d-restricted T cells protects NOD mice from developing diabetes by regulating dendritic cell subsets. Proc Natl Acad Sci USA. 2001;98:13838–43. doi: 10.1073/pnas.251531798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol. 2007;179:5041–53. doi: 10.4049/jimmunol.179.8.5041. [DOI] [PubMed] [Google Scholar]
- 15.Chen YG, Choisy-Rossi CM, Holl TM, et al. Activated NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol. 2005;174:1196–204. doi: 10.4049/jimmunol.174.3.1196. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Cho S, Ueno A, et al. Ligand-dependent induction of noninflammatory dendritic cells by anergic invariant NKT cells minimizes autoimmune inflammation. J Immunol. 2008;181:2438–45. doi: 10.4049/jimmunol.181.4.2438. [DOI] [PubMed] [Google Scholar]
- 17.Diana J, Griseri T, Lagaye S, et al. NKT cell-plasmacytoid dendritic cell cooperation via OX40 controls viral infection in a tissue-specific manner. Immunity. 2009;30:289–99. doi: 10.1016/j.immuni.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 18.Ly D, Mi Q-S, Hussain S, Delovitch TL. Protection from type 1 diabetes by invariant NK T cells requires the activity of CD4+CD25+ regulatory T cells. J Immunol. 2006;177:3695–704. doi: 10.4049/jimmunol.177.6.3695. [DOI] [PubMed] [Google Scholar]
- 19.Parekh VV, Wilson MT, Olivares-Villagomez D, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. 2005;115:2572–83. doi: 10.1172/JCI24762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keir ME, Butte MJ, Freeman GJ, Sharpe AH, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–04. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813–24. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 22.Parekh VV, Lalani S, Kim S, et al. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol. 2009;182:2816–26. doi: 10.4049/jimmunol.0803648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forestier C, Takaki T, Molan A, et al. Improved outcomes in NOD mice treated with a novel Th2 cytokine-biasing NKT cell activator. J Immunol. 2007;178:1415–25. doi: 10.4049/jimmunol.178.3.1415. [DOI] [PubMed] [Google Scholar]
- 24.Ly D, Tohn R, Rubin B, et al. An alpha-galactosylceramide C20:2 N-acyl variant enhances anti-inflammatory and regulatory T cell-independent responses that prevent type 1 diabetes. Clin Exp Immunol. 2009;160:185–98. doi: 10.1111/j.1365-2249.2009.04074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu KO, Im JS, Molano A, et al. Modulation of CD1d-restricted NKT cell responses by using N-acyl variants of alpha-galactosylceramides. Proc Natl Acad Sci USA. 2005;102:3383–8. doi: 10.1073/pnas.0407488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy C, Shepherd D, Fleire S, et al. The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med. 2007;204:1131–44. doi: 10.1084/jem.20062342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Im JS, Arora P, Bricard G, et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity. 2009;30:888–98. doi: 10.1016/j.immuni.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beverly B, Kang SM, Lenardo MJ, Schwartz RH. Reversal of in vitro T cell clonal anergy by IL-2 stimulation. Int Immunol. 1992;4:661–71. doi: 10.1093/intimm/4.6.661. [DOI] [PubMed] [Google Scholar]
- 29.Chang WS, Kim JY, Kim YJ, et al. Programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol. 2008;181:6707–10. doi: 10.4049/jimmunol.181.10.6707. [DOI] [PubMed] [Google Scholar]
- 30.Maldonado-Lopez R, De Smedt T, Michel P, et al. CD8alpha+ and CD8alpha− subclasses of dendritic cells direct the development of distinct T helper cells in vivo. J Exp Med. 1999;189:587–92. doi: 10.1084/jem.189.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pulendran B, Smith JL, Caspary G, et al. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci USA. 1999;96:1036–41. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vremec D, Zorbas M, Scollay R, et al. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J Exp Med. 1992;176:47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson SB, Delovitch TL. Janus-like role of regulatory iNKT cells in autoimmune disease and tumour immunity. Nat Rev Immunol. 2003;3:211–22. doi: 10.1038/nri1028. [DOI] [PubMed] [Google Scholar]
- 34.Zhou D, Cantu C, Sagiv Y, et al. Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science. 2004;303:523–7. doi: 10.1126/science.1092009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sagiv Y, Hudspeth K, Mattner J, et al. Impaired glycosphingolipid trafficking and NKT cell development in mice lacking Niemann–Pick type C1 protein. J Immunol. 2006;177:26–30. doi: 10.4049/jimmunol.177.1.26. [DOI] [PubMed] [Google Scholar]
- 36.Yuan W, Qi X, Tsang P, et al. Saposin B is the dominant saposin that facilitates lipid binding to human CD1d molecules. Proc Natl Acad Sci USA. 2007;104:5551–6. doi: 10.1073/pnas.0700617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrantz N, Sagiv Y, Liu Y, et al. The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J Exp Med. 2007;204:841–52. doi: 10.1084/jem.20061562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dascher CC, Brenner MB. Evolutionary constraints on CD1 structure: insights from comparative genomic analysis. Trends Immunol. 2003;24:412–8. doi: 10.1016/s1471-4906(03)00179-0. [DOI] [PubMed] [Google Scholar]
- 39.Lang GA, Maltsev SD, Besra GS, Lang ML. Presentation of alpha-galactosylceramide by murine CD1d to natural killer T cells is facilitated by plasma membrane glycolipid rafts. Immunology. 2004;112:386–96. doi: 10.1111/j.1365-2567.2004.01896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park YK, Lee JW, Ko YG, et al. Lipid rafts are required for efficient signal transduction by CD1d. Biochem Biophys Res Commun. 2005;327:1143–54. doi: 10.1016/j.bbrc.2004.12.121. [DOI] [PubMed] [Google Scholar]
- 41.Bai L, Sagiv Y, Liu Y, et al. Lysosomal recycling terminates CD1d-mediated presentation of short and polyunsaturated variants of the NKT cell lipid antigen alphaGalCer. Proc Natl Acad Sci USA. 2009;106:10254–9. doi: 10.1073/pnas.0901228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu L, Gabriel CL, Parekh VV, Van Kaer L. Invariant natural killer T cells: innate-like T cells with potent immunomodulatory activities. Tissue Antigens. 2009;73:535–45. doi: 10.1111/j.1399-0039.2009.01256.x. [DOI] [PubMed] [Google Scholar]
- 43.Driver JP, Scheuplein F, Chen YG, et al. Invariant natural killer T-cell control of type 1 diabetes: a dendritic cell genetic decision of a silver bullet or Russian roulette. Diabetes. 2010;59:423–32. doi: 10.2337/db09-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boise LH, Minn AJ, Noel PJ, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 45.Ueda Y, Levine BL, Huang ML, et al. Both CD28 ligands CD80 (B7-1) and CD86 (B7-2) activate phosphatidylinositol 3-kinase, and wortmannin reveals heterogeneity in the regulation of T cell IL-2 secretion. Int Immunol. 1995;7:957–66. doi: 10.1093/intimm/7.6.957. [DOI] [PubMed] [Google Scholar]
- 46.Ward SG, Westwick J, Hall ND, Sansom DM, et al. Ligation of CD28 receptor by B7 induces formation of D-3 phosphoinositides in T lymphocytes independently of T cell receptor/CD3 activation. Eur J Immunol. 1993;23:2572–7. doi: 10.1002/eji.1830231029. [DOI] [PubMed] [Google Scholar]
- 47.Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229:114–25. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blair PJ, Riley JL, Levine BL, et al. CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-X(L) induction. J Immunol. 1998;160:12–5. [PubMed] [Google Scholar]
- 49.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. 2009;9:28–38. doi: 10.1038/nri2451. [DOI] [PubMed] [Google Scholar]
- 51.Cerundolo V, Barral P, Batista FD. Synthetic iNKT cell agonists as vaccine adjuvants – finding the balance. Curr Opin Immunol. 2010;22:417–24. doi: 10.1016/j.coi.2010.04.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.