

Abstract
Our previous study demonstrated that T helper (Th) cells from patients with rheumatoid arthritis (RA) display an altered expression profile of Notch receptors and enhanced activation of Notch signalling. The aim of this study was to investigate the role of distinct Notch receptors and ligands in the activation and differentiation of collagen II (CII)-reactive Th cells upon antigen-specific restimulation. Spleen mononuclear cells (SMNCs) from CII-immunized DBA/1J mice were restimulated by culturing with CII. CII-specific proliferation and differentiation of T cells were determined by tritiated thymidine (3[H]-TdR) incorporation and flow cytometric analysis, respectively. The mRNA expression of Notch receptors and Hes1 was assessed by real-time polymerase chain reaction (PCR). There was a clear increase in the percentage of Th1 cells and Th17 cells after CII restimulation. No significant difference was observed in the percentage of regulation T cells (Treg) in SMNCs with or without CII restimulation. CII restimulation induced up-regulated transcript levels of Hes1 in CII-reactive CD4+ T cells. The mRNA level of Notch3 was also up-regulated significantly, while the levels of the other three Notch receptors were not increased. Inhibition of Notch signalling by N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) and Notch3 antibody decreased the collagen-specific T cell proliferation and attenuated Th1- and Th17-type responses, while treatment with Notch ligand Delta-like 1 promoted such a response. The present study demonstrates that Notch signalling is engaged in CII-specific Th1- and Th17-type expansion in which Notch3 and Delta-like1 were involved. Selective inhibition of Notch signalling mediated by Notch3 or Delta-like1 may offer a new strategy for the treatment of RA.
Keywords: collagen, Notch signalling, Th1 cells, Th17 cells
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease of the synovial joints characterized by leucocyte invasion and synoviocyte activation, leading to cartilage and bone destruction. Although the exact mechanism of RA pathogenesis is not well defined, the infiltration of autoreactive CD4+ T cells into synovium has been thought to be the major instigator of joint inflammation. Type II collagen (CII), which is expressed exclusively in the articular cartilage of joints, has been considered as one of the major autoantigens in human RA as well as in the collagen-induced arthritis (CIA) model [1]. In particular, the higher prevalence of CII-specific T cells noted during the early phase of RA indicates that CII-specific T cell proliferation and differentiation plays an important role in the initiation of inflammation in the articular joints; however, the underlying mechanism remains unknown [1].
Recent studies have identified the Notch pathway as a key regulator of peripheral T cell activation and effector cell differentiation [2–4]. The Notch signalling pathway is highly conserved beyond species and plays a critical role in a variety of cellular functions, including cell proliferation, differentiation and apoptosis [5,6]. To date, four Notch receptors (Notch1–4) and five of their ligands (Delta-like 1, 3, 4; Jagged1, 2) have been identified in mammals. Upon ligand binding, the intracellular domain (ICD) of the receptor is cleaved proteolytically and translocated into the nucleus, where it associates with the recombination signal binding protein (RBP)-Jκ transcription factor and regulates expression of several target genes, such as the basic helix–loop–helix (bHLH) proteins hairy-enhancer of split-like 1 (HES-1) and HES-5 [7,8]. The potential role for Notch signalling in peripheral T cells linked Notch receptor expression to T cell activation, proliferation and cytokine production [9]. Notch receptors are present in both CD4+ and CD8+ peripheral T cells, and the expression pattern of four receptors is changed following T cell activation [10,11]. Stimulation of purified CD4+ T cells with CD3- and CD28-specific antibodies results in Notch receptor cleavage and up-regulation [12]. Upon antigen-specific stimulation in proteolipid protein (PLP)-reactive T cells from an animal model, experimental autoimmune encephalomyelitis (EAE), specific induction of Notch1 and Notch3 transcripts were noted. However, selective inhibition of the Notch3 receptor, but not Notch1, abrogated proliferation, Th1- and Th17-type responses of PLP-reactive T cells [13].
As yet, however, certain aspects of how Notch regulates Th cell differentiation are controversial. Our previous study has demonstrated that Th cells from patients with rheumatoid arthritis (RA) display an altered expression profile of Notch receptors and enhanced activation of Notch signalling compared with those from healthy controls [14]. The aim of this study was to investigate the role of distinct Notch receptors and ligands in the activation and differentiation of collagen-reactive Th cells upon antigen-specific restimulation which may provide useful information for further understanding of Notch signalling-mediated autoimmune diseases, including RA.
Materials and methods
Mice and immunization
Male DBA/1J mice aged 8–10 weeks were supplied by the Model Animal Research Center of Nanjing University (Nanjing). All animal experiments were undertaken in accordance with approval of the Scientific Investigation Board of Jiangsu University.
Two mg/ml bovine type II collagen (Chondrex, Redmond, WA, USA) was emulsified with equal volume of Freund's complete adjuvant (Sigma-Aldrich, St. Louis, MO, USA), and then DBA/1J mice received 100 µg bovine type II collagen by intradermal injection at the base of the tail.
Cell isolation and stimulation
On day 10 after immunization, spleens were collected. Suspension of spleen mononuclear cells (SMNCs) were prepared from spleens of three mice per group in complete RPMI-1640 medium (Gibco-BRL, Grand Island, NY, USA) containing 10% fetal calf serum (FCS), 10 mM HEPES, 2 mM l-glutamine, 0·1 mg/ml penicillin, 0·1 mg/ml streptomycin and 50 µM 2-mercaptoethanol (ME). SMNCs (1 × 106 cells/well) were then incubated with collagen II (CII) at a concentration of 5 µg/ml in the presence or absence of N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) (5 µM; Sigma), α-Notch3 (10 µg/ml; R&D Systems, Minneapolis, MN, USA), Delta-like 1-Fc or Jagged1-Fc fusion proteins (10 µg/ml; R&D). For the determination of Hes1 and four Notch receptors mRNA expression, CD4+ T cells were isolated from SMNCs after varied treatment by depletion of non-CD4+ T cells using a CD4+ T cell isolation kit (Miltenyi Biotec, Auburn, CA, USA).
Proliferation assay
SMNCs from CII-immunized DBA/1J mice were cultured with CII for 3 days in 96-well flat-bottomed plates at 1 × 106 cells/well with or without DAPT (5 µM) or α-Notch3 (10 µg/ml). One µCi of [3H]-thymidine (Shanghai Institute of Atomic Nucleus, Chinese Academy of Sciences, Shanghai) was added into each well 16 h before termination of culturing and isotope incorporation was assayed with a liquid scintillation counter (Pharmacia-LKB, Freiburg, Germany). Results were expressed as mean ± standard deviation (s.d.) of counts per minute (cpm) of triplicates or quadruplicates.
Flow cytometric analysis
For analysis of Th1 and Th17 cells, restimulated SMNCs were suspended in complete culture medium and cultures were stimulated for 5 h using 50 ng/ml phorbol myristate acetate (PMA; Sigma-Aldrich, MO, USA) and 1 µg/ml ionomycin (Sigma-Aldrich) in the presence of 5 µg/ml brefeldin A (Sigma-Aldrich) at 37°C and 5% CO2. Cells were then washed in phosphate-buffered saline (PBS) and surface-labelled with CD4-fluorescein isothiocyanate (FITC) (eBioscience, San Diego, CA, USA). Following surface staining, cells were fixed and permeabilized using IntraPrep Permeabilization Reagent (Beckman Coulter Inc., Fullerton, CA, USA), and then stained with interferon (IFN)-γ-phycoerythrin (PE) or interleukin (IL)-17A-PE. For analysis of Treg cells, restimulated SMNCs were surface-labelled with CD4-PE and CD25-PE-cycanin 5 (Cy5) without PMA and ionomycin stimulation followed by fixation and permeabilization and intracellular staining with forkhead box P3 (FoxP3)-FITC.
Labelled cells were washed and analysed with a fluorescence activated cell sorter (FACS) Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA) using CellQuest software (Becton Dickinson). In each case, staining was compared with that of the appropriately labelled isotype control antibody.
RNA extraction and real-time PCR
Total RNA was extracted from purified CD4+ T cell preparation using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was prepared by reverse transcription with oligo(dT) from total RNA extraction. Real-time PCR for Notch1, Notch2, Notch3, Notch4, Hes1 and a reference gene (β-actin) was performed in a LightCycler instrument (Roche Molecular Diagnostics, Mannheim, Germany) with the SYBRgreen mastermix kit (TaKaRa, Ohtsu, Japan). Each target gene expression was then normalized relative to β-actin. Primers used were: forward (5′-TCCAGAGTGCCACCGATG-3′) and reverse (5′-TCCACCGGCTCACTCTTCAC-3′) for Notch1; forward (5′-ACCCTCCGCCGAGACTCT-3′) and reverse (5′-TCCCAGAACCAATCAGGTTAGC-3′) for Notch2; forward (5′-CAGGCGAAAGCGAGAACAC-3′) and reverse (5′-GGCCATGTTCTTCATTCCCA-3′) for Notch3; forward (5′-TGTCTCCCCCATAGAGTATGCA-3′) and reverse (5′-CTCGAAATCAACTTTGTCCTCTTG-3′) for Notch4; forward (5′-GACTGTGAAGCACCTCCG-3′) and reverse (5′-GTCATGGCGTTGATCTGG-3′) for Hes1; and forward (5′-GAAGTCCCTCACCCTCCCAA-3′) and reverse (5′-GGCATGGACGCGACCA-3′) for β-actin.
Statistical analysis
The two-tailed Student's t-test and analysis of variance (anova) test were used for determining significant differences (P ≤ 0·05) between groups.
Results
Collagen-specific reactivation tends to Th1- and Th17-type expansion
We first explored the characterization of the CII-specific T cell response by flow cytometric analysis of T subsets, including Th1, Treg and Th17 cells. DBA/1J mice were immunized with bovine CII, and 10 days later SMNCs were collected and restimulated by culturing with CII for 3 days in vitro. As shown in Fig. 1a, there was a clear increase in the percentage of IFN-γ-producing CD4+ T cells (Th1 cells) and IL-17-producing CD4+ T cells (Th17 cells) after CII restimulation compared with controls (both P < 0·05). No significant difference was observed in the percentage of Treg in SMNCs with or without CII restimulation (P > 0·05). Figure 1b shows the typical flow cytometric results of three T subsets in dot-plots. These results indicate that CII-specific reactivation tends to Th1- and Th17-type expansion.
Fig. 1.
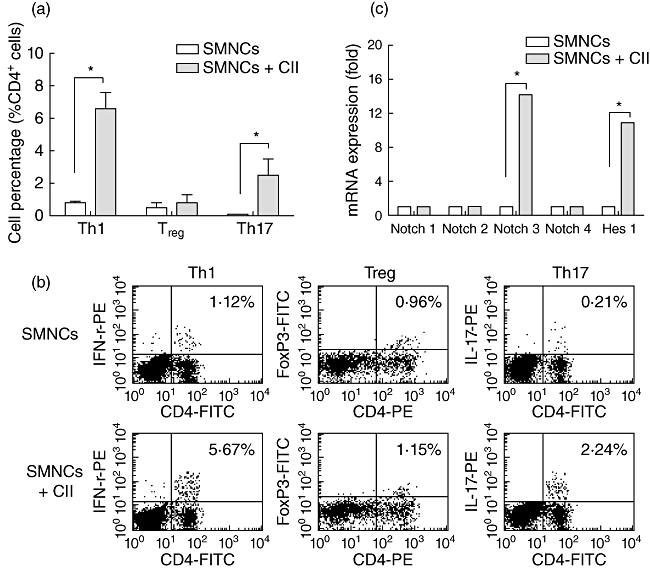
Collagen-specific reactivation tends to T helper type 1 (Th1)- and Th17-type expansion along with activated Notch signalling and increased Notch3 expression. (a) Spleen mononuclear cells (SMNCs) from collagen II (CII)-immunized DBA/1J mice were cultured in vitro with or without CII; 3 days later, cells were collected and the percentage of Th1, regulatory T cells (Treg) and Th17 cells were analysed using flow cytometric intracellular staining, as described in Methods. (b) The representative flow cytometric results summarized in (a) are shown; the percentages of relative cytokine- or transcript factor-expression T cells are indicated in the dot-plots. (c) After 3 days' culture with or without CII, CD4+ T cells were purified from SMNCs by magnetic sorting kits and were assessed for transcript levels of Hes1 and four Notch receptors, including Notch1, Notch2, Notch3 and Notch4 by real-time polymerase chain reaction (PCR). *P < 0·05.
Activation of Notch signalling and increased expression of Notch3 mRNA in collagen-specific T cell response
As recent evidence suggests that Notch signalling is an important modulator of T cell-mediated immune responses, we next wanted to know whether Notch signalling could be activated in the collagen-specific T cell response. To explore this, SMNCs from immunized mice were restimulated by CII for 3 days and then CD4+ T cells were purified by magnetic sorting kits and assessed for increased transcript levels of Hes1 and four Notch receptors, including Notch1, Notch2, Notch3 and Notch4. Hes1 is a downstream target of Notch signalling, and an increase in transcripts of this gene indicates active Notch signalling in cells. As shown in Fig. 1c, CII restimulation induced up-regulated transcript levels of Hes1 in CII-reactive CD4+ T cells. The mRNA level of Notch3 was also up-regulated significantly, while the levels of the other three Notch receptors were not increased. These data indicate the participation of Notch signalling and the potential role of Notch3 receptor in CII-specific Th1- and Th17-type expansion.
Inhibition of Notch signalling by DAPT and Notch3 antibody decrease collagen-specific T cell proliferation and attenuate Th1- and Th17-type responses
Based on the above data, we next used the γ-secretase inhibitor DAPT, which prevents activation of all Notch receptors by inhibiting the final enzymatic cleavage and specific neutralizing antibody to Notch3 to determine the effect of Notch signalling inhibition on collagen-specific T cell responses. Data in Fig. 2a indicate that both DAPT (5 µM) and α-Notch3 (10 µg/ml) could induce suppression for CII-initiated lymphoproliferation well, as expected. As shown in Fig. 2b, addition of DAPT reduced the percentage of Th1 and Th17 cells in SMNCs co-cultured with CII. Similar results were obtained when SMNCs were incubated with CII and α-Notch3. Neither DAPT nor α-Notch3 changed the percentage of Treg cells. No significant difference of suppression effect between DAPT and α-Notch3 was observed. These results demonstrate that activation of Notch signalling through Notch3 receptor mediates collagen-specific Th1- and Th17-type expansion.
Fig. 2.
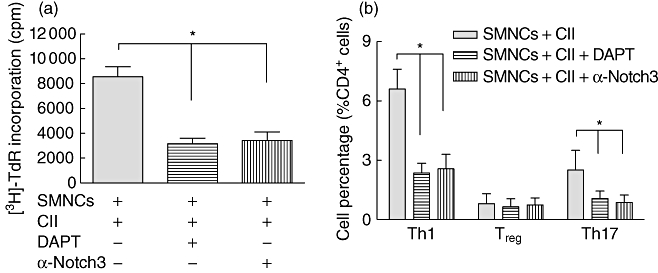
Inhibition of Notch signalling by N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) and Notch3 antibody decrease the collagen-specific T cell proliferation and attenuate T helper type 1 (Th1)- and Th17-type responses. Spleen mononuclear cells (SMNCs) from collagen II (CII)-immunized DBA/1J mice were restimulated with CII in the presence of DAPT (5 µM) or α-Notch3 (10 µg/ml). (a) The proliferation of SMNCs was determined by tritiated thymidine (3[H]-TdR) incorporation. Results were expressed as mean ± standard deviation of counts per minute (cpm) of triplicates or quadruplicates. (b) The percentage of Th1, regulatory T cells (Treg) and Th17 cells in SMNCs were analysed using flow cytometric intracellular staining.
Notch ligand Delta-like 1 promotes collagen-specific Th1- and Th17-type expansion
Notch signalling is initiated by ligand–receptor interaction between neighbouring cells. We next used Delta-like 1-Fc and Jagged1-Fc fusion proteins that bind to Notch receptors and activate the Notch pathway to explore the effect of Notch ligands on this expansion. As depicted in Fig. 3a and b, the addition of Delta-like 1-Fc fusion protein increased the percentage of Th1 and Th17 cells while Jagged1-Fc fusion protein did not change the percentage of Th1 and Th17 cells significantly. The percentage of Treg cells remained low with or without the treatment of two Notch ligands. These results confirm the engagement of Notch signalling and indicate that it should be Delta-like 1 rather than Jagged1 that promotes collagen-specific Th1- and Th17-type expansion.
Fig. 3.
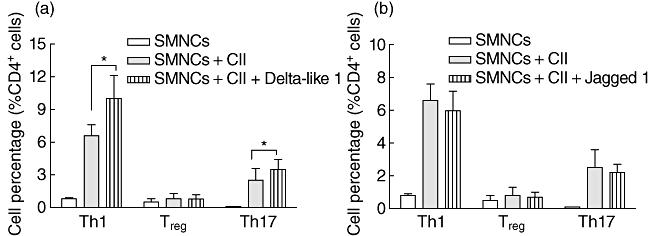
Notch ligand Delta-like 1 promote the collagen-specific T helper type 1 (Th1)- and Th17-type expansion. Spleen mononuclear cells (SMNCs) from collagen II (CII)-immunized DBA/1J mice were cultured in vitro with or without CII in the presence of Notch ligand Delta-like 1 (a) or Jagged1-Fc (b) fusion protein; 3 days later, cells were collected and the percentage of Th1, regulatory T cells (Treg) and Th17 cells were analysed using flow cytometric intracellular staining.
Discussion
A fundamental feature of T cell-dependent immune responses is the necessity for a very small population of CD4+ T cells to undergo clonal expansion and activation following encounter with a specific antigen. In the present study, we established an in vitro collagen-specific proliferation system in which the percentages of three CD4+ T cell subsets were analysed. The increased percentage of Th1 cells and Th17 cells after CII restimulation indicates that collagen-specific reactivation tends to Th1- and Th17-type expansion. T cell responses to CII immunization have been studied extensively in mice with the I-Aq haplotype, which are highly susceptible to CIA (e.g. the DBA/1 strain). Intradermal injection of CII emulsified in complete Freund's adjuvant results in the activation and expansion of antigen-specific CD4+ T cells with the Th1 phenotype, which initiate the harmful response [15]. By using tetrameric human leucocyte antigen D-related 1 (HLA-DR1) with a covalently bound immunodominant CII peptide, Latham et al. also reported that DR1–CII-tetramer+ cells expressed high levels of Th1 and proinflammatory cytokines, including IL-2, IFN-γ, IL-6, tumour necrosis factor (TNF)-α, and especially IL-17 [16]. These data confirm the pathogenic role of CII-specific Th1 and Th17 cells in promoting the development of disease in the arthritis model.
Notch signalling plays an essential role in the development of embryonic haematopoietic stem cells and influences multiple lineage decisions of developing lymphoid and myeloid cells. Moreover, recent evidence suggests that Notch is an important modulator of T cell-mediated immune responses. One of the most intriguing, and perhaps most controversial, functions assigned recently to Notch proteins is that of a regulator of Th cell differentiation. To assess whether Notch signalling is activated in collagen-specific Th1- and Th17-type expansion, we determined the abundance of the Notch target gene Hes-1. Hes-1 is the most well-characterized, γ-secretase-dependent transcriptional target gene of Notch signalling, and up-regulated expression of Hes-1 may be related to activated Notch signalling. As expected, we observed up-regulated transcript levels of Hes1. When we used γ-secretase inhibitor DAPT to block Notch signalling in SMNCs from CII immunized mice co-cultured with CII, we found that DAPT reduced T cell proliferation and the percentage of Th1 and Th17 cells. Palaga et al. also reported that γ-secretase inhibitor (GSI)-mediated inhibition of Notch signalling in peripheral CD4+ T cells stimulated by CD3- and CD28-specific antibodies resulted in decreased T cell proliferation and reduced IFN-γ production [12].
We next determined which Notch receptor mediated the CII-specific Th1 and Th17 cell expansion. After co-culture with CII for 72 h, CD4+ T cells were isolated from SMNCs derived from CII immunized mice and transcript levels of four Notch receptors, including Notch1, Notch2, Notch3 and Notch4, were assessed. We found that CII restimulation up-regulated Notch3 transcription significantly in CD4+ T cells. To further confirm the specific role of Notch3, we added specific neutralizing antibody to Notch3 to the SMNCs restimulation system and found that anti-Notch3 treatment reduced T cell proliferation and the frequency of Th1 and Th17 cells. These results indicate that Notch3 plays an important role in CII-specific T cell proliferation and expansion. Over-expression of the Notch3 intracellular domain in T cells has been reported to induce differentiation of IFN-γ-secreting Th1 but reduced IL-4-secreting Th2 cells. When Notch3 expression was inhibited with anti-sense-DNA, the Th1-type differentiation was also inhibited [17]. Our results were partly different from another research group, which explored the role of Notch signalling in myelin-reactive CD4+ T cells using the EAE model, and found that both Notch1 and Notch3 were up-regulated upon specific antigen restimulation, although Notch1 inhibition did not affect the proliferation and differentiation of autoreactive T cells [13]. These different data may result from the use of different antigens as well as different animal models. Nevertheless, we agree with the important role of Notch3 in antigen-specific Th1 and Th17 cell expansion other than Treg cells.
Notch signalling is initiated by ligand–receptor interaction between neighbouring cells. We next asked which Notch ligands are involved in CII-specific T cell proliferation and differentiation by the addition of Delta-like 1-Fc and Jagged1-Fc fusion proteins into SMNCs co-cultured with CII from CII immunized mice. Our results indicate that it should be Delta-like 1 rather than Jagged1 that promotes the collagen-specific Th1- and Th17-type expansion. In EAE, pathogenic Th1 and Th17 cells develop in the central nervous system, causing autoimmunity. Specific antibodies against Delta-like 1, which attenuated EAE, have opposite effects to antibodies against Jagged1 which exacerbated EAE [18]. Maekawa et al. reported that Delta-like 1 interaction with Notch3 on CD4+ T cells promoted development towards the Th1 phenotype [17]. However, Delta-like 4-expressing dendritic cells (DCs), when activated with Toll-like receptor (TLR) ligands or Mycobacterium antigens, can promote the generation of Th17 cells through activation of the Th17 cell-specific transcription factor retinoic acid-related orphan receptor γ-T (RORγt) [19,20]. The specific interactions of Notch ligands and receptors on T cells may be regulated by the expression pattern of Notch ligands on neighbour cells [17]. In any case, the question of ligand receptor specificity in their ability to induce Th cell differentiation is currently unclear, and will need to be determined in the future [9].
In summary, the present study demonstrates that Notch signalling is engaged in collagen-specific Th1- and Th17-type expansion involving Notch3 and Delta-like1. Selective inhibition of Notch signalling transduced by Notch3 or Delta-like1 may offer a new strategy for the treatment of RA.
Acknowledgments
This study was supported by grants from the Natural Science Foundation of China (30872335), Society Development Foundation of Zhenjiang (SH2008035) and Medical Science and Technology Development Foundation of Jiangsu Province Department of Health (H200950). The authors wish to thank Drs L.W. Lu and L.J. Xin for their helpful suggestions, discussions and excellent technical assistance.
Disclosure
The authors declare that they have no conflict of interest.
References
- 1.Cho YG, Cho ML, Min SY, Kim HY. Type II collagen autoimmunity in a mouse model of human rheumatoid arthritis. Autoimmun Rev. 2007;7:65–70. doi: 10.1016/j.autrev.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Yuan JS, Kousis PC, Suliman S, Visan I, Guidos CJ. Functions of notch signaling in the immune system: consensus and controversies. Annu Rev Immunol. 2010;28:343–65. doi: 10.1146/annurev.immunol.021908.132719. [DOI] [PubMed] [Google Scholar]
- 3.Minato Y, Yasutomo K. Regulation of acquired immune system by notch signaling. Int J Hematol. 2005;82:302–6. doi: 10.1532/IJH97.05095. [DOI] [PubMed] [Google Scholar]
- 4.Hoyne GF. Notch signaling in the immune system. J Leukoc Biol. 2003;74:971–81. doi: 10.1189/jlb.0303089. [DOI] [PubMed] [Google Scholar]
- 5.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi E, Chiba S, Kumano K, et al. Expression of Notch ligands, Jagged1, 2 and Delta1 in antigen presenting cells in mice. Immunol Lett. 2002;81:59–64. doi: 10.1016/s0165-2478(01)00326-1. [DOI] [PubMed] [Google Scholar]
- 7.Allman D, Punt JA, Izon DJ, Aster JC, Pear WS. An invitation to T and more: notch signaling in lymphopoiesis. Cell. 2002;109(Suppl):S1–11. doi: 10.1016/s0092-8674(02)00689-x. [DOI] [PubMed] [Google Scholar]
- 8.Maillard I, Adler SH, Pear WS. Notch and the immune system. Immunity. 2003;19:781–91. doi: 10.1016/s1074-7613(03)00325-x. [DOI] [PubMed] [Google Scholar]
- 9.Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity. 2010;32:14–27. doi: 10.1016/j.immuni.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Adler SH, Chiffoleau E, Xu L, et al. Notch signaling augments T cell responsiveness by enhancing CD25 expression. J Immunol. 2003;171:2896–903. doi: 10.4049/jimmunol.171.6.2896. [DOI] [PubMed] [Google Scholar]
- 11.Laky K, Fowlkes BJ. Notch signaling in CD4 and CD8 T cell development. Curr Opin Immunol. 2008;20:197–202. doi: 10.1016/j.coi.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palaga T, Miele L, Golde TE, Osborne BA. TCR-mediated Notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. J Immunol. 2003;171:3019–24. doi: 10.4049/jimmunol.171.6.3019. [DOI] [PubMed] [Google Scholar]
- 13.Jurynczyk M, Jurewicz A, Raine CS, Selmaj K. Notch3 inhibition in myelin-reactive T cells down-regulates protein kinase C theta and attenuates experimental autoimmune encephalomyelitis. J Immunol. 2008;180:2634–40. doi: 10.4049/jimmunol.180.4.2634. [DOI] [PubMed] [Google Scholar]
- 14.Jiao Z, Wang W, Guo M, et al. Expression analysis of Notch-related molecules in peripheral blood T helper cells of patients with rheumatoid arthritis. Scand J Rheumatol. 2010;39:26–32. doi: 10.3109/03009740903124424. [DOI] [PubMed] [Google Scholar]
- 15.Fournier C. Where do T cells stand in rheumatoid arthritis? Joint Bone Spine. 2005;72:527–32. doi: 10.1016/j.jbspin.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Latham KA, Whittington KB, Zhou R, Qian Z, Rosloniec EF. Ex vivo characterization of the autoimmune T cell response in the HLA-DR1 mouse model of collagen-induced arthritis reveals long-term activation of type II collagen-specific cells and their presence in arthritic joints. J Immunol. 2005;174:3978–85. doi: 10.4049/jimmunol.174.7.3978. [DOI] [PubMed] [Google Scholar]
- 17.Maekawa Y, Tsukumo S, Chiba S, et al. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. 2003;19:549–59. doi: 10.1016/s1074-7613(03)00270-x. [DOI] [PubMed] [Google Scholar]
- 18.Elyaman W, Bradshaw EM, Wang Y, et al. JAGGED1 and delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5990–8. doi: 10.4049/jimmunol.179.9.5990. [DOI] [PubMed] [Google Scholar]
- 19.Mukherjee S, Schaller MA, Neupane R, Kunkel SL, Lukacs NW. Regulation of T cell activation by Notch ligand, DLL4, promotes IL-17 production and Rorc activation. J Immunol. 2009;182:7381–8. doi: 10.4049/jimmunol.0804322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito T, Schaller M, Hogaboam CM, et al. TLR9 regulates the mycobacteria-elicited pulmonary granulomatous immune response in mice through DC-derived Notch ligand delta-like 4. J Clin Invest. 2009;119:33–46. doi: 10.1172/JCI35647. [DOI] [PMC free article] [PubMed] [Google Scholar]