

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motor neurons, leading to muscle weakness, paralysis, and death, most often from respiratory failure. The only FDA approved drug for the treatment of ALS, riluzole, only extends the median survival in patients by 2–3 months. There is an urgent need for novel therapeutic strategies for this devastating disease. Using a high-throughput screening assay targeting an ALS cultured cell model (PC12-G93A-YFP cell line), we previously identified three chemotypes that were neuroprotective. We present a further detailed analysis of one promising scaffold from that group, pyrimidine-2,4,6-triones (PYTs), characterizing a number of PYT analogs using SAR and ADME. The PYT compounds show good potency, superior ADME data, low toxicity, brain penetration, and excellent oral bioavailability. Compounds from this series show 100% efficacy in the protection assay with a good correlation in activity between the protection and protein aggregation assays. The modifications of the PYT scaffold presented here suggest that this chemical structure may be a novel drug candidate scaffold for use in clinical trials in ALS.
Keywords: Pyrimidine-2,4,6-triones (PYT); amyotrophic lateral sclerosis; mutant G93A SOD1; blood-brain barrier penetration; ADME
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the progressive loss of motor neurons, leading to muscle atrophy, paralysis, and death within 3–5 years1. While sporadic (~90% of cases) and familial forms of the disease display similar clinical features, a direct causative pathway to neuronal dysfunction and death has not yet been established. The similarities in the clinical and pathological features of familial ALS and sporadic ALS has led investigators to use the models of genetic mutations associated with familial ALS as a strategy for elucidating disease pathogenesis and defining novel treatments for both forms of the disease. The incidence of ALS is ~2 cases per 100,000 individuals, which corresponds to a 1 in 2000 lifetime risk to develop the disease, with death occurring within 2–5 years of diagnosis2. An increased prevalence of ALS has been observed,3 with the worldwide affected population estimated to reach ~108,000 patients by 2012,4 with one -third residing in the U.S5. Disability is significant and patient care costs in the late stages of ALS can exceed $200,000 per year,6 placing immense emotional and economic burdens on patients and their families. There is no effective treatment for ALS; riluzole, a presumptive anti-glutamatergic agent,7 is the only FDA-approved therapeutic drug for ALS, extending the median survival by only 2–3 months8,9.
While the vast majority of ALS cases occur sporadically, about 10% of ALS cases are familial. ALS pathology is caused by the degeneration of large pyramidal neurons in the motor cortex and associated corticospinal tracts, lower motor neurons originating in the brainstem nuclei and the spinal cord anterior horn. There has been accumulating evidence that ALS is a non-cell-autonomous disease in which glia play a prominent active role in disease progression10. Major neuropathological findings include: (1) neurofilamentous swelling of proximal axons and accumulation of neurofilament and peripherin in axons and cell bodies; (2) perikaryal inclusion bodies containing phosphorylated neurofilament proteins, ubiquitin, and mutant Cu/Zn superoxide dismutase (SOD1) in some cases of familial ALS; (3) fragmentation of the Golgi apparatus and formation of cytoplasmic neuronal inclusion bodies (Bunina bodies); (4) reduced diameter of distal neurons; and (5) axonal Wallerian degeneration and attenuation of dendrites. The genetic linkage of several mutations in the gene for Cu/Zn superoxide dismutase (SOD 1), which promote protein aggregation, and seen in about 20% of the cases of familial ALS,11 provided the first indication of a potential causal factor in the disease process.
Imbalances in protein homeostasis, which can be caused by cell stress or expression of certain mutant proteins, can result in the appearance of alternative conformational states that are able to self-associate to form protein aggregates and inclusion bodies. Aberrant protein aggregation has been reported as a common feature in familial as well as sporadic forms of ALS and mutant SOD1 transgenic mouse models12,13. A cultured cell model, PC12-G93A-YFP cell line, developed by Morimoto and coworkers14, was utilized in a high throughput assay to identify compounds that protect against mutant SOD1-induced cytotoxicity. Two assays were established: a cytotoxicity protection assay, in which compounds were screened for their ability to protect cells from the cytotoxic effects of aggregated mutant SOD1, and a protein aggregation assay, in which compounds that are active in the cytotoxicity protection screen were tested for their ability to reduce the aggregation of mutant SOD1.15
A library of approximately 50,000 small molecules was assembled for screening. The library was constructed to address two goals: maximization of chemical structural diversity and inclusion of biologically well-characterized compounds. Pyrimidine-2,4,6-triones (PYT) were identified among the active compounds and selected as one of the scaffolds for chemistry optimization. To the best of our knowledge, PYT compounds have not been reported in any publication or patent for the treatment of ALS, and, therefore, represent a novel chemical scaffold for this disease. Compounds from this series show 100% efficacy in the protection assay with a good correlation in activity between the protection and protein aggregation assays. Thus, it is reasonable to deduce that these compounds protect PC12 cells against mutant SOD1-induced cytotoxicity, at least in part, by reducing SOD1 protein aggregation, and that these compounds are not themselves cytotoxic.
Chemistry
Parallel synthesis technology was applied to the synthesis of PYT compounds. As shown in Scheme 1, starting from commercially available S,S-dimethyl carbonodithioate and amines, urea intermediates were synthesized in high yields.16 After the solvent was removed under vacuum, without any further purification, the crude urea compounds were treated with malonic acid in acetic acid/acetic anhydride at 60–90 °C to provide PYT compounds in moderate to high yields.17 Most of the final products were easily recrystallized from ether/hexanes. The reactions were carried out in 20 mL vials over a period of two days, and often 8–12 reactions were set up at the same time on pie blocks with an IKA stir plate. This method readily resulted in high yields and excellent purity of diverse PYT analogs for SAR studies.
Scheme 1.

Parallel Synthesis of PYTs
A new synthetic route was designed for the synthesis of PYT analogs with different R1 and R2 groups as shown in Scheme 2. By changing the reaction solvent, temperature, and the amount of starting material, a precursor of the urea compound could be obtained with very high yield after purification, and this reaction could be carried out on a large scale. The precursor reacted with another amine to provide the desired urea intermediate with different R1 and R2 groups. Without any further purification, PYT compounds with different R1 and R2 group could be achieved using parallel synthesis procedures.
Scheme 2.

Synthesis of PYT analogs with different R1 and R 2 groups
Further modifications at the R3 position of PYT were executed as shown in Scheme 3. Following the same parallel synthesis technology, reactions between crude urea intermediates and substituted malonic acid provided the PYT analogs with sp3 hybridized groups, such as methyl and dimethyl groups. Introduction of an sp2 hybridized group on the PYT ring at the R3 position was accomplished by a Knoevenagel condensation between PYT analogs with aldehydes catalyzed by a mixture of benzoic acid and piperidine.18
Scheme 3.

Synthesis of PYT analogs with different types of R 3 groups
Further attempts to modify the PYT ring to enol forms were successfully performed, as shown in Scheme 4.
Scheme 4.

Synthesis of enol forms of PYT analogs
Reactions between urea intermediates, such as 48, with oxalyl dichloride or succinyl dichloride provided the desired five-membered ring products (49, 50, 51) or seven-membered ring product, such as 52 (Scheme 5).
Scheme 5.

Synthesis of other PYT analogs
Screening Results
Compound activity was determined using the previously described cytotoxicity protection assay.15 As shown in Figure 1, PYT compounds contain three carbonyl groups and two nitrogen atoms. From the initial high throughput screen, active compounds had R3 as hydrogen or the carbon between the carbonyls was sp2 hybridized (alkenyl groups). When R3 was alkyl, the compounds were usually inactive. When both R1 and R2 were H, the PYT compounds showed very weak activity. When R1 and R2 were alkyl chains with two or more carbons, the compounds showed good potency. Some compounds with R1 as hydrogen and R2 as alkyl (>2 carbons) also showed moderate to good potency, depending on the R3 group.
Figure 1.

PYT Compounds
When one of the R groups on the PYT ring is H, the compounds (1 and 2) exhibited no activity (Figure 2). When both R groups are alkyl groups, the potency increased with the size of the alkyl chain (compare 3, 4, and 5). The introduction of Ph rings to the side chain of PYTs generally increased the potency. Comparing 6, 7, 8, and 10, it is obvious that the greater the number of carbon atoms in the chain, the higher the potency of the analogs. However, bulkiness in the side chain significantly reduces potency, such as in 11. Changing the phenyl ring to a heterocycle, such as a thiophene ring, did not improve the potency (7 vs 12). Further introduction of other atoms, such as O atoms, to provide additional possible binding sites also did not improve the potency (8 vs 13). However, a phenyl ring resulted in compounds that were generally more potent than those with a much smaller ethyl group (13 vs 14).
Figure 2.

PYT analogs with simple R groups
The introduction of Ph rings resulted in PYT analogs with higher potency and with further opportunities for SAR studies. PYT analogs with different substituted groups on the Ph rings were synthesized by the same parallel synthesis technology (Figure 3). PYT analogs with electron withdrawing groups on the Ph rings showed greater potency than those with electron donating groups. The position of substitution on the Ph ring did not show obvious effects on the potency (compare 15, 16, and 17). Different sized electron withdrawing groups, such as F and Cl, showed similar small effects on the potency (16 vs 18 and 17 vs 19). Additional electron-withdrawing groups on the Ph ring slightly increased the potency of the PYTs (compare 18 and 19 to 20). Extra electron-donating groups on the Ph ring decreased the potency of PYTs (21 vs 22).
Figure 3.

PYT analogs with different substituted groups on the Ph rings
PYT analogs with different R1 and R2 groups did not show any obvious improvement in potency despite several combinations of different side chain groups (Figure 4). The range of potency was approximately 1 to 4 μM. If one side chain was held constant and the other side chain modified, the change in the variable side chain would affect the potency of the compound as discussed above; however, the trends were not followed when both chains were modified. Comparing 23 to 27, the change in potency was very small although the size of the right side chain was modified very differently. The introduction of heterocycles or O atoms on one side chain affected the potency only very slightly (28, 31, 32, and 33). An electron-withdrawing group on one side chain increased the potency slightly (29 vs 7; 28 vs 31) and an electron-donating group decreased the potency (30 vs 7). The introduction of a basic nitrogen into the side chain of the PYT led to an inactive analog (34).
Figure 4.

PYT analogs with different R1and R2 groups
As shown in Figure 5, 37 and 38, with sp3 hybridized groups, had no activity and even 39, with an sp2-hybridized group but no aromatic ring at the end of the double bond, was devoid of activity. An sp2-hybridized group with an aromatic ring was key to good potency. The aromatic ring could be a phenyl ring or other heterocycle. The electron density and substitution pattern on the phenyl ring at R3 did not have much effect on the potency (40–43). A substituted pyrrole ring at R3 enhanced potency, at least in comparison of 44 and 45.
Figure 5.

PYT analogs with different R3 groups
The conformation of the six-membered ring of the PYT core was elucidated by X-ray crystallography of 3 and 43. As shown in Figure 6, the six-membered ring of the PYT core of 3 and 43 is flat, similar to a benzene ring, with both substituents cis. This unusual structure may be responsible for the activity.
Figure 6.

X-ray structure of the PYT core from different angles
Compounds 46 and 47 (Scheme 4), the vinyl chloride and enol ether, respectively, derived from 7, are still planar, but are inactive. Therefore, planarity alone is not sufficient for activity. To test the effect of core ring size, 49–51, having a five-membered ring, and 52, the corresponding seven-membered ring analog, were synthesized (Scheme 5); none was active. Possibly the distance or angle between the carbonyls is critical for activity.
Mutant SOD1-induced cytotoxicity protection assay
PC12 cells expressing G93A SOD1-YFP were employed to develop a high throughput assay to identify compounds that block mutant SOD1 toxicity. Figure 7 is a representative dose-response curve for 7 in this assay.
Figure 7.
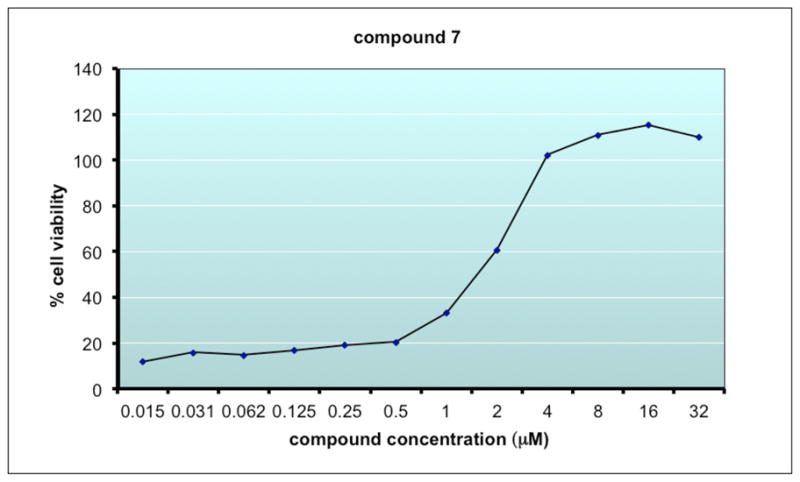
Dose-response curve for 7 in G93A-SOD1 protection assay
Inhibition of high content aggregation of G85R SOD1 by 7
The reduction in the number of aggregates of G85R SOD1 by 7 was found to be concentration dependent (Figure 8). Fluorescence images of cells treated with MG132 and with MG132 plus 10 μM compound 7 showed a significant reduction in aggregates in the 7-treated cells (see Supporting Information).
Figure 8.

Concentration dependent reduction of aggregates by 7 in the high content G85R-SOD1 aggregation assay
In vitro and in vivo ADME studies
In vitro ADME studies were conducted on 7, 10, and 16. As shown in Table 1, compound 7 showed the best overall combination of potency, PBS solubility, and microsomal and plasma stabilities. Fluorine substitution on the phenyl ring of 7 (compound 16) worsened the ADME properties except for plasma stability. Extending the two-carbon side chains of 7 by two carbons to give 10 was beneficial for enhancement of the potency and solubility, but detrimental for microsomal and plasma stability. The plasma protein binding for 7 (at 2 μM concentration) was 95% for rat and 90% for human plasma.
Table 1.
ADME properties for selected PYT compounds
| Potency (EC50) | Solubility (16h) | Human Metabolic Potential (T1/2) | Mouse Metabolic Potential (T1/2) | Plasma Stability (Calc T1/2) | |
|---|---|---|---|---|---|
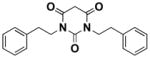 |
1.68 uM | 62.5 uM | 61 min | 39 min | 60.5 min |
| 0.68 uM | 105 uM | 15 min | 11 min | 21.8 min | |
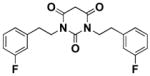 |
3.23 uM | 31.3 uM | 31 min | 22 min | 131.2 min |
Caco-2 permeability assays were carried out on 7, 10, and 16 (Table 2). Permeability for 7 and 16 is very good, with favorable asymmetry ratios. Although permeability for 10 is good, there appears to be a potential efflux problem. The ability of 7 to penetrate the blood-brain-barrier was confirmed, showing a 3:1 blood:brain ratio (150 μM: 50 μM) (Figure 9). Although blood is measured in μL and brain in mg, the density of blood plasma is 1.025 mg/μL.19 Therefore, the weight of blood is roughly equivalent to its volume, and a direct comparison between the amount of compound in blood and brain can be made. This indicates that sufficient compound can be delivered to the brain to attain maximum activity, based on the in vitro cell assay.
Table 2.
Caco-2 permeability assays
| Compound | Concentration (uM) | Assay duration (h) | Mean A→B Papp (10−6 cm s−1) | Mean B→A Papp (10−6 cm s−1) | Asymmetry ratioa |
|---|---|---|---|---|---|
| 7 | 50 | 2 | 73.4 | 26.0 | 0.4 |
| 10 | 50 | 2 | 9.2 | 30.2 | 3.3 |
| 16 | 50 | 2 | 66.3 | 17.1 | 0.3 |
Papp(B→A)/Papp(A→B)
Figure 9.
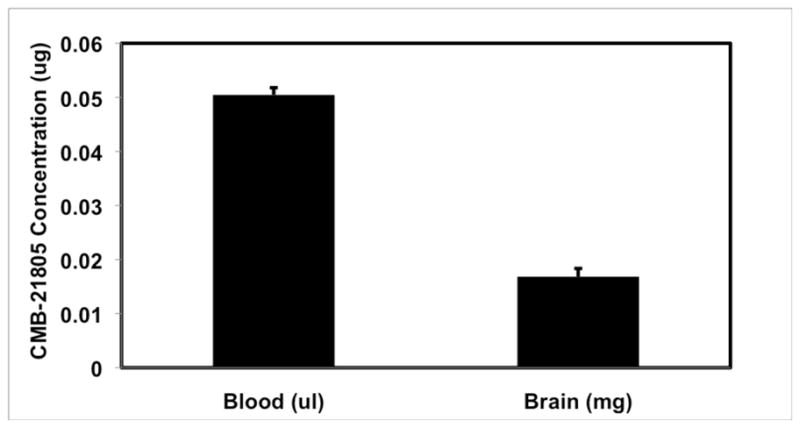
Blood Brain Penetration of 7
The IC50 values for 7 with the major CYP isozymes present in human liver microsomes, CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, were all >50 μM, indicating its lack of interference with major metabolism enzymes in liver.
The human ether-a-go-go related gene (hERG) encodes for a protein that is the inner pore-forming portion of a potassium channel in heart muscle. This potassium ion channel controls the flow of K+ ions across the cell membrane. It has been reported that compounds that inhibit this channel produce heart arrhythmias (acquired long QT syndrome),20 which can lead to death in some cases, and unfortunately in some instances this toxic liability was identified only after clinical introduction of the drug. Therefore, it is imperative that compounds do not block the hERG channel. The in vitro effect of 7 on hERG potassium channel current expressed in human embryonic kidney cells (HEK293) was evaluated at room temperature using a patch-clamp technique, the most definitive in vitro hERG inhibition assay. A positive control (E-4031) was used to confirm the sensitivity of the test system, and the TurboSol evaluation system was performed to confirm that poor compound solubility was not a reason for low hERG inhibition in the test system. As shown in Table 4, there is little hERG channel blockage by 7.
Table 4.
Effect of 7 on the hERG channel
| Compound | n | Concentration (μM) | Mean hERG Inhibition (%) | Standard Deviation | Standard Error |
|---|---|---|---|---|---|
| 7 | 2 | 10 | 4.9 | 1.8 | 1.3 |
| Positive control | 2 | 0.5 | 98.7 | 0.7 | 0.5 |
In vivo PK profiling of 7 at 1 mg/kg bolus intravenous and oral administration doses in Sprague-Dawley rats was determined (Table 5). Although a moderate plasma clearance was observed for both i.v. and p.o. administration, the oral bioavailability was 98%, indicating extremely good oral absorption.
Table 5.
Plasma PK profile of 7 (1 mg/kg i.v. & 1 mg/kg p.o.) in Sprague-Dawley rats administered in PBS
| Route | AUC last (ng · h/mL) | AUC/Dose (ng · h/mL/dose) | T1/2 (h) | Cl (mL/min/kg) | Vss (L/kg) | Cmax (ng/mL) | Tmax (h) | F (%) |
|---|---|---|---|---|---|---|---|---|
| i.v. | 121 | 126 | 0.95 | 133 | 9.0 | - | - | - |
| p.o. | 124 | 124 | 1.4 | - | - | 55 | 0.75 | 98 |
A profiling screen was conducted for 7 with a broad package of 68 in vitro radioligand binding assays covering proteins involved in the most commonly observed adverse CNS, cardiovascular, pulmonary, and genotoxic events in order to detect potential adverse activity, additional unexpected activity, relative selectivity, and specificity (MDS Pharma Services). Compound 7 was tested at a 10 μM concentration in duplicate. No significant inhibitory effect was noted (the four receptors showing the greatest effects [and % inhibition] were dopamine D3 [41%], sodium channel site 2 [21%], estrogen ERα [20%], and GABA transporter [19%]; see Supporting Information for detailed results), suggesting that 7 would be unlikely to introduce adverse effects related to the enzymes and receptorstested.
Conclusions
PYT analogs were identified as potential drug candidates with good potency, ADME properties, and protein profiling properties. Some generalities about the SAR of PYT analogs can be made: 1) The size of the R1/R2 group is critical for good potency; 2) Electron density changes on the Ph ring of the R1/R2 groups do not affect the potency; 3) Other potential pharmacophores on the R1/R2 groups (e.g., oxygen atoms) do not affect potency; 4) R3 groups with sp2 hybridization at the core ring increase potency but increase the molecular weight as well; 5) A six-membered flat ring of PYT is essential for good potency. One PYT compound, 7, has exhibited a desirable combination of potency, ADME properties, low toxicity, brain penetration, oral bioavailability, and protein profiling and has been selected for further animal testing; this may be a novel drug candidate scaffold for use in ALS clinical trials.
Experimental Section
General Procedures
All reactions were carried out in oven- or flame-dried glassware under an atmosphere of air unless otherwise noted. Except as otherwise indicated, all reactions were magnetically stirred and monitored by analytical thin-layer chromatography using Whatman pre-coated silica gel flexible plates (0.25 mm) with F254 indicator or Merck pre-coated silica gel plates with F254 indicator. Visualization was accomplished by UV light (256 nm) or by potassium permanganate and/or phosphomolybdic acid solution as an indicator. Flash column chromatography was performed using silica gel 60 (mesh 230–400) supplied by E. Merck. Yields refer to chromatographically and spectrographically pure compounds, unless otherwise noted. Commercial grade reagents and solvents were used without further purification except as indicated below. Tetrahydrofuran (THF) was distilled from sodium-benzophenone ketyl under an atmosphere of dry nitrogen.
1H NMR and 13C NMR spectra were recorded on a Bruker Avance III (500 MHz 1H, 125 MHz 13C) with DCH Cryo-Probe. Chemical shift values (δ) are reported in ppm relative to CDCl3 [δ 7.26 ppm (1H), 77.16 ppm (13C)]. The proton spectra are reported as follows δ (multiplicity, number of protons). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), h (heptet), m (multiplet) and br (broad). Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA. The C, H, and N analyses are performed by combustion using automatic analyzers, and all of the compounds analyzed showed >95% purity.
Typical procedure for parallel synthesis of PYT analogs
S,S-Dimethyl carbonodithioate (0.5 mmol) was added dropwise to a solution of amine (1.05 mmol) in MeOH (5 mL) with stirring, and the mixture was heated at 60 °C for 24 h. After removal of solvent in vacuo, the crude urea residue and malonic acid (0.5 mmol) were dissolved in 3 mL of glacial acetic acid. The mixture was heated to 60 °C and 2 mL of acetic anhydride was added over 30 min. The reaction mixture was heated with stirring at 90 °C for 4 h. The solvent was removed under vacuum at 60 °C, and the residue was recrystallized from ether/hexanes or purified by silica gel chromatography to provide the desired PYT analog.
1,3-Diphenethylurea (48)
1H NMR (500 MHz, CDCl3) δ 7.27 (m, 4 H), 7.21 (m, 2 H), 7.10 (m, 4 H), 4.46 (br, 2 H), 3.38 (m, 4 H), 2.76 (t, 4 H); 13C NMR (125 MHz, CDCl3) δ 158.1, 139.2, 128.9, 128.6, 126.5, 41.7, 36.5.
1-Butylpyrimidine-2,4,6(1H,3H,5H)-trione (1)
1H NMR (500 MHz, CDCl3) δ 8.25 (s, 1 H), 3.90 (t, 2 H), 2.17 (s, 2 H), 1.60 (m, 2 H), 1.35 (m, 2 H), 0.94 (t, 3 H); 13C NMR (125 MHz, CDCl3) δ 170.6, 161.0, 149.0, 126.8, 40.8, 24.6, 20.1, 13.8. EC50 > 32 μM.
1-Cyclohexylpyrimidine-2,4,6(1H,3H,5H)-trione (2)
1H NMR (500 MHz, CDCl3) δ 7.92 (s, 1 H), 4.66 (m, 1 H), 2.70 (s, 2 H), 2.33 (m, 2 H), 1.84 (m, 2 H), 1.65 (m, 2 H), 1.36 (m, 2 H), 1.25 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 196.4, 171.2, 160.9, 148.9, 96.0, 54.7, 29.1, 26.4, 25.2, 24.7. Elemental Analysis: Comb. Anal. (C10H14N2O3), Calculated: C, 57.13; H, 6.71; N, 13.33; O, 22.83; Found: C, 56.92; H, 6.31; N, 11.21; Purity: >95%. EC50 > 32 μM.
1,3-Dipropylpyrimidine-2,4,6(1H,3H,5H)-trione (3)
1H NMR (500 MHz, CDCl3) δ 3.81 (m, 4 H), 3.64 (s, 2 H), 1.61 (m, 4 H), 0.92 (t, 6 H); 13C NMR (125 MHz, CDCl3) δ 164.7, 151.5, 137.8, 43.6, 39.7, 21.3, 11.3. Elemental Analysis: Anal. (C10H16N2O3), Calculated: C, 56.59; H, 7.60; N, 13.20; O, 22.61; Found: C, 56.30; H, 7.51; N, 13.25; Purity: >95%. EC50 7.68 μM.
1,3-Dicyclohexylpyrimidine-2,4,6(1H,3H,5H)-trione (4)
1H NMR (500 MHz, CDCl3) δ 4.58 (m, 2 H), 3.59 (s, 2 H), 2.25 (m, 4 H), 2.25 (m, 4 H), 1.84 (m, 4 H), 1.63 (m, 6 H), 1.35-1.19 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 165.1, 151.4, 55.4, 41.1, 29.2, 26.4, 25.2. Elemental Analysis: Comb. Anal. (C16H24N2O3), Calculated: C, 65.73; H, 8.27; N, 9.58; O, 16.42; Found: C, 65.86; H, 8.32; N, 9.53; Purity: >95%. EC50 = 2.97 μM.
1,3-Bis(3,3-dimethylbutyl)pyrimidine-2,4,6(1H,3H,5H)-trione (5)
1H NMR (500 MHz, CDCl3) δ 3.88 (m, 4 H), 3.63 (s, 2 H), 1.46 (m, 4 H), 0.97 (S, 9H); 13C NMR (125 MHz, CDCl3) δ 164.5, 151.5, 41.1, 39.8, 39.1, 30.0, 29.2. Elemental Analysis: Comb. Anal. (C16H28N2O3), Calculated: C, 64.83; H, 9.52; N, 9.45; O, 16.19; Found: C, 64.93; H, 9.62; N, 9.42; Purity: >95%. EC50 2.03 μM.
1,3-Dibenzylpyrimidine-2,4,6(1H,3H,5H)-trione (6)
1H NMR (500 MHz, CDCl3) δ 7.41 (m, 4 H), 7.30 (m, 6 H), 5.03 (s, 4 H), 3.69 (s, 2 H); 13C NMR (125 MHz, CDCl3) δ 164.5, 136.0, 129.3, 128.7, 128.2, 45.2, 39.8. EC50 4.58 μM.
1,3-Diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (7)
1H NMR (500 MHz, CDCl3) δ 7.29 (m, 4 H), 7.22 (m, 6 H), 4.08 (m, 4 H), 3.56 (s, 2 H), 2.86 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 151.1, 137.8, 129.0 128.7, 126.8, 43.1, 39.6, 34.1. Elemental Analysis: Comb. Anal. (C20H20N2O3), Calculated: C, 71.41; H, 5.99; N, 8.33; O, 14.27; Found: C, 71.29; H, 5.91; N, 8.27; Purity: >95%. EC50 1.68 μM.
1,3-Bis(3-phenylpropyl)pyrimidine-2,4,6(1H,3H,5H)-trione (8)
1H NMR (500 MHz, CDCl3) δ 7.25 (m, 4 H), 7.18 (m, 6 H), 3.89 (m, 4H) 3.34 (s, 2 H), 2.68 (m, 4H) 1.97 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 164.5, 151.2, 140.9, 128.4, 128.2, 126.0, 41.8, 39.3, 33.0, 28.4. EC50 1.32 μM.
1,3-Bis(3,3-diphenylpropyl)pyrimidine-2,4,6(1H,3H,5H)-trione (9)
1H NMR (500 MHz, CDCl3) δ 7.29-7.27 (m, 16 H), 7.17 (m, 4 H), 4.03 (d, 2 H), 3.90 (m, 4 H), 3.06 (s, 2 H), 2.42 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.6, 151.2, 144.2, 128.7, 127.5, 126.5, 49.7, 41.4, 39.1, 32.7. EC50 1.39 μM.
1,3-Bis(4-phenylbutyl)pyrimidine-2,4,6(1H,3H,5H)-trione (10)
1H NMR (500 MHz, CDCl3) δ 7.28 (m, 4 H), 7.19 (m, 6 H), 3.89 (t, 4 H), 3.61 (s, 2 H), 2.65 (t, 4 H), 1.65 (m, 8 H); 13C NMR (125 MHz, CDCl3) δ 164.6, 151.4, 142.0, 128.5, 128.4, 125.9, 41.9, 39.6, 35.5, 28.6, 27.6. Elemental Analysis: Comb. Anal. (C24H28N2O3), Calculated: C, 73.44; H, 7.19; N, 7.14; O, 12.23; Found: C, 73.29; H, 7.26; N, 7.20; Purity: >95%. EC50 0.68 μM.
1,3-Bis(2,3-dihydro-1H-inden-1-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (11)
1H NMR (500 MHz, CDCl3) δ 7.25 (m, 8 H), 5.46 (m, 2 H), 2.98 (m, 2 H), 2.86 (m, 2 H), 2.59 (m 2 H), 2.02 (m, 2 H), 1.80 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 170.0, 143.5, 143.2, 128.0, 126.8, 124.9, 124.1, 54.8, 34.1, 30.3, 23.6. EC50 > 32 μM.
1,3-Bis(2-(thiophen-2-yl)ethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (12)
1H NMR (500 MHz, CDCl3) δ 7.16 (m, 2 H), 6.94 (m, 2 H), 6.86 (m 2 H), 4.15 (m, 4 H), 3.61 (s, 2 H), 3.11 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 151.1, 139.8, 127.2 125.9, 124.3, 43.1, 39.7, 27.9. Elemental Analysis: Comb. Anal. (C16H16N2O3S2), Calculated: C, 55.15; H, 4.63; N, 8.04; O, 13.78; S, 18.40; Found: C, 55.15; H, 4.54; N, 8.04; Purity: >95%. EC502.16 μM.
1,3-Bis(2-phenoxyethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (13)
1H NMR (500 MHz, CDCl3) δ 7.24 (m, 4 H), 6.94 (m, 2 H), 6.86 (m, 4 H), 4.32 (t, 4 H), 4.19 (m, 4 H), 3.63 (s, 2 H); 13C NMR (125 MHz, CDCl3) δ 164.5, 158.3, 151.4, 129.6, 121.3, 114.6, 64.2, 41.0, 39.7. Elemental Analysis: Comb. Anal. (C20H20N2O5), Calculated: C, 65.21; H, 5.47; N, 7.60; O, 21.72; Found: C, 65.36; H, 5.53; N, 7.59; Purity: >95%. EC503.26 μM.
1,3-Bis(2-ethoxyethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (14)
1H NMR (500 MHz, CDCl3) δ 4.16 (t, 4 H), 3.68 (m, 4 H), 3.56 (s, 2 H), 3.45 (m, 4 H), 1.15 (t, 6H); 13C NMR (125 MHz, CDCl3) δ 167.7, 149.8, 66.8, 66.2, 47.3, 41.5, 39.7, 15.0. EC50 7.75 μM.
1,3-Bis(2-fluorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (15)
1H NMR (500 MHz, CDCl3) δ 7.20 (m, 4 H), 7.05 (m, 4 H), 4.12 (t, 4 H), 3.55 (s, 2 H), 2.91 (t, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 151.1, 131.3, 128.8, 128.7, 125.0, 124.3, 115.5, 41.7, 39.5, 27.6. EC50 3.29 μM.
1,3-Bis(3-fluorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (16)
1H NMR (500 MHz, CDCl3) δ 7.30 (m, 2 H), 7.04-6.96 (m, 6 H), 4.10 (m, 4 H), 3.64 (s, 2 H), 2.89 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.3, 151.1, 140.3, 130.2, 124.7, 116.0, 115.9, 113.9, 42.9,39.6, 33.8. EC50 3.23 μM.
1,3-Bis(4-fluorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (17)
1H NMR (500 MHz, CDCl3) δ 7.20 (m, 4 H), 7.00 (m, 4 H), 4.06 (m, 4 H), 3.60 (s, 2 H), 2.84 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.3, 151.2, 130.5, 115.9, 115.6, 115.4, 43.1, 39.6, 33.3 Elemental Analysis: Comb. Anal. (C20H18F2N2O3), Calculated: C, 64.51; H, 4.87; F, 10.20; N, 7.52; O, 12.89; Found: C, 63.23; H, 4.56; N, 6.85; Purity: >95%. EC50 3.45 μM.
1,3-Bis(3-chlorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (18)
1H NMR (500 MHz, CDCl3) δ 7.25 (m, 6 H), 7.15 (m, 2 H), 4.09 (m, 4 H), 3.65 (s, 2 H), 2.86 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.3, 151.1, 139.8, 134.4, 130.0, 129.2, 127.1, 42.9, 39.6, 33.7. EC50 2.97 μM.
1,3-Bis(4-chlorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (19)
1H NMR (500 MHz, CDCl3) δ 7.28 (m, 4 H), 7.17 (m, 4 H), 4.05 (m, 4 H), 3.61 (s, 2 H), 2.83 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.3, 151.1, 136.2, 132.7, 130.4, 128.8, 42.9, 39.6, 33.4. EC50 3.26 μM.
1,3-Bis(2,4-dichlorophenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (20)
1H NMR (500 MHz, CDCl3) δ 7.37 (m, 2 H), 7.18 (m, 4 H), 4.11 (t, 4 H), 3.60 (s, 2 H), 2.98 (t, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.3, 151.1, 135.0, 134.5, 133.5, 131.9, 129.5, 127.4, 41.4, 39.6, 31.3. EC50 1.87 μM.
1,3-Bis(4-methoxyphenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (21)
1H NMR (500 MHz, CDCl3) δ 7.15 (m, 4 H), 6.84 (m, 4 H), 4.05 (m, 4 H), 3.77 (s, 6 H), 3.57 (s, 2 H), 2.81 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 158.5, 151.2, 130.6, 129.8 128.7, 114.0, 55.3, 43.3, 39.6, 33.2. Elemental Analysis: Comb. Anal. (C22H24N2O5), Calculated: C, 66.65; H, 6.10; N, 7.07; O, 20.18; Found: C, 66.50; H, 5.98; N, 7.06; Purity: >95%. EC50 3.36 μM.
1,3-Bis(3,4-dimethoxyphenethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (22)
1H NMR (500 MHz, CDCl3) δ 6.78 (m, 3 H), 4.07 (m, 4 H), 3.89 (s, 6 H), 3.84 (s, 6 H), 3.61 (s, 2 H), 2.81 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 151.2, 149.0, 147.9, 130.2, 121.0, 112.0, 111.2, 56.0, 43.3, 39.7, 33.8. EC50 11.59 μM.
5-Methyl-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (37)
1H NMR (500 MHz, CDCl3) δ 7.29 (m, 4 H), 7.22 (m, 6 H), 4.11 (m, 4 H), 3.34 (q, 1 H), 2.90 (m, 4 H), 1.45 (d, 3 H); 13C NMR (125 MHz, CDCl3) δ 168.7, 150.9, 137.8, 129.1 128.6, 126.8, 44.1, 43.1, 34.0, 14.7. Elemental Analysis: Comb. Anal. (C21H22N2O3), Calculated: C, 71.98; H, 6.33; N, 7.99; O, 13.70; Found: C, 69.98; H, 6.17; N, 7.84; Purity: >95%. EC50 > 32 μM.
5,5-Dimethyl-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (38)
1H NMR (500 MHz, CDCl3) δ 7.29-7.21 (m, 10 H), 4.10 (m, 4 H), 2.29 (t, 4 H), 1.33 (s, 6 H); 13C NMR (125 MHz, CDCl3) δ 172.4, 150.5, 137.8, 129.2, 128.6, 126.8, 47.3, 42.9, 34.0, 24.9. EC50 > 32 μM.
Typical procedure for parallel synthesis of PYT analogs with different R1 and R2 groups
S,S-Dimethyl carbonodithioate (0.5 mmol) was added dropwise to a solution of R1NH2 (0.75 mmol) in H2O (5 mL) with stirring at room temperature, and the mixture was stirred for 24 h. After removal of solvent in vacuo, the residue was recrystallized from ether/hexanes to afford R1NHCOSMe.
A solution of R1NHCOSMe (0.5 mmol) and R2NH2 (0. 5 mmol) in MeoH (5 mL) was heated at 60 °C with stirring for 24 h. After removal of solvent in vacuo, the crude urea residue and malonic acid (0.5 mmol) were dissolved in 3 mL of glacial acetic acid. The mixture was heated to 60 °C and 2 mL of acetic anhydride was added over 30 min. The reaction mixture was heated with stirring at 90 °C for 4 h. The solvent was removed under vacuum at 60 °C, and the residue was recrystallized from ether/hexanes or purified by silica gel chromatography to provide the desired PYT analog.
S-Methyl 4-fluorophenethylcarbamothioate
1H NMR (500 MHz, CDCl3) δ 7.13 (m, 2 H), 6.99 (m, 2 H), 5.41 (br, 1 H), 3.51 (m, 2 H), 2.80 (m, 2 H), 2.34 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 167.9, 162.7, 160.8, 134.2, 130.3, 130.2, 115.6, 115.5, 42.7, 35.2, 12.4.
1-Cyclohexyl-3-phenethylpyrimidine-2,4,6(1H,3H,5H)-trione (23)
1H NMR (500 MHz, CDCl3) δ 7.29 (m, 2 H), 7.24 (m, 3 H), 4.59 (m, 1 H), 4.08 (m, 2 H), 3.58 (s, 2 H), 2.89 (m, 2 H), 2.22 (m, 2 H), 1.84 (m, 2 H), 1.67-1.57 (m, 3 H), 1.34-1.22 (m, 3 H); 13C NMR (125 MHz, CDCl3) δ 164.9, 164.8, 151.2, 137.9, 129.0 128.6, 126.8, 55.4, 43.1, 40.3, 34.1, 29.1, 26.4, 25.2. EC50 1.67 μM.
1-Octyl-3-phenethylpyrimidine-2,4,6(1H,3H,5H)-trione (24)
1H NMR (500 MHz, CDCl3) δ 7.30-7.23 (m, 5 H), 4.12 (m, 2 H), 3.83 (t, 2 H), 3.61 (s, 2 H), 2.90 (t, 2 H), 1.54 (m, 2 H), 1.28 (m, 10 H), 0.88 (t, 3 H); 13C NMR (125 MHz, CDCl3) δ 164.5, 151.3, 137.9, 129.1 128.7, 126.8, 43.1, 42.2, 39.7, 34.1, 31.9, 29.3, 28.0, 26.9, 22.7, 14.2. EC50 0.71 μM.
1-(2-Cyclohexenylethyl)-3-phenethylpyrimidine-2,4,6(1H,3H,5H)-trione (25)
1H NMR (500 MHz, CDCl3) δ 7.32-7.25 (m, 5 H), 5.39 (s, 1 H), 4.13 (m, H), 3.97 (t, 2 H), 3.61 (s, 2 H), 2.91 (m, 2 H), 2.19 (m, 2 H), 1.96-1.86 (m, 4 H), 1.64-1.54 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.5, 164.4, 151.2, 137.9, 134.6, 129.0 128.7, 126.8, 123.9, 43.1, 40.6, 39.6, 36.2, 34.1, 28.1, 25.4, 22.9. 22.3. EC50 1.74 μM.
1-Benzyl-3-phenethylpyrimidine-2,4,6(1H,3H,5H)-trione (26)
1H NMR (500 MHz, CDCl3) δ 7.44-7.22 (m, 10 H), 5.05 (s, 2 H), 4.12 (m, 2 H), 3.63 (s, 2 H), 2.92 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 164.6, 164.3, 151.3, 137.7, 136.1, 129.2, 129.0 128.7, 128.6, 128.1, 126.8, 45.0, 43.2, 39.6, 34.0. EC50 1.90 μM.
1-Phenethyl-3-(3-phenylpropyl)pyrimidine-2,4,6(1H,3H,5H)-trione (27)
1H NMR (500 MHz, CDCl3) δ 7.30-7.20 (m, 10 H), 4.07 (m, 2 H), 3.96 (t, 2 H), 3.49 (s, 2 H), 2.90 (m, 2 H), 2.70 (t, 2 H), 1.99 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 164.6, 164.4, 151.2, 141.1, 137.8, 129.0, 128.7, 128.5, 128.2, 126.8, 126.1, 43.1, 42.0, 39.5, 34.1, 33.1, 28.6. EC50 1.88 μM.
1-Phenethyl-3-(2-(thiophen-2-yl)ethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (28)
1H NMR (500 MHz, CDCl3) δ 7.30-7.14 (m, 6 H), 6.94 (m, 2 H), 4.17 (m, 2 H), 3.90 (m, 2 H), 3.64 (s, 2 H), 3.17 (m, 2 H), 2.73-2.67 (m, 4 H), 1.67 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 196.5, 169.4, 160.8, 151.2, 142.0, 139.8, 128.5, 127.1, 125.9, 125.7, 124.3, 96.0, 43.0, 42.7, 42.5, 41.4, 29.6, 28.8, 27.9, 24.9. EC50 3.39 μM.
1-(4-Chlorophenethyl)-3-phenethylpyrimidine-2,4,6(1H,3H,5H)-trione (29)
1H NMR (500 MHz, CDCl3) δ 7.26 (m, 7 H), 7.20 (m, 2 H), 4.09 (m, 4 H), 3.61 (s, 2 H), 2.87 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 164.3, 151.1, 137.8, 136.2, 132.7, 130.4, 129.0, 128.8, 128.7, 126.9, 43.2, 42.9, 39.6, 34.1, 33.4. Elemental Analysis: Comb. Anal. (C20H19ClN2O3), Calculated: C, 64.78; H, 5.16; Cl, 9.56; N, 7.55; O, 12.94; Found: C, 64.96; H, 5.19; N, 7.55; Purity: >95%. EC50 1.19 μM.
1-(4-Methoxyphenethyl)-3-phenethylpyrimidine-2,4,6(1H,3H,5H)-trione (30)
1H NMR (500 MHz, CDCl3) δ 7.31 (m, 2 H), 7.26 (m, 3 H), 7.17 (m, 2 H), 6.86 (m, 2 H), 4.08 (m, 4 H), 3.81 (s, 3 H), 3.60 (s, 2 H), 2.87 (t, 2 H), 2.83 (t, 2 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 158.5, 151.2, 137.8, 130.0, 129.8, 129.0 128.7, 126.8, 114.0, 55.3, 43.3, 43.1, 39.6, 34.1, 33.2. Elemental Analysis: Comb. Anal. (C21H22N2O4), Calculated: C, 68.84; H, 6.05; N, 7.65; O, 17.47; Found: C, 68.65; H, 6.00; N, 7.59; Purity: >95%. EC50 2.94 μM.
1-(4-Fluorophenethyl)-3-(2-(thiophen-2-yl)ethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (31)
1H NMR (500 MHz, CDCl3) δ 7.20-7.15 (m, 3 H), 7.00-6.94 (m, 3 H), 6.86 (d, 1 H), 4.16 (t, 2 H), 4.05 (m, 2 H), 3.60 (s, 2 H), 3.12 (t, 2 H), 2.84 (t, 4 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 162.8, 160.9, 151.1, 139.6, 133.5, 130.5, 127.2, 125.9, 124.3, 115.6, 115.4, 43.2, 43.0, 39.6, 33.3, 27.9. EC50 2.16 μM.
1-(4-Fluorophenethyl)-3-(2-phenoxyethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (32)
1H NMR (500 MHz, CDCl3) δ 7.30 (m, 2 H), 7.21 (m, 2 H), 6.97 (m, 3 H), 6.89 (m, 2 H), 4.35 (t, 2 H), 4.19 (t, 2 H), 4.09 (m, 2 H), 3.64 (s, 2 H), 2.89 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 164.4, 164.3, 162.8, 160.9, 158.3, 151.3, 133.5, 130.5, 129.6, 121.3, 115.6, 115.4, 114.5, 64.2, 43.2, 41.0, 39.6, 33.2. EC50 1.32 μM.
1-(4-Fluorophenethyl)-3-(2-isopropoxyethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (33)
1H NMR (500 MHz, CDCl3) δ 7.21 (m, 2 H), 6.99 (m, 2 H), 4.07 (m, 4 H), 3.64 (s, 2 H), 3.62-3.56 (m, 3 H), 3.28 (t, 2 H), 1.12 (d, 6 H); 13C NMR (125 MHz, CDCl3) δ 164.6, 162.8, 160.9, 151.3, 133.5, 130.5, 115.6, 71.6, 64.1, 43.2, 41.4, 39.7, 33.3, 22.1. Elemental Analysis: Comb. Anal. (C17H21FN2O4), Calculated: C, 60.70; H, 6.29; F, 5.65; N, 8.33; O, 19.03; Found: C, 60.56; H, 6.23; N, 8.24; Purity: >95%. EC50 2.97 μM.
1-(4-Fluorophenethyl)-3-(2-morpholinoethyl)pyrimidine-2,4,6(1H,3H,5H)-trione (34)
1H NMR (500 MHz, CDCl3) δ 7.19 (m, 2 H), 6.96 (m, 2 H), 4.08 (m, 4 H), 3.75-3.66 (m, 4 H), 2.87 (m, 4 H), 2.78-2.61 (m, 6 H); 13C NMR (125 MHz, CDCl3) δ 196.4, 162.7, 160.8, 134.8, 130.5, 130.4, 115.5, 115.3, 96.0, 53.7, 42.4, 33.5. EC50 > 32 μM.
1-(4-Fluorophenethyl)-3-(4-phenylbutyl)pyrimidine-2,4,6(1H,3H,5H)-trione (35)
1H NMR (500 MHz, CDCl3) δ 7.29 (m, 3 H), 7.20 (m, 4 H), 6.90 (m, 2 H), 4.01 (m, 2 H), 3.90 (m, 2 H), 3.54 (s, 2 H), 2.80 (m, 2 H), 2.56 (m, 2 H), 1.55 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 164.5, 162.8, 160.9, 151.3, 142.0, 133.5, 130.5, 128.5, 126.0, 115.5, 115.4, 43.1, 42.0, 39.6, 35.5, 33.3, 28.7, 27.6. Elemental Analysis: Comb. Anal. (C22H23FN2O3), Calculated: C, 69.09; H, 6.06; F, 4.97; N, 7.33; O, 12.55; Found: C, 68.92; H, 6.55; N, 7.19; Purity: >95%. EC50 3.55 μM.
1-(2,2-Diphenylethyl)-3-(4-phenylbutyl)pyrimidine-2,4,6(1H,3H,5H)-trione (36)
1H NMR (500 MHz, CDCl3) δ 7.31-7.19 (m, 15 H), 4.60 (m, 2 H), 4.55 (t, 1 H), 3.81 (m, 2 H), 3.43 (s, 2 H), 2.64 (m, 2 H), 1.63-1.54 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 196.3, 164.6, 164.4, 151.3, 142.0, 141.4, 141.0, 128.8, 128.6, 128.5, 128.4, 128.1, 127.1, 126.8, 126.0, 48.6, 45.6, 41.8, 39.5, 35.5, 28.5, 27.5. EC50 3.61 μM.
Typical procedure for synthesis of PYT analogs with R3 groups by Knoevenagel condensation
Into a mixture of 7 (0.5 mmol), piperidine (0.05 mmol), benzoic acid (0.05 mmol) and benzene (10 mL), the aldehyde (0.5 mmol) was added slowly. The reaction apparatus was equipped with a Dean-Stark trap, and the reaction mixture was refluxed for 8 h. After cooling the mixture to room temperature and washing it with water, the solvent was removed in vacuo and the residue was purified by silicagel chromatography to provide the desired PYT analog.
5-(Cyclohexylmethylene)-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (39)
1H NMR (500 MHz, CDCl3) δ 7.27 (m, 4 H), 7.22 (m, 6 H), 4.16 (m, 2 H), 4.03 (m, 2 H); 3.37 (m, 1 H), 2.87 (m, 4 H), 1.66 (m, 5H), 1.16-0.48 (m, 6 H); 13C NMR (125 MHz, CDCl3) δ 168.8, 151.1, 137.8, 129.1 128.6, 126.8, 47.3, 42.9, 39.3, 34.7, 34.0, 32.8,, 26.3, 26.0. EC50 > 32 μM.
5-(2,6-Dichlorobenzylidene)-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (40)
1H NMR (500 MHz, CDCl3) δ 8.28 (s, 1 H), 7.30-7.14 (m, 13 H), 4.11 (m, 2 H), 4.00 (s, 2 H), 2.86 (m, 2 H), 2.76 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 160.3, 158.9, 151.7, 150.3, 138.0, 137.9, 133.1, 132.9, 129.1, 129.0, 128.7, 128.6, 127.9, 126.8, 126.7, 123.4, 43.7, 43.1, 34.1, 34.0. EC50 3.10 μM.
5-(2-Methylbenzylidene)-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (41)
1H NMR (500 MHz, CDCl3) δ 8.66 (s, 1 H), 7.43-7.14 (m, 14 H), 4.12 (m, 2 H), 4.04 (m, 2 H), 2.87 (m, 2 H), 2.80 (m, 2 H), 2.29 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 161.7, 159.6, 158.8, 150.6, 138.5, 138.2, 132.8, 131.6, 130.2, 130.0, 129.1, 128.7, 128.6, 126.8, 126.7, 125.3, 118.8, 43.7, 43.0, 34.2, 20.4. EC50 2.23 μM.
5-(4-(Benzyloxy)benzylidene)-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (42)
1H NMR (500 MHz, CDCl3) δ 8.28 (s, 1 H), 8.02 (d, 2 H), 7.22 (m, 5 H), 7.09 (m, 10 H), 6.84 (d, 2 H), 4.97 (s, 2 H), 3.97 (m, 4 H), 2.70 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 163.5, 162.8, 160.6, 159.0, 150.7, 138.4, 137.9, 135.9, 129.2, 129.1, 128.9, 128.6, 128.5, 127.6, 126.7, 126.6, 125.8, 114.9, 114.6, 70.4, 43.8, 43.1, 34.3. EC50 2.84 μM.
1,3-Dicyclohexyl-5-(2,4-dimethoxybenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (43)
1H NMR (500 MHz, CDCl3) δ 8.51 (s, 1 H), 8.39 (m, 1 H), 6.56 (m, 1 H), 6.42 (m, 1 H), 4.75 (m, 2 H), 3.88 (s, 6 H), 2.34 (m, 4 H), 1.82 (m, 4 H), 1.66 (m, 6 H), 1.32 (m, 6 H); 13C NMR (125 MHz, CDCl3) δ 165.9, 163.4, 162.4, 161.3, 153.0, 151.1, 135.6, 115.7, 104.9, 97.5, 55.9, 55.7, 54.8, 29.3, 26.6, 25.4. Elemental Analysis: Comb. Anal. (C25H32N2O5), Calculated: C, 68.16; H, 7.32; N, 6.36; O, 18.16; Found: C, 67.98; H, 7.36; N, 6.36; Purity: >95%. EC50 2.58 μM.
5-((1H-Pyrrol-2-yl)methylene)-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (44)
1H NMR (500 MHz, CDCl3) δ 8.34 (s, 1 H); 7.56 (m, 1 H), 7.29 (m, 8 H), 7.22 (m, 2 H), 7.15 (m, 1 H), 6.56 (m, 1 H); 4.20 (m, 4 H), 2.92 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 163.4, 163.0, 150.8, 143.1, 138.5, 131.5, 129.7, 129.1, 128.6, 126.7, 126.6, 114.8, 105.8, 43.6, 43.2, 34.3. EC50 3.55 μM.
5-((2,5-Dimethyl-1-phenyl-1H-pyrrol-3-yl)methylene)-1,3-diphenethylpyrimidine-2,4,6(1H,3H,5H)-trione (45)
1H NMR (500 MHz, CDCl3) δ 8.62 (s, 1 H), 7.55 (m, 4 H), 7.31 (m, 7 H), 7.21 (m, 4 H), 4.18 (m, 4 H), 2.94 (m, 4 H), 2.32 (s, 3 H), 2.05 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 164.0, 161.4, 151.3, 149.5, 138.9, 138.8, 137.1, 129.9, 129.1, 128.6, 128.5, 127.7, 126.5, 118.8, 111.2, 107.6, 43.6, 42.8, 34.4, 12.9, 11.9. EC50 0.87 μM.
Synthesis of 6-chloro-1,3-diphenethylpyrimidine-2,4(1H,3H)-dione (46)
A mixture of 7 (0.5 mmol) and POCl3 (2 mL) was heated at 100 °C with stirring for 8 h. After cooling the mixture to room temperature, the mixture was poured into 20 mL of EtOAc. After being washed with water and dried over Na2SO4, the solvent was removed in vacuo, and the residue was purified by silica gel chromatography, to provide 46 in 85% yield. 1H NMR (500 MHz, CDCl3) δ 7.27 (m, 6 H), 7.21 (m, 4 H), 5.90 (s, 1 H), 4.24 (t, 2 H), 4.13 (t, 2 H), 2.95 (t, 2 H), 2.89 (t, 2 H); 13C NMR (125 MHz, CDCl3) δ 160.5, 150.6, 145.6, 138.3, 137.1, 129.1, 129.0, 128.9, 128.6, 127.1, 126.7, 102.3, 48.3, 43.1, 34.9, 33.6. Elemental Analysis: Comb. Anal. (C20H19ClN2O3), Calculated: C, 67.70; H, 5.40; Cl, 9.99; N, 7.89; O, 9.02; Found: C, 66.95; H, 5.55; N, 7.82; Purity: >95%. EC50 > 32 μM.
Synthesis of 6-methoxy-1,3-diphenethylpyrimidine-2,4(1H,3H)-dione (47)
A mixture of 46 (0.5 mmol) and NaOEt (0.05 mmol) was refluxed in MeOH (5 mL) with stirring for 6 h. After cooling the mixture to room temperature, the mixture was poured into 20 mL EtOAc. After being washed with water and dried over Na2SO4, the solvent was removed in vacuo, and the residue was purified by silica gel chromatography to provide compound 47 in about 50% yield. 1H NMR (500 MHz, CDCl3) δ 7.24-7.08 (m, 10 H), 4.96 (s, 1 H), 4.05 (m, 4 H), 3.62 (s, 3 H), 2.82 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 162.9, 161.2, 150.8, 138.8, 138.2, 129.2, 129.0 128.6, 128.5 126.7, 126.5, 56.8, 43.6, 42.6, 34.8, 34.0. EC50 > 32 μM.
Typical procedure for the synthesis of PYT analogs with 5- or 7-membered ring cores
Into a mixture of 48 (0.5 mmol), Et3N (1.1 mmol), THF (5 mL), oxalyl dichloride (0.5 mmol for 5-membered core) or succinyl dichloride (0.5 mmol for 7-membered core) was added slowly. After being stirred at room temperature for 10 h, the reaction was quenched with water and poured into 20 mL of EtOAc. The mixture was washed with water and brine and dried over Na2SO4. The solvent was removed in vacuo, and the residue was purified by silica gel chromatography to provide the desired PYT analog.
1,3-Diphenethylimidazolidine-2,4,5-trione (49)
1H NMR (500 MHz, CDCl3) δ 7.24-7.09 (m, 10 H), 3.79 (m, 4 H), 2.86 (m, 4 H); 13C NMR (125 MHz, CDCl3) δ 156.4, 153.5, 136.8, 128.9, 128.8, 127.2, 40.5, 33.9. Elemental Analysis: Comb. Anal. (C19H18N2O3), Calculated: C, 70.79; H, 5.63; N, 8.69; O, 14.69; Found: C, 71.03; H, 5.75; N, 8.73; Purity: >95%. EC50 > 32 μM.
1-Cyclohexylimidazolidine-2,4,5-trione (50)
1H NMR (500 MHz, CDCl3) δ 8.08 (s, 1 H), 4.04 (m, 1 H), 2.07 (m, 2 H), 1.88 (m, 2 H), 1.70 (m, 3 H), 1.31 (m, 3 H); 13C NMR (125 MHz, CDCl3) δ 156.6, 156.1, 152.2, 52.7, 29.6, 25.7, 24.8. EC50 >32 μM.
1,3-Dicyclohexylimidazolidine-2,4,5-trione (51)
1H NMR (500 MHz, CDCl3) δ 4.01 (m, 2 H), 2.07 (m, 4 H), 1.86 (m, 4 H), 1.66 (m, 6 H), 1.30 (m, 6 H); 13C NMR (125 MHz, CDCl3) δ 156.6, 153.6, 52.5, 29.6, 25.7, 24.9. Elemental Analysis: Comb. Anal. (C15H22N2O3), Calculated: C, 64.73; H, 7.97; N, 10.06; O, 17.24; Found: C, 64.80; H, 7.99; N, 9.94; Purity: >95%. EC50 > 32 μM.
1,3-Diphenethyl-1,3-diazepane-2,4,7-trione (52)
1H NMR (500 MHz, CDCl3) δ 7.24-7.09 (m, 10 H), 3.59 (m, 4 H), 2.82 (m, 4 H), 2.55 (s, 4H); 13C NMR (125 MHz, CDCl3) δ 176.2, 159.3, 139.3, 128.7, 128.3, 127.1, 44.8, 39.9, 26.4. EC50 > 32 μM.
Mutant SOD1-induced cytotoxicity protection assay
Cells were seeded at 15,000 cells/well in 96-well plates and incubated 24 h prior to compound addition. Compounds were assayed in twelve-point dose response experiments to determine potency and efficacy. The highest compound concentration tested was 32 μM, which was decreased by one-half with each subsequent dose. After 24 h incubation with the compounds, MG132 was added at a final concentration of 100 nM. MG132 is a well-characterized proteasome inhibitor, which would be expected to enhance the appearance of protein aggregation by blocking the proteosomal clearance of aggregated proteins. Cell viability was measured 48 h later using the fluorescent viability probe, Calcein-AM (Molecular Probes). Briefly, cells were washed twice with PBS, Calcein-AM was added at a final concentration of 1 μM for 20 min at room temperature, and fluorescence intensity was read in a POLARstar fluorescence plate reader (BMG). Fluorescence data were coupled with compound structural data, then stored and analyzed using the CambridgeSoft Chemoffice Enterprise Ultra software package.
In vitro and in vivo ADME studies
In vitro ADME properties of PYT compounds were tested at Apredica, Inc. (Watertown, MA), a contract research organization. To assess levels of drug present in blood and brain tissue specimens, a 50 mg/kg dose was peritoneally administered to wild type B6SJL mice (n = 6). An untreated group (n = 6) was used as a negative control. Blood and brain were removed 1 h after injection and rapidly quenched at −80 °C. Specimens were subsequently mixed with extraction buffer (100% ice cold methanol), sonicated on ice for 20 sec, then spun at 15,000 rpm at 4 °C for 30 min. Supernatant (100 μL) was then analyzed using a Ceas 16 channel EC-HPLC. Experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Radioligand binding assays for activity profiling
These studies were performed at MDS Pharma Services (Taipei, Taiwan).
Supplementary Material
Acknowledgments
We thank the National Institutes of Health [1R43NS057849], the ALS Association (TREAT program), the Department of Defense [AL093052], the ALS Therapy Alliance, and the Veterans Administration at the Edith Nourse Rogers Memorial Veterans Hospital, Bedford, MA for their generous support of the research project. We are grateful to Biogen Idec (Cambridge, MA) for financial assistance related to the DMPK assays and to Dr. Robert Scannevin (Biogen Idec) for coordinating that effort.
ABBREVIATIONS
- ADME
absorption, distribution, metabolism, excretion
- ALS
amyotrophic lateral sclerosis
- PYT
pyrimidine 2,4,6-trione
- SAR
structure-activity relationship
- SOD1
Cu/Zn superoxide dismutase
Footnotes
Supporting Information Available: X-ray analysis of 3 and 43, in vitro pharmacokinetic studies experimentals, and radioligand assay data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brown RWJ. The Motor Neuron Diseases. In: Fauchi AS, Braunwald E, Isselbacher KJ, WIlson JD, Martin JB, Kasper DL, Hauser SL, Longo DL, editors. Harrison’s Principles of Internal Medicine. McGraw-Hill; New York: 1998. pp. 2368–2371. [Google Scholar]
- 2.Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009:S3–9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- 3.Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci. 2001;191:3–9. doi: 10.1016/s0022-510x(01)00630-x. [DOI] [PubMed] [Google Scholar]
- 4.Waldmeier PC, Tatton WG. Interrupting apoptosis in neurodegenerative disease: potential for effective therapy? Drug Discov Today. 2004;9:210–218. doi: 10.1016/S1359-6446(03)03000-9. [DOI] [PubMed] [Google Scholar]
- 5.Gitkin A, Harris G, Birchenough J, Boylan BC, Whitesell C. Nervous Breakdown: A Detailed Analysis of the Neurology Market. UBS Warburg LLC; New York: 2001. [Google Scholar]
- 6.Mitsumoto H, Rabkin JG. Palliative care for patients with amyotrophic lateral sclerosis: “prepare for the worst and hope for the best”. J Am Med Assoc. 2007;298:207–216. doi: 10.1001/jama.298.2.207. [DOI] [PubMed] [Google Scholar]
- 7.Jimonet P, Audiau F, Barreau M, Blanchard JC, Boireau A, Bour Y, Coléno MA, Doble A, Doerflinger G, Do Huu C, Donat MH, Duchesne JM, Ganil P, Guérémy C, Honoré E, Just B, Kerphirique R, Gontier S, Hubert P, Laduron PM, Le Blevec J, Meunier M, Miquet J-M, Nemecek C, Pasquet M, Piot O, Pratt J, Rataud J, Reibaud M, Stutzmann J-M, Mignani S. Riluzole series. Synthesis in vivo “antiglutamate” activity of 6-substituted-2-benzothiazolamines, and 3-substituted-2-imino-benzothiazolines. J Med Chem. 1999;42:2828–2843. doi: 10.1021/jm980202u. [DOI] [PubMed] [Google Scholar]
- 8.Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 9.Francis K, Bach JR, DeLisa JA. Evaluation and rehabilitation of patients with adult motor neuron disease. Arch Phys Med Rehabil. 1999;80:951–963. doi: 10.1016/s0003-9993(99)90089-8. [DOI] [PubMed] [Google Scholar]
- 10.Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187(6):761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusela JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 12.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 13.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 14.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22(11):1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benmohamed R, Arvanites AC, Kim J, Ferrante RJ, Silverman RB, Morimoto RI, Kirsch DR. Identification of compounds protective against G93A SOD1 toxicity for the treatment of Amyotrophic Lateral Sclerosis. Amyotroph Lat Scler. 2010:11. doi: 10.3109/17482968.2010.522586. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Artuso E, Carvoli G, Degani L, Fochi R, Magistris C. A general, facile, and safe procedure for the preparation of S-methyl N-alkylthiocarbamates by methylthiocarbonylation of primary aliphatic amines with S,S-dimethyl dithiocarbonate. Synthesis. 2007:1096–1102. [Google Scholar]
- 17.Ingle VN, Gaidhane PK, Dutta SS, Naha PP, Sengupta MS. Synthesis of novel galactopyranosyl-derived spiro barbiturates. J Carbohyd Chem. 2006:661–671. [Google Scholar]
- 18.Ragoussis N. Modified Knoevenagel condensations: Synthesis of (E)-3-alkenoic acids. Tetrahedron Lett. 1987:93–96. [Google Scholar]
- 19.Robertson JS. Blood pressure maxima in humans. J Clin Basic Cardiol. 2006;9:37–40. [Google Scholar]
- 20.(a) Towart R, Linders JTM, Hermans AN, Rohrbacher J, van der Linde HJ, Ercken M, Cik M, Roevens P, Teisman A, Gallacher DJ. Blockade of the IKs potassium channel: An overlooked cardiovascular liability in drug safety screening? J Pharmacol Toxicol Meth. 2009;60(1):1–10. doi: 10.1016/j.vascn.2009.04.197. [DOI] [PubMed] [Google Scholar]; (b) Du L, Li M, You Q. The interactions between hERG potassium channel and blockers. Curr Top Med Chem. 2009;9(4):330–338. doi: 10.2174/156802609788317829. [DOI] [PubMed] [Google Scholar]; (c) Mitcheson JS. hERG Potassium Channels and the Structural Basis of Drug-Induced Arrhythmias. Chem Res Toxicol. 2008;21(5):1005–1010. doi: 10.1021/tx800035b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.