

Abstract
Using a large set of high mass accuracy and resolution ETD tandem mass spectra, we characterized ETD-induced neutral losses. From these data we deduced the chemical formula for 20 of these losses. Many of them have been previously observed in electron-capture dissociation (ECD) spectra, such as losses of the side chains of arginine, aspartic acid, glutamic acid, glutamine, asparagine, leucine, histidine, and carbamidomethylated cysteine residues. With this information, we examined the diagnostic value of these amino acid-specific losses. Among 1285 peptide–spectrum matches, 92.5% have agreement between neutral loss-derived peptide amino acid composition and the peptide sequences. Moreover, we show that peptides can be uniquely identified by using only the accurate precursor mass and amino acid composition based on neutral losses; the median number of sequence candidates from an accurate mass query is reduced from 21 to 8 by adding side chain loss information. Besides increasing confidence in peptide identification, our findings suggest the potential use of these diagnostic losses in ETD spectra to improve false discovery rate estimation and to enhance the performance of scoring functions in database search algorithms.
Keywords: Electron transfer dissociation, Electron capture dissociation, Neutral loss, Side chain loss, Accurate mass
Introduction
Peptide cation dissociation generally induces backbone fragmentation to generate either b- and y-type or c- and z-type fragment ions (for collision- [1, 2] or electron-based [3–6] methods, respectively). Losses of amino acid side chains, however, are also observed [1–3, 7–12]. These processes have been well characterized for collisional activation and, more recently, for electron-capture dissociation [8–12]. ECD work has characterized not only general amino acid side chain loss, but also side chain loss specific to aspartic/isoaspartic [13–15], glutamic/γ-glutamic acid [16], O-fucosylation [17], SO2 [18], isoleucine/leucine [19], and alkylated cysteine residues [11]. Combined, these studies reveal about 20 common ECD-specific losses [8–12, 20]. Beyond extending our fundamental knowledge of how dissociation occurs, such research holds promise to help in the correlation of spectrum to sequence [21].
Over the last few years electron-transfer dissociation (ETD)—the ion/ion analog of electron-capture dissociation (ECD)—has become increasingly widespread for a variety of proteomic applications [6, 22–30]. In general, ETD and ECD spectra share many similarities; however, differences have been documented [31]. Direct comparison of ECD and ETD is confounded by extreme differences in the pressure at which dissociation occurs [13]. Unlike ECD, which occurs under high vacuum conditions (~10−9 Torr) after capture of a free electron by a multiply protonated peptide cation, ETD follows from transfer of an electron from an anion to the peptide cation at much higher pressures (~10−3 Torr). To date, ETD has been typically analyzed on low-resolution trapping instruments, while ECD is acquired primarily on high-resolution Fourier transform instruments. Despite these differences, those who have compared ETD and ECD have seen value in doing so [13]. To our knowledge, there has been no reported systematic study of ETD-induced side chain losses. With the recent coupling of ETD to the high resolution and high mass accuracy Orbitrap mass analyzer [26, 32], such an analysis is possible. Using an extensive dataset, we catalog 20 amino acid side chain losses following ETD. Five of these have not been reported in either ETD experiments or in ECD spectra contained within the SwedECD database. Further, we examine the frequency and specificity of each loss. With this information, we investigate possible diagnostic value for targeted and large-scale protein sequence analysis.
Experimental
LC-MS/MS Sample Preparation, Data Collection, and Database Searching
Human embryonic stem cells were lysed and digested with LysC (Wako Chemicals, Osaka, Japan). Generated peptides were desalted, of which 1.5 μg were loaded onto a capillary column, separated over a 90 min gradient, and analyzed on a Thermo Scientific (San Jose, CA, USA) ETD-enabled LTQ Orbitrap XL. Both MS1 and MS2 scans were acquired at a resolving power of 60,000. Fluoranthene reagent cations were introduced into both MS1 and MS2 scans, generating an internal calibrant peak having an m/z value of 202.07770 [33]. Raw data files were cleaned by DTA Generator [34, 35], and subsequently searched against a concatenated target-decoy human database (IPI v3.63) using the Open Mass Spectrometry Search Algorithm (OMSSA ver. 2.1.4) [36], with cysteine carbamidomethylation as a fixed modification, methionine oxidation as a variable modification, and three missed cleavages allowed. The peptide–spectrum matches (PSMs) were then filtered to a false discovery rate of 1%. Further details of this software can be found in Wenger et al. [35].
Synthetic Peptide Samples
Synthetic Peptides were purchased from Thermo Fisher Scientific. Peptides with c-terminal lysine were heavy labeled (13C6, 15N2) while internal cysteine residues were carbamidomethylated. Synthetic peptides were dissolved in 50% methanol, 2% acetic acid. The peptide solutions were directly infused via pico tip (New Objective, Woburn, MA, USA). The ETD spectra were collected on a Thermo Scientific ETD-enabled LTQ Orbitrap Velos. MS2 and MS3 scans were acquired at a resolving power of either 15,000 or 30,000.
Neutral Loss Extraction
Information about precursor neutral loss peaks was extracted from the raw data with software developed in-house (“PepGate”) using Visual C# with the Microsoft .NET Framework 3.5 and Thermo XRawfile COM library. The extracted peak information contains the mass of the loss, the precursor's m/z, charge state, and signal-to-noise ratio. The neutral loss masses were then converted to elemental composition. Next, the theoretical masses of these elemental compositions were matched back to the neutral losses with a mass tolerance of ±5 ppm. A FileMaker Pro Advanced 9 database was then generated, containing the PSMs, neutral losses, and summary of the frequency of the amino acids.
MySQL Peptide Database Construction
We created a peptide database from human IPI ver. 3.63 by digesting proteins in silico with LysC enzymatic specificity, allowing up to three missed cleavages. The resulting peptide database was imported into MySQL (ver. 5.1.19-community). The final MySQL database included one “peptides” table, which consisted of three fields: “proID” (unique protein identifier), “sequence” (peptide sequence), and “mass” (peptide mass). The “mass” field was indexed using a binary tree structure.
Perl Programs for MySQL Database Query
From the FileMaker database file, a tab delimited text file was exported with each line containing information from each scan, such as the scan number, the precursor peptide mass tolerance (±3 ppm), and the amino acid content derived from neutral loss peaks. We wrote programs in Perl (ActivePerl ver. 5.10.0.1003) to build two SQL query statements for each scan, one with and one without the amino acid content information. The program then connected to and queried the MySQL database using the DBI Perl package through a DBD MySQL driver installed from Active State Perl Package Manager (ver. 4.02). The query results were written to a tab-delimited text file, which was then imported to FileMaker Pro Advanced 9 for further analysis. R (ver. 2.8.1) was used to plot the histograms of the SQL query results.
Results and Discussion
To obtain a large dataset of high resolution and high mass accuracy ETD tandem mass spectra, we obtained a complex mixture of proteins from lysed human cells. The mixture was digested with LysC and the resulting peptides were loaded onto a nanoflow reverse-phased capillary LC column and gradient eluted into an ETD-enabled LTQ Orbitrap XL mass spectrometer. Fluoranthene anions were used to induce ETD; fluoranthene cations, a byproduct of the chemical ionization process, were used as an internal standard to provide high mass accuracy [33]. Following the 2 h separation, database searching confirmed a total of 2699 PSMs (1% FDR).
We wrote custom software, PepGate, to identify en masse any generated neutral losses from these data. Neutral loss masses were then compiled and mapped to the corresponding chemical formulas. From these formulas, theoretical masses were generated and mapped back to all PSMs with a mass tolerance of ±5 ppm. Table 1 presents a comprehensive report containing all identified amino acid side chain losses along with a summary of sensitivity and specificity of each. Fifteen out of the 20 amino acid side chain losses presented in Table 1 also occur in ECD tandem mass spectra found within the SwedECD database [3, 8–12, 20, 21, 37]. The SwedECD database comprises a collection of ECD tandem mass spectra collected under typical ECD reaction conditions. Under alternate ECD conditions, we found evidence for three more of the ETD observed neutral losses that were missing from SwedECD [10, 15, 19, 38]. Falth et al. used the SwedECD database to calculate the sensitivity and specificity of ECD neutral losses. We note an overall excellent agreement with the calculations of Falth and co-workers for sensitivity and specificity of losses overlapping between ECD and ETD [12]. This high degree of overlap confirms that ECD and ETD are similar. However, distinct differences exist; for example, the loss from lysine side chain that has been reported in ECD was not observed in our ETD data [3, 8–12, 20, 21, 37]. Below we detail the five side chain neutral losses observed in ETD spectra that are not observed in the SwedECD database: CH2O2, C2H5NO, C2H7N2O, C4H7N2, and C4H8N2.
Table 1.
Summary table of the mass, chemical formula, amino acid composition, sensitivity, and specificity of each detected neutral loss. A column indicating whether or not the loss has been observed in the SwedECD database is also included [12]
| Neutral Loss (Da) | Chemical Formula | Amino Acid | Observed in SwedECD Data base | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|---|
| 17.02655 | NH3 | N-terminal amine | √ | 88 | N/A |
| 27.99491 | CO | D/E | √ | 31 | 91 |
| 34.05310 | N2H6 | R | √ | 55 | 99 |
| 44.03745 | CH4N2 | R | √ | 21 | 99 |
| 45.02146 | CH3NO | Q/N | √ | 62 | 96 |
| 46.00548 | CH2O2 | D/E | 13 | 96 | |
| 59.03711 | C2H5NO | Q/N | 32 | 80 | |
| 59.04835 | CH5N3 | R | √ | 50 | 97 |
| 60.02113 | C2H4O2 | R | √ | 50 | 97 |
| 60.08132 | C3H10N | L | √ | 2 | 95 |
| 73.08915 | C4H11N | L | √ | 2 | 95 |
| 75.05584 | C2H7N2O | C | 67 | 67 | |
| 82.05310 | C4H6N2 | H | √ | 11 | 100 |
| 83.06092 | C4H7N2 | H | 23 | 99 | |
| 84.06875 | C4H8N2 | H | 12 | 98 | |
| 89.14768 | C3H7NO2 | E | √ | 1 | 75 |
| 90.00136 | C2H4NOS | C | √ | 41 | 98 |
| 101.09530 | C4H11N3 | R | √ | 3 | 100 |
| 107.02831 | C3H9NOS | C | √ | 30 | 98 |
| 108.05751 | C7H8O | Y | √ | 1 | 60 |
Loss of CH2O2 (46.00548)
The loss of CH2O2 (46.00548) has been widely accepted as originating from the side chain of either aspartic acid or glutamic acid in collision-activated dissociation [39]. The loss of a 46 Da side chain observed in ECD studies was attributed to the loss of a side chain carboxyl group plus one hydrogen (HCOO) in an early study [9], but was not associated with any specific amino acids at the time. Recent work of Li and co-workers suggests that partial side chain loss via α-hydrogen abstraction could explain this result [31]. To rule out the possibility that this loss arises from low-level, unintended collisional activation during the ion preparation and storage process for ETD, we analyzed a synthetic peptide containing an aspartic acid residue, YYNDVILHK. Triply protonated cations of the peptide were subjected to an ETD reaction with and without the fluoranthene reagent anions present. The neutral loss of CH2O2 was observed in the ETD MS/MS spectrum, but was not present when reagent ions were prevented from entering the trap (data not shown). These data suggest that the loss of CH2O2 arises from an ETD reaction.
Loss of C2H5NO (59.03711)
Zubarev et al. proposed that the loss of C2H5NO (59.03711) originates from glutamine or asparagine side chains [9]; however, they did not observe this from their large-scale analysis of the SwedECD database [12]. The loss of 58 Da has been reported in ECD spectra of Gln-containing peptides, which was explained by a C2H4NO loss from Gln [15]. Here we observe that the neutral loss detected in ETD is associated with the loss of an additional 1 Da (either H-atom or H+). We have found that 32% of peptides containing a Gln or Asn residue exhibit this loss, and 80% of spectra containing this loss were matched to peptides having a Gln or Asn residue.
Loss of C2H7N2O (75.05584 Da)
Presented in Figure 1 is the ETD spectrum of a triply protonated peptide having the sequence RQVLIRPCSK. By comparing the neutral loss regions of the singly and doubly charged ETnoD products, we notice that there is a neutral loss of 75.05542 from singly charged ETnoD product ([M + 3H]+••), but not from the doubly charged ETnoD product ([M + 3H]++•). At a 10 ppm mass tolerance a mass of 75.05542 Da unambiguously matches to the chemical formula of C2H7N2O. We postulate this loss follows from two sequential electron transfer events (Figure S-1,<MMC ID = 1> which can be found in the electronic version of this article). The first transfer to the primary amine at the N-terminus cleaves the N–Cα bond and results in loss of NH3, a well-established process in ECD [40]. This doubly charged product ion can then engage in the capture of a second electron, which in the presence of a carbamidomethylated cysteine side chain, can induce loss of 58.02929 Da (C2H4NO). The loss of 58.02929 Da is also observed in ECD spectra of peptides that were carbamidomethylated [17, 38, 41]. This two-step process also explains the presence of the loss of C2H7N2O only in the singly charged ETnoD product region (Figure 1b). To confirm this sequence, the [M + 3H − NH3]++• ETnoD product was isolated after an initial ETD reaction was performed on the [M + 3H]3+ precursor of a synthetic peptide containing two carbamidomethylated cysteine residues, LFVHNVVCHACK. The [M + 3H − NH3]++• ETnoD product was then subjected to another ETD reaction which produced the [M + 3H − C2H7N2O]+•• product (data not shown). This is further evidence that the loss of C2H4NO only occurs after the loss of NH3 and subsequent capture of an electron. While each event in this two-step process has been reported separately in ECD spectra, to our knowledge this is the first explanation of the mechanism of a 75.05584 Da neutral loss in ETD. Evidence for loss of 75.05584 Da was present in 67% of all Cys-containing peptides (n = 196).
Figure 1.

ETD spectrum of the triply protonated peptide RQVLIRPCSK with the assignment of product ion peaks and neutral losses in Da; (a) m/z range from lock mass to the doubly charged precursor ion; (b) m/z range from the doubly charged precursor ion to the singly charged precursor ion
Loss of C4H7N2 and C4H8N2 (83.05969 and 84.06875 Da)
In our dataset, we observed the loss of C4H6N2 (82.05310 Da) from His residues with similar sensitivity and specificity (11% and 100%, respectively), to that previously reported in ECD (38% and 99%, respectively) [8, 9, 12]. We found, however, the additional loss of 1 Da also occurred. The sum of the histidine side chain loss and the additional 1 Da (either H-atom or H+) losses maps to either C4H7N2 or C4H8N2. As shown in Table 1, these two types of losses are specific to histidine-containing peptides. Figure 2 demonstrates an example of such losses from the peptide YELHLK. In Figure 2b, an abundant peak shows a neutral loss of 83.05969 from the singly charged ETnoD product. The His82 loss is rather weak; the loss of 83.05969 appears only after two ion/ion reactions (Figure 2b), and not after the initial ion/ion reaction (Figure 2a). We speculate that the loss of His side chain in ETD occurs as a result of sequential ion/ion reactions. One transfer event initiates a H-atom loss (or proton abstraction) and another event induces cleavage of the side chain for a combined loss of 83 Da (C4H7N2) (Figure 2b) rather than 82 Da.
Figure 2.

ETD spectrum of triply protonated peptide YELHLK with the assignment of product ion peaks and neutral losses in Da; (a) m/z range from lock mass to the doubly charged precursor ion; (b) m/z range from the doubly charged precursor ion to the charge-reduced precursor ion
To offer a more direct comparison with ECD we synthesized the peptide ALANGFARSHALL and collected high-resolution ETD spectra of this peptide. Cooper et al. reported a loss of 82 Da from [M + 2H]2+ following ECD [8]. Interestingly, we did not observe the loss of 82 Da from either the [M + 2H]2+ or [M + 3H]3+ precursor after a single ion/ion reaction. A loss of 83 Da, however, was clearly present following two ion/ion reactions of the triply protonated precursor (Figure 3). To further confirm our supposition we collected ETD spectra of nine other His-containing peptides. Table 2 lists the His losses from various precursors of the total ten peptides. Nine out of 10 (90%) triply charged precursors showed a loss of 83 Da from [M + 3H]+••. Contrarily, only two of the 10 (20%) doubly charged precursors showed a loss of 82 Da from [M + 2H]+•. These observations confirm that the loss of 83 Da can be interpreted as the sum of initial proton or hydrogen atom loss and a subsequent His side chain loss.
Figure 3.

ETD spectra of ALANGFARSHALL with assignment of: (a) losses from [M + 2H]+• in the spectrum of [M + 2H]++,(b) losses from [M + 3H]++• in the spectrum of [M + 3H]+++, and (c) losses from [M + 3H]+•• in the spectrum of [M + 3H]+++. Each spectrum is the average of 500 scans. Note that although this peptide does not contain Asp/Glu, the loss of CO (27.995 Da) is clearly present. Loss of CO from a peptide not containing Asp or Glu has been observed by Pitteri et al. [52]
Table 2.
Summary table of losses around 82 Da from synthetic histidine-containing peptides. “Loss” refers to the measured mass difference between the charge-reduced precursor and the most intense isotopic peak
| Peptide | Charge of precursor | Loss from +1 charge reduced | Loss from +1 charge reduced | Loss from +1 charge reduced in MS3* |
|---|---|---|---|---|
| AGAHPLESHYR | 2 | |||
| 3 | 83.060 | 81.044 | 83.060 | |
| 4 | 84.067 | 82.054 | 84.067 | |
| ALANGFARSHALL | 2 | |||
| 3 | 83.060 | 83.04a | ||
| AWLNEHR | 2 | |||
| 3 | 83.060 | 83.060 | ||
| GFSQNTSLTQHQR | 2 | |||
| 3 | 83.060 | 83.060 | ||
| GYYYLHR | 2 | 82.055 | ||
| 3 | 83.060 | 83.060 | ||
| HAAAKAAAH | 2 | 81.044 | ||
| 3 | 83.060 | 81.046 | 83.060 | |
| HAAAKAAK | 2 | |||
| 3 | 83.060 | |||
| HFLTLAPIK | 2 | |||
| 3 | 83.059 | 83.054 | ||
| HTGTPLGDIPYGK | 2 | |||
| 3 | ||||
| LPISAAHpSpSKTR | 2 | |||
| 3 | 83.058 |
An ion trap was used to collect MS3 ETD scans of ALANGFARSHALL [M + 3H]++•
MS3 scans were carried on doubly charged ETnoD products
Loss of N2H6, CH4N2, CH5N3, and C4H11N3 (34.05310, 44.03745, 59.04835, and 101.09530 Da)
Among the neutral losses observed in all ETD spectra examined in this experiment, the arginine side chain is one of the most prevalent and abundant. We observed the four major types of neutral losses from arginine residues previously reported in ECD spectra [8–12, 20, 21], namely Arg34 (N2H6, 34.05310), Arg44 (CH4N2, 44.03745), Arg59 (CH5N3, 59.04835), and Arg101 (C4H11N3, 101.09530). Figure 4 summarizes the detected frequencies of these four neutral loss types. The 16 rows in the table exhaust the possibilities of all combinations of these four arginine neutral loss types. This table shows that Arg34 and Arg59 losses are the two most frequent from the side chain of arginine following ETD. In addition, the combination of these two peaks shows the highest detected frequency among all 15 possible combinations of these four neutral losses, considering any one of the four neutral losses is present in a given spectrum. The presence of Arg44 and Arg101 losses alone is rather rare (with detection frequencies of 19 and 0, respectively).
Figure 4.

The detected frequency of arginine side chain neutral losses. Each row sum represents the number of PSMs with the corresponding combination of arginine side chain neutral losses. Each column sum represents the frequency of each specific neutral loss
Loss of NH3 (17.02655 Da)
The loss of 17.02655 appears in the vast majority of the ETD spectra. Within ±10 ppm mass tolerance, the numeric value of 17.02655 exclusively matches to NH3. It is debatable whether this loss of NH3 is from the N-terminal amine or from a residue side chain [8, 9]. In a number of cases, where the peptides do not terminate with a basic residue, a strong neutral loss of NH3 is still detected, suggesting that the NH3 is not from the C-terminal lysine. Moreover, ETD of the N-terminal acetylated peptide (ac) SDKPDMAEIEKFDK (Figure S-2)<MMC 1> does not produce a detectable NH3 loss from the charge-reduced precursor, [M + 3H]+••. Instead, an observed loss of 17.006 corresponds to the cleavage of a hydroxyl (−OH) group from the serine residue. This evidence suggests that the loss of NH3 is from the serine residue. This evidence suggests that the loss of NH3 is from N-terminal amine; therefore, the presence of 17.02655 has no diagnostic value that would indicate the amino acid content of the peptide. However, the absence of NH3 loss may indicate the existence of N-terminal modification.
Agreement of Amino Acid Content from Neutral Losses with PSMs
Note that while the specificity of many losses is fairly high (Table 1), the highest sensitivity of the amino acid-specific losses is only 67%. Therefore, the diagnostic value of these losses lies in confirmation, rather than exclusion, of the presence of certain amino acids in peptide precursors. There have been previous attempts to take advantage of neutral loss phenomena that occur in ECD experiments to improve database searching through post-acquisition data filtering [42]. This report, however, concluded that use of such losses was not advantageous for database searching. In our dataset, 1285 PSMs contained amino acid-specific neutral losses. Among them, 1189 (92.5%) agree with the database search result. The 7.5% disagreement could have a number of causes: modified peptides, false positive peptide identifications, false positive neutral loss peak detection, etc. Nonetheless, this high percentage of agreement suggests the potential use of these diagnostic losses to increase confidence in peptide identification from database searching.
We reasoned that the specificity of neutral losses in ETD spectra may be of use to control false discovery rate (FDR) following database searching. Incorporation of the loss information into our FDR-based filtering procedures, however, only produced marginal results, partially because of the absence or low abundance of losses in many spectra (data not shown). Further work in programming the ETD loss information into search algorithms, thereby improving scoring functions, may prove more fruitful.
Peptide Identification Using Accurate Mass and Neutral Losses
It has been demonstrated that peptides can be directly identified by accurate precursor mass measurement [43–51]. To further explore the use of these amino acid-specific neutral losses listed in Table 1, we asked whether combining high mass accuracy information with amino acid contents as indicated by these specific neutral losses could be used to uniquely identify peptide sequences. Towards this goal, we first performed an in silico LysC-digestion of human protein database with up to three missed cleavages. We then transformed this peptide list into a MySQL database. For example, Figure 5 shows the candidate peptide list of a SQL query using only mass accuracy criteria (±3 ppm) of a precursor having mass of 1622.71992 Da. From the MS/MS data we concluded the peptide must contain aspartic acid (D), cysteine (C), and either glutamine (Q) or asparagine (N). Applying these additional amino acid content criteria resulted in the unique identification of the peptide SVSGTDVQEECREK. This example demonstrates the potential use of high mass accuracy of peptide precursors and the diagnostic value of neutral loss to either draw conclusion about the peptide sequence or help filter out the false positive identifications.
Figure 5.
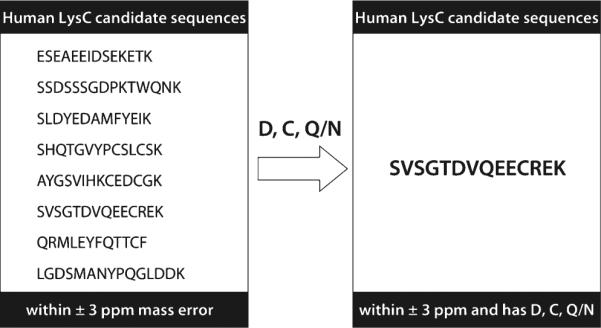
Amino acid content information derived from neutral losses in ETD spectrum resolves the ambiguity of peptide identification from accurate mass-based protein database searching [43–51]. There are eight candidate sequences (left panel) from human LysC peptide database that pass the accurate mass filtering (1622.71992 ± 3 ppm). From the ETD tandem mass spectrum (scan number 1286), we conclude that the precursor peptide contains aspartic acid, cysteine, and also must contain either glutamine or asparagine. Applying this information filters out seven peptides and leaves only one candidate – SVSGTDVQEECREK (right panel), which is the same peptide identified by database searching
To gain a more complete understanding of the value of neutral loss peaks for identifying peptides, we performed a large-scale SQL query in which we used 1474 MS/MS spectra to generate the SQL query from each. A Perl script was written to connect to and query the human LysC peptide database. Approximately 7% of these SQL queries lead to unique peptide identification. Figure 6 displays the frequency histograms of number of distinct peptides from SQL queries against the human LysC peptide database. Comparing with the SQL queries using only mass accuracy with the same level of tolerance (±3 ppm), we observe a significant shift towards a much narrower list of candidate sequences as a result of the extra information amino acid contents derived from the neutral losses provides. By adding side chain loss information, the median number of sequence candidates from accurate mass query is reduced from 21 to 8.
Figure 6.

Histograms showing the frequency of sequence candidates from SQL query against the human LysC in silico digested peptide database using information either only from accurate mass (red) or from accurate mass and amino acid composition automatically derived from ETD neutral losses (blue)
Conclusion
The neutral losses that occur following ETD were characterized by use of an Orbitrap mass analyzer with high resolving power in a large-scale manner. Most of the observed neutral losses match with those previously reported within the SwedECD database. With the high mass accuracy achieved with the Orbitrap mass analyzer, we identified a unique chemical formula for many neutral losses in ETD spectra and linked them to the side chains of specific amino acids. We also explored the efficiency of combining the accurate mass and amino acid content derived from neutral losses in ETD spectra to query human protein database. We showed that unique peptides could be identified by using accurate mass and side chain loss information; the specificity of accurate mass queries were improved significantly by adding side chain loss information (the median of sequence candidates reduced from 21 to 8). This may provide a useful method for either filtering out false positive identifications or identifying candidate sequences directly from information about their mass and amino acid contents.
Supplementary Material
Acknowledgments
The authors thank Graeme McAlister, Danielle Swaney, and Jason Russell for helpful discussions. The authors acknowledge funding for this work by the Beckman Foundation, the Wisconsin Center of Excellence in Genomics Science (P50 HG004952), and the National Institutes of Health (NIH R01GM080148 and PO1GM081629 to J.J.C.).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s13361-010-0029-0) contains supplementary material, which is available to authorized users.
References
- 1.McLafferty FW, Venkataraghavan R, Irving P. Determination of amino acid sequences in peptide mixtures by mass spectrometry. Biochem. Biophys. Res. Commun. 1970;39:274–278. doi: 10.1016/0006-291x(70)90789-8. [DOI] [PubMed] [Google Scholar]
- 2.Hunt DF, Yates JR, Shabanowitz J, Winston S, Hauer CR. Protein sequencing by tandem mass-spectrometry. Proc. Natl. Acad. Sci. U.S.A. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 1998;120:3265–3266. [Google Scholar]
- 4.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein pequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S. A. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coon JJ. Collisions or electrons? protein sequence analysis in the 21st century. Anal. Chem. 2009;81:3208–3215. doi: 10.1021/ac802330b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wipf HK, Irving P, McCamish M, Venkataraghavan R, McLafferty FW. Mass-spectrometric studies of peptides. 5. Determination of amino-acid sequences in peptide mixtures by mass-spectrometry. J. Am. Chem. Soc. 1973;95:3369–3375. doi: 10.1021/ja00791a048. [DOI] [PubMed] [Google Scholar]
- 8.Cooper HJ, Hudgins RR, Hakansson K, Marshall AG. Characterization of amino acid side chain losses in electron capture dissociation. J. Am. Soc. Mass Spectrom. 2002;13:241–249. doi: 10.1016/S1044-0305(01)00357-9. [DOI] [PubMed] [Google Scholar]
- 9.Haselmann KF, Budnik BA, Kjeldsen F, Polfer NC, Zubarev RA. Can the (M· –X) region in electron capture dissociation provide reliable information on amino acid composition of polypeptides? Eur. J. Mass Spectrom. 2002;8:461–469. doi: 10.1255/ejms.555. [DOI] [PubMed] [Google Scholar]
- 10.Cooper HJ, Hakansson K, Marshall AG, Hudgins RR, Haselmann KF, Kjeldsen F, Budnik BA, Polfer NC, Zubarev RA. Letter: the diagnostic value of amino acid side chain losses in electron capture dissociation of polypeptides. comment on: “can the (M-center dot-X) region in electron capture dissociation provide reliable information on amino acid composition of polypeptides?”. Eur. J. Mass Spectrom. 2002;8:461–469. doi: 10.1255/ejms.555. Eur. J. Mass Spectrom. 2003, 9, 221–222. [DOI] [PubMed] [Google Scholar]
- 11.Chalkley RJ, Brinkworth CS, Burlingame AL. Side chain fragmentation of alkylated cysteine residues in electron capture dissociation mass spectrometry. J. Am. Soc. Mass Spectrom. 2006;17:1271–1274. doi: 10.1016/j.jasms.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 12.Falth M, Savitski MM, Nielsen ML, Kjeldsen F, Andren PE, Zubarev RA. Analytical utility of small neutral losses from reduced species in electron capture dissociation studied using SwedECD database. Anal. Chem. 2008;80:8089–8094. doi: 10.1021/ac800944u. [DOI] [PubMed] [Google Scholar]
- 13.O'Connor PB, Cournoyer JJ, Pitteri SJ, Chrisman PA, McLuckey SA. Differentiation of aspartic and isoaspartic acids using electron transfer dissociation. J. Am. Soc. Mass Spectrom. 2006;17:15–19. doi: 10.1016/j.jasms.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 14.Sargaeva NP, Lin C, O'Connor PB. Identification of aspartic and isoaspartic acid residues in amyloid β peptides, including A β 1–42, using electron-ion reactions. Anal. Chem. 2009;81:9778–9786. doi: 10.1021/ac901677t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cournoyer JJ, Pittman JL, Ivleva VB, Fallows E, Waskell L, Costello CE, O'Connor PB. Deamidation: differentiation of aspartyl from isoaspartyl products in peptides by electron capture dissociation. Protein Sci. 2005;14:452–463. doi: 10.1110/ps.041062905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li XJ, Lin C, O'Connor PB. Glutamine deamidation: differentiation of glutamic acid and γ-Glutamic Acid in peptides by electron capture dissociation. Anal. Chem. 2010;82:3606–3615. doi: 10.1021/ac9028467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mormann M, Macek B, de Peredo AG, Hofsteenge J, Peter-Katalinic J. Structural studies on protein O-fucosylation by electron capture dissociation. Int. J. Mass Spectrom. 2004;234:11–21. [Google Scholar]
- 18.Chrisman PA, Pitteri SJ, Hogan JM, McLuckey SA. SO2-electron transfer ion/ion reactions with disulfide linked polypeptide ions. J. Am. Soc. Mass Spectrom. 2005;16:1020–1030. doi: 10.1016/j.jasms.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kjeldsen F, Haselmann KF, Sorensen ES, Zubarev RA. Distinguishing of Ile/Leu amino acid residues in the PP3 protein by (Hot) electron capture dissociation in fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2003;75:1267–1274. doi: 10.1021/ac020422m. [DOI] [PubMed] [Google Scholar]
- 20.Fung YM, Chan TW. Experimental and theoretical investigations of the loss of amino acid side chains in electron capture dissociation of model peptides. J. Am. Soc. Mass Spectrom. 2005;16:1523–1535. doi: 10.1016/j.jasms.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Savitski MM, Nielsen ML, Zubarev RA. Side chain losses in electron capture dissociation to improve peptide identification. Anal. Chem. 2007;79:2296–2302. doi: 10.1021/ac0619332. [DOI] [PubMed] [Google Scholar]
- 22.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta. 2006;1764:1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swaney DL, McAlister GC, Coon JJ. Decision tree-driven tandem mass spectrometry for shotgun proteomics. Nat. Methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, McAlister GC, Thomson JA, Coon JJ. Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 2008;105:4093–4098. doi: 10.1073/pnas.0710515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McAlister GC, Berggren WT, Griep-Raming J, Horning S, Makarov A, Phanstiel D, Stafford G, Swaney DL, Syka JE, Zabrouskov V, Coon JJ. A proteomics grade electron transfer dissociation-enabled hybrid linear ion trap-orbitrap mass spectrometer. J. Proteome Res. 2008;7:312–3136. doi: 10.1021/pr800264t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swaney DL, Wenger CD, Thomson JA, Coon JJ. Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc. Natl., Acad. Sci. U.S.A. 2009;106:995–1000. doi: 10.1073/pnas.0811964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zee BM, Garcia BA. Electron transfer dissociation facilitates sequencing of adenosine diphosphate-ribosylated peptides. Anal. Chem. 2010;82:2–31. doi: 10.1021/ac902134y. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Udeshi ND, O'Malley M, Shabanowitz J, Hunt DF, Hart GW. Enrichment and site mapping of O-Linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell. Proteom. 2010;9:153–160. doi: 10.1074/mcp.M900268-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimsrud PA, den Os D, Wenger CD, Swaney DL, Schwartz D, Sussman MR, Ane JM, Coon JJ. Large-scale phosphoprotein analysis in medicago truncatula roots provides insight into in vivo kinase activity in legumes. Plant Physiol. 2010;152:19–28. doi: 10.1104/pp.109.149625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Lin C, Han L, Costello CE, O'Connor PB. Charge remote fragmentation in electron capture and electron transfer dissociation. J. Am. Soc. Mass Spectrom. 2010;21:646–656. doi: 10.1016/j.jasms.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McAlister GC, Phanstiel D, Good DM, Berggren WT, Coon JJ. Implementation of electron-transfer dissociation on a hybrid linear ion trap-orbitrap mass spectrometer. Anal. Chem. 2007;79:3525–3534. doi: 10.1021/ac070020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wenger CD, McAlister GC, Xia Q, Coon JJ. Sub-part-per-million precursor and product mass accuracy for high-throughput proteomics on an electron transfer dissociation-enabled orbitrap mass spectrometer. Mol. Cell. Proteom. 2010;9:754–763. doi: 10.1074/mcp.M900541-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Good DM, Wenger CD, McAlister GC, Bai DL, Hunt DF, Coon JJ. Post-acquisition ETD spectral processing for increased peptide identifications. J. Am. Soc. Mass Spectrom. 2009;20:1435–1440. doi: 10.1016/j.jasms.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wenger CD, Phanstiel DH, Lee MV, Coon JJ. COMPASS: An Open-Source Software Suite for High-Throughput Quantitative Proteomics with OMSSA. Proteomics. 2010 Accepted. [Google Scholar]
- 36.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang XY, Shi WY, Bryant SH. Open mass spectrometry search algorithm. J. Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 37.Fung YME, Duan LF, Chan TWD. A comparative study of the collision-induced dissociation and the electron capture dissociation of model peptides using electrospray ionization fourier-transform mass spectrometry. Eur. J. Mass Spectrom. 2004;10:449–457. doi: 10.1255/ejms.648. [DOI] [PubMed] [Google Scholar]
- 38.Adamson JT, Hakansson K. Infrared multiphoton dissociation and electron capture dissociation of high-mannose type glycopeptides. J. Proteome Res. 2006;5:49–501. doi: 10.1021/pr0504081. [DOI] [PubMed] [Google Scholar]
- 39.Bandu ML, Grubbs T, Kater M, Desaire H. Collision induced dissociation of ↦ hydroxy acids: evidence of an ion-neutral complex intermediate. Int. J. Mass Spectrom. 2006;251:40–46. [Google Scholar]
- 40.Li XJ, Cournoyer JJ, Lin C, O'Connor PB. The effect of fixed charge modifications on electron capture dissociation. J. Am. Soc. Mass Spectrom. 2008;19:1514–1526. doi: 10.1016/j.jasms.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chowdhury SM, Munske GR, Ronald RC, Bruce JE. Evaluation of low energy CID and ECD fragmentation behavior of mono-oxidized thio-ether bonds in peptides. J. Am. Soc. Mass Spectrom. 2007;18:493–501. doi: 10.1016/j.jasms.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweet SMM, Jones AW, Cunningham DL, Heath JK, Creese AJ, Cooper HJ. Database search strategies for proteomic data sets generated by electron capture dissociation mass spectrometry. J. Proteome Res. 2009;8:5–5484. doi: 10.1021/pr9008282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zubarev RA, Demirev PA, Hakansson P, Sundqvist BUR. Approaches and limits for accurate mass characterization of large biomolecules. Anal. Chem. 1995;67:3793–3798. [Google Scholar]
- 44.Zubarev RA, Hakansson P, Sundqvist B. Accuracy requirements for peptide characterization by monoisotopic molecular mass measurements. Anal. Chem. 1996;68:4060–4063. [Google Scholar]
- 45.Zubarev RA, Hakansson P, Sundqvist B. Accurate monoisotopic mass measurements of peptides: possibilities and limitations of high resolution Time-of-Flight particle desorption mass spectrometry. Rapid Commun. Mass Spectrom. 1996;10:1386–1392. [Google Scholar]
- 46.Conrads TP, Anderson GA, Veenstra TD, Pasa-Tolic L, Smith RD. Utility of accurate mass tags for proteome-wide protein identification. Anal. Chem. 2000;72:3349–3354. doi: 10.1021/ac0002386. [DOI] [PubMed] [Google Scholar]
- 47.Smith RD, Anderson GA, Lipton MS, Pasa-Tolic L, Shen Y, Conrads TP, Veenstra TD, Udseth HR. An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics. 2002;2:513–523. doi: 10.1002/1615-9861(200205)2:5<513::AID-PROT513>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 48.Spengler B. De novo sequencing, peptide composition analysis, and composition-based sequencing: a new strategy employing accurate mass determination by fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 2004;15:703–714. doi: 10.1016/j.jasms.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 49.An Thieu V, Kirsch D, Flad T, Muller C, Spengler B. Direct protein identification from nonspecific peptide pools by high-accuracy MS data filtering. Angew. Chem. Int. Ed Engl. 2006;45:3317–3319. doi: 10.1002/anie.200503787. [DOI] [PubMed] [Google Scholar]
- 50.Spengler B. Accurate mass as a bioinformatic parameter in data-to-knowledge conversion: fourier transform ion cyclotron resonance mass spectrometry for peptide de novo sequencing. Eur. J. Mass. Spectrom. 2007;13:83–87. doi: 10.1255/ejms.840. [DOI] [PubMed] [Google Scholar]
- 51.Spengler B, Hester A. Mass-based classification (MBC) of peptides: highly accurate precursor ion mass values can be used to directly recognize peptide phosphorylation. J. Am. Soc. Mass Spectrom. 2008;19:1808–1812. doi: 10.1016/j.jasms.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Pitteri SJ, Chrisman PA, McLuckey SA. Electron-transfer ion/ion reactions of doubly protonated peptides: effect of elevated bath gas temperature. Anal. Chem. 2005;77:5662–5669. doi: 10.1021/ac050666h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.