

Abstract
Epicardium-derived cells (EPDCs) contribute to formation of coronary vessels and fibrous matrix of the mature heart. Nuclear factor of activated T-cells cytoplasmic 1 (NFATC1) is expressed in cells of the proepicardium (PE), epicardium and EPDCs in mouse and chick embryos. Conditional loss of NFATC1 expression in EPDCs in mice causes embryonic death by E18.5 with reduced coronary vessel and fibrous matrix penetration into myocardium. In osteoclasts, calcineurin-mediated activation of NFATC1 by receptor activator of NFκB ligand (RANKL) signaling induces cathepsin K (CTSK) expression for extracellular matrix degradation and cell invasion. RANKL/NFATC1 pathway components also are expressed in EPDCs, and loss of NFATC1 in EPDCs causes loss of CTSK expression in the myocardial interstitium in vivo. Likewise, RANKL treatment induces Ctsk expression in PE-derived cell cultures via a calcineurin-dependent mechanism. In chicken embryo hearts, RANKL treatment increases the distance of EPDC invasion into myocardium, and this response is calcineurin dependent. Together, these data demonstrate a crucial role for the RANKL/NFATC1 signaling pathway in promoting invasion of EPDCs into the myocardium by induction of extracellular matrix-degrading enzyme gene expression.
Keywords: Heart development, EPDC, NFATC1, Chicken, Mouse
INTRODUCTION
During cardiac morphogenesis, cells of the PE migrate onto and envelop the myocardium forming the epicardium (Gittenberger-de Groot et al., 2010; Lie-Venema et al., 2007; Reese et al., 2002). A subset of epicardial cells undergo epithelial-to-mesenchymal transformation (EMT) and invade the subepicardial space and myocardium as EPDCs. EPDCs contribute to the formation of coronary vessels and the collagen-rich extracellular matrix (ECM) supporting scaffold for the cardiomyocytes called the fibrous matrix. EPDC invasion into subepicardium and myocardium is vital to proper coronary vessel and fibrous matrix formation; however, molecular mechanisms that regulate this process remain largely unknown.
The PE is an outgrowth of extracardiac mesothelial cells overlying the septum transversum (mouse)/sinus venosus (chick) that migrates onto the myocardium thus forming epicardium. Within the epicardium, the orientation of cell division differentiates cells that will remain in the epicardium from cells that delaminate from the epicardium and undergo EMT to become mesenchymal EPDCs (Wu et al., 2010). The PE and its derivative epicardium and EPDCs express transcription factors including Wilms' tumor 1 (WT1) and TBX18, which are characteristic of mesothelial cell lineages (Gittenberger-de Groot et al., 2010; Lavine and Ornitz, 2009; Lie-Venema et al., 2007; Olivey and Svensson, 2010; Wu et al., 2010). After invasion of EPDCs into myocardium, expression of WT1 and TBX18 is downregulated as the cells differentiate into coronary smooth muscle, endothelia and interstitial fibroblasts. In the mature heart, EPDC derivatives are present in the ECM-rich fibrous matrix component of the myocardium, as well as in the smooth muscle and endothelial cells of the coronary vasculature (Gittenberger-de Groot et al., 2010; Lavine and Ornitz, 2009; Lie-Venema et al., 2007; Reese et al., 2002).
NFATC1 is a transcription factor of the NFAT family that is regulated by calcium and the phosphatase calcineurin (Crabtree and Olson, 2002). During cardiac development, NFATC1 is expressed by endocardium as well as endocardial cushion and valve endothelial cells (de la Pompa et al., 1998; Ranger et al., 1998). Mice lacking NFATC1 expression have normal endocardial cushion formation and EMT, but lack the ECM remodeling and Ctsk expression that are necessary for valve maturation, causing embryonic lethality at embryonic day (E)12.5-E14.5 (Combs and Yutzey, 2009b; de la Pompa et al., 1998; Lange and Yutzey, 2006; Ranger et al., 1998). NFATC1 is also required in osteoclasts where it promotes ECM remodeling and invasion (Aliprantis et al., 2008; Negishi-Koga and Takayanagi, 2009). To stimulate osteoclast function, RANKL (TNFSF11 – Mouse Genome Informatics) binds to the RANK receptor, leading to activation of calcineurin and nuclear translocation of NFATC1 (Negishi-Koga and Takayanagi, 2009; Sitara and Aliprantis, 2010). Activated NFATC1 promotes expression of an array of potent ECM-degrading enzymes, including CTSK (Negishi-Koga and Takayanagi, 2009). NFATC1-dependent ECM degradation and osteoclast invasion are necessary for vascularization of bone and collagen deposition for new bone formation (Motyckova and Fisher, 2002; Raggatt and Partridge, 2010). Likewise, EPDC invasion is dependent upon ECM remodeling. However, the role of NFATC1 in EPDC invasion and maturation has not been investigated previously.
Here, we examine the function of NFATC1 in EPDC invasion of myocardium. NFATC1 is expressed by a subset of cells in the PE, epicardium, EPDCs and coronary vessels. The WT1-Cre mouse line (Zhou et al., 2008) was used for conditional loss of NFATC1 expression in EPDCs and their derivatives. The initial stages of epicardium formation and EMT are apparently normal in Wt1-Cre(+);Nfatc1(fl/fl) mutant embryos, but these animals do not survive after birth. At late fetal stages, there is an overall decrease of coronary vessel formation in the deep myocardium and reduced penetration of interstitial fibroblasts and fibrous matrix, which is indicative of a lack of EPDC invasion. Expression of the ECM-remodeling enzyme CTSK is reduced in hearts of mice that lack NFATC1 expression in EPDCs. Supporting studies in cultured chicken embryo hearts and isolated PE cells demonstrate that RANKL/NFATC1 signaling promotes CTSK expression and EPDC invasion into myocardium. Together, these studies support a mechanism whereby RANKL-mediated activation of NFATC1 in EPDCs promotes expression of the ECM-degrading enzyme CTSK and cell invasion into the myocardium.
MATERIALS AND METHODS
Chicken and mouse embryo collection
Nfatc1−/− and Nfatc1(fl/fl) mouse lines were obtained from Dr Laurie Glimcher (Brigham and Women's Hospital, Boston, MA, USA) (Aliprantis et al., 2008; Ranger et al., 1998). Wt1-Cre mice were obtained from Dr William Pu (Children's Hospital, Boston, MA, USA) (Zhou et al., 2008). GATA5-Cre mice were obtained from Dr Vesa Kaartinen (University of Michigan, MI, USA) (Merki et al., 2005). Mouse embryos were generated via timed matings with observation of a copulation plug designated as E0.5. Embryos that were alive and morphologically comparable with littermates were collected at E9.5-E17.5. Genotyping for NFATC1 mutation and/or WT1-Cre expression was performed by PCR as previously described (Aliprantis et al., 2008; Ranger et al., 1998; Zhou et al., 2008). Fertilized white leghorn chicken eggs (Charles River Laboratories, CT, USA) were incubated at 38°C under high humidity, and embryos were sacrificed at E4, E5, E7 and E14. All animal procedures were approved and performed in accordance with institutional guidelines.
Immunofluorescence and laser scanning confocal microscopy
Mouse and chick embryos were collected, fixed, dehydrated and paraffin-embedded as previously described (Shelton and Yutzey, 2007). Sections (5 μm) were deparaffinized and rehydrated, and antigen retrieval was performed using Antigen Unmasking Solution (#H3300; Vector Labs, CA, USA). Sections were prepared for immunofluorescence and laser scanning confocal microscopy (ICLSM) as previously described using the following primary antibodies: NFATC1 (1:100) (BD Biosciences, CA, USA), WT1 (1:100) (Calbiochem, CA, USA), CTSK (1:100) (Santa Cruz, CA, USA), CX40 (1:100) (Santa Cruz), COL1A1(1:100) (Millipore, MA, USA), MF20 (1:100) (Developmental Studies Hybridoma Bank, IA, USA), phosphohistone H3 (1:100) (Millipore), Alexa Fluor-conjugated secondary antibodies (Invitrogen Life Tech, CA, USA) and ToPro3 iodide nuclear stain (Invitrogen) (Combs and Yutzey, 2009b).
Preparation and processing of chick heart sections for ICLSM with TBX18 antibody (Santa Cruz) (1:250) was performed as previously described (Christoffels et al., 2009). Primary antibody labeling and tyramide signal amplification (TSA) was performed with Renaissance TSA Fluorescence Systems Tyramide Signal Amplification Kit (Perkin Elmer, MA, USA) according to the manufacturer's instructions.
For each independent experiment, immunofluorescence was detected using a Zeiss LSM 510 confocal microscope, and images were captured using Zeiss LSM version 3.2 SP2 software in parallel using identical confocal laser settings, and constant PMT filters and integration levels.
In situ hybridization
In situ hybridization (ISH) for mouse Nfatc1, Rankl and Rank, and chick Nfatc1, Rankl and Ctsk mRNAs was performed as previously described (Combs and Yutzey, 2009b; Lange and Yutzey, 2006).
Mouse embryo RNA isolation and real time RT-PCR
E14.5 mouse ventricle and forelimb tissue was collected in 200 μl Trizol (Invitrogen), the RNA was isolated and 1 μg of total RNA was used for cDNA synthesis with SuperScript II (Invitrogen) according to the manufacturer's protocols. Real-time PCR was performed using Taqman gene expression assays (Applied Biosystems Life Tech, CA, USA) for Sma (Assay ID: Mm01546133_m1) and Ctsk (Assay ID: Mm00484039_m1) on StepOnePlus Real-Time PCR System (ABI) according to the manufacturer's instructions. The gene encoding β2-microglobulin (Assay ID: Mm00437762_m1) was used for normalization, and results were confirmed by normalization with the gene encoding β-actin (Assay ID: Mm02619580_g1). Data were collected for four Wt1-Cre(+);Nfatc1(fl/fl) embryos and four Wt1-Cre(−);Nfatc1(fl/wt) control littermates (n=4).
Quantification of ventricular COL1A1 immunofluorescence and percentage myocardial penetration
For quantification of the area of COL1A1 immunofluorescence per ventricular area, pictomicrographs of right ventricle, left ventricle, apex, interventricular septum and atrioventricular valves were generated from at least three comparable non-consecutive sections of six Wt1-Cre(+);Nfatc1(fl/fl) embryos and six control littermates (n=6) using methods described for ICLSM. Ventricular area (in pixels) and the pixel area above 42 fluorescent units (green fluorescent pixels) were measured using Image J64. The fluorescent area was divided by the ventricular area to determine fluorescence per ventricular area. The fluorescent area per ventricular area for control embryos was set to one to determine the fold change for Wt1-Cre(+);Nfatc1(fl/fl) embryos.
To determine percentage myocardial penetration of COL1A1 fluorescence, ventricular and apex myocardial wall diameters were measured in an upper (basal), middle and lower (apical) region of each image using Image J64. The images were converted to binary (black and white). The distance from epicardium to the deepest point of COL1A1 reactivity (distance penetration) along the ventricle/apex myocardial wall diameter was measured for the upper, middle and lower points on each image. The distance of penetration divided by the total ventricular/apex diameter was calculated as the percentage myocardial penetration.
Immunohistochemical analysis of intramyocardial vessel penetration and myocardial spaces
Immunohistochemistry (IHC) was performed using ImmunoPure ABC Ultra-Sensitive Peroxidase IgG Staining Kit (Fisher Scientific, MA, USA) according to manufacturer's instructions. The following primary antibodies were applied overnight at 4°C; NFATC1, SMA (1:200) (Sigma-Aldrich, MO, USA), CX40, MF20. Horseradish peroxidase (HRP) detection was performed using 3,3-Diaminobenzidine(DAB) Enhanced Metal Substrate Kit (Pierce Biotech, IL, USA) according to the manufacturer's protocol.
Pictomicrographs were obtained with an Olympus BX51 microscope using Digital Camera Systems Spot software version 4.5 and filter and exposure settings were unchanged for comparison between Wt1-Cre(+);Nfatc1(fl/fl) and control animals. The distance from the epicardium to the closest vessel wall (vessel penetration) was measured, and percentage intramyocardial penetration was calculated as vessel penetration divided by ventricular diameter. Coronary vessels 2.33 μm2 or greater were used for quantification. Vessel penetration and percentage myocardial space per ventricular area were measured using ImageJ 64. Images for quantification were collected from at least four non-consecutive comparable sections from six Wt1-Cre(+);Nfatc1(fl/fl) embryos and six control littermates (n=6).
Chicken PE cell cultures
Aggregated PE cells, identified based on cell morphology and morphological landmarks, were dissected from PE/atrioventricular groove region of day 4.5 chicken embryos using tungsten needles (Schulte et al., 2007). The cell aggregates from four embryos were transferred to a 0.01% rat-tail collagen-coated chamber slide. Cells were cultured for 4 days in culture media [M199 (Cellgro Mediatech, VA, USA), 10% fetal bovine serum (Fisher), 1% chick embryo extract (Sera Labs International, West Sussex, UK) and 1% penicillin-streptomycin (Invitrogen)]. The culture media contained the following treatments; 800 ng/ml recombinant human (rh) RANKL (R&D Systems, MN, USA), 1 μg/ml rhOPG (R&D), 1 μg/ml cyclosporin A (CsA) (Novartis International AG, Basel, Switzerland) and 800 ng/ml BSA (Sigma) (vehicle control), and was refreshed on day 3 of culture. Cells were collected, RNA isolated and real time RT-PCR for Ctsk expression was performed as previously described (Combs and Yutzey, 2009b). Cultured primary chick PE cells were collected and processed for ICLSM as previously described (Combs and Yutzey, 2009b). Four independent experiments were performed in biological duplicate for each treatment type (n=4).
Chicken whole heart cultures and assessment of cell migration
E7 chick whole hearts were collected and labeled with 25 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen) in M199 media for 1 hour. Hearts were cultured 18 hours in 0.01% BSA-coated chamber slides containing culture media (described above) with treatments described above. Four independent experiments were performed in biological triplicate for each treatment group (n=4). Cultured chicken whole hearts were collected and paraffin embedded as above with the exception that d-Limonene (Hemo-D, Fisher) was used instead of xylene. Sections (5 μm) were blocked using ImmunoPure ABC Ultra-Sensitive Peroxidase Mouse IgG Staining Kit (Fisher) according to the manufacturer's instructions. Primary antibody was applied overnight at 4°C (CTSK). Secondary antibody was applied according to the manufacturer's protocol and streptavidin anti-568 (Invitrogen) (1:100) was applied for 30 minutes.
For quantification of EPDC migration, pictomicrographs were collected and a separate binary image was made. The distance each CFSE-labeled cell migrated away from the epicardium into the apex myocardium was measured in two comparable non-serial sections for each treatment group per independent experiment using Image J64. Average distance of EPDC migration was determined by the total distance of cell migration divided by total number of cells that migrated away from the epicardium for each treatment group. Four independent experiments were performed in biological duplicate (n=4).
Statistical analysis
Statistical significance was determined by Student's t-test with P≤0.01 as indicated. Data are reported as a mean with standard error of the mean (s.e.m.).
RESULTS
NFATC1 is expressed in the PE, epicardium, EPDCs, and mature coronary vessels in vivo
NFATC1 expression in the epicardium and epicardium-derived structures was visualized by immunofluorescence with confocal laser scanning microscopy (ICLSM) of E9.5, E10.5, E11.5, E14.5 and E17.5 mouse embryos, as well as E4, E7 and E14 chick embryo heart sections. During epicardial formation and proliferation, NFATC1 protein is expressed in mouse PE cells as well as cells of the septum transversum and endocardium at E9.5-E10.5 (Fig. 1A,A′,D-E″). In E4 chick, NFATC1 protein is localized to epithelial cells of the PE, liver bud, epicardium and sinus venosus (see Fig. S1A in the supplementary material). Later, NFATC1 is expressed by a subset of cells of the epicardium and EPDCs of the subepicardial space, beginning to invade the myocardium, at E11.5-E14.5 in mouse and E7 in chick (Fig. 1B,B′,F-F‴ and see Fig. S1B in the supplementary material). NFATC1 also is expressed by endothelial cells of coronary vessels and aortic valve endocardium at E17.5, just prior to birth (Fig. 1C,C′ and see Fig. S1C in the supplementary material). This pattern of NFATC1 protein expression by cells of the epicardium and EPDCs is consistent with mRNA expression in both chick and mouse hearts, as determined by in situ hybridization (ISH) (see Fig. S2 in the supplementary material). Significantly, NFATC1 is co-expressed with the transcription factors Wilms' tumor 1 (WT1) (Fig. 1D-F‴) and TBX18 (see Fig. S1D-F in the supplementary material), which are expressed by PE, epicardium and EPDCs (Haenig and Kispert, 2004; Zhou et al., 2008). At E14.5, NFATC1 is localized primarily to the cytoplasm of epicardial epithelial cells. This is in contrast to the nuclear NFATC1 localization indicative of its activation in PE and EPDCs that invade the subepicardial space and myocardium (Fig. 1D-F‴). Together, these data demonstrate that NFATC1 is expressed by the PE, epicardium, EPDCs and mature coronary endothelial cells. In addition, NFATC1 activation and nuclear localization occurs in PE and in EPDCs invading the subepicardium and myocardium.
Fig. 1.

NFATC1 colocalizes with WT1 in the PE, epicardium and EPDCs and is expressed by coronary endothelial cells. (A-C′) Immunofluorescence and laser scanning confocal microscopy (ICLSM) was performed on mouse heart sections using anti-NFATC1 antibody (green) and ToPro3 (blue). (A,A′) NFATC1-positive cells are apparent in the PE (arrow) and septum transversum (arrowhead) at E10.5. A′-C′ are magnified views of the boxed regions in A-C. (B,B′) NFATC1-positive epicardial cells (arrows) and endocardial cells (arrowhead) at E11.5 are indicated. (C,C′) Anti-NFATC1 labeling of coronary endothelial cells (arrow) is shown. (D-F‴) ICLSM performed on E9.5 and E14.5 mouse heart sections using anti-NFATC1 antibody (green), anti-WT1 antibody (red) and ToPro3 (blue) is shown. (D-E″) NFATC1 (green) is nuclear and colocalizes with WT1 (red) in cells of the PE at E9.5. (E-E″) Magnified views of the boxed region in D. (F-F‴) NFATC1-positive (green) and WT1-positive (red) cells of the epicardium (arrow) and EPDCs of the AVC (arrowhead) and right ventricle (*) are indicated. (F‴) Merged image with a magnified view of the AVC to highlight nuclear NFATC1 (arrows) and colocalization with WT1 in EPDCs. Inset in F‴ is a higher magnification view of the boxed regions in F-F‴. Arrows in the inset indicate NFATC1 nuclear localization. AoV, aortic valve; At, atrium; AVC, atrioventricular canal; Clm, coelom; CV, coronary vessel; EC, endocardial cushions; Endo, endocardium; Epi, epicardium; PE, proepicardium; RV, right ventricle; ST, septum transversum; Vt, ventricle.
Wt1-Cre(+);Nfatc1(fl/fl) mice have fewer NFATC1-positive epicardial cells, fewer NFATC1-positive EPDCs and reduced or absent myocardial spaces
NFATC1 function in epicardium and EPDCs was examined in mice with conditional loss of NFATC1. Systemic loss of NFATC1 does not obviously affect the initial stages of PE migration or epicardium formation (see Fig. S3 in the supplementary material). However, systemic loss of NFATC1 results in embryonic lethality at E12.5-E14.5 due to defects in heart valve remodeling (de la Pompa et al., 1998; Ranger et al., 1998; Chang et al., 2004). Therefore, mice with conditional loss of NFATC1 in EPDCs were generated to determine the requirements for NFATC1 in EPDC maturation and formation of coronary vessels and fibrous matrix. WT1-Cre mice [Wt1-Cre(+)] were bred with NFATC1-flox mice [Nfatc1(fl/fl)] for conditional loss of NFATC1 function in cells of the WT1 lineage (Aliprantis et al., 2008; Zhou et al., 2008).
Wt1-Cre(+);Nfatc1(fl/fl) embryos are grossly indistinguishable from control littermates of the genotypes Wt1-Cre(+);Nfatc1(fl/wt), Wt1-Cre(−);Nfatc1(fl/fl) or Wt1-Cre(−); Nfatc1(fl/wt) throughout development (see Fig. S4A-C,E-G in the supplementary material). At a macroscopic level, the hearts of Wt1-Cre(+);Nfatc1(fl/fl) embryos are comparable with controls in size and structure. However, there is evidence for compromised heart function in retention of a W-shaped (bifid) apex beyond E14.5 (see Fig. S4G,H in the supplementary material, asterisk). Whereas Wt1-Cre(+);Nfatc1(fl/fl) embryos are collected at the expected Mendelian ratio of 25% at E13.5-E14.5, Wt1-Cre(+);Nfatc1(fl/fl) embryos are under-represented in those collected at E17.5-E18.5 (19%) and make up only 3% of pups genotyped postnatally (see Fig. S4I in the supplementary material). In utero echocardiograms demonstrated that the majority of Wt1-Cre(+);Nfatc1(fl/fl) embryos have no detectable heartbeat at E17.5-E18.5 (data not shown). Similar results were obtained with Gata5-Cre(+);Nfatc1(fl/−) mice generated in parallel (data not shown) (Merki et al., 2005). Like Wt1-Cre(+);Nfatc1(fl/fl) mice, Gata5-Cre(+);Nfatc1(fl/−) mice die prenatally, with 0% being present for genotyping at postnatal day (P)21. These data indicate that loss of NFATC1 in EPDCs results in loss of cardiac function and prenatal lethality at E17.5-E18.5.
Successful deletion of NFATC1 expression in epicardial cells/EPDCs of Wt1-Cre(+); Nfatc1(fl/fl) mice is demonstrated by lack of anti-NFATC1 antibody labeling of epicardial cells and subepicardial EPDCs, as well as fewer NFATC1-positive intramyocardial EPDCs (Fig. 2B,B′). By contrast, NFATC1-positive EPDCs are seen in the subepicardium and in interstitial cells of the myocardium of control embryos (Fig. 2A,A′). NFATC1 protein expression is apparent in non-epicardium-derived tissues such as the atrioventricular valve in both Wt1-Cre(+); Nfatc1(fl/fl) and control embryos as previously described (Fig. 2; see Fig. S5 in the supplementary material) (de la Pompa et al., 1998; Ranger et al., 1998). A few NFATC1-positive interstitial cells are noted in the myocardium in Wt1-Cre(+);Nfatc1(fl/fl) mice, owing to incomplete Cre-mediated recombination or NFATC1 expression in WT1 non-expressing cells. NFATC1 deletion also occurs throughout the epicardium and in a subset of cells of the sinus venosus region in Wt1-Cre(+);Nfatc1(fl/fl) embryos at E10.5 (data not shown). These data demonstrate that Wt1-Cre(+);Nfatc1(fl/fl) mice have loss of NFATC1 expression specifically in cells of the WT1 lineage, including EPDCs, and can be used to determine the role of NFATC1 in EPDC function.
Fig. 2.

Wt1-Cre(+);Nfatc1(fl/fl) embryos have reduced NFATC1-positive epicardial cells and EPDCs with reduced myocardial spaces. Immunohistochemistry was performed on E17.5 mouse heart sections with anti-NFATC1 antibody (brown). (A,A′) Control Wt1-Cre(−);Nfatc1(fl/wt) embryo section with NFATC1-positive cells of the epicardium and EPDCs of the subepicardium and myocardium (arrows in A′; magnified view of boxed area in A) indicated. NFATC1-positive mitral valve (MV) endothelium (arrowhead) is shown. Abundant myocardial spaces (asterisks) are seen throughout the control myocardium, as indicated. (B,B′) Wt1-Cre(+);Nfatc1(fl/fl) embryo section with reduced (black arrow in B′) or absent (red arrow B′) NFATC1 staining of the epicardium and EPDCs is shown. NFATC1 reactivity is apparent in mitral valve endothelium (arrowhead in B) of Wt1-Cre(+);Nfatc1(fl/fl) embryo heart section. Myocardial spaces are greatly reduced or absent in Wt1-Cre(+);Nfatc1(fl/fl) myocardium (B′). (C) Quantification of the percentage myocardial area occupied by spaces was determined in six embryos of each genotype (n=6). *P≤0.01.
At E17.5 normal ventricular myocardium is characterized by abundant myocardial spaces and blood vessels in both the shallow (outer) and deep (inner) myocardium. By contrast, the ventricular myocardium of Wt1-Cre(+);Nfatc1(fl/fl) embryos is dense with cardiomyocytes, and has a significant reduction in percentage myocardial spaces (Fig. 2). Previous studies have indicated a role for epicardium signaling in myocardium compaction (Olivey and Svensson, 2010). However, a dense compacted myocardium is apparent in hearts of Wt1-Cre(+);Nfatc1(fl/fl) embryos at E17.5, indicating that this process is unaffected by loss of NFATC1 function in cells of the WT1-Cre lineage (Figs 2 and 3; see Fig. S4H in the supplementary material). Coronary vessels in Wt1-Cre(+);Nfatc1(fl/fl) embryos are formed in the atrioventricular canal (AVC), but are reduced in the deep myocardium (Fig. 2B,B′). Therefore, loss of NFATC1 expression in EPDCs of the WT1-Cre lineage results in reduced or absent myocardial spaces with dense myocardium, limited coronary vessel penetration and prenatal lethality.
Fig. 3.

Wt1-Cre(+);Nfatc1(fl/fl) embryos lack interstitial fibrous matrix and have reduced penetration of COL1A1-expressing cells. (Aa-Af) Trichrome staining of E17.5 control and Wt1-Cre(+);Nfatc1(fl/fl) embryo heart sections. (a,c,e) Fibrillar collagen (blue) is seen throughout the myocardium and tricuspid valve (arrowhead) of a control Wt1-Cre(−); Nfatc1(fl/fl) embryo. (b,d,f) The Wt1-Cre(+);Nfatc1(fl/fl) embryo lacks blue fibrillar collagen staining in myocardium and has a bifid apex (asterisk). Fibrillar collagen in the tricuspid valve (TV; arrowhead) is indicated. (Ba-Bf) Immunofluorescence and laser scanning confocal microscopy performed with anti-COL1A1 antibody (green) and ToPro3 (blue) on E17.5 mouse heart sections. (a,c,e) COL1A1-positive matrix is present throughout the myocardium (bracket) and in the tricuspid valve (arrowhead) of a control Wt1-Cre(+);Nfatc1(fl/wt) embryo. (b,d,f) COL1A1-positive matrix extends into the shallow myocardium as indicated by solid bracket. Deeper myocardial areas lack COL1A1 reactivity (broken bracket) in Wt1-Cre(+);Nfatc1(fl/fl) embryos. COL1A1 reactivity in the tricuspid valve (arrowhead) is retained and epicardial blebbing is noted (d, arrow) in the Wt1-Cre(+);Nfatc1(fl/fl) embryo. (C,D) Quantification of fold change in fluorescence per area (C) and percentage myocardial penetration of COL1A1 florescence (D) was determined in six embryos of each genotype (n=6). *P≤0.01. RV, right ventricle; LV, left ventricle; IVS, interventricular septum; AV, atrioventricular; TV, tricuspid valve.
Loss of NFATC1 expression in EPDCs results in decreased cardiac fibrous matrix formation
EPDCs that migrate into the myocardium represent the majority of cells populating the ventricular wall with interstitial fibroblasts that synthesize the ECM of the fibrous matrix (Gittenberger-de Groot et al., 2010; Lie-Venema et al., 2007; Reese et al., 2002). The formation of cardiac fibrous matrix was examined in E17.5 mouse hearts lacking epicardial NFATC1. Trichrome staining demonstrates that the fibrous matrix (blue) of the heart is essentially absent in Wt1-Cre(+);Nfatc1(fl/fl) embryos (Fig. 3Ab,Ad,Af; see Fig. S4H in the supplementary material). In addition, the apex in Wt1-Cre(+);Nfatc1(fl/fl) hearts is bifid, consistent with loss of fibrous matrix collagens in other mouse models (Fig. 3Af; see Fig. S4H in the supplementary material) (Lincoln et al., 2006). By contrast, abundant fibrous matrix (blue) is seen throughout the myocardial interstitium in control littermates (Fig. 3Aa,Ac,Ae; see Fig. S4D in the supplementary material). As expected, collagen deposition (blue) in the atrioventricular valves of Wt1-Cre(+);Nfatc1(fl/fl) embryos is apparently unaffected (see Fig. S4H in the supplementary material), indicative of normal NFATC1 function in valves. Together, these data indicate that NFATC1 function in EPDCs is necessary for formation of the fibrous matrix during cardiac development.
Because collagen 1 is a major component of the cardiac fibrous matrix, anti-collagen 1a1 (COL1A1) antibody labeling and ICLSM were used to further demonstrate a specific loss of fibrillar COL1A1 in the ventricular myocardium of Wt1-Cre(+);Nfatc1(fl/fl) embryos (Ott et al., 2008). Wt1-Cre(+);Nfatc1(fl/fl) embryos have COL1A1-positive matrix deposition that extends from the surface epicardium into the shallow myocardium, but have significantly reduced COL1A1 reactivity in deeper myocardial interstitium (Fig. 3Bb,Bd,Be). By contrast, control embryos have anti-COL1A1-positive matrix that extends from the epicardium through the inner myocardium to the endothelium of the trabeculae (Fig. 3Ba,Bc,Be). Quantification of percentage penetration of COL1A1 immunoreactivity in the myocardium demonstrates that, in Wt1-Cre(+);Nfatc1(fl/fl) embryos, COL1A1 protein extends to only 22.5% of the distance that spans the ventricular free wall from epicardium to trabeculae, and to 17.4% at the apex (Fig. 3D). By contrast, control littermates had COL1A1 penetration of 89.0% and 90.4% of total ventricular free wall and apex thickness, respectively. Quantification of total COL1A1 protein per area also demonstrated Wt1-Cre(+);Nfatc1(fl/fl) embryo hearts have a significant reduction in COL1A1 protein in the right ventricle, left ventricle, apex and interventricular septum compared with control littermates (Fig. 3C). Importantly, atrioventricular valves express high levels of COL1A1 and there are no significant differences in COL1A1-positive immunofluorescence per atrioventricular valve area among genotypes. These data demonstrate that loss of NFATC1 expression in EPDCs results in decreased fibrous matrix synthesis and decreased fibrous matrix penetration into the myocardium.
Loss of NFATC1 expression in EPDCs results in decreased penetration of coronary vessels and fewer activated fibroblasts within the myocardium
EPDC migration into myocardium is required for intramyocardial coronary vessel development and NFATC1 is expressed in both EPDCs and maturing coronary vessels (Fig. 1; see Figs S1 and S5 in the supplementary material) (Gittenberger-de Groot et al., 2010; Lie-Venema et al., 2007; Majesky, 2004). EPDC invasion is also required for investment of the ventricular myocardium with interstitial fibroblasts (Taylor et al., 2003). Wt1-Cre(+);Nfatc1(fl/fl) embryos were examined in order to determine the effects of loss of NFATC1 expression in EPDCs on interstitial fibroblast investment and coronary vessel formation. During cardiac morphogenesis, EPDC-derived interstitial cells express markers associated with fibroblast activation, such as smooth muscle α-actin (SMA) (Haudek et al., 2009). IHC with anti-SMA antibody was used to investigate the distribution of interstitial and coronary smooth muscle cells in ventricular myocardium of Wt1-Cre(+);Nfatc1(fl/fl) and control embryos. Deep ventricular areas, towards the trabeculae, in Wt1-Cre(+);Nfatc1(fl/fl) embryos have greatly reduced or absent activated fibroblasts and coronary smooth muscle cells (Fig. 4B,F). However, both of these cell types are observed in the shallow myocardium of the right ventricular free wall. By contrast, SMA-positive cells are prevalent throughout right ventricular and interventricular septum myocardium of control embryos at E17.5 (Fig. 4A,E). Thus, Wt1-Cre(+);Nfatc1(fl/fl) embryos have reduced interstitial fibroblast and coronary smooth muscle cell penetration into myocardium, whereas cell differentiation is apparently unaffected. Real-time RT-PCR demonstrates that, as early as E14.5, Wt1-Cre(+);Nfatc1(fl/fl) embryos have reduced SMA expression in ventricular myocardium, compared with control littermates, whereas SMA expression in the forelimbs is unaffected (Fig. 4J). Together, these data demonstrate that loss of NFATC1 function in EPDCs results in reduced fibroblast activation throughout the myocardium and is consistent with the observed reduction in fibrous matrix deposition.
Fig. 4.

Wt1-Cre(+);Nfatc1(fl/fl) embryos have reduced investment of activated fibroblasts and reduced intramyocardial vessel penetration. Immunohistochemistry performed on E17.5 mouse heart sections using anti-SMA (A,B,E,F) or anti-CX40 (C,D,G,H) antibodies (brown). (A,E) SMA-positive coronary vessels (A, arrows) and activated fibroblasts (E, inset arrowheads) in the control Wt1-Cre(+);Nfatc1(fl/wt) embryo are indicated. (B,F) SMA-positive coronary vessels (B, arrows) in shallow but not deep myocardium and lack of intramyocardial activated fibroblasts (F, inset) are apparent in the Wt1-Cre(+);Nfatc1(fl/fl) embryo. (C,G) CX40-positive coronary vessels (C, arrows and inset) and IVS vessels (arrowhead, G inset) in a control Wt1-Cre(+);Nfatc1(fl/wt) embryo. (D,H) Shallow CX40-positive vessels (D, arrows and inset) and lack of IVS vessels (H, inset) are depicted in a Wt1-Cre(+);Nfatc1(fl/fl) embryo. IVS, interventricular septum; RV, right ventricle. (I) Quantification of percentage intramyocardial penetration of coronary vessels was determined in six embryos of each genotype (n=6). (J) Real-time RT-PCR quantification of fold change in SMA expression for four E14.5 embryos of each Wt1-Cre(−);Nfatc1(fl/wt) control and Wt1-Cre(+);Nfatc1(fl/fl) genotype (n=4). *P≤0.01.
The majority of differentiated coronary vessel cells arise from EPDCs; therefore, EPDC invasion into myocardium is required for coronary vessel formation and is vital to proper heart development and myocyte survival (Gittenberger-de Groot et al., 2010; Lavine and Ornitz, 2009; Lie-Venema et al., 2007). Coronary vessel formation was assessed in mice lacking NFATC1 expression in EPDCs. IHC and ICLSM, in conjunction with antibody labeling with anti-SMA and anti-connexin 40 (CX40), was used to detect smooth muscle (SMA) and endothelial (CX40) cells of the coronary vessels. Coronary vessels containing smooth muscle and endothelial components are apparent in the shallow myocardium of Wt1-Cre(+);Nfatc1(fl/fl) embryos at E17.5. However, penetrant vessels are not apparent in deeper myocardial areas (Fig. 4B,D,F,H). By contrast, coronary vessels comprised of both smooth muscle and endothelial cells are found in both shallow and deep myocardial areas in control littermates (Fig. 4A,C,E,G). Quantification of intramyocardial vessel penetration demonstrates that Wt1-Cre(+);Nfatc1(fl/fl) embryos have significantly shallower placement of coronary vessels within the myocardium compared with control littermates (Fig. 4I). There are no significant differences in the total number of coronary vessels assessed per heart section among genotypes. It is important to note that although location is affected in Wt1-Cre(+);Nfatc1(fl/fl) embryos, loss of NFATC1 expression in EPDCs does not prevent differentiation of coronary smooth muscle or endothelial cells (Fig. 4; see Fig. S5 in the supplementary material). In addition, there are no differences in the numbers of apoptotic cells among genotypes at E14.5 or E17.5, as determined by TUNEL assay (data not shown). There are also no significant differences in epicardial cell/EPDC proliferation among genotypes at E11.5, the peak proliferative period for these cells (see Fig. S6 in the supplementary material) (Wu, 2010). Together, these data demonstrate that loss of NFATC1 in EPDCs leads to reduced penetration of EPDCs into myocardium necessary for coronary vessel and fibrous matrix formation. However, loss of NFATC1 does not appear to affect total coronary vessel number, EPDC cell differentiation, cell survival or proliferation.
Wt1-Cre(+);Nfatc1(fl/fl) embryos have significantly reduced CTSK expression in the myocardial interstitium in vivo, and RANKL treatment increases Ctsk expression in cultured PE cells via a calcineurin-dependent mechanism
The RANKL/NFATC1 signaling pathway is active in osteoclasts where it is required for cell invasion via induction of the ECM-degrading enzyme Ctsk (Negishi-Koga and Takayanagi, 2009). To determine whether RANKL/NFATC1 pathway components are expressed in a manner consistent with function in EPDC invasion into the myocardium, in situ hybridization using probes for Nfatc1, Rankl, Rank and Ctsk mRNA was performed on mouse and chick heart sections. Nfatc1, Rankl and Rank mRNAs are expressed by EPDCs in the subepicardium and myocardium of E13.5 mouse (Fig. 5D-F). Additionally, Nfatc1, Rankl and Ctsk mRNAs are expressed by EPDCs in the subepicardium and myocardium of E7 chick (Fig. 5A-C). EPDC expression of RANKL/NFATC1 pathway components is seen in addition to previously reported expression by endocardial cushion endothelial cells (Fig. 5) (Combs and Yutzey, 2009b; Lange and Yutzey, 2006). This spatiotemporal expression pattern is consistent with RANKL/NFATC1 function in EPDC invasion of the subepicardial space and myocardium.
Fig. 5.

RANKL/NFATC1 pathway components are expressed in mouse and chick embryos during EPDC invasion. (A-F) In situ hybridization was performed on E7 chick (A-C) and E13.5 mouse (D-F) heart sections. (A,D) Nfatc1 mRNA expression in EPDCs (arrowheads) and valve endothelial cells (arrows) is indicated. (B,E) Rank ligand (B,E) and receptor (F) are expressed by EPDCs (arrowheads) and valve endothelial cells (arrows). (C) Ctsk mRNA is expressed by avian EPDCs (arrowheads) and valve endothelial cells (arrow). RV, right ventricle; LV, left ventricle.
NFATC1 function is required for CTSK expression in endocardial cushion endothelial cells and osteoclasts (Lange and Yutzey, 2006; Negishi-Koga and Takayanagi, 2009). Anti-CTSK antibody labeling and ICLSM was used to determine that CTSK expression is greatly reduced or absent in the myocardium of Wt1-Cre(+);Nfatc1(fl/fl) embryos (Fig. 6B,D). By contrast, CTSK is abundantly expressed in the ventricular myocardial interstitium of control embryos during EPDC invasion at E14.5 (Fig. 6A,C). Real-time RT-PCR also demonstrates significantly reduced Ctsk transcript levels in ventricles of Wt1-Cre(+);Nfatc1(fl/fl) embryo hearts, compared with control littermates at E14.5, whereas Ctsk expression in forelimbs is not significantly different among genotypes (Fig. 6E). The co-expression of RANKL/NFATC1 pathway components during EPDC invasion, combined with the loss of CTSK expression in mice lacking NFATC1 in EPDCs, is evidence for RANKL/NFATC1 pathway function as a molecular mechanism promoting EPDC invasion into myocardium.
Fig. 6.

Wt1-Cre(+);Nfatc1(fl/fl) embryos have reduced CTSK expression in the myocardial interstitium. Immunofluorescence and laser scanning confocal microscopy was performed on E14.5 mouse heart sections with anti-CTSK antibody (green). (A,C) CTSK expression in control Wt1-Cre(+);Nfatc1(fl/wt) embryo (arrows) is depicted. (B,D) CTSK expression is reduced in a Wt1-Cre(+);Nfatc1(fl/fl) embryo (arrow). (E) Fold change in Ctsk mRNA expression was quantified using real-time RT-PCR for four E14.5 embryos of each Wt1-Cre(−); Nfatc1(fl/wt) control and Wt1-Cre(+);Nfatc1(fl/fl) genotype (n=4). (F) Ctsk expression was assessed in primary chick PE cell cultures treated with rhRANKL, CsA or rhRANKL+CsA. Fold change in Ctsk was quantified using real-time RT-PCR from four independent experiments performed in duplicate (n=4). *P≤0.01. LV, left ventricle.
The ability of RANKL to activate NFATC1 and promote Ctsk expression was assessed in cultured primary avian PE-derived cells. PE cells from E4 chick embryos were explanted and subjected to treatment with recombinant human (rh) RANKL, cyclosporin A (CsA), a pharmaceutical calcineurin inhibitor, bovine serum albumin (BSA) as a vehicle control or rhRANKL+CsA. Cultured PE cells maintain expression of epicardial markers such as WT1, TBX18 and NFATC1 over the 4-day culture period, and few contaminant MF20-positive myocytes were observed (see Fig. S7 in the supplementary material). Treatment of cultured PE cells with rhRANKL increases NFATC1 nuclear localization compared with BSA-treated controls, while addition of CsA or the RANKL inhibitor osteoprotegrin (OPG) to cultures inhibits RANKL-induced NFATC1 nuclear translocation (see Fig. S8 in the supplementary material). RANKL treatment of PE-derived cells also significantly increases Ctsk mRNA expression, as detected by real-time RT-PCR (Fig. 6F). In these experiments, rhRANKL treatment leads to a fivefold increase in Ctsk expression; however, rhRANKL-induced Ctsk expression is blocked by addition of CsA. Addition of CsA alone to PE cells significantly reduces Ctsk expression below the level of transcription detected in control BSA-treated cells, suggesting that PE-derived cells have a basal level of calcineurin-induced Ctsk expression. In addition, NFATC1 and CTSK proteins are co-expressed in RANKL-treated PE cells (see Fig. S9C,C′ in the supplementary material). Thus, rhRANKL induces NFATC1 nuclear localization and activates CTSK gene expression in cultured PE cells by a calcineurin-dependent mechanism. Together with the observation that CTSK expression and EPDC invasion is reduced in mice with EPDC-specific loss of NFATC1, these data support a mechanism whereby calcineurin-mediated RANKL activation of NFATC1 promotes CTSK expression required for ECM degradation and EPDC invasion into myocardium.
RANKL treatment promotes EPDC invasion into myocardium and CTSK expression via a calcineurin-dependent mechanism in cultured chick whole hearts
The ability of RANKL to promote NFATC1 activation and EPDC invasion in vivo was examined using cultured chicken embryo whole hearts. Whole hearts were isolated at E7 and the epicardium labeled with CFSE. Whole hearts were then cultured for 18 hours in the presence of rhRANKL, rhOsteoprotegrin (rhOPG) a soluble RANK receptor (RANK antagonist), CsA, BSA as a vehicle control, rhRANKL+rhOPG or rhRANKL+CsA. Hearts were then paraffin-embedded and sectioned for antibody labeling and ICLSM. EPDC invasion was quantified by measuring the distance CFSE-labeled EPDCs migrated away from the epicardium and into the myocardium. Addition of rhRANKL to the media of cultured hearts significantly increases the distance of EPDC invasion into the myocardium (Fig. 7B-B″). Addition of rhRANKL+ CsA to the media inhibits the effects of rhRANKL-induced EPDC invasion (Fig. 7D-D″), demonstrating that rhRANKL promotes EPDC invasion via a calcineurin-dependent mechanism. Addition of rhRANKL+OPG, CsA alone or OPG alone significantly decreases EPDC invasion compared with BSA-treated controls. This suggests a basal level of endogenous RANKL/NFAT signaling occurs in control hearts that is required for EPDC invasion. Although rhRANKL increases EPDC migration distance, there was no difference among treatments in the number of EPDCs that migrated away from the epicardium. Therefore, manipulation of RANKL/NFAT signaling in cultured hearts does not affect epicardium cell EMT or proliferation. These data demonstrate that rhRANKL promotes EPDC invasion into the myocardium via a calcineurin-dependent mechanism.
Fig. 7.

RANKL increases EPDC migration distance and CTSK expression via a calcineurin/NFAT-dependent mechanism. (A-D″) E7 chick whole hearts were treated with CFSE to label the epicardium then cultured with addition of BSA (A-A″), RANKL (B-B″), CsA (C-C″) or RANKL+CsA (D-D″) to the media. Immunofluorescence and laser scanning confocal microscopy was performed to detect CFSE (green), CTSK (red) and ToPro3 (blue). CFSE-labeled EPDCs (arrows) invade the myocardium. CTSK expression by CFSE-labeled EPDCs (yellow color on merged section in B″) is indicated (arrowheads). (E) Quantification of average distance of EPDC migration into myocardium (n=4). *P≤0.01.
In order to determine whether manipulation of RANKL/NFAT signaling in EPDCs affects CTSK expression, ICLSM with anti-CTSK antibody was performed on CFSE-labeled cultured chick hearts subjected to altered RANKL/calcineurin signaling. Addition of rhRANKL to the media of cultured hearts results in increased CTSK expression compared with BSA-treated controls (Fig. 7B′). Addition of CsA in combination with rhRANKL restores CTSK immunoreactivity to levels comparable with BSA-treated controls (Fig. 7D′). Addition of CsA alone to cultured hearts results in CTSK protein expression below basal levels (Fig. 7C′). Thus, rhRANKL treatment increases CTSK expression via a calcineurin-dependent mechanism in intact hearts. Taken together, these data support a mechanism whereby RANKL/NFATC1 signaling promotes EPDC invasion via CTSK gene induction.
DISCUSSION
Here, we demonstrate that RANKL activation of NFATC1 promotes CTSK expression and invasion of EPDCs into myocardium (Fig. 8). Loss of NFATC1 in EPDCs results in decreased coronary vessel and fibrous matrix penetration into ventricular myocardium, leading to embryonic death in mice. By contrast, NFATC1 expression in PE and epicardium is not necessary for epicardium formation, EMT or EPDC differentiation into coronary smooth muscle, coronary endothelium or interstitial fibroblasts. This study is the first to report a molecular mechanism for promoting EPDC invasion of myocardium necessary for coronary vessel and fibrous matrix development.
Fig. 8.
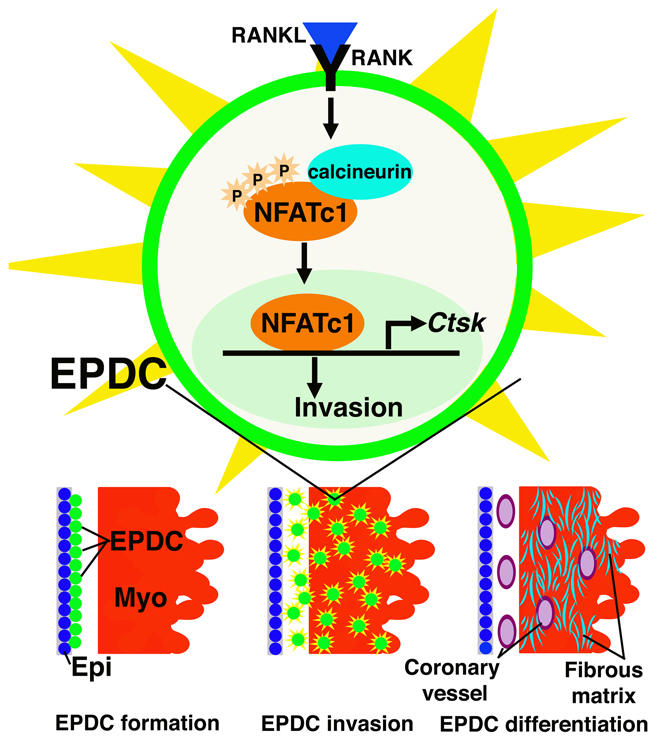
Model of NFATC1-induced EPDC invasion. RANKL induces NFATC1 nuclear localization to promote CTSK expression for ECM degradation and EPDC invasion into myocardium. EPDCs that differentiate into cells of the coronary vasculature and interstitial fibroblasts, which produce the fibrous matrix, require NFATC1 for invasion. NFATC1 expression in PE and EPDCs is not necessary for epicardium formation or for EPDC differentiation.
In addition to being expressed by PE and epicardial cells that will undergo EMT to generate EPDCs, NFATC1 also is expressed endocardial cushion endothelial cells that will undergo EMT to populate the endocardial cushions (de la Pompa et al., 1998; de Lange et al., 2004; Lincoln et al., 2004; Ranger et al., 1998). Previous studies have demonstrated that NFATC1 function is not required for endocardial cushion formation or EMT; however, expression of NFATC1 by endocardial cushion endothelial cells is required for later processes of valve remodeling (de la Pompa et al., 1998; Lange and Yutzey, 2006; Ranger et al., 1998). Likewise, data reported here demonstrate that NFATC1 function is not required for initial stages of epicardium formation or EMT, but is necessary for ECM remodeling associated with EPDC invasion.
WT1-expressing EPDCs differentiate into coronary endothelia, coronary smooth muscle and interstitial fibroblasts (Zhou et al., 2008; Zhou et al., 2010). Coronary endothelial, smooth muscle and interstitial fibroblast differentiation is observed in mice lacking NFATC1 expression in cells of the WT1 lineage. Therefore, NFATC1 function is not required for differentiation of these cell lineages during heart development. The observed loss of NFATC1 expression in endothelial cells of the coronary vessels in Wt1(+);Nfatc1(fl/fl) embryos demonstrates that WT1-expressing cells contribute to this lineage in vivo. There is controversy as to the specific origins of coronary endothelial cells, and it is possible that the WT1-expressing coronary endothelial cells observed may be from a non-PE origin, as suggested by Red-Horse and colleagues (Norden et al., 2010; Red-Horse et al., 2010). Previous studies by Zeini et al. have demonstrated a requirement for calcineurin-NFAT signaling in endothelial cells for coronary angiogenesis during vascular plexus formation (Zeini et al., 2009). Our data demonstrate that NFATC1 is not specifically required for this process. However, our observations do not preclude the necessity for other calcineurin targets, including other NFAT family members, in vascular plexus formation. Our studies, taken together with previous work, demonstrate a conserved NFATC1-dependent mechanism for invasion of myocardium by multiple EPDC progenitor cell types, including those of coronary vasculature.
RANKL/NFATC1 pathway function promotes EPDC invasion into myocardium via induction of ECM-remodeling enzymes such as CTSK. RANKL/NFATC1 pathway function is required for ECM remodeling by a variety of cell types during development, including osteoclasts, cardiac valve endothelial cells and EPDCs (Combs and Yutzey, 2009a; Negishi-Koga and Takayanagi, 2009). In osteoclasts, NFATC1 activation via RANKL signaling upregulates several ECM-degrading enzymes, including MMP9, MMP13 and CTSK, which leads to cell invasion by ECM remodeling (Karsenty et al., 2009; Sitara and Aliprantis, 2010). As mice lacking Ctsk expression have no reported cardiac abnormalities, it is likely that multiple ECM-remodeling enzymes are regulated by NFATC1 in EPDCs (Chang et al., 2004; Funicello et al., 2007; Saftig et al., 1998). This is supported by the observation that loss of NFATC1 in EPDCs or valve endothelial cells leads to ECM remodeling defects resulting in embryonic lethality (de la Pompa et al., 1998; Ranger et al., 1998). Together, these studies indicate that NFATC1 is a major regulator of ECM remodeling and cell invasion in multiple cell types.
CTSK is a cysteine protease linked to physiological and pathological cell migration/invasion via ECM degradation (Negishi-Koga and Takayanagi, 2009; Onishi et al., 2010; Rapa et al., 2006). CTSK is a downstream target of calcineurin/NFATC1 activation in multiple cell types, including osteoclasts, valve endothelial cells and EPDCs (Lange and Yutzey, 2006; Negishi-Koga and Takayanagi, 2009). Cathepsins play an important role in ECM degradation and vascular cell invasion during development, wound healing and cancer metastasis (Lutgens et al., 2007; Onishi et al., 2010; Saftig et al., 1998). The mode of action for CTSK is well described in bone, where osteoclasts secrete CTSK to degrade collagen-rich ECM, forming lacunae through which vasculogenic progenitor cells migrate (Karsenty et al., 2009; Motyckova and Fisher, 2002). EPDC migration through lacunae in the myocardial interstitium has been previously described, but the origin of these intramyocardial spaces is unknown (Gittenberger-de Groot et al., 2010; Lie-Venema et al., 2007; Reese et al., 2002). Lack of intramyocardial spaces in Wt1-Cre(+);Nfatc1(fl/fl) embryos suggests that these lacunae could be formed via NFATC1-dependent ECM remodeling during EPDC invasion. Together, these studies support a mechanism whereby remodeling enzymes, including CTSK, create spaces in the myocardium necessary for EPDC migration and investment during coronary vessel and fibrous matrix development.
EPDCs are a heterogeneous population of cells that form coronary vessels and fibrous matrix, are necessary for Purkinje fiber differentiation and support cardiomyoctyes by secreting promitotic factors (Gittenberger-de Groot et al., 2010; Lepilina et al., 2006; Smart et al., 2007). Recent evidence also indicates that a subset of EPDCs differentiate into cardiomyocytes during development (Cai et al., 2008; Zhou et al., 2008). EPDC dysfunction has been linked to adult cardiac disease, Ebstein's malformation, arrhythmias and cardiomyopathies in humans (Gittenberger-de Groot et al., 2010). Adult EPDCs have the potential to reactivate patterns of developmental gene expression and aid in neovascularization and cardiomyocyte survival after ischemic injury (Smart et al., 2007). Because of their regenerative potential, EPDCs hold promise as a source of progenitor cells in adult human hearts and may be useful therapeutically. Data presented here demonstrate that, regardless of terminal cell fate, EPDCs share a common NFATC1-dependent mechanism required for invasion into myocardium. Therefore, this signaling mechanism could potentially be exploited in the development of EPDC-based therapies by increasing EPDC migration into myocardium for cardiomyocyte support and formation of EPDC derivatives.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01HL082716 and R01HL094319 (K.E.Y.) and an American Heart Association-Great Rivers Pre-doctoral Fellowship (M.D.C.). Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.060996/-/DC1
References
- Aliprantis A. O., Ueki Y., Sulyanto R., Park A., Sigrist K. S., Sharma S. M., Ostrowski M. C., Olsen B. R., Glimcher L. H. (2008). NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J. Clin. Invest. 118, 3775-3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C. L., Martin J. C., Sun Y., Cui L., Wang L., Ouyang K., Yang L., Bu L., Liang X., Zhang X., et al. (2008). A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. P., Neilson J. R., Bayle J. H., Gestwicki J. E., Kuo A., Stankunas K., Graef I. A., Crabtree G. R. (2004). A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell 118, 649-663 [DOI] [PubMed] [Google Scholar]
- Christoffels V. M., Grieskamp T., Norden J., Mommersteeg M. T., Rudat C., Kispert A. (2009). Tbx18 and the fate of epicardial progenitors. Nature 458, E8-E9; discussion E9-10 [DOI] [PubMed] [Google Scholar]
- Combs M. D., Yutzey K. E. (2009a). Heart valve development: regulatory networks in development and disease. Circ. Res. 105, 408-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs M. D., Yutzey K. E. (2009b). VEGF and RANKL regulation of NFATc1 in heart valve development. Circ. Res. 105, 565-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G. R., Olson E. N. (2002). NFAT signaling: choreographing the social lives of cells. Cell 109 Suppl., S67-S79 [DOI] [PubMed] [Google Scholar]
- de la Pompa J. L., Timmerman L. A., Takimoto H., Yoshida H., Elia A. J., Samper E., Potter J., Wakeham A., Marengere L., Langille B. L., et al. (1998). Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 392, 182-186 [DOI] [PubMed] [Google Scholar]
- de Lange F. J., Moorman A. F., Anderson R. H., Manner J., Soufan A. T., de Gier-de Vries C., Schneider M. D., Webb S., van den Hoff M. J., Christoffels V. M. (2004). Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 95, 645-654 [DOI] [PubMed] [Google Scholar]
- Funicello M., Novelli M., Ragni M., Vottari T., Cocuzza C., Soriano-Lopez J., Chiellini C., Boschi F., Marzola P., Masiello P., et al. (2007). Cathepsin K null mice show reduced adiposity during the rapid accumulation of fat stores. PLoS One 2, e683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittenberger-de Groot A. C., Winter E. M., Poelmann R. E. (2010). Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 14, 1056-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenig B., Kispert A. (2004). Analysis of TBX18 expression in chick embryos. Dev. Genes Evol. 214, 407-411 [DOI] [PubMed] [Google Scholar]
- Haudek S. B., Gupta D., Dewald O., Schwartz R. J., Wei L., Trial J., Entman M. L. (2009). Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc. Res. 83, 511-518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G., Kronenberg H. M., Settembre C. (2009). Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 25, 629-648 [DOI] [PubMed] [Google Scholar]
- Lange A. W., Yutzey K. E. (2006). NFATc1 expression in the developing heart valves is responsive to the RANKL pathway and is required for endocardial expression of cathepsin K. Dev. Biol. 292, 407-417 [DOI] [PubMed] [Google Scholar]
- Lavine K. J., Ornitz D. M. (2009). Shared circuitry: developmental signaling cascades regulate both embryonic and adult coronary vasculature. Circ. Res. 104, 159-169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepilina A., Coon A. N., Kikuchi K., Holdway J. E., Roberts R. W., Burns C. G., Poss K. D. (2006). A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127, 607-619 [DOI] [PubMed] [Google Scholar]
- Lie-Venema H., van den Akker N. M., Bax N. A., Winter E. M., Maas S., Kekarainen T., Hoeben R. C., deRuiter M. C., Poelmann R. E., Gittenberger-de Groot A. C. (2007). Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. ScientificWorldJournal 7, 1777-1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln J., Alfieri C. M., Yutzey K. E. (2004). Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev. Dyn. 230, 239-250 [DOI] [PubMed] [Google Scholar]
- Lincoln J., Florer J. B., Deutsch G. H., Wenstrup R. J., Yutzey K. E. (2006). ColVa1 and ColXIa1 are required for myocardial morphogenesis and heart valve development. Dev. Dyn. 235, 3295-3305 [DOI] [PubMed] [Google Scholar]
- Lutgens S. P., Cleutjens K. B., Daemen M. J., Heeneman S. (2007). Cathepsin cysteine proteases in cardiovascular disease. FASEB J. 21, 3029-3041 [DOI] [PubMed] [Google Scholar]
- Majesky M. W. (2004). Development of coronary vessels. Curr. Top. Dev. Biol. 62, 225-259 [DOI] [PubMed] [Google Scholar]
- Merki E., Zamora M., Raya A., Kawakami Y., Wang J., Zhang X., Burch J., Kubalak S. W., Kaliman P., Belmonte J. C., et al. (2005). Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. USA 102, 18455-18460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motyckova G., Fisher D. E. (2002). Pycnodysostosis: role and regulation of cathepsin K in osteoclast function and human disease. Curr. Mol. Med. 2, 407-421 [DOI] [PubMed] [Google Scholar]
- Negishi-Koga T., Takayanagi H. (2009). Ca2+-NFATc1 signaling is an essential axis of osteoclast differentiation. Immunol. Rev. 231, 241-256 [DOI] [PubMed] [Google Scholar]
- Norden J., Grieskamp T., Lausch E., van Wijk B., van den Hoff M. J., Englert C., Petry M., Mommersteeg M. T., Christoffels V. M., Niederreither K., et al. (2010). Wt1 and retinoic acid signaling in the subcoelomic mesenchyme control the development of the pleuropericardial membranes and the sinus horns. Circ. Res. 106, 1212-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivey H. E., Svensson E. C. (2010). Epicardial-myocardial signaling directing coronary vasculogenesis. Circ. Res. 106, 818-832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi T., Hayashi N., Theriault R. L., Hortobagyi G. N., Ueno N. T. (2010). Future directions of bone-targeted therapy for metastatic breast cancer. Nat. Rev. Clin. Oncol. 7, 641-651 [DOI] [PubMed] [Google Scholar]
- Ott H. C., Matthiesen T. S., Goh S. K., Black L. D., Kren S. M., Netoff T. I., Taylor D. A. (2008). Perfusion-decellularized matrix: using nature's platform to engineer a bioartificial heart. Nat. Med. 14, 213-221 [DOI] [PubMed] [Google Scholar]
- Raggatt L. J., Partridge N. C. (2010). Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 285, 25103-25108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger A. M., Grusby M. J., Hodge M. R., Gravallese E. M., de la Brousse F. C., Hoey T., Mickanin C., Baldwin H. S., Glimcher L. H. (1998). The transcription factor NF-ATc is essential for cardiac valve formation. Nature 392, 186-190 [DOI] [PubMed] [Google Scholar]
- Rapa I., Volante M., Cappia S., Rosas R., Scagliotti G. V., Papotti M. (2006). Cathepsin K is selectively expressed in the stroma of lung adenocarcinoma but not in bronchioloalveolar carcinoma. A useful marker of invasive growth. Am. J. Clin. Pathol. 125, 847-854 [DOI] [PubMed] [Google Scholar]
- Red-Horse K., Ueno H., Weissman I. L., Krasnow M. A. (2010). Coronary arteries form by developmental reprogramming of venous cells. Nature 464, 549-553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese D. E., Mikawa T., Bader D. M. (2002). Development of the coronary vessel system. Circ. Res. 91, 761-768 [DOI] [PubMed] [Google Scholar]
- Saftig P., Hunziker E., Wehmeyer O., Jones S., Boyde A., Rommerskirch W., Moritz J. D., Schu P., von Figura K. (1998). Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc. Natl. Acad. Sci. USA 95, 13453-13458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte I., Schlueter J., Abu-Issa R., Brand T., Manner J. (2007). Morphological and molecular left-right asymmetries in the development of the proepicardium: a comparative analysis on mouse and chick embryos. Dev. Dyn. 236, 684-695 [DOI] [PubMed] [Google Scholar]
- Shelton E. L., Yutzey K. E. (2007). Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev. Biol. 302, 376-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitara D., Aliprantis A. O. (2010). Transcriptional regulation of bone and joint remodeling by NFAT. Immunol. Rev. 233, 286-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart N., Risebro C. A., Melville A. A., Moses K., Schwartz R. J., Chien K. R., Riley P. R. (2007). Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 445, 177-182 [DOI] [PubMed] [Google Scholar]
- Taylor P. M., Batten P., Brand N. J., Thomas P. S., Yacoub M. H. (2003). The cardiac valve interstitial cell. Int. J. Biochem. Cell Biol. 35, 113-118 [DOI] [PubMed] [Google Scholar]
- Wu M., Smith C. L., Hall J. A., Lee I., Luby-Phelps K., Tallquist M. D. (2010). Epicardial spindle orientation controls cell entry into the myocardium. Dev. Cell 19, 114-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeini M., Hang C. T., Lehrer-Graiwer J., Dao T., Zhou B., Chang C. P. (2009). Spatial and temporal regulation of coronary vessel formation by calcineurin-NFAT signaling. Development 136, 3335-3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., Ma Q., Rajagopal S., Wu S. M., Domian I., Rivera-Feliciano J., Jiang D., von Gise A., Ikeda S., Chien K. R., et al. (2008). Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., von Gise A., Ma Q., Hu Y. W., Pu W. T. (2010). Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev. Biol. 338, 251-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}