

Abstract
Overexpression of several aquaporins has been reported in different types of human cancer but the role of AQPs in human carcinogenesis has not yet been clearly defined. Here, we demonstrate that ectopic expression of human AQP5 (hAQP5), a water channel expressed in lung, salivary glands, and kidney, induces many phenotypic changes characteristic of transformation both in vitro and in vivo. Furthermore, the cell proliferative ability of AQP5 appears to be dependent upon the phosphorylation of a cAMP-protein kinase (PKA) consensus site located in a cytoplasmic loop of AQP5. In addition, phosphorylation of the PKA consensus site was found to be phosphorylated preferentially in tumors. These findings altogether indicate that hAQP5 plays an important role in human carcinogenesis and, furthermore, provide an attractive therapeutic target.
Keywords: AQP5, Aquaporins, Overexpression
1. Introduction
Aquaporin 5 (AQP5) is a member of the aquaporin (AQP) family of transmembrane water channel proteins which are widely distributed in various tissues throughout the body and play a major role in transcellular and transepithelial water movement [1,2]. Similar to the other AQPs, AQP5 consists of six transmembrane domains separated by five connecting loops, A–E [3] (Fig. 1). The N-terminal and C-terminal halves are sequence-related, and loop B and E each contain the Asn-Pro-Ala (NPA) motif seen in the other aquaporins [3]. However, the cytoplasmic loop D contains a cAMP-protein kinase phosphorylation consensus site (Ser-Arg-Arg-Thr-Ser) which is seen only in AQP2 [3–5]. In normal tissues, AQP5 is expressed strongly in the salivary glands and the eye but expression is also seen in the lacrimal gland, trachea, and lung [3]. Furthermore, examination of the distribution of AQP5 in lacrimal gland determined that a selective defect in the trafficking of AQP5 in the lacrimal gland may contribute to the pathogenesis of Sjogren syndrome [5].
Fig. 1.
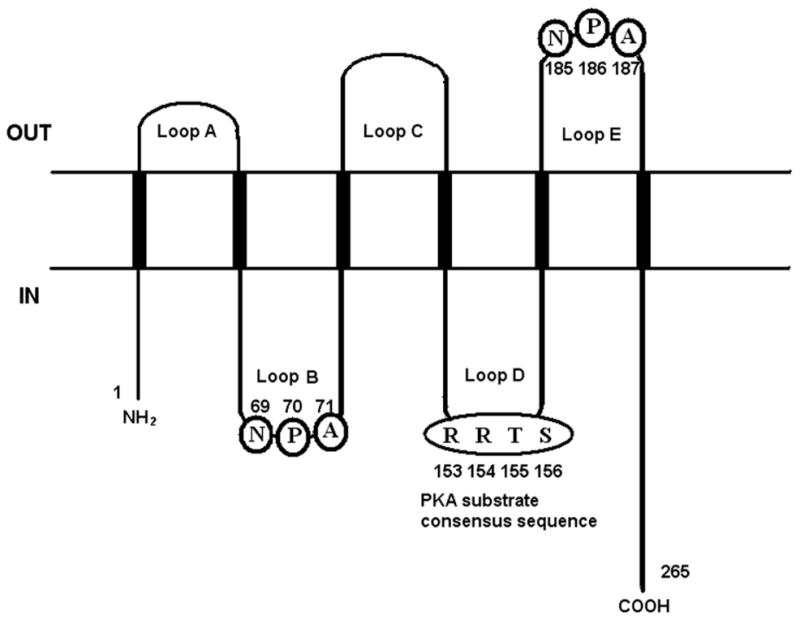
Membrane topology of hAQP5. AQP5 consists of six transmembrane domains connected by five loops (A–E). Two NPA motifs have been identified, one in loop B (amino acids 69–71) and the other in loop E (amino acids 185–187). Loop D contains a cAMP-protein kinase phosphorylation consensus sequence (amino acids 153–156).
Only recently has the role of these various hAQPs in human carcinogenesis become an area of active research. Expression of hAQP1 is frequently associated with brain tumors, colon cancers, and microvessels of multiple myeloma (MM) paralleling angiogenesis [6–8]. Other reports have alluded to the role of other AQPs in the development of human cancer [6,8,9]. For example, expression of AQP3 was increased in renal cell cancer, as was AQP5 in pancreatic cancer [9,10]. AQP1 appears to be involved in cell cycle control [8,11], and expression of hAQP1, hAQP3, and hAQP5 have been shown to be simultaneously induced during lymphocyte activation [12]. Most recently, based on the novel oncogenic properties of AQP1, increased levels of AQP1 were suggested to play a role in tumorigenesis [13]. Also, we have previously reported the induction of AQP5 expression in colon and colorectal cancer cell lines and its association with an early stage of colon cancer development [8]. However, none of these studies have provided convincing evidence of the oncogenic properties of AQPs and, therefore, are still preliminary. To elucidate the role hAQPs in human carcinogenesis, we examined the role of hAQP5 [3] as a potential oncoprotein by conducting a series of in vitro and in vivo experiments [3,14]. Here, we report that the overexpression of AQP5 induced phenotypic changes characteristic of transformation both in vitro and in vivo. Furthermore, the oncogenic ability of AQP5 may be dependent upon phosphorylation of its cAMP-protein kinase phosphorylation consensus site.
2. Materials and methods
2.1. Cell lines, culture conditions, and human tissues
All cell lines were obtained from the American Type Tissue Collection (Rockville, MD). The lung cancer cell line A431 was grown in Dulbecco’s Modified Eagle Medium (DMEM) in the presence of 10% fetal bovine serum (FBS). The lung cancer cell lines A549, H1299, H1437, H1650, H1838, H1974, H2030, H23, and H838 and the colorectal cancer cell line DLD1 were grown in RPMI1640 in the presence of 10% FBS. The colorectal cancer cell line HCT116 was grown in McCoy’s 5a Medium in the presence of 10% FBS. The murine fibroblast cell line NIH3T3 was cultured in DMEM in the presence of 10% FBS. The normal lung cell line BEAS-2B was grown in LHC-9 medium in the presence of 0.5 ng/ml recombinant epidermal growth factor (EGF), 500 ng/ml hydrocortisone, 0.005 mg/ml insulin, 0.035 mg/ml bovine pituitary extract, 500 nM ethanolamine, 500 nM phosphoethanolamine, 0.01 mg/ml transferrin, 6.5 ng/ml 3,3′,5-triiodothyronine, 500 ng/ml epinephrine, 0.1 ng/ml retinoic acid (Clonetics Corporation, Walkersville, MD). All cells were cultured in 5% CO2 balanced air at 37 °C. Three normal human lung tissues and three NSCLC tissues were obtained from Asan Medical Center, Seoul, Korea.
2.2. Plasmid constructs and production of stable cell lines
Human cDNA from HEK 293 was amplified by polymerase-chain reaction (PCR) using primers for AQP5 wild-type and deletion mutants and then inserted into EcoRI and XhoI site of pcDNA 3.1(+). Cloned genes were confirmed by restriction analysis and by DNA sequencing of both strands. Serine 156 or Asparagine 185 are replaced with alanine or aspartic acid, respectively, by PCR-based site-directed muatgenesis to produce the S156A or N185D AQP5 mutant based on the AQP5 pcDNA3.1 construct. Wild-type AQP5 and two mutants were inserted into the EcoRI and BamHI sites of p3XFLAG-CMV-14 (SIGMA) and verified by DNA sequencing of both strands of the mutants. Expression constructs were transfected into NIH3T3 cells with FuGENE 6 according to manufacturer’s recommendations. All transfectants were selected with 800 μg/mL G418 for 3 weeks and selected clones were screened with Western blot using AQP5 antibody and/or RT-PCR. When confluent, cells were subcultured by trypsinization (0.05% trypsin, 0.53 mM EDTA in Hanks’ balanced salt solution). To generate BEAS-2B stable clones, p3XFLAG-CMV-14 expression constructs mixed with FuGENE 6 was used according to manufacturer’s recommendations. Twenty-four hours after transfection, cells were selected with 300 μg/mL G418 and fed every 2– 3 days for 3 weeks in supplemented LHC-9 media containing G418. Selected clones were screened with Western blot with FLAG antibody (SIGMA) and/or RT-PCR.
2.3. Western blot analysis
Lysates from cultured cells and tissues were prepared in ice-cold NP-40 lysis buffer (10 mM Tris–Cl (pH7.4), 137 mM NaCl, 10% glycerol and 0.1% Nonidet P-40) containing an inhibitor cocktail of 10 mM β-glycerol phosphate, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 10 mM Na orthovanadate, 4.5 U/ml aprotinin (Sigma), and 1 μg/ml leupeptin (Sigma). Crude protein lysates (30 μg) were separated by 12% SDS–PAGE (BIORAD), transferred to nitrocellulose membranes (BIORAD), and blocked for 1 h with 5% nonfat dry milk in Tris-buffered saline with 0.05% Tween 20. The rabbit anti-AQP5 antibody (Alpha Diagnostic) was used as a 1:200 dilution and the mouse anti-beta actin antibody (Sigma) was used at a 1:5000 dilution. Appropriate anti-rabbit and anti-mouse horseradish peroxidase-conjugated secondary antibodies (1:3000; Amersham) were used. Immunoreactive bands were detected by enhanced chemiluminescence (Pierce).
2.4. Cell proliferation assay and colony formation assay
For the proliferation assay, cell lines were grown in six well plates at a density of 1 × 104 cells per well for 5 days. Measurements were made in triplicate for each of the cell lines and the experiments were repeated three times using MTT measurements and additional time by counting cells with a hemacytometer. For the colony formation assay, NIH3T3 cells stably expressing each plasmid were seeded at a density of 200 cells per plate in 100 mm plates. After 14 days of cultivation, dishes were stained with 0.5% crystal violet solution to visualize colony formation and counted foci of more than 50 cells in three dishes triplicate.
2.5. siRNA treatment
siRNAs against AQP5 were generated using the Silencer siRNA Construction Kit (Ambion) according to the manufacturer’s recommendations. For siRNA #1, the oligonucleotide sequences were 5′-AAAACTCTGCGAACACGGCCCCTGTCTC-3′ (sense) and 5′-AAGGCCGTGTTCGCAGAGTTCCTGTCTC-3′ (antisense). For siRNA #2, the oligonucleotide sequences were 5′-AAGAGCAGCCAGTGAAGTAGACCTGTCTC-3′ (sense) and 5′-AATCTACTTCACTGGCTGCTCCCTGTCTC-3′ (antisense). The HCT116 colon cancer cell line expressing AQP5 was seeded at a density of 5 × 105 cells/plate in 35 mm plates and incubated overnight. Cells were transfected with 75 nM of siRNA using FuGENE 6 transfection reagent (Roche) in Opti-MEM I reduced serum medium (Invitrogen) at 37 °C in a 5% CO2 atmosphere for 24 h. The medium was removed and replaced with fresh culture medium supplemented with 10% FBS. Control cells were similarly transfected with scrambled siRNA or without siRNAs (i.e. vehicle only). All the experiments were performed at least three times.
2.6. Tumor xenografts
Cells (1 × 106) were injected subcutaneously into athymic nude mice (CD-1-nuBR, Charles River), which were killed after 11 weeks. Tumor size was measured weekly, and tumor volume (V) was estimated from the length (L) and width (W) of the tumors using the formula V = (Π/6) × ((L + W)/2) (Weinberg R.A.). Animal experiments were approved by the Central Animal Facility at the Johns Hopkins Medical Institute. Multiple observations (>5) in each group of mice were used to generate means and standard deviations in tumor volume.
2.7. Immunoprecipitation
The indicated cell lines or tissues were lysed in NP-40 lysis buffer (10 mM Tris–Cl (pH7.4), 137 mM NaCl, 10% glycerol and 0.1% Nonidet P-40) in an inhibitor cocktail containing 10 mM β-glycerol phosphate, 1 mM phenyl-methylsulfonyl fluoride, 10 mM NaF, 10 mM Na orthovanadate, 4.5 U/ml aprotinin (Sigma), and 1 μg/ml leupeptin (Sigma). Lysates were then incubated with anti-AQP5 antibody (Santa Cruz) at 4 °C overnight and protein G-agarose was added, washed three times with lysis buffer in 2 h. Proteins were visualized by standard SDS–PAGE and immunoblotted with anti-phospho-(Ser/Thr) PKA substrate antibody (Cell signaling) or AQP5 antibody.
2.8. Immunofluorescence
Cells were fixed for 20 min at 4 °C with 4% paraformaldehyde (Sigma) and were permeabilized with 0.1% Triton X-100 for 90 s. Nonspecific binding was blocked by incubating fixed cells with 10% horse serum in PBS containing 0.02% Triton X-100 for 1 h. Cells were stained with anti-FLAG mouse monoclonal antibody (Sigma) overnight at 4 °C followed by fluorescent-labeled secondary antibody Alexa 594 (Molecular Probes) for 1 h. After washing, monolayers were excised with razor blade, mounting media including DAPI was added, and cover-slips were mounted.
3. Results
3.1. Treatment with AQP5 siRNA can decrease cell proliferation in cancer cell lines overexpressing AQP5
A previous study by our group reported that, along with AQP1 and AQP3, AQP5 was expressed in colon and colorectal cancer and its expression was associated with an early stage of colon carcinogenesis [8]. We sought to examine the role that AQP5 may play in carcinogenesis and, thus, we examined the expression of AQP5 in both nonsmall-cell lung cancer (NSCLC) and colon cancer cell lines by Western blotting (Fig. 2A). Consistent with our previous report, the two colon cancer cell lines examined, HCT116 and DLD1, expressed AQP5 [8] (Fig. 2A). Of the 10 NSCLC cell lines examined, AQP5 expression was observed in five of the cell lines (Fig. 2A).
Fig. 2.

(A) Expression of AQP5 in NSCLC cell lines and colon cancer cell lines. Western blot analysis was performed using antibodies for AQP5 for 2 colon cancer cell lines (HCT116, DLD1) and 10 NSCLC cell lines (H1975, H1437, H1650, H1838, H838, H23, H1299, H2030, A549, and A431). β-Actin was used as the loading control. (B) Treatment with AQP5 siRNA decreases cell proliferation. HCT116 cells were transfected with one of the AQP5 siRNA at 75 nM (siRNA #1 and siRNA #2) with various controls. The MTT assay demonstrated that cells transfected with AQP5 siRNA exhibited decreased cell proliferation as compared with the controls. The experiment was performed three times. Statistically significant difference were noted between cells expressing wild-type AQP5 and mutants/mock (t test, p < 0.001).
To examine whether AQP5 may have a role in tumorigenesis, we first designed siRNA against hAQP5 (AQP5 siRNA). HCT116 cells were treated with 75 nM of one of the two siRNA designed against AQP5 for 6 days. While similar rates of proliferation were observed among all the control cell lines, by day 6 a profound decrease in cell proliferation was observed in the cells treated with AQP5 siRNA (Fig. 2B). This initial observation suggested a potential role of AQP5 in cell proliferation.
3.2. Overexpression of AQP5 induces phenotypic changes in vivo
To further elucidate this observation, we have constructed two deletion mutants [3,4] based on the membrane topology of hAQP5 (Fig. 1). The first deletion mutant (Δ162–263) carries amino acids from N-terminus to the Loop D domain (residues 1–161). This construct does not contain the second NPA motif [1,15], a water channel-forming unit, although it still carries the protein kinase A (PKA) substrate sequence in loop D. The second deletion mutant (Δ148–263) carries amino acids from N-terminus to the fourth transmembrane domain (residues 1–147) and does not contain either second NPA motif or the PKA substrate sequence. NIH 3T3 cells, which do not express AQP5, were transfected with wild-type human AQP5 construct, one of the deletion mutants, or the empty vector (MOCK) (Fig. 3A). In the cell proliferation assay (Fig. 3B), stable cells with wild-type hAQP5 exhibited a significantly increased proliferation rate compared to MOCK. Likewise, stable cells expressing the Δ162–263 construct showed an increased proliferation rate compared to MOCK, although not as strong as the wild-type AQP5 cells. However, stable cells expressing Δ148–263 showed no significant difference in cell proliferation as compared to MOCK. The result of the colony formation assay using the same stable cell lines were consistent with those of the cell proliferation assays (Fig. 3C and D). In the colony formation assay (Fig. 3C), stable cells with wild-type hAQP5 exhibited a significantly higher rate of colony formation as compared to MOCK. Likewise, stable cells expressing the Δ162–263 construct showed a higher rate of colony formation as compared to MOCK, although not as high as cells with wild-type AQP5. However, stable cells expressing the Δ148–263 construct showed no significant difference in colony formation as compared to MOCK. From these two experiments and based on prior reports [3,4,14], we have hypothesized that this difference in proliferative ability between the Δ162–263 cells and the Δ148–263 cells was due to absence of the PKA substrate sequence in the Δ148–263 construct. Several reports have demonstrated that AQP5 expression is regulated by cAMP through a PKA pathway. Yang et al. reported the induction of AQP5 message and proteins by cAMP in cultured mouse epithelial cells [16], while Sidhaye et al. had reported a biphasic effect on AQP5 expression by cAMP: decreased membrane expression by short-term exposure and subsequent recovery by long-term exposure [17]. Based on these two prior reports and the known role of cAMP-mediated cell proliferation pathways [18], we suspected the role of this particular site in cell proliferation.
Fig. 3.

Overexpression of AQP5 increases cell proliferation. (A) Selection of stable cell lines. RT-PCR was used to identify NIH3T3 cells stably transfected with the mammalian expression vector pcDNA3 with full-length AQP5 cDNA and empty pcDNA3 vector (MOCK). Two selected clones are shown as an example. There is no detectable AQP5 in MOCK cells. Similar studies were performed in selecting stable cells expressing the deletion constructs or the point mutation constructs. (B) Cell proliferation assay. Stable cell expressing wild-type AQP5 or the Δ162–263 deletion construct exhibited a significantly increased proliferation rate compared to MOCK cells. However, stable cells expressing the Δ148–263 deletion construct failed to show a significant difference in cell proliferation as compared to MOCK cells. The experiment was performed three times. Statistically significant difference were found between cells expressing wild-type AQP5 and mutants/mock (t test, p < 0.001). (C–D) Colony formation assay. The number of colonies per each stable cell line was measured. Stable cells expressing wild-type AQP5 and the Δ162–263 deletion construct exhibited a significantly increased proliferation rate compared to MOCK cells. However, stable cells expressing the Δ148–263 deletion construct failed to show a significant difference in cell proliferation as compared toMOCK cells. Representative picture of the colonies are shown in (D). The experiment was performed three times. Statistically significant difference were observed between cells expressing wild-type AQP5 and mutants/mock (t test, p < 0.001).
3.3. Expression of AQP5 enhances cell proliferation in a phosphorylation-dependent manner
To examine the role of PKA-mediated phosphorylation of serine/threonine consensus sites of AQP5 in cell proliferation, we constructed two site-directed mutants [4,19], one at the site of second NPA motif (N185D), and the other at the site of the phosphorylation consensus sequences (S156A) (Fig. 1). Consistent with the data using the deletion constructs, both the stable cells with wild-type AQP5 and cells expressing the N185D mutant demonstrated a significant increase in cell proliferation while cells expressing the S156A mutant showed no significant difference compared with MOCK (Fig. 4A).
Fig. 4.

Overexpression of AQP5 appears to increase cell proliferation in a phosphorylation-dependent manner. (A) Cell proliferation assay. Cell proliferation is significantly greater in stable cells with wild-type AQP5 or the N185D mutant than in stable cells with the S156A mutant or the empty vector. The experiment was performed three times. We found a statistically significant difference between cells expressing wild-type AQP5 and mutants/mock (t test, p < 0.001). (B–C) Tumor growth in athymic mice. Tumor growth was observed within 8 weeks for athymic mice subcutaneously injected with stable cells with wild-type AQP5 or the N185D mutant. No tumor growth is observed in mice injected with stable cells with the S156A cells or MOCK. Means and standard deviations of tumor volume were calculated from multiple observations in two groups of mice. Statistically significant difference were noted between cells expressing wild-type AQP5 and mutants/mock (t test, p < 0.001). Photographs of athymic mice 8 weeks after subcutaneous injection of stable cells with various hAQP5 constructs and MOCK are shown in (C). (D) AQP5 is phosphorylated in cancer cell lines and NSCLC tissues. AQP5 is phosphorylated in the two NSCLC cell lines examined, H1975 and H1838, and in 2 of 3 primary NSCLC tissues. No phosphorylation is detected in the three primary normal tissues.
To study this effect in vivo, we performed xenoplantation in athymic mice with these stable cell lines. Athymic mice injected with stable cells with wild-type AQP5 formed a tumor nodule 4 weeks after injection, while mice injected with stable cells with N185D mutant formed a tumor nodule 6 weeks after injection. In contrast, athymic mice injected with stable cells with S156A mutant or MOCK did not form any detectable tumor, even after 26 weeks of follow-up (Fig. 4B and C).
The association of AQP5 phosphorylation and carcinogenesis was confirmed by our examination of NSCLC cancer cell lines and primary lung tissue samples. AQP5 protein was isolated from the cell lines and tissue samples by immunoprecipitation using an AQP5 antibody and then probed for a phosphorylated S156 site using an (Ser/Thr) PKA substrate antibody. AQP5 was found to be phosphorylated in the NSCLC cell lines H1975 and H1838 and in two of three NSCLC primary tissue samples, all of which showed a clear pattern of AQP5 expression (Fig. 4D). The NSCLC tissue sample which did not demonstrate AQP5 phosphorylation did not express AQP5. In contrast, AQP5 was not phosphorylated in the three normal lung tissues examined (Fig. 4D).
To exclude the possibility of these in vitro and in vivo effects coming from impaired intracellular trafficking of the AQP5 protein itself, immunofluorescence analysis was performed. The human bronchial epithelium cell line BEAS-2B was stably transfected with either wild-type AQP5 or one of the mutant constructs. As seen in Fig. 5, proper membrane trafficking of AQP5 was observed in cells expressing wild-type AQP5 or the S156A mutant construct, demonstrating that S156 phosphorylation does not play a key role in membrane trafficking. Additionally, it was concluded that the partial decrease of cell proliferation and colony formation seen in cells expressing the Δ148–263 deletion construct could have come from incomplete membrane trafficking due to a result of improper protein folding as reported before (Fig. 5) [4,19]. These results altogether confirmed a crucial role of phosphorylation in serine/threonine consensus sequences of loop D in AQP5-mediated cell proliferation.
Fig. 5.

Immunofluorescence assay of stable cell lines. To exclude the possibility that these oncogenic properties are due to impaired intracellular trafficking of the AQP5 protein itself, immunofluorescence analysis was performed on stable cell lines expressing the various AQP5 constructs. S156 phosphorylation was shown not to play a key role in membrane trafficking.
4. Discussion
There has been increasing evidence that aquaporins may play a role in carcinogenesis. Various aquaporins have been shown to be overexpressed in tumors and ectopic expression of AQP1 induced phenotypics changes in NIH 3T3 cells which are characteristic of transformation [13]. However, the evidence has not been convincing. For example, a recent report that had shown that the role of AQP1 in tumorigenesis depends on its contribution to angiogenesis questions the direct role of AQPs in human carcinogenesis [3]. In addition, Saadoun et al. reported that AQP1 deletion impairs tumor microvessel proliferation, producing extensive tumor necrosis [7].
In this report, we provide the first compelling data that AQP5 can play a key role in human carcinogenesis. We demonstrate that a clear expression of AQP5 in NSCLC and colon cancer cell lines and that while silencing of AQP5 can decrease cell proliferation, ectopic expression of AQP5 leads to cell proliferation and colony formation in vitro and tumor growth in mice. Furthermore, mutation of the PKA consensus site led to a decrease in cell proliferation which was comparable to the controls. In fact, in athymic mice, no detectable tumor was observed even after following the mice for 26 weeks. In addition, mutation of the PKA consensus site led to proper membrane trafficking of AQP5, suggesting that observed differences in cell proliferation are mediated by phosphorylation of this site rather than improper trafficking of the protein. If AQP5 phosphorylation plays a key role in cell proliferation, it is possible that AQP5 may be involved with key pathways of carcinogenesis, such as the Ras signaling pathway, and should be investigated further. Furthermore, the potential importance of AQP5 phosphorylation was demonstrated by the different phosphorylation status of AQP5 in normal lung tissues and NSCLC tissues. In both normal and cancer tissues, AQP5 expression was seen; however, AQP5 phosphorylation was observed in NSCLC tissues but not in normal lung tissues. If AQP5 plays a role in carcinogenesis, the contrasting phosphorylation status between normal and cancer tissues suggests that it may not be expression of AQP5 which is key to carcinogenesis but rather phosphorylation of AQP5. Thus, AQP5 phosphorylation should be examined in a greater number of tissues and cell lines.
Interestingly, only two out of three NSCLC tissues exhibited an expression of AQP5, while all three normal lung tissues expressed AQP5 and expression level seems to be higher in cancer tissue samples with AQP5 expression as compared to normal. Likewise, 5 out of 10 tested cell lines did not express AQP5 which can be due to secondary event like promoter methylation or inhibition of promoter activity by binding repressor element to AQP5 promoter. For the expression pattern observed from tissue samples, the interpretation can be intriguing as, although we can not rule out the possibility that AQP5 expression can be reduced during NSCLC carcinogenesis, this does not necessarily mean that AQP5 can be down-regulated inNSCLC, since we were not able to obtain any pair samples that have normal and corresponding cancer tissues. Our immunohistochemistry data with matched normal lung and NSCLC tissue microarray samples (Jang et al., unpublished results) suggested that the expression level of AQP5 is higher in tumors than in normal tissues. For general pattern of AQP5 expression in NSCLC, we have reported that AQP5 is expressed in 35.3% (IHC score of 1+, 144/407) of the resected NSCLC tissue samples (Chae et al., manuscript submitted). This expression level of AQP5 can be comparable to that of Erb/HER2 in breast cancer which is 25–30% at maximum.
It should be noted, however, that a mutation in the NPA motif also led to a decrease in cell proliferation compared to cells expressing wild-type AQP5, though it was not as profound as the mutation of the PKA consensus site. Improper trafficking of AQP5 was observed in cells expressing constructs containing a mutation of the NPA motif. These results suggest that proper trafficking of the protein may be needed to function properly and thus meet the increased metabolic needs of cancer cells. However, we cannot exclude the possibility that the decrease in cell proliferation of the AQP5 mutants may result from disruption of protein folding or oligomerization that affects membrane translocation [1,19].
Based on these observations, we believe that AQP5 provides a unique opportunity as a novel therapeutic target. The significant decrease in cell proliferation observed in the S156A mutant cells suggests that designing small molecules to block this particular site of phosphorlyation may have therapeutic benefits. In addition, the observation that AQP5 is not phosphorylated at S156 in normal tissues but is phosphorylated in tumors may provide a unique opportunity in designing tumor-specific molecular inhibitors. Also, based on our expression study, designing a therapeutic monoclonal antibody will be another approach we can take. Both AQP1 and AQP5 has six membrane-spanning domains and carries three exofacial loops, which will provide potential sites against which therapeutic antibodies can be targeted.
In conclusion, this report provides the first molecular evidence of the oncogenic properties of AQP5 and suggests that AQP5 may play a direct role in human carcinogenesis, in contrast with the prior report that AQP1 plays a role in tumorigenesis indirectly through its contribution to angiogenesis [20]. While further characterization of the pathways by which AQP5 is involved in human carcinogenesis is warranted, we believe that AQP5 has potential as a novel therapeutic target in human cancers and that the other AQPs should be similarly investigated.
Acknowledgments
The study was supported in part by the SPORE grant P50 CA96784-01 (to C.M.), Fondation de France, AP-HP and Lilly Fondation Grant (to J.-C.S.), Cancer Research Grant from Pyung-Ya Foundation (to C.M.), and KOSEF research grant (R01-2004-000-10670-0 to S.J.J.).
References
- 1.King LS, Agre P. Pathophysiology of the aquaporin water channels. Annu Rev Physiol. 1996;58:619–648. doi: 10.1146/annurev.ph.58.030196.003155. [DOI] [PubMed] [Google Scholar]
- 2.Verkman AS, van Hoek AN, Ma T, Frigeri A, Skach WR, Mitra A. Water transport across mammalian cell membranes. Am J Physiol. 1996;270:C12–C30. doi: 10.1152/ajpcell.1996.270.1.C12. [DOI] [PubMed] [Google Scholar]
- 3.Raina S, Preston GM, Guggino WB, Agre P. Molecular cloning and characterization of an aquaporin cDNA from salivary, lacrimal, and respiratory tissues. J Biol Chem. 1995;270:1908–1912. doi: 10.1074/jbc.270.4.1908. [DOI] [PubMed] [Google Scholar]
- 4.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem. 1997;272:14800–14804. doi: 10.1074/jbc.272.23.14800. [DOI] [PubMed] [Google Scholar]
- 5.Tsubota K, Hirai S, King LS, Agre P, Ishida N. Defective cellular trafficking of lacrimal gland aquaporin-5 in Sjogren’s syndrome. Lancet. 2001;357:688–689. doi: 10.1016/S0140-6736(00)04140-4. [DOI] [PubMed] [Google Scholar]
- 6.Vacca A, Frigeri A, Ribatti D, Nicchia GP, Nico B, Ria R. Microvessel overexpression of aquaporin 1 parallels bone marrow angiogenesis in patients with active multiple myeloma. Br J Haematol. 2001;113:415–421. doi: 10.1046/j.1365-2141.2001.02738.x. [DOI] [PubMed] [Google Scholar]
- 7.Saadoun S, Papadopoulos MC, Davies DC, Bell BA, Krishna S. Increased aquaporin 1 water channel expression in human brain tumours. Br J Cancer. 2002;87:621–623. doi: 10.1038/sj.bjc.6600512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moon C, Soria JC, Jang SJ, Lee J, Hoque MO, Sibony M. Involvement of aquaporins in colorectal carcinogenesis. Oncogene. 2003;22:6699–6703. doi: 10.1038/sj.onc.1206762. [DOI] [PubMed] [Google Scholar]
- 9.Kageyama Y, Sasaki S, Yamamura Y, Oshima H, Ikawa Y. Water channel protein subtype suggests the origin of renal cell carcinoma. J Urol. 1996;156:291–295. [PubMed] [Google Scholar]
- 10.Burghardt B, Elkaer ML, Kwon TH, Racz GZ, Varga G, Steward MC. Distribution of aquaporin water channels AQP1 and AQP5 in the ductal system of the human pancreas. Gut. 2003;52:1008–1016. doi: 10.1136/gut.52.7.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moon C, Williams JB, Preston GM, Copeland NG, Gilbert DJ, Nathans D. The mouse aquaporin-1 gene. Genomics. 1995;30:354–357. doi: 10.1006/geno.1995.0029. [DOI] [PubMed] [Google Scholar]
- 12.Moon C, Rousseau R, Soria JC, Hoque MO, Lee J, Jang SJ. Aquaporin expression in human lymphocytes and dendritic cells. Am J Hematol. 2004;75:128–133. doi: 10.1002/ajh.10476. [DOI] [PubMed] [Google Scholar]
- 13.Hoque MO, Soria JC, Woo J, Lee T, Lee J, Jang SJ. Aquaporin 1 is overexpressed in lung cancer and stimulates NIH-3T3 cell proliferation and anchorage-independent growth. Am J Pathol. 2006;168:1345–1353. doi: 10.2353/ajpath.2006.050596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 15.Preston GM, Smith BL, Zeidel ML, Moulds JJ, Agre P. Mutations in aquaporin-1 in phenotypically normal humans without functional CHIP water channels. Science. 1994;265:1585–1587. doi: 10.1126/science.7521540. [DOI] [PubMed] [Google Scholar]
- 16.Yang F, Kawedia JD, Menon AG. Cyclic AMP regulates aquaporin 5 expression at both transcriptional and post-transcriptional levels through a protein kinase A pathway. J Biol Chem. 2003;278:32173–32180. doi: 10.1074/jbc.M305149200. [DOI] [PubMed] [Google Scholar]
- 17.Sidhaye V, Hoffert JD, King LS. cAMP has distinct acute and chronic effects on aquaporin-5 in lung epithelial cells. J Biol Chem. 2005;280:3590–3596. doi: 10.1074/jbc.M411038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 19.Jung JE, Karoor V, Sandbaken MG, Lee BJ, Ohama T, Gesteland RF. Utilization of selenocysteyl-tRNA[Ser]Sec and seryl-tRNA[Ser]Sec in protein synthesis. J Biol Chem. 1994;269:29739–29745. [PubMed] [Google Scholar]
- 20.Saadoun S, Papadopoulos MC, Hara-Chikuma M, Verkman AS. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature. 2005;434:786–792. doi: 10.1038/nature03460. [DOI] [PubMed] [Google Scholar]