

1. Introduction
For over a century, ketenes have intrigued organic chemists with their unusual physical properties and unique spectrum of chemical reactivity; it is not an exaggeration to say that they represent one of the keystone reactive intermediates of physical organic chemistry. Their synthetic potential, originating in part from their high reactivity, has proven to be powerful as well, from the Staudinger synthesis of β-lactams to the photochemical Wolff rearrangement. It is perhaps no surprise that the past several decades have also shown that ketenes are excellent precursors for catalytic asymmetric reactions, creating chiral centers mainly through addition across their C=C bonds. Due to their electrophilic nature, ketenes readily react with nucleophiles to form zwitterionic enolates that form the basis for much asymmetric chemistry. Through catalytic, asymmetric methodology, researchers have successfully utilized ketenes in a range of reaction manifolds, including [2+2] and [4+2] cycloadditions, reductive couplings, nucleophilic SN2 substitutions, as well as both electrophilic and nucleophilic additions. In this review, these reaction pathways are discussed in terms of such diverse reactions as ketene alcoholysis and aminolysis, α-halogenation, α-amination, α-hydroxylation, and the formation of β-, γ-, and δ-lactones and β-and γ-lactams.
1.1. Ketenes and their properties
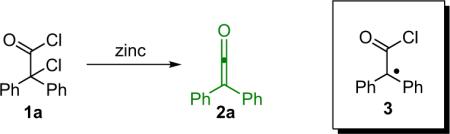
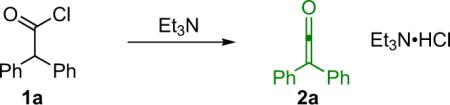
In 1905 at the University of Strasbourg, Hermann Staudinger undertook an attempted synthesis of radical species 3 (Eq. 1.1) inspired by the work of Gomberg on the triphenylmethyl radical. To his surprise, upon the reaction of α-chlorodiphenylacetyl chloride (1a) with zinc, an entirely unanticipated closed shell compound was formed, one that he eventually identified as diphenylketene (2a, Eq. 1.1).1 Since this serendipitous discovery, ketenes have been the source of countless scholarly texts and research papers.2 Due to their unusual properties, ketenes are versatile starting materials for a wide variety of compounds of interest and have been used as precursors for everything from industrially produced acetic acid to antibiotics and more. As can be expected for a `marquee' class of compounds that has been so heavily studied, much research has been done to illuminate ketene reactivity and its dependence on structure and substitution patterns. What follows is meant as an abridged introduction only, as the topic has been thoroughly covered elsewhere, especially in the work of Tidwell.3
 |
(1.1) |
1.2. Structure and reactivity of ketenes
Ketenes are characterized by an unusual `heteroallenic' bond structure that is the source of the their distinctive reactivity. The highest occupied molecular orbital (HOMO) of a ketene is located perpendicular to the plane of the ketene while the lowest unoccupied molecular orbital (LUMO) lies in the plane. This orientation places significant negative charge on both the oxygen and the β-carbon, while placing a similarly substantial positive charge on the α-carbon. (Fig. 1.1). The electropositive nature of the α-carbon in part explains why nucleophiles readily add at this position.
Figure 1.1.
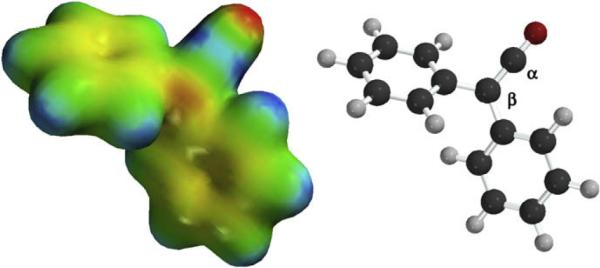
Electron density map of diphenylketene; red=e− rich, blue=e− deficient.
The ketene's substituents play a major role in its reactivity. Electronically, species that are capable of donating electron density into the carbonyl group through s–p conjugation as well as those that can act as p-acids stabilize the ketene, while electronegative groups and p-donors destabilize the ketene. Similarly, steric bulk on the β-carbon stabilizes the ketene through shielding of the reactive sites; the impact of these substituents can be understood by considering a few examples. Diphenylketene is quite stable, and it can be stored neat at room temperature for extended periods of time and can even be distilled. On the other hand, phenylketene must be kept in solution at low temperatures to avoid degradation, while fluoro- and difluoroketenes are so reactive that they have not yet been directly observed in solution.2b,c
1.3. Methods of ketene generation
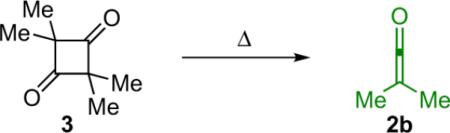
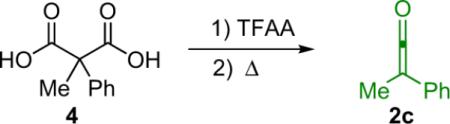
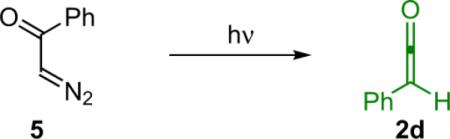
In light of the wide variability in ketene reactivity and substituent tolerance, it is not surprising that numerous methods of generation have been developed over the years. Thermolysis is a useful method for generating relatively stable disubstituted ketenes. For example, dimethylketene (2b) can be obtained by thermal cracking of its dimer (Eq. 1.2),4 and methylphenylketene can be generated by treating methylphenylmalonic acid (4) with trifluoroacetic anhydride (TFAA) followed by pyrolysis (Eq. 1.3).5 To a lesser extent, photolysis of dimers is also possible, although yields are low due to competing decarbonylation reactions.6 A more viable light (or heat) based method of ketene generation, one that has seen extensive use in synthetic chemistry, is the Wolff rearrangement. For instance, photolysis of phenyldiazoacetate (5) produces a clean solution of phenylketene (Eq. 1.4).7 One of the advantages of using the Wolff rearrangement is that it can be used to produce a dilute solution of ketene over an extended period of time; this can be helpful in reducing unwanted side reactions such as ketene dimerization. While these are effective ways to generate ketenes, they are made somewhat less accessible because they involve specialized equipment or unstable starting materials.
 |
(1.2) |
 |
(1.3) |
 |
(1.4) |
 |
(1.5) |
A more popular method for ketene generation in synthetic chemistry is the dehydrohalogenation of inexpensive acid chlorides with tertiary amines (Eq. 1.5). This method works well for the more stable, distillable disubstituted ketenes.2a Unfortunately, the more synthetically useful monosubstituted ketenes are generally much more reactive and must therefore be made in situ, where they suffer from contamination with ammonium salt byproducts. Generally, the presence of trialkylammonium byproducts presents no problem; in some cases, these species can potentially erode selectivity in asymmetric reactions by acting either as Brønsted acids or, through equilibrium with the free amine, nucleophilic catalysts.8 To some extent, these difficulties can be overcome by employing resin-bound bases that leave behind a clean solution of ketene after filtration. For example, Lectka has employed the powerful, resin-bound phosphazene base BEMP9 for this purpose. Passing a solution of acid chloride through an addition funnel containing at least 1 equiv of BEMP at −78 ° C has been shown to give good results in several systems.10 While BEMP affords ketene solutions without contaminants, there are disadvantages to this method: BEMP is quite moisture sensitive and can be difficult to handle for larger scale reactions; it is also very expensive when compared to other methods of ketene generation.
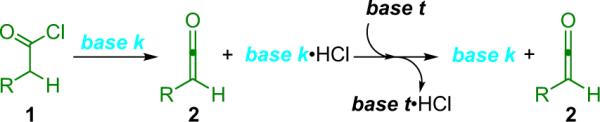
In an effort to overcome these challenges, a simple and accessible method of in situ ketene generation known as `shuttle deprotonation' was developed by the Lectka group in 2000.11 Under these conditions, a catalytic amount of a strong kinetic base (base k) dehydrohalogenates an acid chloride and subsequently transfers the proton to a strong thermodynamic base (base t, Scheme 1.1). By choosing a non-nucleophilic base to sequester the protons and a kinetic base that will act as the chiral catalyst in later steps, namely benzoylquinine (BQ, 6a), the problem of the competitive, achiral nucleophile can be overcome.
Scheme 1.1.
Shuttle deprotonation.
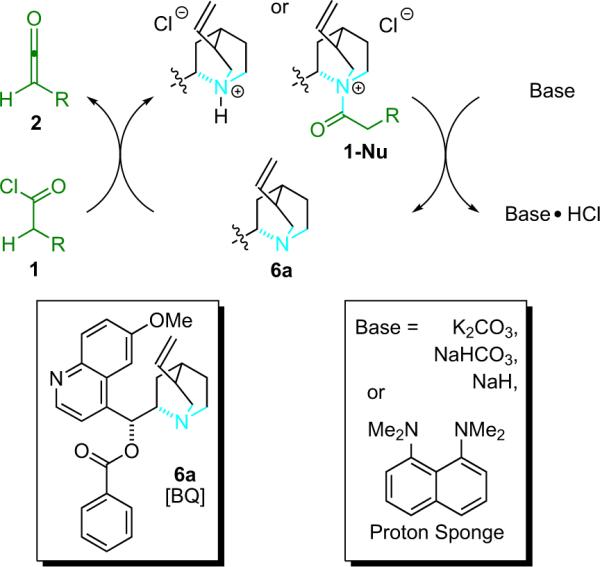
The shuttle deprotonation reaction can be performed with any number of different thermodynamic bases. In most cases, the mechanism presumably remains the same (Fig. 1.2). Potentially, BQ can generate the ketene through two routes: (1) by direct dehydrohalogenation; or (2) by the generation of an acyl ammonium intermediate that is subsequently deprotonated to give a zwitterionic ketene enolate, which is in equilibrium with free ketene. It is believed that both pathways to enolate formation may operate simultaneously, but which one predominates depends on the properties of the acid chloride.
Figure 1.2.
Shuttle deprotonation cycle for BQ as the kinetic base.
While proton sponge has been used effectively in many applications, its drawbacks include hydrochloride salt solubility in more polar solvents, and reactivity with potential oxidants, for example, in the conditions for asymmetric α-halogenation (see Section 5). For such applications the use of potassium carbonate, sodium bicarbonate, or sodium hydride has been shown to be effective when paired with catalytic quantities of crown ether and BQ.12 Additionally, the heterogeneous nature of these reactions has been exploited to allow for nearly pure solutions of ketene to be produced by simple filtration at low temperature following a ketene preformation step.13 To simplify this procedure, special glassware has been designed (Fig. 1.3). The apparatus consists of two round-bottom flasks linked by a fritted disc; ketene is formed heterogeneously on one side and then filtered through a frit onto the other.
Figure 1.3.
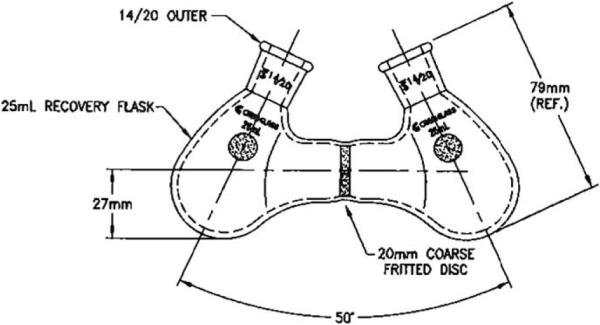
The double reaction flask.
Evidence for the preformation of ketene can be obtained from spectroscopic experiments. When BEMP is utilized, ketenes are undoubtedly formed from acid chlorides. On the other hand, the shuttle deprotonation systems that utilize proton sponge, bicarbonate, or Hünig's base do not require the generation of free ketene for the reaction to proceed as the zwitterionic intermediate enolate is often formed reversibly under these conditions. The situation is more opaque when sodium hydride is used in situ—the presence of catalyst is necessary for the production of phenylketene, but it is not clear whether the BQ catalyst was acting as a nucleophile or a base, or both (Eq. 1.6).
 |
(1.6) |
1.4. Cinchona alkaloids as catalysts
Cinchona alkaloids are abundant natural products that are one of the more popular chiral organocatalysts,14 and they constitute the most often used catalysts for asymmetric ketene reactions. They exist as pseudoenantiomeric pairs, for instance quinine (6b) and quinidine (7b) are a complimentary diastereomeric pair that generally provide opposite optical induction. Among their many attributes, cinchona alkaloids are readily derivatized, most often at the chiral alcohol, to provide reactivity and selectivity that is amenable to a wide variety of reaction conditions (Fig. 1.4).
Figure 1.4.
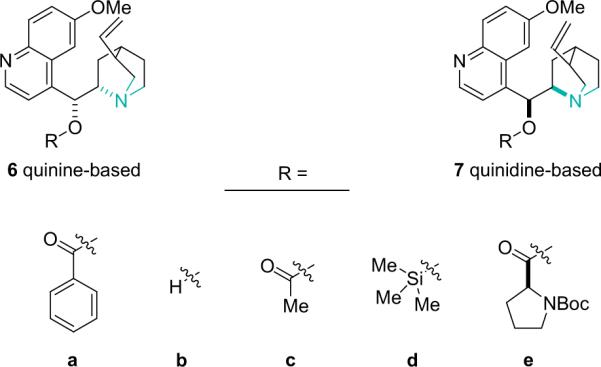
Cinchona alkaloid catalysts.
Lectka has employed molecular modeling calculations to account for (and predict) the sense of induction that is consistently observed in cinchona alkaloid catalyzed ketene reactions. For example, molecular mechanics (MM) calculations using a modified MMFF force field on a model BQ–phenylketene complex (1d-Nu) reveal a low energy structure in which the re face is exposed to approach of the electrophile, whereas si-face approach is 2.64 kcal/mol higher in energy (depending on the force field employed, Fig. 1.5).15
Figure 1.5.
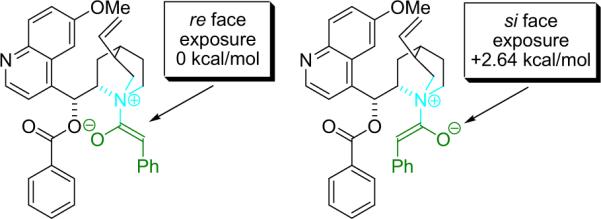
Stereochemical models of the putative zwitterionic intermediate of BQ and phenylketene (1d-Nu).
2. Asymmetric alcoholysis and aminolysis
In 1960, Pracejus reported one of the earliest catalytic asymmetric reactions of ketenes. He employed chiral trialkylamines to produce chiral ester products from ketenes and alcohols.16 At −110 °C in toluene, acetylquinine (6c) catalyzes the methanolysis of phenylmethylketene (2c); the α-phenylpropionate ester (8a) was obtained in up to 74% ee, representing a tremendous breakthrough in ketene chemistry (Eq. 2.1). He proposed that a complex between the catalyst and ketene had formed and that this was responsible for optical induction during alcoholysis. The complex proposed is what we now refer to as a chiral ammonium zwitterionic ketene enolate, and is the reactive intermediate most often implicated in asymmetric ketene reactions. This pioneering work set the stage for the modern asymmetric reactions of ketenes.17
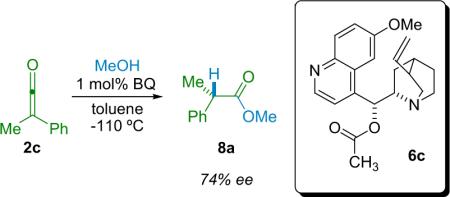 |
(2.1) |
2.1. Ketene alcoholysis
Despite the promise of Pracejus's early work, it was not until decades later that researchers sought to adapt and expand upon his methodology. A study by Calmes et al. in 1997 closely examined the effect of the nature of the catalyst in the alcoholysis of in situ generated disubstituted ketenes by chiral alcohols.18 In the late 1990s Fu et al.19 re-examined the potential of catalyzed asymmetric ester formation from ketenes, employing their highly selective planar-chiral ferrocene catalysts (9)20 (Scheme 2.1).
Scheme 2.1.
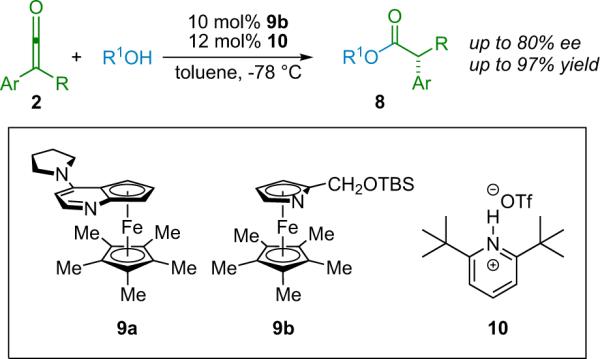
Fu's stereoselective ester synthesis.
Their early experiments with Pracejus's substrates gave disappointing results, with ee's topping out at 55% with 10 mol % catalyst loading. However, careful consideration of their proposed reaction mechanism provided clues to help improve the selectivity (Scheme 2.2). If their proposed mechanism were correct and the abstraction of a proton by complex 2-Nu was the chirality determining step, then the use of an alternate proton source might improve the enantioselectivity of their reactions. Brønsted acid additive screening found that the bulky di-tert-butylpyridinium salt 10 successfully raised the ee to 80%.
Scheme 2.2.
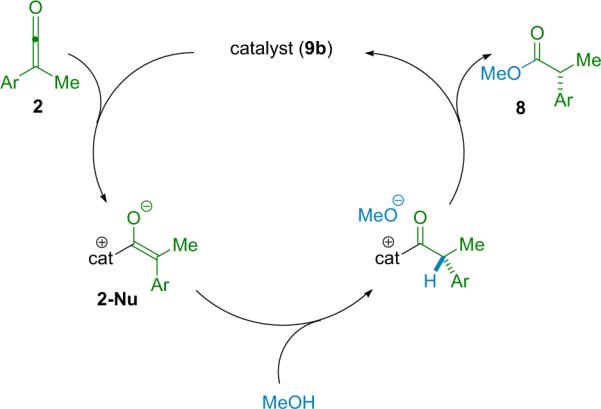
Proposed mechanism for catalyzed ester synthesis.
At the same time, the fact that the bulky Brønsted acid additive was able to provide increased enantioselectivity corroborated their working hypothesis. However, it was not until after expanding their methodology to include amide products (see Section 2.2) that an alternate mechanism presented itself, indicating potentially beneficial modification of this system toward high selectivity (Scheme 2.3).21
Scheme 2.3.
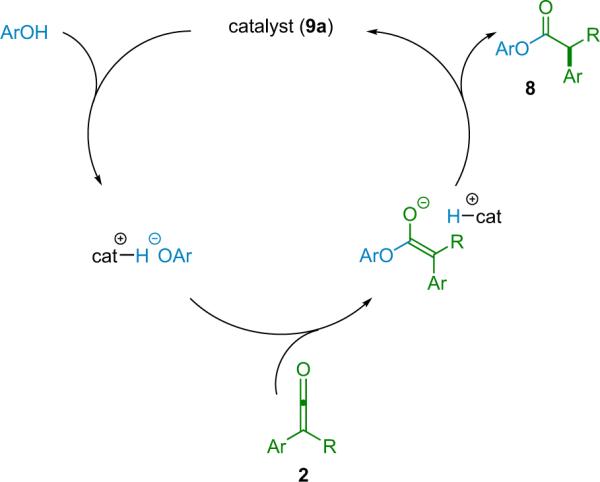
Alternate mechanism for catalyzed ester synthesis.
In this new mechanism, while the abstraction of a proton by an enolate complex is still the chirality determining step, the source of that proton is now the chiral catalyst instead of an achiral acid or alcohol. Such an arrangement could theoretically place the catalyst much closer to the forming chiral center, thus enhancing its effectiveness. Catalyst 9b quantitatively deprotonates phenol in solution, whereas methanol gave no evidence of deprotonation under the same conditions. The use of a bulky phenol instead of methanol leads to enhanced selectivity, in some cases to above 90% ee (Table 2.1), while both obviating the need for the acid additive and lowering the catalyst loading to 3 mol %.
Table 2.1.
A selection of esters synthesized.
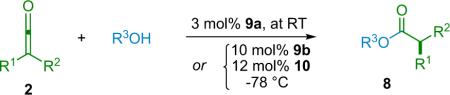
| Entry | R1 | R2 | R3 | Cat | ee (%) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | Me | Ph | Me | 9b | 77 | 87 |
| 2 | Et | Ph | Me | 9b | 68 | 92 |
| 3 | Me | m-PhO-Ph | Me | 9b | 74 | 96 |
| 4 | Me | 4-i-Bu-Ph | Me | 9b | 77 | 88 |
| 5 | Ph | Me | 2-t-Bu-Ph | 9a | 79 | 87 |
| 6 | Ph | i-Bu | 2-t-Bu-Ph | 9a | 84 | 79 |
| 7 | o-Anisyl | Me | 2-t-Bu-Ph | 9a | 94 | 78 |
| 8 | p-Cl-Ph | i-Pr | 2-t-Bu-Ph | 9a | 89 | 97 |
| 9 | 3-Thienyl | i-Pr | 2-t-Bu-Ph | 9a | 79 | 94 |
Interestingly, Fu expanded this methodology into the realm of enolizable aldehydes (11) in 2005 (Scheme 2.4).22 This alternative method, employing catalyst 9a, produces enol esters in excellent yield and ee up to 98%. As with the simple alcoholysis method above, the authors' initial mechanistic queries gave results consistent with two mechanisms, analogous to Schemes 2.2 and 2.3, respectively.
Scheme 2.4.
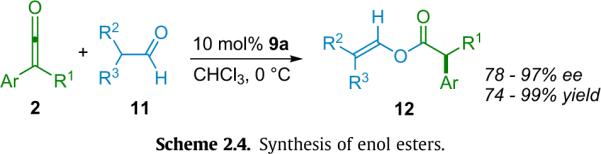
Synthesis of enol esters.
2.2. Ketene aminolysis
Following their success in the catalytic asymmetric methodology for the synthesis of chiral esters, the Fu research group turned their sights on a logical follow up, chiral amides.23 In theory, by simply employing an amine as the nucleophile instead of an alcohol the desired products should be readily obtained. Practically, the problem is that amines are too nucleophilic, giving high background rates even at low temperature. It is likely for this reason that Pracejus, who investigated this route to chiral amides concomitant with his early work on chiral esters, chose to focus instead on using chiral amines to impart selectivity in his products.24 Fu et al. sought to render the reaction more manageable by choosing a less reactive amine nucleophile. An initial screen showed pyrrole to be a promising candidate as it gave no background reaction, however, the product of the reaction showed disappointing selectivity. This prompted a further screen of a number of pyrrole derivatives to examine the effect of various substituents on product selectivity (Scheme 2.5).
Scheme 2.5.
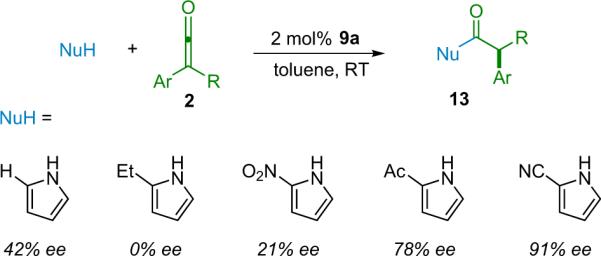
Screening of pyrrole derivatives.
The screen demonstrated that pyrroles possessing electron-withdrawing groups perform better, and 2-cyanopyrrole provided the best enantioselectivity. By combining 2-cyanopyrrole and any of a number of aryl/alkyl ketenes in the presence of catalyst 9a they were able to produce the desired amide products (13) in excellent yield and with high ee (Table 2.2). These products can be derivatized in a variety of ways, with negligible racemization and in generally high yield. The respective carboxylic acid, ester, or amide can be produced easily, and with the appropriate reducing agent, the corresponding aldehyde or alcohol can be made selectively.
Table 2.2.
A selection of amides synthesized.
| Entry | R | Ar | ee (%) | Yield (%) |
|---|---|---|---|---|
| 1 | Et | Ph | 90 | 93 |
| 2 | i-Pr | Ph | 95 | 96 |
| 3 | Et | o-Tolyl | 98 | 95 |
| 4 | Me | o-Anisyl | 94 | 94 |
| 5 | Bn | 3-(N-methylindolyl) | 86 | 80 |
Remarkably, in the mechanism of the reaction, the catalyst is believed to act as a Brønsted acid/base (instead of acting as a nucleophile as previously thought). Deprotonation of the pyrrole facilitates its nucleophilic attack on the ketene substrate; then the resulting protonated catalyst acts as a chiral proton source, selectively donating its proton to the pyrrole-ketene enolate (Scheme 2.6).
Scheme 2.6.
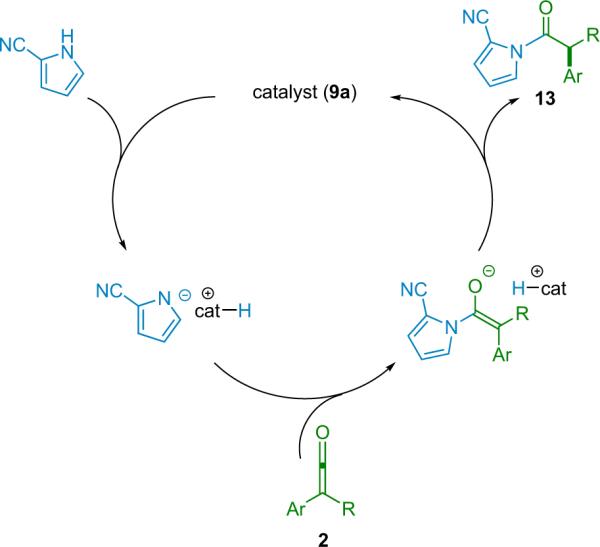
Catalytic cycle for the aminolysis of disubstituted ketenes.
Fu next sought to expand the utility of the reaction by employing azide as an alternative nucleophile.25 By combining hydrazoic acid with aryl/alkyl disubstituted ketenes in the presence of catalyst 9c, Fu et al. were able to produce acyl azides (13, Scheme 2.7), allowing them to exploit the Curtius rearrangement to produce isocyanates (14) that can then be converted into either chiral amines (15) or carbamates (16) through hydrolysis or alcoholysis, respectively (Scheme 2.7).
Scheme 2.7.
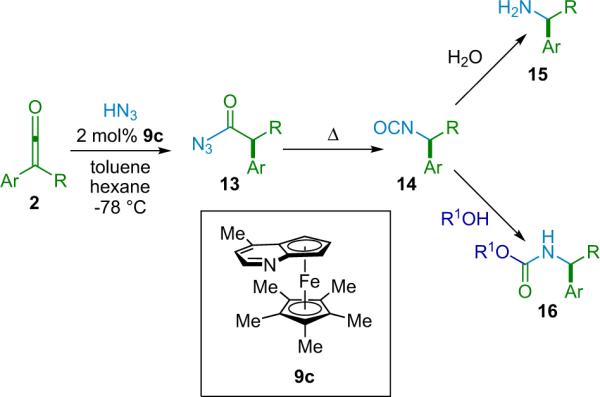
Reaction of hydrazoic acid with ketenes.
Much like the related reactions in this series, this catalytic cycle is believed to proceed through a Brønsted acid/base pathway, analogous to that depicted in Scheme 2.6. The method is fairly robust, working effectively for a variety of disubstituted ketenes, as long as they possess significant steric hindrance (Table 2.3). However, when bulky ketenes are employed, the reaction proceeds smoothly, giving the desired products in excellent yield and very high selectivity. The authors report the production of carbamates from methanol,26 and they show one example of an amine product included (from ethyl o-tolyl ketene, 93% ee, 97% yield). However, the reaction sequence seems capable of efficiently producing a range of chiral amines and their carbamate equivalents.
Table 2.3.
Synthesis of carbamates.
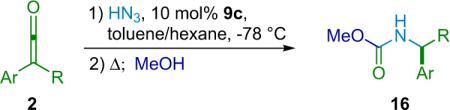
| Entry | R | Ar | ee (%) | Yield (%) |
|---|---|---|---|---|
| 1 | i-Pr | Ph | 96 | 93 |
| 2 | i-Pr | p-MeO-Ph | 97 | 94 |
| 3 | i-Pr | 3-Thienyl | 94 | 92 |
| 4 | t-Bu | Ph | 76 | 94 |
| 5 | cy | Ph | 96 | 92 |
3. Asymmetric β-lactone synthesis
Interest in the synthesis of β-lactones has a long history, and rightly so as these compounds possess structures combining the gross atom connectivity of an aldol adduct with bond-strain activation approaching that of an epoxide. An additional feature that the β-lactone holds over simple aldol products is the ability to be cleaved selectively at the acyl–oxygen or the alkyl–oxygen bond as desired, leading to a myriad of potential products from a single source. Romo's recent review provides an excellent introduction to the synthetic and medicinal chemistry of β-lactones.27
3.1. Cinchona alkaloid catalyst systems
While β-lactones were investigated by Staudinger soon after his discovery of ketenes,28 it was not until over 70 years later, with the pioneering work of Wynberg, that a catalytic, enantioselective route to their synthesis was found. Wynberg and Staring employed cinchona alkaloids as nucleophilic catalysts to mediate the coupling of unsubstituted ketene with activated ketones and aldehydes.29 In their initial report, they demonstrated the synthesis of both the (R) and the (S) forms of malic acid through the β-lactone intermediate.29b Scheme 3.1 shows the synthetic route for (S)-malic acid (20), obtained by forming the initial β-lactone (18) via quinidine (7b) catalysis. The non-natural (R)-malic acid is obtained by the same means, but employing quinine (6b) as the catalyst.
Scheme 3.1.
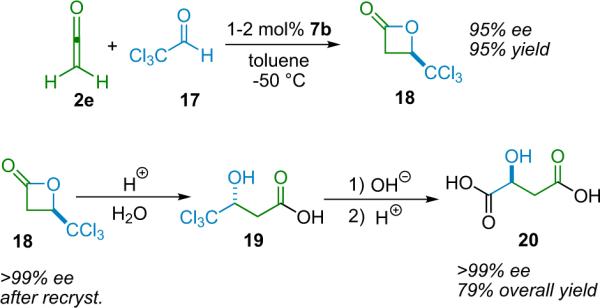
A catalytic, asymmetric synthesis of (S)-malic acid.30
In their follow-up publication, they expanded upon their β-lactone synthesis, exploring a range of aldehyde reactants (21), and providing a simple and powerful means of directly accessing chiral β-lactones (22).29a As with the initial reaction, employing the diastereomeric alkaloids quinine and quinidine as catalysts allowed for the synthesis of either enantiomer of a given product as desired, maintaining the flexibility of this method (Table 3.1).
Table 3.1.
Wynberg's β-lactone synthesis.
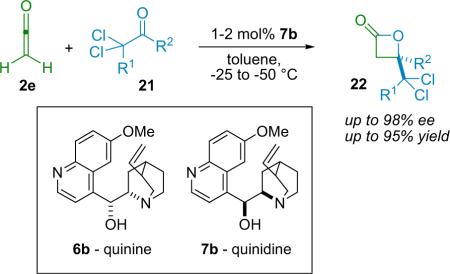
| Entry | R1 | R2 | ee (%) | Yield (%) |
|---|---|---|---|---|
| 1 | Cl | H | 76a | 89 |
| 2 | Cl | H | 98 | 89 |
| 3 | H | H | 45 | 67 |
| 4 | Cl | Me | 94 | 72 |
| 5 | Cl | p-Cl-Ph | 90 | 68 |
| 6 | Cl | p-NO2-Ph | 89 | 95 |
Runs with catalyst 6b, yields opposite enantiomer.
Wynberg's work forged the way for other researchers, guiding the development of a new and robust range of catalytic methodology for the synthesis of β-lactones (24). As can be expected from a reaction that is so effective within a limited scope, other groups have been inspired to develop the means to make it more general, and thus increase its utility. In a very nice extension, Romo has made use of shuttle deprotonation (see Section 1.3) to generate the unsubstituted ketene substrate in situ from acetyl chloride (1e),31 obviating the need to add it as a gas and thus avoiding the large excess of ketene necessitated by the Wynberg method (Table 3.2). In his original publication, Wynberg proposed that the tertiary amine catalysts he studied promote stereoselectivity by complexation with the ketene substrate.29b While he did not expand upon the catalyst mode of action, Romo has proposed a logical catalytic mechanism (Table 3.2).31
Table 3.2.
Romo's β-lactone synthesis.
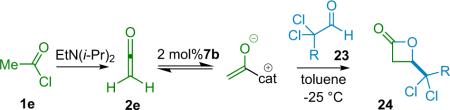
| Entry | R | ee (%) | Yield (%) |
|---|---|---|---|
| 1 | Bn | 94 | 85 |
| 2 | C6H13 | 93 | 73 |
| 3 | Piv-O(CH2)2 | 94 | 80 |
| 4 | i-Pr | 98 | 40 |
3.2. Bifunctional systems
The Nelson research group has developed a variant of the Wynberg method using shuttle deprotonation for the in situ generation of ketenes.32 Nelson's optimized reaction employs a silylmodified cinchona alkaloid catalyst (6d, TMS-Q) and a variable amount of LiClO4 (amount depends on substrate and ranges from 15 to 300 mol %). This effective combination allows for the use of unsubstituted or methyl-substituted ketenes and unactivated aldehydes, greatly enhancing the scope of the reaction while maintaining high enantioselectivity and good yield, as well as producing the disubstituted products (26) with excellent diastereoselectivity (Table 3.3).
Table 3.3.
Nelson's β-lactone products.
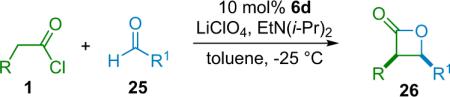
| Entry | R | R1 | ee (%) | dr (%) | Yield (%) |
|---|---|---|---|---|---|
| 1 | H | Cy | 94a | - | 85 |
| 2 | H | BnOCH2 | 84 | - | 70 |
| 3 | Me | Ph(CH2)2 | >99 | 96 | 84 |
| 4 | Me | p-F-Ph | >99 | >96 | 85 |
| 5 | Me | o-Cl-Ph | >99a | 96 | 80 |
| 6 | Me | o-Tol | >99 | >96 | 76 |
Runs with catalyst 7d, yields opposite enantiomer.
In his proposal of the reaction mechanism, Nelson suggests that the reactants combine in a six-membered transition state wherein the metal coordinates the catalyst-bound enolate to the aldehyde, both activating the components and generating a tightly stereo-controlled environment for bond formation (Scheme 3.2).
Scheme 3.2.
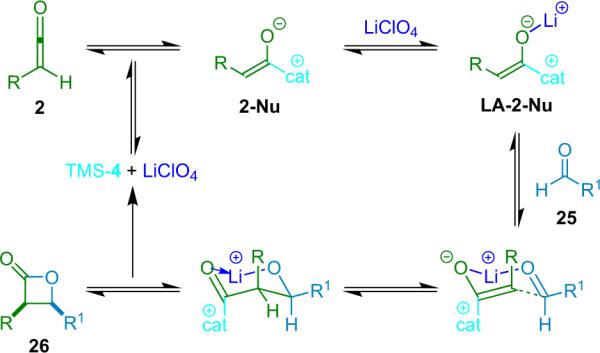
Proposed reaction mechanism for a bifunctional system.
Calter has found that certain metal salt cocatalysts can invert the diastereoselectivity of the reaction, yielding products favoring the trans-diastereomer.33 By pairing one of their silylated cinchona alkaloid catalysts with 15 mol % Sc(OTf)3 instead of the LiClO4 cocatalyst employed by Nelson, the Calter group has been able to cyclize a number of alkyl-substituted ketene and aromatic aldehyde pairs with excellent diastereo- and enantioselectivity, as well as good yield (Table 3.4).
Table 3.4.
A trans-selective β-lactone reaction.
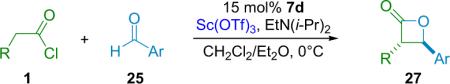
| Entry | R | Ar | ee (%) | dr | Yield (%) |
|---|---|---|---|---|---|
| 1 | Me | Ph | 92 | 91:9 | 75 |
| 2 | Me | p-NO2-Ph | 96 | 91:9 | 82 |
| 3 | Me | p-CN-Ph | 99 | 92:8 | 80 |
| 4 | Et | p-CN-Ph | 99 | 95:5 | 80 |
In the same communication, Calter reports that lanthanide metal triflates are effective for forming cis-β-lactones when paired with alkoxy-substituted ketenes. In particular Er(OTf)3 was found to give high diastereoselectivity (Table 3.5). Calter proposes that the change in diastereoselectivity in the reaction is due to a change in transition state prompted by an interaction between the metal cocatalyst and the substrate. He suggests that, initially, each substrate is bound to a separate metal center; the aldehyde is activated by one while the ketene enolate is bound to the other.
Table 3.5.
cis-β-Lactone formation with lanthanide cocatalyst.
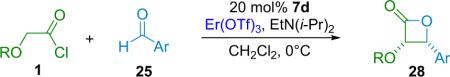
| Entry | R | Ar | ee (%) | dr | Yield (%) |
|---|---|---|---|---|---|
| 1 | Ph | Ph | >99 | 88:12 | 58 |
| 2 | Ph | p-Br-Ph | >99 | 88:12 | 55 |
| 3 | Ph | m-Cl-Ph | >99 | 92:8 | 88 |
| 4 | Bn | p-CN-Ph | >99a | 87:13 | 68 |
Runs with catalyst 6d, yields opposite enantiomer.
The authors believe that when these two complexes associate with each other, one of two things can happen: either one metal fragment is lost and the remaining metal complex helps coordinate the new complex into a closed transition state (TS) resulting in a cis-β-lactone; or both metals are retained, and an open TS results, leading to the trans-β-lactone product. The open TS is seen to be more favorable both sterically and electronically, and thus the trans products are favored, in the case of aliphatic ketenes (scenario A, Fig. 3.1). The extra binding center in the alkoxyketenes creates a favorable open-gauche TS (scenario B, Fig. 3.1).
Figure 3.1.
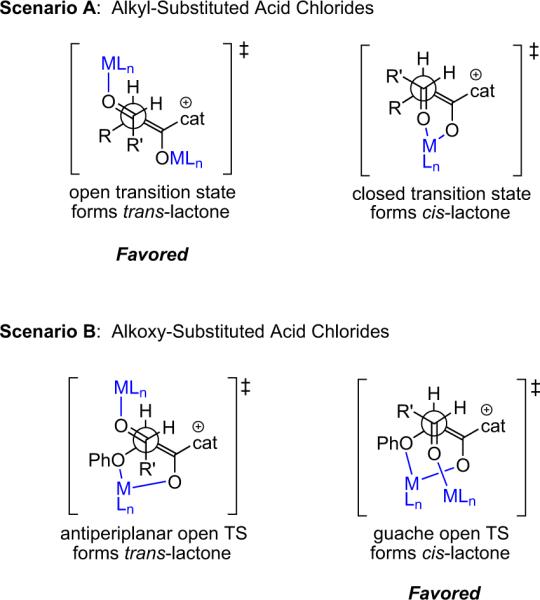
Proposed transition states in Calter's catalytic system.
Calter distinguishes these two proposed complexes as antiperiplanar open and gauche open transition states, leading to trans and cis products, respectively. The involvement of two molecules of metal is supported by the observation that in going from 7.5 to 15 mol % loading of Sc(OTf)3 in the reaction of propionyl chloride with p-cyanophenylaldehyde (entry 3, Table 3.4), the diastereomeric ratio (dr) is seen to increase from 56:44 to 92:8 (trans/cis).
Lin et al. have developed a bifunctional catalyst that incorporates a cinchona alkaloid nucleophile to activate the ketene to a nucleophilic enolate and a salen-bound CoII that putatively binds to and activates the aldehyde.34 The reaction between ethenone and benzyloxyacetaldehyde proceeds in high yield (91%) and ee (>99%) only when the two catalyst functional sections are directly attached, yet the stereochemistry of the diaminopropanoic acid linker proved to be inconsequential (Fig. 3.2).
Figure 3.2.
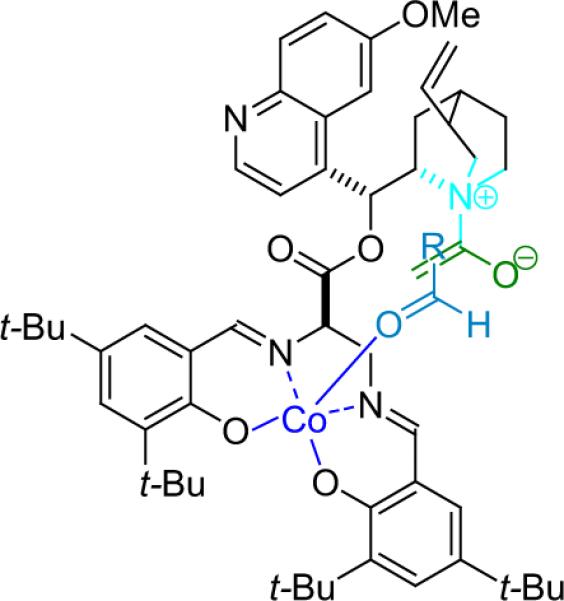
Proposed activated complex in Lin's bifunctional cyclization.
3.3. Lewis acid based catalyst systems
The Miyano group, after an initial report detailing the enantioselective synthesis of β-lactones promoted by a stoichiometric amount of an aluminum–BINOL complex,35 rapidly published a modified catalyst system (30a) that allows them to produce their products in generally good yield and modest enantioselectivity (up to 74% ee, Scheme 3.3).36 This method has the advantage of being effective with a range of aldehydes.
Scheme 3.3.
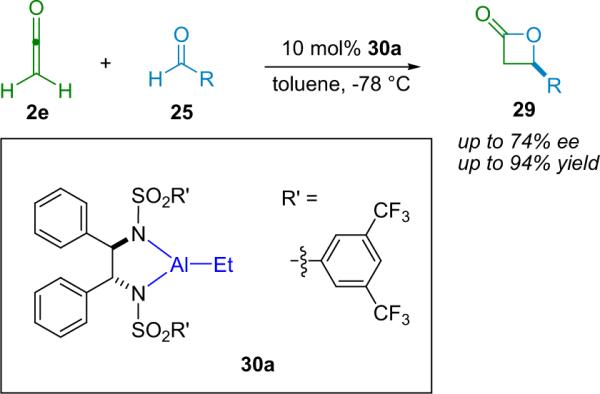
The Miyano β-lactone synthesis.
Building on Miyano's research, but working with the more stable silylketene (2f), the research groups of Kocienski and Pons employed variants of Miyano's catalysts (30b) to produce silylated β-lactones in moderate to good yield and enantioselectivity and with excellent diastereoselectivity (Scheme 3.4).37 The authors found that the silylketene reactions require 30% catalyst loading to ensure maximum conversion and selectivity.
Scheme 3.4.
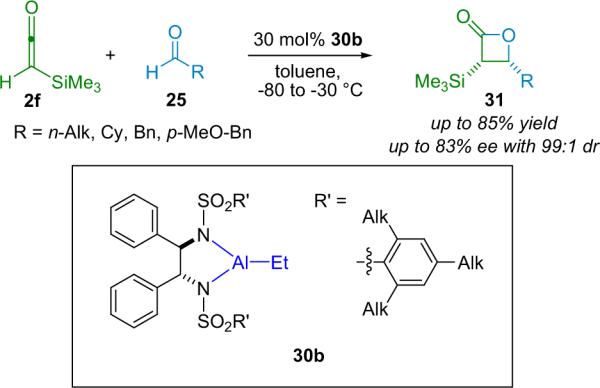
The Kocienski and Pons β-lactone synthesis.
The Romo group has also employed a variety of clever means to synthesize β-lactones stereoselectively. Among their early work38 was a chelation-controlled, highly diastereoselective synthesis of benzyloxy-substituted lactones mediated by magnesium dibromide etherate. Having successfully developed this diastereoselective, Lewis acid catalyzed reaction, the Romo group moved on to chiral Lewis acids,39 employing two different catalyst systems in the pursuit of an efficient asymmetric method (Scheme 3.5). While their TiIV–TADDOL catalyst39a was effective in this reaction, its performance was generally surpassed by their later developed AlIII–diol catalyst (32).39b
Scheme 3.5.
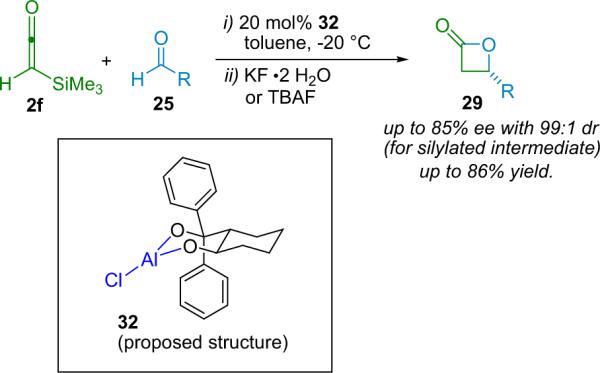
The Romo β-lactone synthesis.
Note that the structure of 32 has not definitively been determined, and Romo reports that it may actually exist as a dimeric species. The structure shown in Scheme 3.5 is believed to represent the catalytically active configuration of the Lewis acid. The authors demonstrated the benefit of using catalyst 32 over the TiIV catalyst; in most cases the ee is more than double, and when R=benzyl (25), the previously almost non-selective reaction is improved to 75% ee when 32 is used. The observed dr of the silylated intermediate is also enhanced by the aluminum-based Lewis acid, as is the overall yield of the two-step reaction sequence.
Early work by the Nelson group on β-lactone synthesis focused on the synthesis of racemic products40 that could then be resolved by the action of an enzyme and an alcohol,41 leading to a mixture of one enantiomer of the lactone and the alcoholysis product of the other enantiomer. While their early catalyst gave the β-lactone in good yield, the resolution step is, of course, inherently limiting to the yield of enantiopure product. Nelson then shifted research to chiral AlIII catalysis,42 employing catalyst 33 to produce β-lactone products (29) in good ee and yield (Table 3.6), and good dr for the analogous method43 using substituted ketenes.
Table 3.6.
Nelson's chiral β-lactone synthesis.
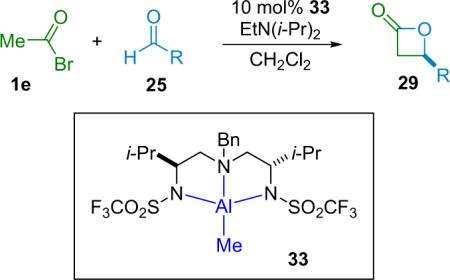
| Entry | R | ee (%) | Yield (%) |
|---|---|---|---|
| 1 | PhCH2CH2 | 92 | 93 |
| 2 | i-Bu | 93 | 80 |
| 3 | BnOCH2C≡C | 93 | 86 |
| 4 | t-Bu-C≡C | 85 | 91 |
In nearly all of his work on β-lactone synthesis, Nelson places emphasis on the practical uses of his products. He has developed efficient processes for converting these β-lactones (29) into β-hydroxy esters (34),42 β-disubstituted carboxylic acids (35),44 and β-azido acids (36) that are readily converted into the corresponding β-amino acids (37).45 In demonstrating these derivatizations, Nelson emphasizes the flexibility and utility of β-lactones as synthetic intermediates (Scheme 3.6).
Scheme 3.6.
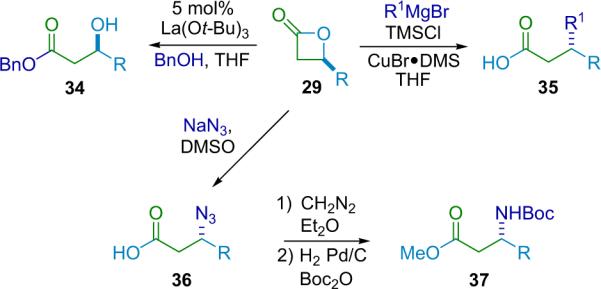
β-Lactone transformations.
Evans has reported a similar Lewis acid catalyzed β-lactone forming reaction. This system involves the [2+2] cycloaddition reaction between an α-silylketene (2f) and α-keto esters (38). The reaction is catalyzed by a bis(oxazoline) CuII complex (39), forming various β-disubstituted lactones in good yield (80% average) and enantioselectivity (83–95% ee, Scheme 3.7), after cleavage of the α-silyl group by KF.46 The cycloaddition is believed to benefit from an orbital interaction between the LUMO of the glyoxylate substrate (activated by the Lewis acid) and the HOMO of the ketene; in essence, Evans regards the ketene as a weak nucleophile in this case.
Scheme 3.7.
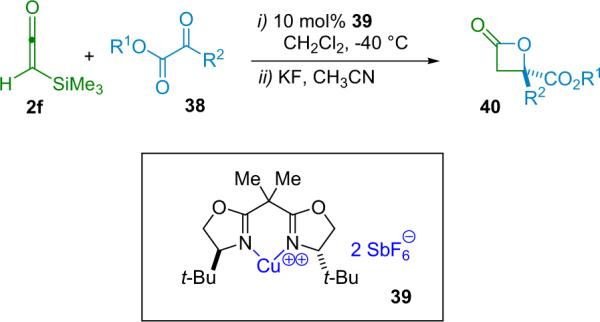
Copper(II) catalyzed β-lactone formation.
In an interesting study of his chiral oxazaborolidine catalyst (40), Corey reported the condensation of ethenone (2e) with several aldehydes (25) to produce the corresponding lactone (29) in good yield and ee (Scheme 3.8).47 Catalyst optimization studies led them to add tributyltin triflate to catalyst 40 to produce more activated catalyst 41. This second catalyst is proposed to act by insertion of the ketene into the phenoxy–tin bond (42) followed by coordination of the aldehyde to the boron and subsequent `intramolecular' cyclization of the ternary complex 43.
Scheme 3.8.
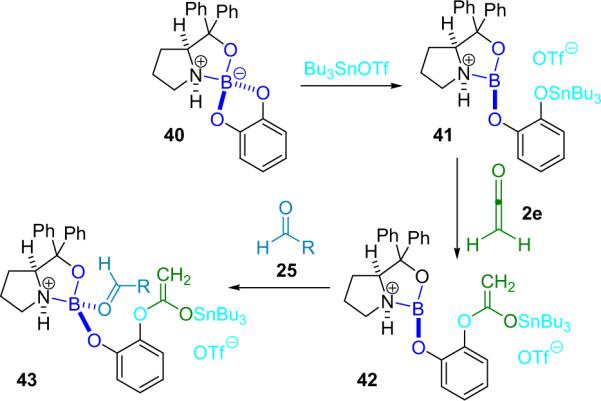
Proposed mechanism of oxazaborolidine catalyzed β-lactone formation.
3.4. Trisubstituted β-lactones
Expanding on his work with disubstituted ketenes, Fu has developed the synthesis of trisubstituted β-lactones (43), employing his planar-chiral catalyst (9).48 This system is capable of catalyzing the cycloaddition of aromatic aldehydes with alkyl/ alkyl disubstituted ketenes (Table 3.7). In his analysis of the reaction, Fu suggests that the catalyst in this case acts in analogous fashion to his ketene methanolysis (see Scheme 2.2), namely by generating a transient chiral ketene enolate intermediate. He has also shown that the β-lactone products, despite their increased bulk, can still undergo the various transformations known for mono- and disubstituted β-lactones in good yield and with retention of ee.
Table 3.7.
Trisubstituted β-lactone synthesis.
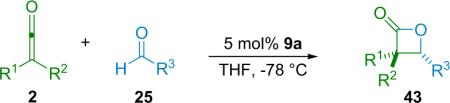
| Entry | R1 | R2 | R3 | ee (%) | Yield (%) |
|---|---|---|---|---|---|
| 1 | Et | Et | Ph | 91 | 92 |
| 2 | Et | Et | 2-Napthyl | 89 | 77 |
| 3 | Et | Et | p-CF3-Ph | 80 | 74 |
| 4 | −(CH2)6− | Ph | 82 | 71 | |
| 5a | i-Pr | Me | Ph | 91 | 48 |
dr 4.2:1; ee is for the cis diastereomer, yield of both diastereomers.
More recently, Ye et al. reported that chiral N-heterocyclic carbenes (NHC, 44) are efficient catalysts for the reaction of disubstituted ketenes with 2-oxoaldehydes (Scheme 3.9).49 The NHC catalysts were used putatively to activate disubstituted ketenes in a manner similar to nucleophilic tertiary amines—by condensing to create a nucleophilic, zwitterionic ketene enolate as the reactive intermediate (2-Nu). In this way, catalyst 44 provides a variety of trisubstituted lactones from alky/aryl disubstituted ketenes in good yield (63–99%) with excellent enantioselectivity (94–99% ee) and good diastereoselectivity as well. The NHC catalysts are easily prepared in situ by stirring the corresponding salt with cesium carbonate, but must be used immediately.
Scheme 3.9.
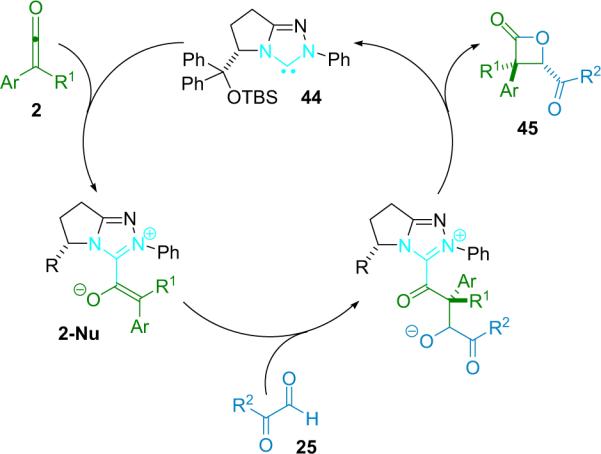
Proposed mechanism of NHC catalyzed β-lactone formation.
3.5. Asymmetric ketene dimerization
A perennial difficulty in dealing with ketenes is their tendency to undergo side reactions that is ironically a consequence of the reactivity that makes them useful substrates. The most common of these is dimerization (Eq. 3.1). Whereas the tendency of ketenes to dimerize can be controlled by factors such as solution concentration and temperature, it remains an issue in many ketene-based reactions. However, the structures of ketene dimers reveal both molecular complexity and chirality, suggesting that this often unwanted side reaction can be useful and interesting in itself.
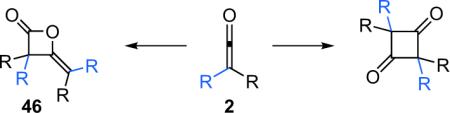 |
(3.1) |
3.5.1. Dimerization of preformed ketenes
The Calter group has sought to exploit the ketene dimerization reaction by rendering it enantioselective. With an eye toward utilizing the chiral dimers as synthetic intermediates,50 his early work focused on the cinchona alkaloid catalyzed dimerization of pregenerated methylketene (Scheme 3.10).51 He screened a number of cinchona alkaloid derivatives, finding that quinidine (7b), or its silylated derivative 7d, was most effective, with the two giving the same high selectivity (98% ee). Quinine (6b) gave only moderate results (70% ee) but its silylated derivative 6d proved to be effective (90% ee). The drop in ee observed for quinine was explained by partial O-acylation—propionylquinine gave only 50% ee when used as the catalyst.
Scheme 3.10.
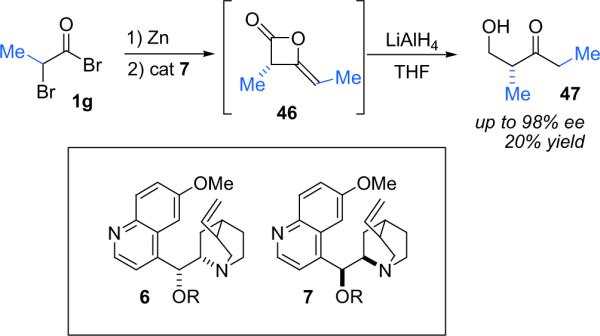
Ketene dimerization with preformed ketene.
Dimer 46 could not be isolated due to a combination of reactivity and volatility, requiring its derivatization. As this method was employed to synthesize polypropionate intermediates, the Calter group moved away from the zinc promoted ketene formation and began to employ a thermolytic ketene generation from propionic anhydride and catalyst 6d or 7d to form dimer 46 at a constant, reproducible rate with high enantioselectivity (Scheme 3.11).52
Scheme 3.11.
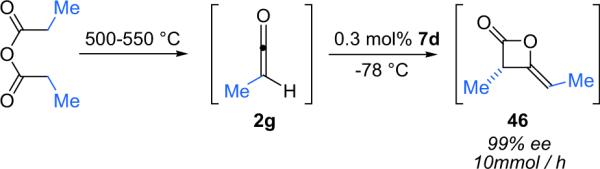
Asymmetric dimerization of a pyrolytically generated ketene.
To generate the desired products, the crude β-lactone mixture was treated with the lithium amide of N,O-dimethylhydroxylamine to open the ring, affording a lithium enolate (47) that reacts directly with a variety of aldehydes giving aldol products with full retention of optical purity and high diastereoselectivity (Table 3.8).
Table 3.8.
Polypropionates from β-lactones.
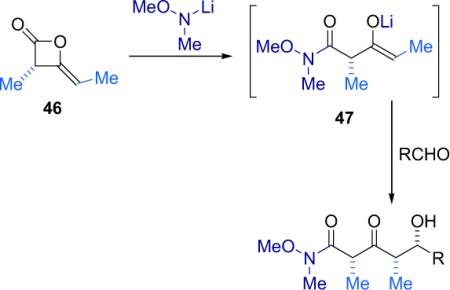
syn,syn:anti,syn ratio.
Yield of syn,syn product only.
Going a step further, Calter employed (S)-2-methylpentanal to obtain a precursor (48) to the class of natural products known as siphonarienes: siphonarienedione (49), siphonarienolone (50), and siphonarienal (51) (Scheme 3.12).52 The 2-methylpentanal can also be used in its racemic form because the diastereomeric purification is relatively simple.
Scheme 3.12.

Synthesis of siphonariene natural products.
Ye et al. have also applied their NHC catalyst (52) to the dimerization of disubstituted ketenes.53 Slight catalyst modification from their previous system (44, Scheme 3.9) allowed the authors to obtain the dimers selectively (Scheme 3.13). A variety of alkyl and aryl groups were accommodated by altering reaction time. As before, the NHC catalyst is produced in situ by stirring the corresponding tetrafluoroborate salt with Cs2CO3 immediately prior to the addition of ketene.
Scheme 3.13.
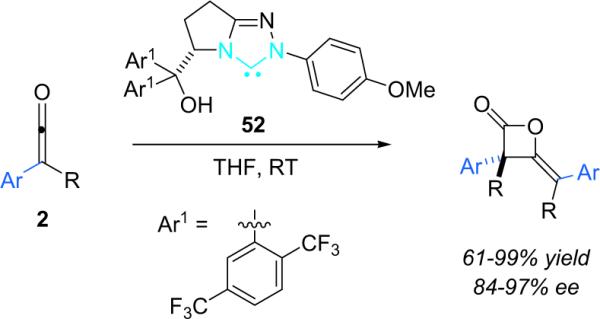
NHC-catalyzed dimerization of disubstituted ketenes.
3.5.2. Dimerization of in situ generated ketenes
The Calter group later sought to expand upon this synthesis by making use of shuttle deprotonation (see Section 1.3) to generate a selection of ketene substrates in situ.54 By employing a combination of TMS-Q (6d) and Hünig's base, the desired dimers were produced with high enantioselectivity and in good overall yield (Table 3.9). The authors note that while the reaction is tolerant of increasing steric bulk the larger substituents require longer reaction times and higher concentrations to produce the desired products. They also remark that the reaction can be performed with silylated quinidine 7d with similar, or even improved, results to obtain the opposite enantiomer (97% ee and 79% yield for R=Me).
Table 3.9.
Ketene dimers generated by shuttle deprotonation.
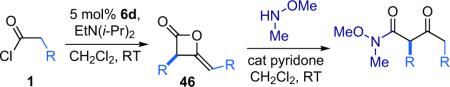
| Entry | R | ee (%) | Yield (%) |
|---|---|---|---|
| 1 | Me | 94 | 72 |
| 2 | Et | 92 | 82 |
| 3 | i-Pr | 96 | 65 |
| 4 | t-Bu | 93 | 58 |
| 5 | TIPS-OCH2 | 91 | 88 |
| 6 | MeO2CCH2 | 92 | 64 |
While most of the dimer products formed by this reaction were not isolable, the esterified ketene substrate (entry 6) was purified by rapid filtration through silica gel prior to step two. The authors also undertook a series of mechanistic studies to elucidate the rate limiting and optical induction steps for this reaction. They determined that the rate limiting step in the reaction of propionyl chloride (1g) with catalyst 6d and Hünig's base was the dehydrohalogenation of propionyl chloride to form methylketene (2g, Eq. 3.2).
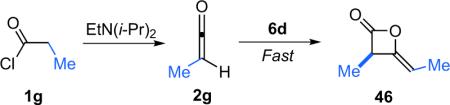 |
(3.2) |
For their studies on the stereochemistry determining step of the reaction, they considered two possible pathways to the product (Scheme 3.14). Pathway A could potentially be followed whether or not the ketene was preformed, however, pathway B was only possible if the ketene was formed in situ. A series of experiments followed wherein both preformed and in situ generated ketene were reacted with a variety of catalysts. In all cases, the products were observed to possess identical selectivity (within experimental error) and thus Calter favors a single reaction pathway for both sets of conditions (pathway A, Scheme 3.14).
Scheme 3.14.
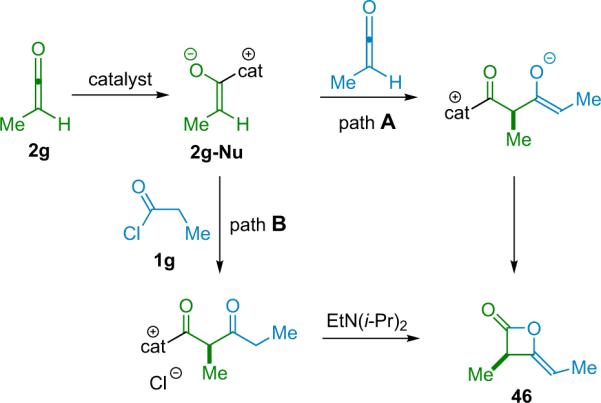
Proposed reaction pathways in the catalyzed formation of ketene dimers.
The Romo research group has also made use of ketene dimerization. Their initial work in this area focused on the epoxidation of racemic dimers to produce unexpectedly stable spiroepoxy β-lactones that can serve as precursors to a variety of useful products.55 During this research, the Romo group discovered that they were able to purify their dimers via flash chromatography. Without purification, the yield and diastereoselectivity in their epoxidation step were low. The resulting disubstituted β-lactones were then derivatized to produce trisubstituted β-lactones capable of acting as fatty acid synthetase (FAS) inhibitors.56 They had found that conducting the two-step process without purification of the dimer intermediate again led to degradation of the dimer with concomitant losses in yield and enantiopurity in the products. With purification, however, they were able to obtain moderate overall yield and high enantioselectivity for the initial sequence in their FAS inhibitor syntheses (Table 3.10).
Table 3.10.
β-Lactones via dimerization/reduction.
| Entry | R | Yield (%), step 1 | Yield (%), step 2 | ee (%) |
|---|---|---|---|---|
| 1 | n-Bu | 75 | 90 | 96 |
| 2 | Cyclopentyl-CH2 | 54 | 89 | 94 |
| 3 | Cyclohexyl-CH2 | 55 | 89 | 90 |
| 4 | Bn | 48 | 85 | 96 |
| 5 | MeO2CCH2 | 60 | 94 | 92 |
4. Asymmetric β-lactam synthesis
Intensive research has generated numerous methods of synthesizing the β-lactam skeleton.57 Commonly, the lactam ring is formed through either ketene–imine cyclizations (another ketene application pioneered by Staudinger)58 or ester enolate–imine condensations59 (the Gilman–Speeter reaction). However, other notable methods are sometimes employed, including photoinduced rearrangements60 and radical cyclizations.61 Despite all of these synthetic methods for obtaining achiral or racemic β-lactams, until the last decade asymmetric methodology has remained scarce, largely limited to chiral auxiliary based systems.62 A more general methodology based on asymmetric catalysis for the synthesis of optically enriched β-lactam was desirable.10a
4.1. Organocatalytic methodology
The Lectka group chose to focus their efforts on a catalytic modification of the highly effective Staudinger method, due to the ready availability of substituted imines, and the relative ease of generating ketenes from commercially available acid halides. Many of the new, clinically active β-lactam serine protease inhibitors being studied contain a carboalkoxy substituent at the β-carbon and could be described as non-natural derivatives of aspartic acid. These could be formed from a Staudinger-type cyclization of ketenes and α-imino esters, which Lectka previously had used fruitfully in the asymmetric synthesis of amino acid derivatives catalyzed by chiral Lewis acids.63
The Staudinger reaction is known as a high background rate process64 normally no catalyst is needed to initiate smooth reaction, even at low temperatures. In order for a catalytic reaction to work, the classical Staudinger pathway (the imine nitrogen acts as a nucleophile toward the electrophilic ketene) must be `broken'. Subsequently, the reaction polarity must be reversed (umpolung) so that the imine and ketene switch roles; the imine must become electrophilic and the ketene nucleophilic. Alteration of the imine polarity was accomplished through the addition of an electron-withdrawing group to the imine nitrogen and a carboalkoxy substituent to the imine α-carbon. By pairing this electrophilic imine with a ketene rendered nucleophilic through catalytic attack of a nucleophile, the desired lactam product can be obtained (Scheme 4.1).
Scheme 4.1.
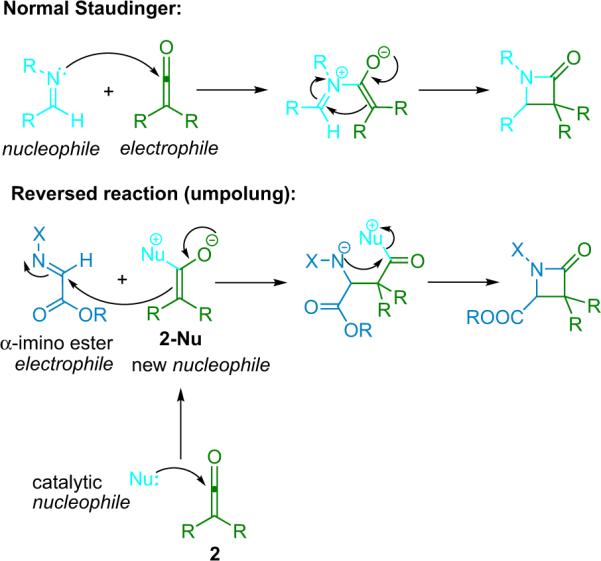
Staudinger reaction `umpolung'.
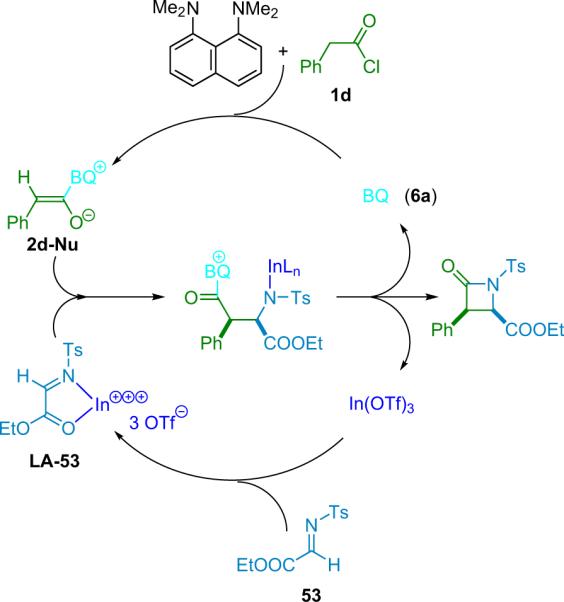
After successful early development of simple diastereoselective amine catalysts,65 the Lectka group was prompted to screen optically active cinchona alkaloid derivatives as potential candidates for enantioselective and diastereoselective catalysts.66 Catalyst development introduced an acyl derivative of quinine, namely benzoylquinine (BQ, 6a). When BQ was applied to the reaction of diphenylketene with N-tosyl imino ester 53, the corresponding β-lactam was produced with high enantioselectivity (99% ee) albeit in only modest (36%) yield.11 One of the most challenging aspects to this new chemistry, aside from the design of the catalytic system, was the development of techniques for using highly reactive monosubstituted ketenes.67 As mentioned previously, these intermediates usually must be formed in situ and at reduced temperatures to prevent unwanted side reactions, most commonly dimerization and polymerization.
While hindered amine bases such as Hünig's base were often inadequate, the combination of catalytic BQ and the non-nucleophilic amine base proton sponge (PS) as a thermodynamic proton sink worked well, and the β-lactams were formed with very high enantio- and diastereoselectivity from a variety of acid chloride substrates (Scheme 4.2).68 The β-lactam forming reaction is now compatible with a wide variety of ketenes, including aryl, alkyl, alkenyl, halo, azo, and oxy-substituted (Fig. 4.1).69 Of particular note, Lectka was able to synthesize both phthalimido and benzyl-substituted β-lactams that have been identified as precursors to cytomegalovirus protease inhibitors and human leukocyte elastase inhibitors, respectively.70
Scheme 4.2.
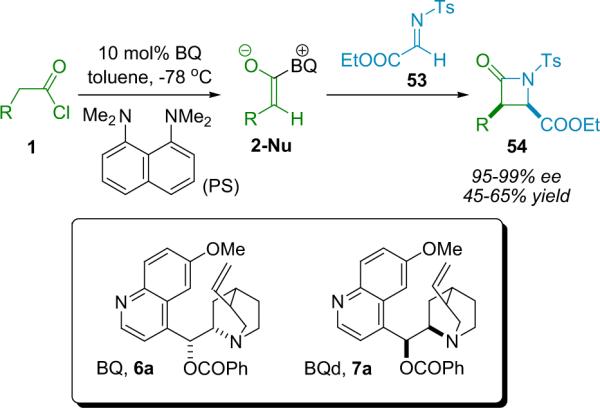
β-Lactam synthesis with BQ and proton sponge.
Figure 4.1.
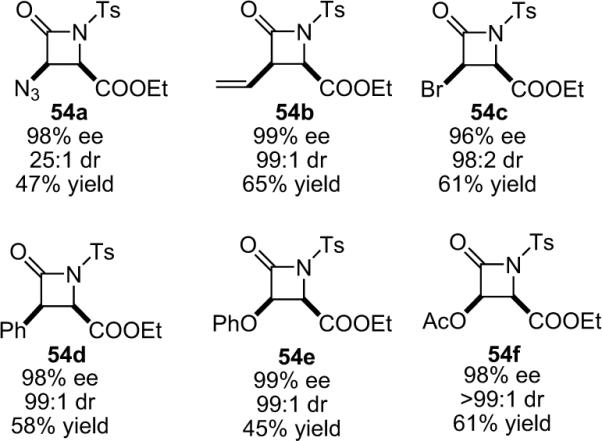
β-lactams synthesized by asymmetric cycloaddition reaction.
The mechanism of this reaction was examined through a series of kinetics studies. For the reaction of phenylacetyl chloride with imino ester 53 catalyzed by BQ (6a) and using PS as the stoichiometric base, the rate determining step is the reaction between BQ and the acid chloride; this is followed by a series of fast cyclization steps with the imino ester. In some cases, the rate of product formation exceeds that of ketene formation when measured independently. This surprising discovery suggests that enolate generation in these cases occurs directly from the acid chloride; discrete ketene formation is consequently circumvented. This ketene-free mechanistic path is shown as scenario A in Scheme 4.3. On the other hand, acid chlorides with electron-withdrawing substituents tend to follow scenario B when proton sponge is the stoichiometric base, initially forming free ketene. When other stoichiometric bases are used (K2CO3, NaH, etc.), discrete ketenes must be synthesized in a `preformation' step to avoid racemization of the product.
Scheme 4.3.

Mechanism of β-lactam formation.
Dehydrohalogenation of the acid chloride substrate is accomplished through the shuttle deprotonation mechanism (see Section 1.3). Various bases can be employed, yielding similar results (Scheme 4.4). In these studies, Lectka discovered that with the stronger bases (NaH and K2CO3) a ketene preformation step was required, necessitating the use of the double reaction flask (see Fig. 1.3). However, both proton sponge and NaHCO3, as weaker bases, not only required no preformation step but might, as shown above, bypass ketene formation altogether.
Scheme 4.4.
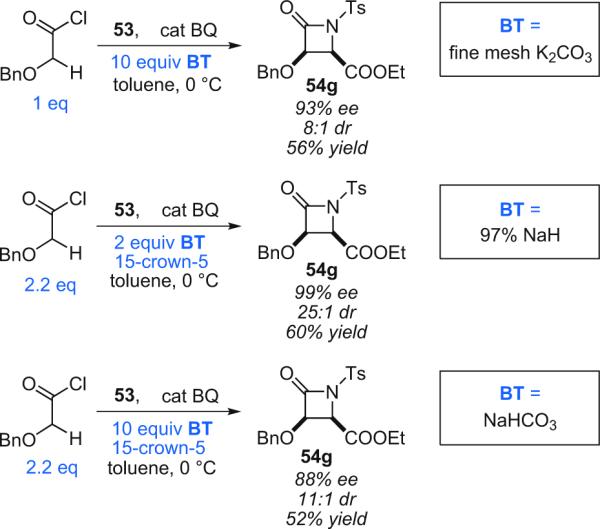
Alternate shuttle deprotonation methods for β-lactam synthesis, BT=thermodynamic base.
4.2. Bifunctional catalytic methodology
4.2.1. cis-β-Lactams
The Lectka group's interest in the field of bifunctional catalysis arose out of long-standing frustration with the low to moderate yields obtained with previous β-lactam methods (see Section 4.1). While BQ (6a) gives exceptional enantioselectivity,66 the authors believe that the desired reaction of the ketene enolate and the tosyl imine (53) was sluggish enough to allow slow side reactions to proceed. Based on previous successes with activated imine alkylations,71 Lectka reasoned that addition of a Lewis acid cocatalyst would render the imine more electrophilic, thereby increasing the rate of the desired [2+2] cycloaddition pathway and suppressing the unwanted secondary reactions.
Initial bifunctional studies tested 10 mol % of the metal complexes that worked best for imine alkylation reactions,72 including Rh(PPh3)3OTf and Cu(PPh3) ClO4,73 to the standard reaction conditions to form β-lactams.74 Unfortunately, the overall yield decreased in the presence of these cocatalysts. In an attempt to discern the reason that known imine binding complexes were detrimental to the cycloaddition reaction, the authors monitored the CuI reaction by UV–vis spectroscopy and found evidence of metal–BQ binding; this is an example of cocatalyst quenching that must be avoided in bifunctional systems (Scheme 4.5).
Scheme 4.5.
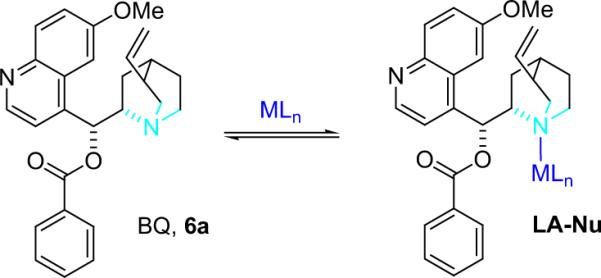
Cocatalyst quenching.
On the other hand, Aggarwal et al. found that group III and lanthanide triflates significantly enhanced their DABCO catalyzed Baylis–Hillman reaction, whereas `traditional' Lewis acids lead to diminished yield.75 These findings suggested that harder, less azaphilic metals would minimize the cocatalyst quenching previously observed. A full spectrum of metal salts was screened with this in mind, demonstrating that triflates of AlIII,ScIII,ZnII, and InIII resulted in increased reaction rates and significantly enhanced chemical yield. Having firmly established In(OTf)3 as the best overall Lewis acid cocatalyst for this reaction, Lectka applied this system to various substrates to determine the scope of its influence (Fig. 4.2). The yield increases in every case, generally by 1.5- to 2-fold with the InIII cocatalyst, and the excellent enantioselectivity remains unaffected.74
Figure 4.2.
Sample of β-lactams synthesized employing cocatalysts BQ and In(OTf)3.
4.2.2. Mechanistic inquiry
There are three potential mechanistic scenarios that could account for the observed effect of the Lewis acid. The original postulate was that InIII binds to the imino ester and increases its reactivity toward the nucleophilic ketene enolate (A, Fig. 4.3). Another possibility is that the metal binds to the zwitterionic enolate, making it more thermodynamically stable (B, Fig. 4.3). This would have a multifaceted effect: (1) the more stable ketene enolate would be more chemoselective, eliminating undesired side reactions; and (2) assuming equilibrium between the ketene and the enolate, metal binding is expected to increase the relative quantity of the enolate, thereby accounting for the increased rate of reaction. A third scenario involves the simultaneous operation of both alternatives, with the metal organizing a termolecular activated complex (C, Fig. 4.3). Based on substantial mechanistic evidence, the metal–enolate binding scenarios (B and C) were eliminated.74b Notably, several seemingly similar bifunctional systems have been reported in which the Lewis acid acts in a different manner: Calter has found that a Lewis acid acts by binding and organizing a chelating version of the ketene enolate instead of activating the imine (see Section 4.2.3).76
Figure 4.3.
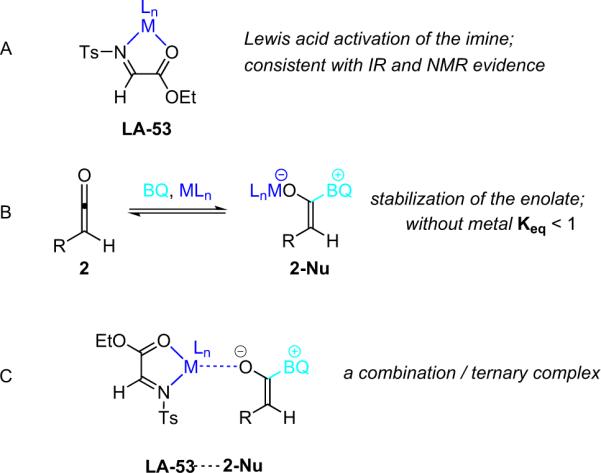
Possible roles of the lewis acid.
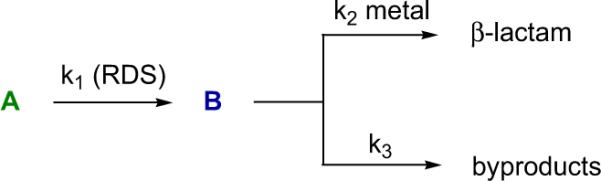
The rate determining step (RDS) of the metal-free reaction involves acylation of the catalyst by the acid chloride and/or ketene formation. In the bifunctional system, it was found that the metal cocatalyst does not participate in the RDS but has a substantial effect on the chemoselectivity of a process after the RDS (Scheme 4.6). By using a `knock-out' strategy where the original RDS is circumvented by preformation of the ketene,12b it was determined that InIII enhances the rate of the C–C bond forming step.74b
Scheme 4.6.
Metal mediated chemoselectivity of the bifunctional pathway.
Lectka also established that metal binding to the imino ester plays a pivotal role in the bifunctional reaction. Coordination and complexation of metals with imino esters are well precedented,77 and the authors found IR and NMR evidence that InIII does bind to the imino ester in this system. However, a more direct (and more elegant) test for productive metal binding was designed, employing a modified imino ester that contains an additional metal binding moiety (55, Scheme 4.7).
Scheme 4.7.
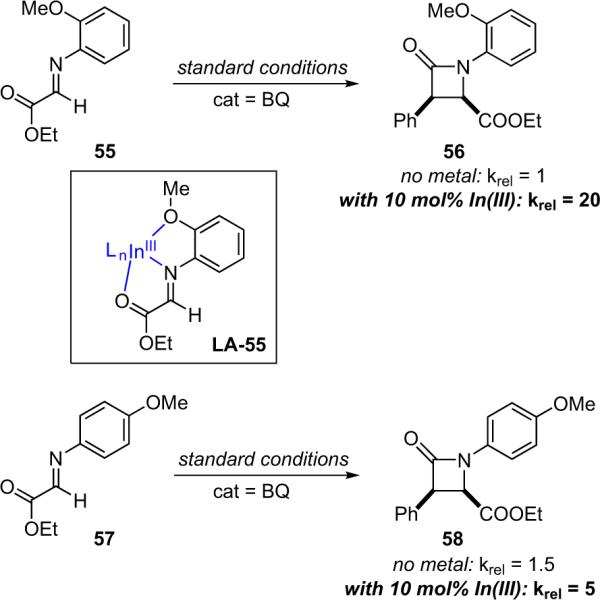
PMP- versus OMP-imine reaction rates.
In the absence of a metal cocatalyst, 57 reacts slightly faster (1.5×) than 55, so the proximity of the methoxy group has the expected effect. However, when In(OTf)3 is added as a cocatalyst, the reactivity is reversed—dimino ester 55 reacts four times faster than substrate 57. Both substrates react faster with the metal than without; while imino ester 57 receives only a 3-fold rate increase from the metal cocatalyst, 55 experiences a tremendous 20-fold rate increase. The difference is clearly due to the tridentate nature of 55. The o-methoxy group helps hold the metal in place so it spends relatively more time activating the imine. Combining all of these data allows formulation of a mechanism for the bifunctional reaction of ketenes with imino esters (Scheme 4.8). Acylation of BQ forms the non-metal-coordinated zwitterionic enolate, activation of the imino ester by the InIII cocatalyst facilitates the C–C bond forming addition reaction, and a transacylation then forms the β-lactam and regenerates the catalyst.
Scheme 4.8.
Proposed mechanism of bifunctional [2+2] cycloaddition.
4.2.3. α-Phenoxy-β-aryl-β-lactams
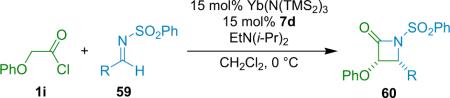
The Calter group has recently applied a silylated quinidine catalyst (7d), which they had used successfully in other ketene reactions, to the synthesis of α-phenoxy-β-aryl-β-lactams.76 They found that electron-withdrawing substituents on the nitrogen are essential to promote clean catalysis, settling on a phenylsulfonyl group for their screening after observing little or no yield with substituents such as methoxy and benzyloxy. Metal screening showed that ScIII and YbIII salts provided the highest conversions, and in further diastereoselectivity optimization, Yb(N(TMS)2)3 was a clear `winner'. For instance, the ytterbium counterion choice improved the cis/trans dr from 6:1 for triflate to 25:1 for hexamethyldisilazide. The corresponding ScIII salts gave 2:1 and 18:1 dr. The combination of catalyst 7d and Yb(N(TMS)2)3 gave moderate to good yield and very good enantioselectivity for a variety of imines at loadings of 15 mol % (Table 4.1).
Table 4.1.
Synthesis of α-phenoxy-β-aryl-β-lactams.
| Entry | R | dr | ee (%) | Yield (%) |
|---|---|---|---|---|
| 1 | Ph | 25:1 | 94 | 83 |
| 2 | p-F-Ph | 19:1 | 97 | 47 |
| 3 | p-Cl-Ph | 28:1 | 97 | 57 |
| 4 | p-CN-Ph | 12:1 | 94 | 52 |
| 5 | p-NO2-Ph | 16:1 | 94 | 55 |
| 6 | p-Tol | 19:1 | 95 | 69 |
4.3. Trisubstituted β-lactams
4.3.1. Planar-chiral catalysts
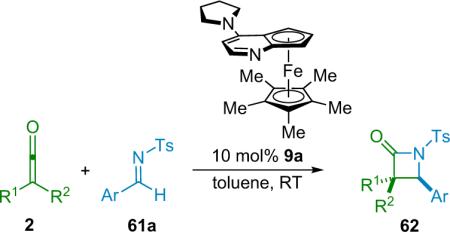
Fu has applied his planar-chiral catalyst system to the asymmetric synthesis of trisubstituted β-lactams,78 using pregenerated, disubstitued ketenes and a selection of imines as substrates. While this system also requires a strong electron-withdrawing group on the imine nitrogen, it is tolerant of a range of cyclic substituents on the α-carbon. His catalyst (9a) provides β-lactam products (62) in good yield and enantioselectivity (Table 4.2). Under the same reaction conditions, and with the same catalyst, Fu was also able to employ unsymmetrical disubstituted ketenes in his reactions, obtaining good cis-diastereoselectivity (8–15:1) with a range of substrates while maintaining high yield and enantioselectivity.
Table 4.2.
Synthesis of trisubstituted cis-β-lactams from disubstituted ketenes.
| Entry | R1 | R2 | Ar | dr | ee (%) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | −(CH2)5− | Ph | - | 81 | 84 | |
| 2 | −(CH2)5− | 1-Furyl | - | 92 | 90 | |
| 3 | −(CH2)5− | β-Styryl | - | 91 | 82 | |
| 4 | i-Bu | Ph | Ph | 8:1 | 98 | 88 |
| 6 | Et | Ph | 1-Furyl | 9:1 | 95 | 97 |
| 7 | i-Bu | Ph | β-Styryl | 10:1 | 98 | 95 |
Some years after this initial communication, Fu et al. reported a variation on their reaction system.79 By changing the electron withdrawing substituent on imine nitrogen from tosyl to triflyl, the diastereoselectivity of the reaction favored trans over cis. Both good yield (80% average) and diastereoselectivity (5–50:1) were obtained, along with moderate to good enantioselectivity (63–85% ee, Eq. 4.1).
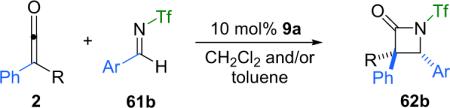 |
(4.1) |
In an effort to explain this interesting diastereomeric reversal, the Fu group examined the interaction between this new imine substrate and catalyst 9a. For example, NMR studies of a mixture of the tosyl imine (61a) and 9a show no significant interaction between the two components, whereas a mixture of triflylimine 61b and 9a appears to exist almost entirely as a bound, zwitterionic species (61b-Nu). With this evidence in hand, they propose that when N-tosyl imine 61a is employed, the lactam is formed in the now familiar cycle of nucleophilic attack on the ketene by the catalyst followed by attack on the imine by the chiral, zwitterionic, catalyst-bound ketene enolate. The cycle is concluded by closure of the β-lactam ring to produce the product and regenerate the catalyst (scenario A, Scheme 4.9). However, Fu believes that if N-triflyl imine 61b is employed, the catalytic cycle is instead initiated by catalyst attack on the imine, leading to an alternate catalyst-bound, zwitterionic intermediate. The cycle would then complete in an analogous fashion by attack of the intermediate on the ketene followed by ring closure with concomitant catalyst regeneration (scenario B, Scheme 4.9).
Scheme 4.9.

Alternate reaction mechanisms.
4.3.2. N-Heterocyclic carbene based catalysis
β-Lactam formation can also be accomplished by chiral N-heterocyclic carbene (NHC) catalysis, as shown by Ye et al. in 2008.80 In this system, the catalyst is formed in situ from the corresponding tetrafluoroborate salt (44-H) by reaction with cesium carbonate (Scheme 4.10). The resulting chiral NHC then acts similarly to the tertiary amine catalysts we have seen so far, by creating a nucleophilic, zwitterionic ketene enolate from disubstituted ketenes. In this way, the reaction is very similar to their system for producing chiral trisubstituted lactones (see Section 3.4).
Scheme 4.10.
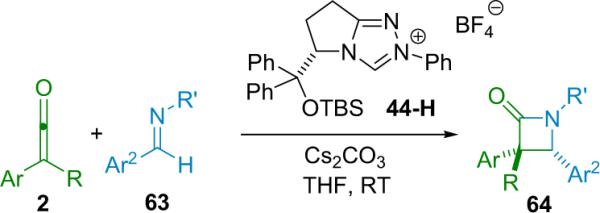
Chiral NHC catalyzed formation of β-lactones.
Initial catalyst optimization studies were done with N-tosyl aryl imines and disubstituted ketenes, and while yields were excellent, diastereo- and enantioselectivity suffered. Digging further, they found that their NHC catalyst could react with tosyl substutited imines, potentially following a mechanism similar to Fu's N-triflyl imines (scenario B, Scheme 4.9). This serendipitous discovery lead Ye to use less activated N-Boc imines for high selectivity, albeit with slightly lower yield (Scheme 4.11). The authors believe that the mechanism follows a stepwise pathway that begins with formation of a zwitterionic enolate (2-Nu).
Scheme 4.11.
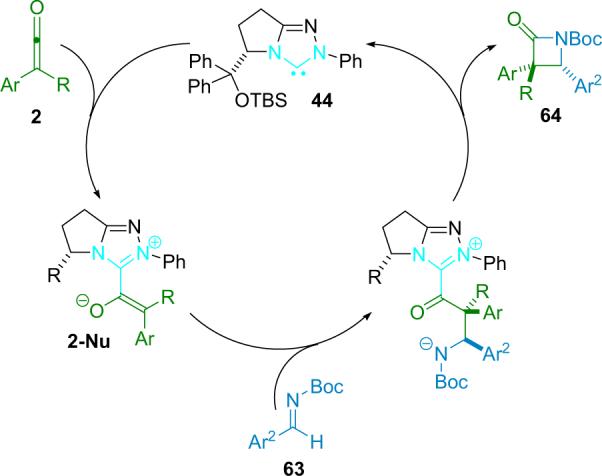
Proposed mechanism of NHC catalyzed reaction.
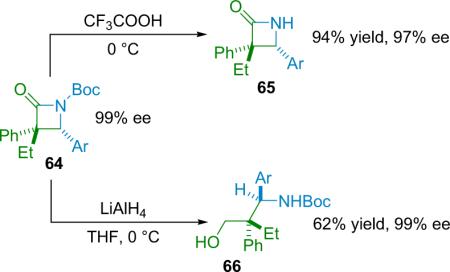
The method is robust and works for a variety of N-Boc aryl imines (63), as well as p-substituted aryl-ethyl disubstituted ketenes, and the products are isolated in good yield with good dr and excellent ee (98% ee on average for the reaction in THF at rt). The N-Boc protection scheme provides simple derivatization: the Boc group can be removed by TFA, affording the free lactam (65) in high yield without significant loss of ee; and the lactam (64) can be reductively opened by LiAlH4, producing the protected γ-amino alcohol (66) without loss of ee (Eq. 4.2).
Independently, Smith et al. reported a similar NHC catalyzed system for the production of chiral β-lactams from N-tosyl aryl imines and disubstituted ketenes.81 Their yields were excellent with several NHC catalysts, including two chiral versions (Fig. 4.4) that unfortunately could not produce higher than 75% ee (diphenylketene and N-tosyl-2-naphthyl imine). However, they were able to raise the ee of this product to >99% by selective crystallization of the minor enantiomer.
 |
(4.2) |
Figure 4.4.
N-Heterocyclic carbene catalysts utilized by smith.
4.4. Aza-β-lactams
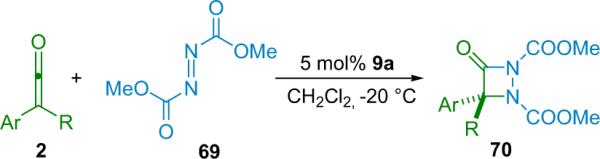
The Fu group has expanded their trisubstituted β-lactam methodology to include aza-β-lactams (70).82 This system is again based on their planar-chiral catalyst 9a in the [2+2] cycloaddition reaction between disubstituted ketenes and azodicarboxylates (Scheme 4.12). Dimethylazodicarboxylate (69) was found to be the best substrate, and its reaction with various alkyl/aryl disubstituted ketenes was thoroughly screened. Most alkyl/aryl combinations worked well and produced the aza-β-lactam in good to excellent yield (53–90%) and good ee (87% on average, though the products recrystallize to >99% ee with little loss of the major enantiomer). Notably, while the authors believe that the reaction goes through the same mechanism as N-tosyl imine reactions (scenario A, Scheme 4.9), they note that the resulting stereochemistry at the newly generated quaternary center results from the opposite sense of induction.
Scheme 4.12.
Catalytic, asymmetric synthesis of aza-β-lactams.
4.5. Oxo-β-lactams
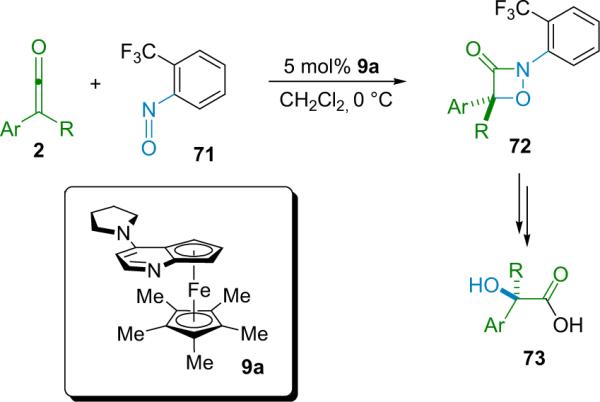
More recently, the Fu group discovered that disubstituted ketenes could react selectively with nitrosoarenes in a [2+2] cycloaddition reaction catalyzed by their planar-chiral catalyst, 9a (Scheme 4.13).83 The 1,2-oxazetidin-3-one products (72), here dubbed oxo-β-lactams, are produced regioselectively when the nitrosoarene is o-CF3-Ph (71). The cycloadducts are formed generally in high yield (87% average) with good enantioselectivity (78% to >98% ee). The authors also use the oxo-β-lactams to make chiral α-hydroxy acids (73) by a multi-step, high-yielding process.
Scheme 4.13.
[2+2] Cycloaddition between ketenes and nitrosoarenes.
5. Asymmetric halogenation
Catalytic enantioselective halogenation reactions can result in compounds that serve as useful intermediates in the construction of functionalized molecules.84 In the case of asymmetric fluorination, the products are medicinally and biochemically valuable in their own right. Halide substituents are amenable to SN2 displacement under a variety of conditions, leading to chiral alcohols, amines, amino acid derivatives, and sulfides. Asymmetric α-halogenations of carbonyl compounds are particularly appealing,85 and can be realized with a variety of substrates, catalysts, and halogenating agents.86 While diatomic halides are known to be highly reactive, they are generally non-selective species and prove to be incompatible with a variety of functional groups within complex molecules, as well as being unsuitable reagents for enantioselective reactions. In order to accomplish asymmetric halogenations of ketenes and derived enolates under suitable conditions, mild sources of electrophilic halogenating agents, tuned precisely to the reactivity of the system, are needed.
In 2001, Lectka became interested in the possibility of catalytic, enantioselective α-halogenation reactions and subsequently published a number of papers on the subject.87 Historically, stereoselective halogenations have been achieved using either chiral halogenating agents or substrates adorned with chiral auxiliaries.88 Today, however, much of the focus lies in developing catalytic, asymmetric methods for generating halogenated compounds. Ultimately, the design of a catalytic, enantioselective halogenation reaction would most effectively utilize a very mild source of halogen that could be easily tuned. The first pioneering step in this field was reported by Togni, who developed a halogenation procedure employing a titanium–TADDOL catalyst (74, Eq. 5.1).89 This protocol is only effective for the halogenation of α-alkyl-1,3-dicarbonyl compounds that can readily form titanium-based enolates in situ.
 |
(5.1) |
5.1. α-Chlorination of monosubstituted ketenes
After Togni's initial report, the catalytic, asymmetric chlorination of acid chlorides was reported by Lectka, who envisioned a process in which cinchona alkaloid derivatived ketene enolates react with a mild electrophilic halogen source in a tandem halogenation/esterification reaction (Scheme 5.1).87c The electrophilic halogen would add to the α-position of the ketene enolate, producing an acyl ammonium intermediate that subsequently transacylates with the leaving group of the electrophile. This sequence produces a versatile series of α-chloroesters (75) that serve as intermediates for the conversion to optically active amines, amides, ethers, and sulfides. Through the use of a selective halogenating reagent with a nucleophilic leaving group, this process could be adapted to react with ketene enolates enantioselectively.
Scheme 5.1.
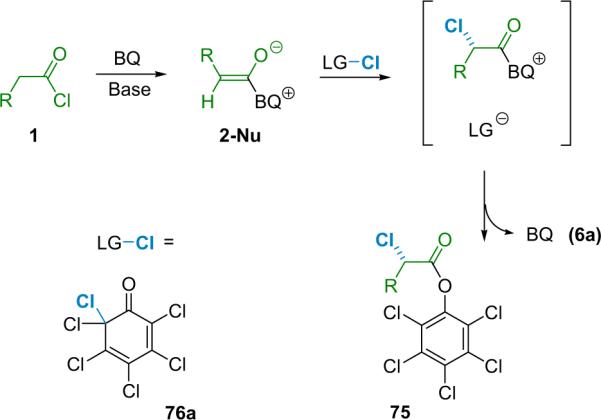
Tandem catalytic asymmetric chlorination/esterification.
The most important issue in the early stages concerned the choice of chlorinating agent. Diatomic chlorine was too reactive, whereas most other agents proved to be completely unreactive. Ironically, sources of halogen that are too mild pose the biggest problem, as they are unable to react with the weakly nucleophilic neutral zwitterionic enolate. It was clear that only a very narrow window of reactivity existed in which a desirable chlorinating agent (76) could operate selectively in a catalytic cycle. Nevertheless, Lectka began by screening sources of electrophilic halogen such as N-chlorosuccinimide (NCS), alkylhypochlorites,90 and various N-chloroamides, in a reaction with in situ generated phenylketene using BQ (6a) as a catalyst. For these tests, phenylacetyl chloride was added to a solution of 10 mol % BQ and 1.1 equiv of proton sponge at −78 °C to generate the ketene. N-Halosuccinimides and N-chloroamides were unsuccessful, as were a bevy of other candidates such as chlorinated pyridones, iodanes, and sulfonamides. On the other hand, tert-butylhypochlorite was too reactive, chlorinating almost anything in the reaction mixture, including solvent. Attention turned to polyhaloquinone-derived reagents such as 76a,91 which have a long history as often unanticipated byproducts of aromatic halogenation reactions. Hundreds of them are known in the literature; they are often easy to make (or in certain cases purchase). For example, pentachlorophenol reacts readily with tert-butylhypochlorite to produce quinone 76a in quantitative yield (Eq. 5.2).
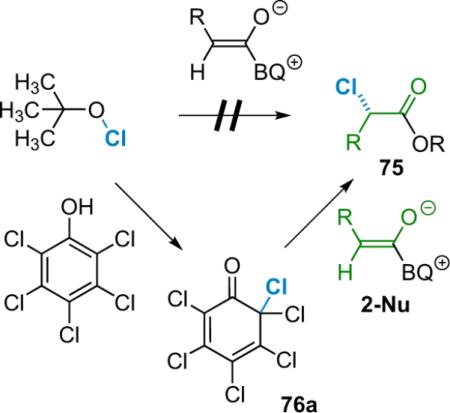 |
(5.2) |
Halogen transfers involving polyhaloquinones are expected to release a stabilized aromatic phenolate anion in a thermodynamically favorable process. This phenolate could then react with the resulting acyl ammonium salt (Scheme 5.1), regenerating the catalyst while generating the final product. Initial reactions of the perchlorinated quinone 76a used phenylacetyl chloride with 10 mol % BQ as catalyst and 1.1 equiv proton sponge. This process affords the corresponding α-chloroester 75 in moderate yield (40%) but with high enantioselectivity (95% ee). Also isolated from the reaction mixture, however, was a fair amount of the non-chlorinated ester, the product resulting from the alcoholysis of phenylketene (2d) by pentachlorophenol. Further investigation revealed that pentachlorophenol was being generated in situ by an undesired side reaction, namely the chlorination of electron rich proton sponge by quinone 76a (Scheme 5.2). The authors attempted to overcome this issue by using various halogenated versions of proton sponge as stoichiometric bases because they should be resistant to further halogenation.92 However, these deactivated bases afforded products in low yield, although the amount of undesired byproduct did drop considerably.
Scheme 5.2.
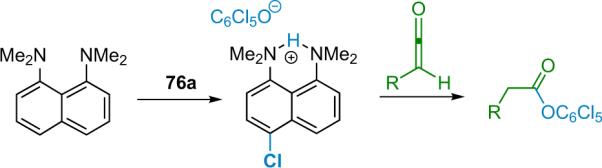
Ketene phenolysis with proton sponge.
In light of these difficulties Lectka decided to focus on pregeneration of ketene solutions with the polymer-bound phosphazene base BEMP.9 When a solution of phenylacetyl chloride in THF is passed through an addition funnel containing at least 1 equiv of BEMP at −78 °C, phenylketene is produced quantitatively.10c,d The ketene solution was allowed to drip slowly into a flask (−78 °C) containing BQ (6a, 10 mol %) and to this was added a solution of 76a. This process yields pentachlorophenyl α-chlorophenylacetate in 80% yield and 99% ee (entry 1, Table 5.1).87d A number of other acid chlorides were screened using this procedure and those results are summarized in Table 5.1.
Table 5.1.
α-Chlorination using BEMP as a dehydrohalogenating agent.
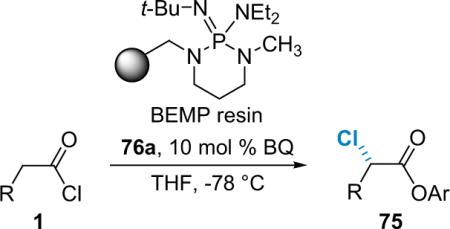
| Entry | R | ee (%) | Yield (%) |
|---|---|---|---|
| 1 | Ph | 99 | 80 |
| 2 | 2-Thiophene | 80 | 66 |
| 3 | Br | 97 | 51 |
| 4 | 1-Np | 95 | 57 |
| 5 | 2-Np | 94 | 63 |
| 6 | PhOCH2 | 97 | 57 |
While this procedure works well, it is somewhat tedious and BEMP resin is expensive. It was the attempt to overcome the need for the BEMP system that led to the discovery discussed in Section 1.3, namely that the shuttle deprotonation method can be performed with heterogeneous bases such as NaH. In this case, the ketene is generated through a system that employs BQ and 15-crown-5 as a phase-transfer catalyst. Phenylacetyl chloride (1d) is added to a stirred suspension of NaH and catalysts in THF at −78 −C. After a ketene pregeneration period, a solution of the chlorinating agent (76a) can then be added slowly to the reaction mixture. This way, the α-chlorinated product 75a is recovered in 63% yield and 95% ee.12b Importantly, the authors were able to apply this method to large scale reactions.
Although cost effective, carbonate salts and sodium hydride present some disadvantages as stoichiometric bases. For example, both require a ketene preformation step that can be difficult to manage; should the concentrations of ketene formed become too high, they are prone to dimerization and other side reactions. Clearly, a method that would involve very slow, `time-release' ketene or ketene enolate generation might be more appealing. It was this reasoning that led us to the use of sodium bicarbonate as a thermodynamic base. During optimization, the authors discovered that an excess of sodium bicarbonate (>10 equiv), in the presence of catalytic amounts of BQ and 15-crown-5 cocatalyst at −35 °C in chlorobenzene, afforded α-haloester 75a in 68% yield with 90% ee.
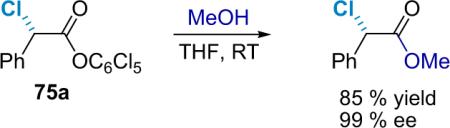
The fact that the products of the asymmetric halogenations are active esters allows facile derivatization to other esters and amides. Amidation (Eq. 5.3) and esterification (Eq. 5.4) of α-chloropentachlorophenyl esters proceeds well with primary amines and alcohols, affording products through room temperature reactions that have retained their optical purity. However, more vigorous conditions are required for amidation with secondary amines and anilines; slight racemization may result in these cases.
 |
(5.3) |
 |
(5.4) |
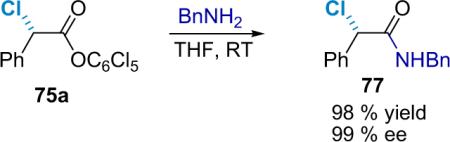
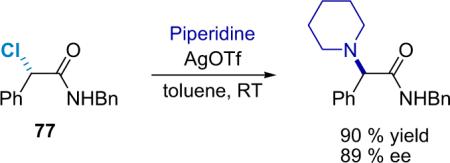
As α-chloro acid derivatives should serve as useful synthetic precursors to more complex molecules, a general method for the stereoselective displacement of the halide group is desirable. D'Angeli et al. have published a report on the stereoselective nucleophilic substitution of 2-bromoamides by amines in the presence of Ag+ or Ag2O.93 The authors observed full inversion of configuration in the presence of AgOTf or powdered Ag2O as a promoter under sonication. Taking their cue, the Lectka group applied a similar procedure to α-chloroamides. For example, when optically enriched amide 77 is mixed with AgOTf and an amine such as piperidine, the corresponding α-amino amide is produced in high yield with stereochemical inversion, although about 10% of optical purity was lost (Eq. 5.5).
 |
(5.5) |
5.2. α-Bromination of monosubstituted ketenes
While the α-chlorination method is effective, the synthetic utility of the product is somewhat limited by the aformentioned tendency toward racemization in some displacement reactions. This led to an expansion of the methodology to include alkyl bromides,94 which are generally believed to be the most synthetically useful of the halides as they often react through a pure SN2 mechanism during nucleophilic displacements under mild conditions.95 As the research progressed, it became clear that interesting mechanistic differences existed between the chlorination and bromination reactions. For example, the initial bromination conditions worked well on a small scale, but increasing the scale of the reaction resulted in exponential loss of yield and enantioselectivity. The chlorination reaction, on the other hand, displays no such erosion upon scale up. It became clear that in order to solve this scalability problem, a clear understanding of the mechanistic differences between these reactions was needed. A series of crossover experiments, ion-pairing tests, and kinetic resolution studies allowed the exploration of the mechanism of the α-bromination reaction in direct comparison to the asymmetric α-chlorination.
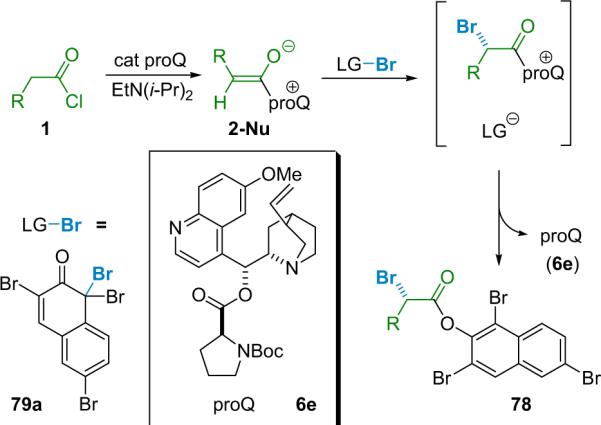
The postulated reaction sequence (Scheme 5.3) matches that for the α-chlorination reaction, proceeding from the acid chloride starting material (1) to a zwitterionic enolate (2-Nu) formed by the action of the nucleophilic catalyst (6e) and a stoichiometric base.86 The authors believe that this transient, chiral nucleophile abstracts electrophilic bromine from the brominating agent (79a), producing an ion-paired intermediate that undergoes transacylation to produce the desired product (78) and regenerate the catalyst for another cycle. The resulting optically enriched α-bromoesters are produced in high ee and moderate to good chemical yield.
Scheme 5.3.
Optimized catalytic, asymmetric bromination/esterification.
The initial bromination conditions were optimized to eliminate problems with loss of selectivity and yield upon scale up. These optimization procedures involved: (1) development, through de novo design, of a more selective catalyst, proQ (6e); (2) using ortho-brominating agent 79a (Scheme 5.3) instead of para-brominating agent 79b (Scheme 5.4); and (3) changing the stoichiometric base from heterogeneous carbonates to homogenous Hünig's base for aliphatic acid chlorides and solubilized NaH for arylacetyl chlorides.94a Catalyst design employed molecular mechanics (MM) calculations to guide substitution of the cinchona alkaloid core, a method that had served Lectka well in the past.15 Eventually it was discovered that esterifying quinine with N-Boc-L-proline gave a catalyst (proQ, 6e) for which MM calculations revealed a significant increase in the gap between the low energy re- and si-face exposed confirmations relative to BQ. This prediction of improved selectivity was confirmed experimentally as proQ gives an average of 10% increase in ee.94b
Scheme 5.4.
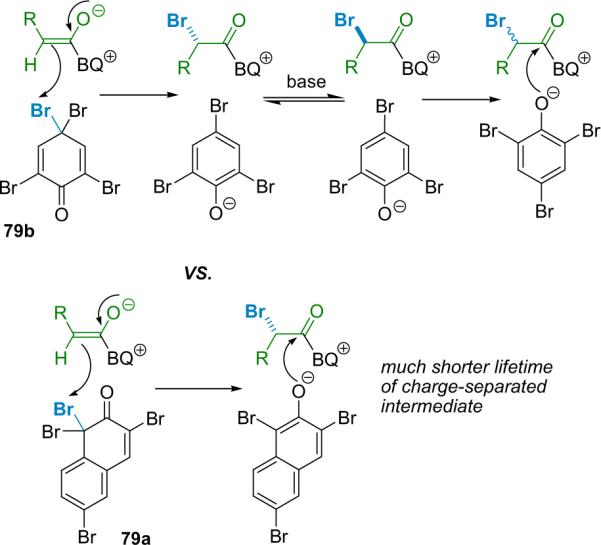
The halogenating agent's effect on intermediate lifetime.
Early experiments revealed that racemization was occurring during the reaction process. The species most susceptible to racemization is likely the acyl ammonium intermediate in the finite time prior to transacylation. Comparison of the initial α-bromination reaction to its analogous α-chlorination led to the realization that this intermediate may be longer-lived in the α-bromination reaction because the phenolate counterion of the para-brominating agent (79b) must `wheel around' to capture the acyl ammonium salt (Scheme 5.4). This led to the use of ortho-brominating agent 79a, which does improve selectivity for every substrate, especially in larger scale reactions.94b
Interesting mechanistic questions continued to come to light, not the least of which is exactly why this reaction differs so significantly from the corresponding α-chlorination. One intriguing issue concerns the extent that free phenolate ions dissociate from the acyl ammonium salt after the halogen transfer step. For example, conducting an asymmetric halogenation of phenylacetyl chloride (1d) with a 1:1 mixture of 76a and 79b yields products 75a and 78a exclusively at all levels of conversion (Scheme 5.5). In general, the brominating agent (79b) is about 10 times as reactive as the chlorinating agent (76a) in THF at −78 °C. No crossover products that would arise from phenolate/acyl ammonium salt dissociation were observed.
Scheme 5.5.
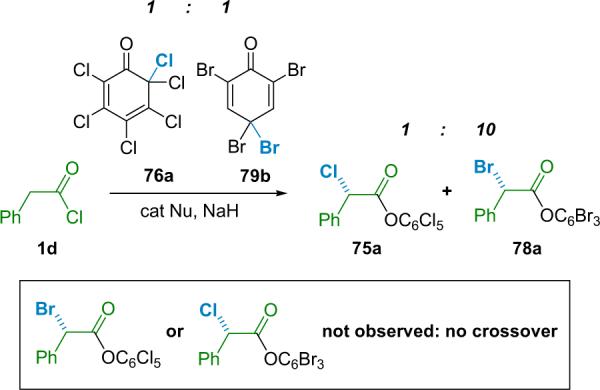
Crossover experiments.
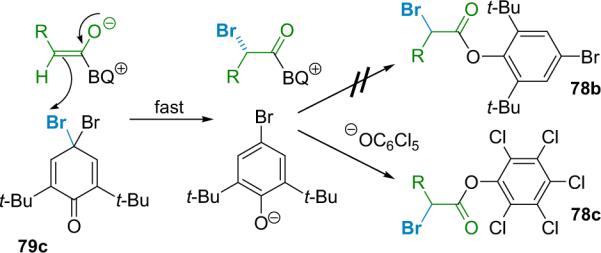
To address the role that ion pairing and/or dissociation plays in racemization, Lectka used another brominating agent derived from 2,6-di-tert-butylphenol, which should provide quick bromination and slow transacylation.96 This would lead to a long-lived ion pair intermediate (Scheme 5.6). However, brominating agent 79c provided little or no desired product (78b) in a standard reaction, but adding 1 equiv of sodium pentachlorophenolate produces a satisfactory yield of product 78c by promoting transacylation and catalyst turnover. This product was virtually racemic; the intervening time between bromination (by 79c) and transacylation is necessarily long compared to reactions that use brominating agent 79b, so racemization does occurs in the ion-paired intermediate. Control experiments revealed that sodium pentachlorophenolate neither catalyzes the reaction nor racemizes an optically pure version of the product.97
Scheme 5.6.
The effect of a hindered brominating reagent.
The displacement of bromine by a variety of nucleophiles provides a distinct advantage as the reaction often proceeds through a pure SN2 mechanism under mild conditions. A practical use for α-bromoester products would be the straightforward synthesis of optically active epoxides. Chiral epoxide chemistry has been a topic of intense interest for a generation, and some of the most notable advances in asymmetric catalysis have been made in this area.98 Several challenges remain; among the most notable is the catalytic, asymmetric synthesis of terminal epoxides, although Jacobsen has addressed the problem from one angle by a catalytic, asymmetric hydrolytic resolution of chiral epoxides.99
The strategy is simple—a reduction of the optically enriched α-bromoester is followed by base-promoted cyclization to the epoxide.86 As a proof of principle, this reaction sequence was performed on α-bromoester 78d. After conversion to the corresponding alcohol 80 by NaBH4, displacement of bromine, via an intramolecular cyclization to form the chiral epoxide 81, was accomplished by treatment of the alcohol with KOH under ambient conditions.100 The reaction proceeded in 80% yield and with full retention of enantioselectivity (Eq. 5.6).
 |
(5.6) |
5.3. α-Chlorination of disubstituted ketenes
Recently, Fu et al. published interesting results on the asymmetric halogenations of disubstituted ketenes.101 An initial screen showed that many commonly employed chlorinating agents (76) were unsatisfactory, though hexachloroacetone (76c) gave nearly complete conversion with promising enantioselectivity (Scheme 5.7). Reasoning that a conformationally constrained variant of 76c might provide improved selectivity, the Fu group turned to 2,2,6,6- tetrachlorocyclohexanone (76d) with excellent results. The use of this compound as a halogenating agent produces α-halogenated esters from disubstituted ketenes in good yield and moderate to high enantioselectivity.
Scheme 5.7.
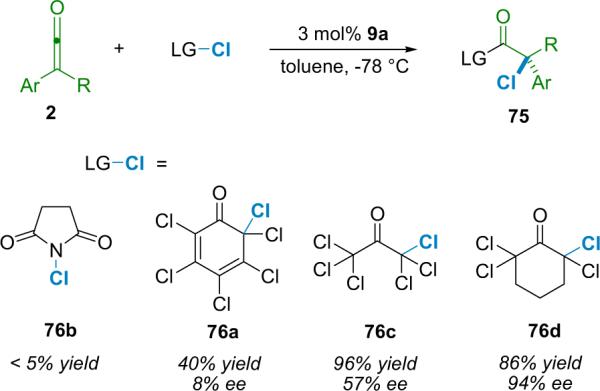
Selection of the chlorinating agent.
At this time, the mechanism of this reaction is still unclear. Fu favors two possible scenarios. The first is similar to the catalytic cycle proposed by Lectka (see Scheme 5.1); the reaction is initiated by catalyst attack on the ketene generating a chiral ketene enolate (2-Nu) that abstracts chlorine from the chlorinating agent (76d). The chlorinated intermediate is subsequently converted to the product (75) via catalyst displacement by the enol byproduct of chlorine abstraction from the chlorinating agent, simultaneously regenerating the catalyst for another cycle (scenario A, Scheme 5.8).
Scheme 5.8.

Possible catalytic cycles for the α-chlorination of disubstituted ketenes.
The second scenario Fu proposes is analogous to the cycle he has put forth for his catalyzed coupling of disubstituted ketenes with phenols (see Scheme 2.3). In this scenario, the catalyst initiates the reaction by direct attack on the chlorinating agent while the byproduct acylates the ketene, generating the enolate and a catalytic source of chiral, electrophilic chlorine. The enolate then abstracts Cl+ from the catalyst complex, simultaneously generating the desired product (75) and regenerating the catalyst for another cycle (scenario B, Scheme 5.8). A key component of the utility of α-haloesters is the flexibility of the ester functionality; the Fu group activates the enol ester with bromine, allowing the conversion to simple alcohols (82) or esters (75c), via a two-step processes in good yield and without loss of ee (Scheme 5.9).
Scheme 5.9.
Transformations of enol ester products.
5.4. α-Fluorination of dually activated ketenes
Ketene enolates have also been found by the Lectka group to react with an electrophilic source of fluorine, N-fluorodibenzenesulfonimide (NFSi, 83).87a Fluorinations of any kind have been highly sought after in the last decade, not least because the number of pharmaceutically active compounds or drugs on the market containing fluorine is growing rapidly every year. In addition, only a very small number of currently known natural products contain fluorine; by the year 1999, only about a dozen fluorinated natural products had been isolated 50 years after the discovery of the first.102 In the 10 years since, researchers have enthusiastically sought fluorinated natural products because of their medicinal promise, yet only a score more have been characterized; the number of fluorinated drugs on the market now exceeds the number of known fluorinated natural products by an order of magnitude103 if a fluorine is desired for a drug candidate, it generally must be installed synthetically. The selective installation of fluorine, particularly at chiral centers, is of crucial importance to medicinal chemistry. Because of the unique properties of the fluorine atom, based on its high electronegativity, small atomic radius, and high C–F bond strength, strategic fluorination can drastically alter the metabolism of a drug, its bioavailability, activity, or a host of other relevant properties.104 In any case, a general catalytic method for the asymmetric α-fluorination of carboxylic acid derivatives (85) is highly desirable.
Lectka's fluorination method is based on a dual-activation strategy where the ketene (generated in situ from acid chlorides) is activated by a chiral catalytic nucleophile (BQd) to make a nucleophilic ketene enolate, the reactivity of which is tuned by an achiral [d8] metal complex. This dual-activation strategy was investigated mechanistically in another reaction (see Section 6.1.2),105 but preliminary evidence suggests that the mechanism of PdII or NiII catalysis is the same in both systems, that is, by formation of a dually activated enolate complex (LA-2-Nu, Scheme 5.10).
Scheme 5.10.
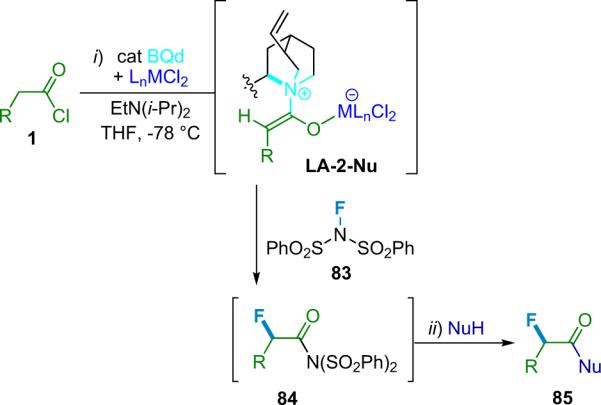
Fluorination of dually activated ketenes.
In the fluorination system, it seems that the fluorinated acyl ammonium intermediate is resolved by the bis-sulfonimide anion, regenerating the BQd and making the semi-stable intermediate 84. This intermediate was not isolable, but reacted smoothly in situ with any number of mild nucleophiles, serendipitously making this fluorination reaction far more versatile. Acid halides containing aromatic as well as heterocyclic substituents proved to be good substrates, producing products in very high ee and good to excellent yields (Fig. 5.1). Protected aminoalkyl-substituted acid chlorides also work well, leading to α-fluorinated β-amino acid derivatives. Other examples, such as the fluorination of an aldol adduct to form 85c shows that the reaction is also compatible with isomerizable carbon-carbon double bonds. A number of acid chloride and quenching nucleophile combinations were screened in this fluorination system, and products were consistently isolated in good yield and excellent ee.
Figure 5.1.
Selected fluorinated products.
The power of this new method is demonstrated in the broad range of different derivatives that can be synthesized by simple modification of the work up conditions; α-fluorinated carboxylic acids, amides, esters, and even peptides are all accessible depending on the nucleophile employed to quench the reaction. For example, a water work up affords α-fluoro carboxylic acids, compounds that potentially are of broad utility as derivatizing agents. Along with an alcohol quench that provides esters, an amine-based work up affords amides; accordingly, thioesters can be readily accessed. In a representative example, work up with L-phenylalanine ethyl ester produces the fluorinated peptide 85d in 68% yield and >99% diastereomeric excess (de). Either enantiomer at the fluorinated carbon can be produced in similarly high ee with the simple selection of BQ or BQd (for instance, 85b was produced by BQ while the rest of the products in Fig. 5.1 were made by BQd catalysis).
One of the best aspects of this method is the ease with which drugs, natural products, and other `exotic' nucleophiles can be functionalized, as the reaction initially provides chiral, α-fluorinated acylsulfonimides that can be quenched `in flask.' For example, the isoquinoline alkaloid106 natural product emetine can be used to quench the α-fluorination reaction of 3-phthalimidopropionyl chloride (1j) (91% yield, >99% de, Eq. 5.7). Given the variety of natural products that can be coupled with the chiral, fluorinated intermediates, large numbers of potentially useful α-fluorinated derivatives are accessible.
 |
(5.7) |
For a complimentary example of the utility of this α-fluorination method, the Lectka group demonstrated the derivatization of indomethacin (an anti-inflammatory drug, Eq. 5.8). Oxalyl chloride cleanly produces the acid chloride 86 from indomethacin, which is then subjected to standard reaction conditions (using trans-(PPh3)2PdCl2) and quenched with the appropriate nucleophile (85f is formed in 85% overall yield and 95% ee when NuH=MeOH). This process should be applicable to a vast array of biologically relevant carboxylic acid derivatives.
 |
(5.8) |
6. Asymmetric [4 + 2] cycloaddition reactions
6.1. o-Benzoquinone derivatives
Catalytic, enantioselective cycloaddition reactions of p-quinones are well documented.107 The products of these reactions are potentially very useful in the realm of natural product synthesis; however, until recently, little analogous work has been done on the catalytic, asymmetric reactions of o-quinones and derivatives. o-Quinones have a potentially much richer reactivity spectrum—dcycloaddition reactions may involve the ring carbons (as in the well-studied p-quinone isomers), but more interestingly, reactions may proceed through the o-heteroatoms to provide useful bicyclic products (Scheme 6.1).108 For example, the α-hydroxy esters obtained from o-quinones have found application as inhibitors of amyloid-β (Aβ) protein production, and thus show promise in the treatment of Alzheimer's disease.109 Chiral α-oxygenated carboxylic acid products can also be derivatized in a variety of useful ways. o-Quinone imides can provide easy access to invaluable, non-natural α-amino acids (both protected and unprotected), which find uses in all areas of biochemical study. Quinone imides also provide access to oxazinones and oxazines, bicyclic products that are useful as antitumor agents, topoisomerase inhibitors, and antibiotics.110 o-Benzoquinone diimides produce quinoxalinones and their quinoxaline derivatives,111 all of which exhibit a wide range of biological activity including antiviral effects, particularly against retroviruses such as HIV.112
Scheme 6.1.
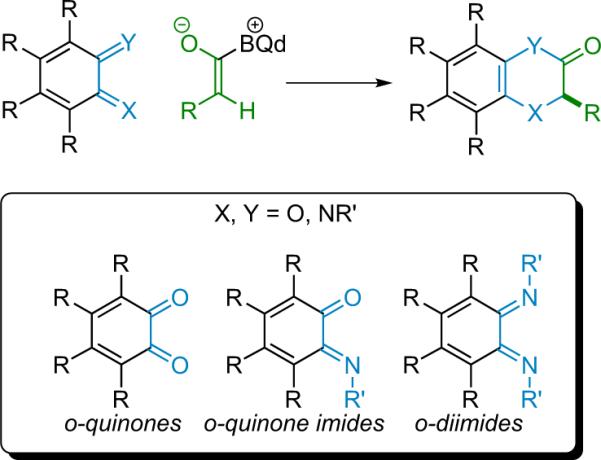
o-Benzoquinone derivatives in [4+2] cycloadditions with ketene enolate.
Lectka has recently discovered that catalytically generated ketene enolates can be used to initiate a cycloaddition reaction with a wide variety of o-benzoquinone derivatives to give [4+2] bicycloadducts in good to excellent yield and very good to þexcellent enantiomeric excess (up to >99% ee, Scheme 6.1). While the products of these cycloadditions are interesting and useful as chiral building blocks for organic synthesis, both they and their readily obtained derivatives show even greater utility and promise in a variety of physiological applications. These reactions are discussed in following subsections.
6.1.1. o-Quinone reactions
The redox chemistry of o-quinones has been extensively outlined.113 Many isolable o-quinones are known in the literature, and many more can be made through straightforward catechol and aminophenol based oxidations.114 Recently, Pettus et al. have reported a very useful synthesis of o-quinones from phenols using hypervalent iodine oxidants.115 In many instances o-quinones can be isolated, but they are also frequently used in situ. o-Quinone cycloadditions give rise to several different product classes, depending on the reaction partners.116 For example, reactions with acetylenes may afford bridged bicyclic adducts,117 whereas cycloadditions with nucleophilic alkenes, such as enol ethers and enamines, are anecdotally noted to proceed through the quinone oxygens.118 Since ketene enolates are somewhat similar in structure and reactivity to these substrates, they should react through oxygen atoms as well.
The initial screen by Lectka et al. employed o-chloranil (87a), butyryl chloride, and BQd (7b, 10 mol %) in the presence of Hünig's base (1 equiv) in THF at −78 °C.119 The yield of the reaction was an excellent 91% with 99% ee (88a, Scheme 6.2). Other o-quinone substrates were also screened, for instance o-bromanil (87b) was found to form product 88b in high ee (95%), and 90% yield. 9,10-Phenanthrenequinone (87c) was screened using similar conditions; its reactivity proved to be much lower than o-chloranil. However, when the reaction temperature was raised to 0 °C, reaction occurred sluggishly to afford product (88c) in reasonable yield. On the other hand, 4,5-dimethoxy-o-quinone failed to provide appreciable product under any conditions.
Scheme 6.2.
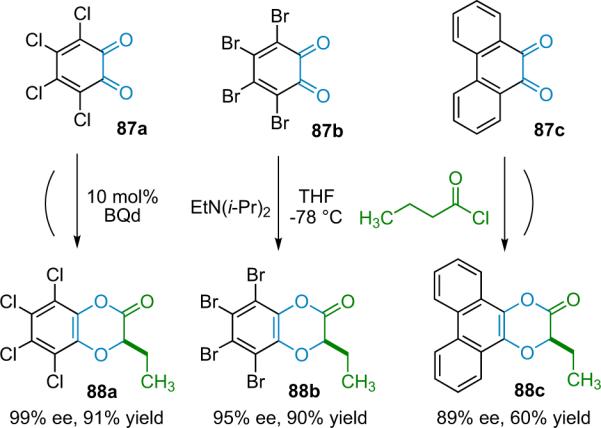
Preliminary o-quinone reactions.
Given the superiority of o-chloranil in the initial tests, the authors decided to investigate its reaction with a variety of acid chlorides. For example, dihydrocinnamoyl chloride performed well, affording cycloadduct 88e (Fig. 6.1) in high ee (99%). Additionally, α-arylacetyl chlorides proved to be excellent substrates. For example, phenylacetyl chloride generated product 88d in high yield (90%) and good ee (90%). Using BQd as catalyst, the (R)-enantiomer (assignment based on R priority as if aliphatic) formed preferentially, while the (S)-enantiomer can be obtained in similarly high enantioselectivity when BQ (6a) is used. This sense of induction held for all quinone derivative substrates, as well as all acid chlorides, and is consistent with other asymmetric reactions that have employed these cinchona alkaloid derivatives to catalytically generate ketene enolates.120
Figure 6.1.
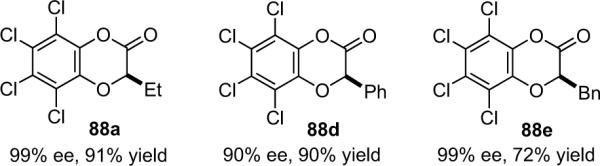
A selection of o-chloranil-derived cycloadducts.
The o-chloranil-derived cycloadducts can be further modified to produce chiral, α-oxygenated carboxylic acid derivatives (Scheme 6.3). For example, methanolysis of benzodioxinone 88d (Fig. 6.1) followed by ceric ammonium nitrate (CAN) oxidation affords (+)-methyl mandelate (90c, Fig. 6.2) in excellent yield (95%) and ee (90%). For maximum utility, reaction with the nucleophile (H2O, ROH, or RNH2) may occur in the same pot as the cycloaddition. Thus, the quinone can be thought of as a template for α-hydroxycarboxylic acid and ester synthesis. Several other cycloadducts were likewise converted to optically active α-hydroxy esters (Fig. 6.2). In each case, the reaction proceeds in high yield and with full retention of optical activity.
Scheme 6.3.
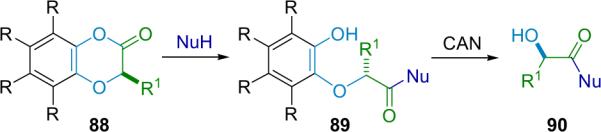
Derivatization of benzodioxinone cycloadducts.
Figure 6.2.
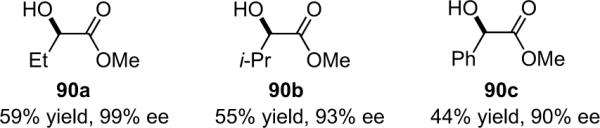
A selection of α-hydroxy esters, from o-chloranil and acid chlorides.
6.1.2. Bifunctional catalytic o-quinone reactions
Despite efforts to increase the yields of the α-hydroxy ester products by conventional means, Lectka's cycloaddition reaction simply proceeds sluggishly, resulting in lower than optimal yields. As he had success in the past with bifunctional catalytic systems, he turned once again to Lewis acid cocatalysis. From the group's earlier work on a bifunctional β-lactam forming system (see Section 4.2), and two other inverse-electron-demand hetero-Diels-Alder reactions of ketene enolates (discussed in Sections 6.1.3 and 6.1.4), each which favored a similar set of metal cocatalysts,120a the authors set out to use these bifunctional principals to add a productive metal cocatalyst to the o-quinone cycloaddition reaction (Fig. 6.3). In other words, they sought to increase the electrophilicity of the quinone by coordinating it with a Lewis acid, thereby amplifying its reactivity with the mildly nucleophilic ketene enolate.105 Unfortunately, this line of screening was met by frustration as oxophilic metal salts were unsuccessful. Surprisingly, it was azaphilic [d8] metal complexes that were able to provide increased rate and yield; bis-triphenylphosphine dichloride salts of nickel, palladium and platinum each provided increased yields of the cycloaddition reaction (and hence the conversion to α-hydroxy esters) up to a 60% increase caused by trans-(PPh3)2PdCl2, along with a corresponding twofold rate increase. Intrigued, Lectka set out to interpret this apparent divergence from the precedent bifunctional catalytic cycloaddition reactions (Scheme 6.3). FTIR and NMR data gave no evidence of coordination between any metal salt and the o-chloranil. It seems that classic Lewis acid action is not the way to understand palladium catalysis in this system, which in turn explains the mechanistic divergence from other analogous cycloaddition reactions.
Figure 6.3.
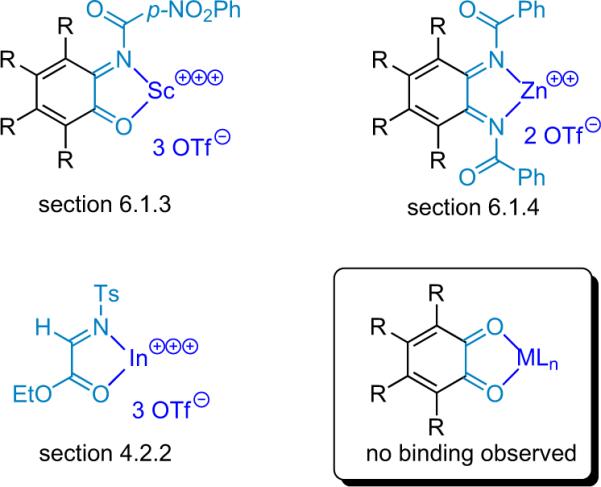
The Lewis acid in bifunctional catalytic systems.
Using density functional theory (DFT) to calculate the electrostatic charges at the carbonyl carbons for each putative structure of Figure 6.4, we find that ScIII (B) should withdraw electron density from the chloranil relative to bare chloranil (A). This means that if this coordination actually occurred, the ScIII would act as a classical Lewis acid, making the electrophilic quinone more reactive, and should therefore make the cycloaddition reaction faster. Since ScIII actually inhibits the reaction progress, we know that it does not bind in this manner, supporting the binding experiments mentioned earlier. The numbers for the two putative PdII bound structures (C) and (D) are more interesting. They show that if palladium were to bind to the chloranil in either a six- or four-coordinate fashion, it would not be productive; in both cases PdII would decrease the electrophilicity of the quinone.
Figure 6.4.

DFT (B3LYP/LANL2DZ) calculations of putative o-chloranil (87a) bound metal complexes.
These DFT calculations (along with several other calculations not discussed here, including transition state models) are in line with the spectroscopic data and support the conclusion that the palladium does not act by coordination to the quinone but must play some other role. After controlling for any possibility of Pd0 involvement, it seemed that the most likely remaining scenario involved PdII coordination to the ketene enolate (Eq. 6.1). Intuitively, it seems that this type of coordination would stabilize the ketene enolate rather than making it more nucleophilic. However, as the ketene enolate is only present in solution in equilibrium with free ketene, such stabilization would theoretically increase the concentration of reactive species, thereby increasing the reaction rate. An additional effect could also have to do with the reactivity/selectivity principle, where increased yields could also be explained by higher chemoselectivity—marked by fewer unproductive side reactions.
 |
(6.1) |
An experiment designed to shed light on this hypothesis necessitated the use of a more stable ketene; phenylethylketene (PEK, 2k) was chosen because it does not dimerize readily or participate in any other side reactions. In a UV–vis experiment at −78 °C, the amount of ketene enolate was increased by almost 40% when PdII was added to solution of PEK and BQ. With this data, backed by substantial theoretical calculation work, the authors proposed that the way to understand PdII cocatalysis in this cycloaddition reaction is through its binding with the ketene enolate, thereby increasing the reaction rate and chemoselectivity (Scheme 6.4). (After this mechanism was clarified, the Lectka group revisited the α-fluorination of acid chlorides, and obtained the results discussed in Section 5.4.)87a
Scheme 6.4.

Palladium(II) cocatalyzed cycloaddition reaction mechanism.
With this improved method for the synthesis of α-hydroxy acid derivatives in hand, the authors demonstrated the ability to access a variety of important, biologically relevant classes of compounds in high yield and enantioselectivity, by simply modifying the acid chloride. As an example, by using 4-chlorobutyryl chloride with BQ (6a), γ-lactam (S)-93 can be made in 82% yield (four steps) and >99% ee (Scheme 6.5). This intermediate is the key component in the synthesis of (l)-vasicinone, a drug that serves as a remedy for bronchitis and asthma, and its formation here constitutes a formal total synthesis of (S)-(−)-vasicinone, the most efficient to date.121 γ-Lactams are also known to be potent antibacterial agents,122 HIV protease inhibitors,123 and thrombin inhibitors.124
Scheme 6.5.

Synthesis of α-hydroxy γ-lactones and lactams.
Analogously, γ-lactones have been employed as novel antibacterial and antitumor agents.125 This structural unit frequently occurs in natural products including classes of alkaloids,126 macrocyclic antibiotics,127 and pheromones.128 This valuable entity can be made by using 4-benzyloxybutyryl chloride to produce γ-lactone 92 in 88% yield (from o-chloranil) and >99% enantiomeric excess (Scheme 6.5).
6.1.3. o-Quinone imide reactions
o-Benzoquinone imides (94) are intriguing for several reasons; the products of their cycloaddition with ketene enolates are chiral benzoxazinones (95) and benzoxazines (97, Scheme 6.6), interesting compounds with biological potential for which few asymmetric syntheses exist. For example, they have been investigated as antitumor agents, as selective inhibitors of drug metabolism and topoisomerase,110 and have been shown to possess promising neuroprotective antioxidant activity.129 Prescription drugs, such as the new generation antibiotic Levaquin (levofloxacin), for example, are also benzoxazines.110a Perhaps most importantly, o-benzoquinone imides can function as a scaffold for the synthesis of optically active non-natural α-amino acid derivatives (98, Scheme 6.6).
Scheme 6.6.
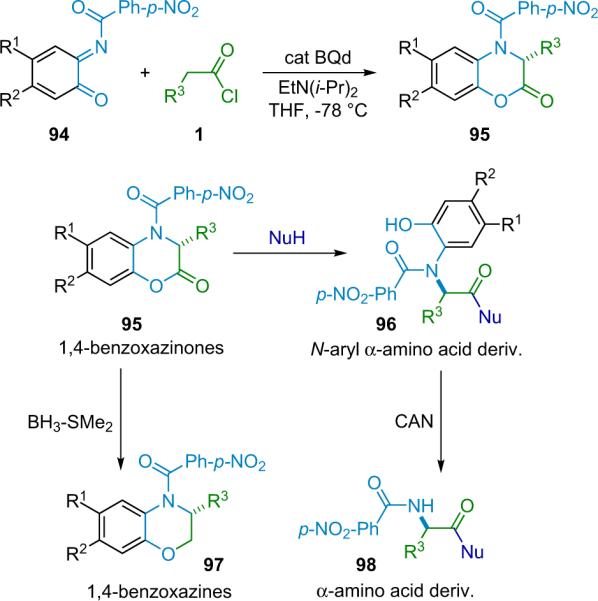
Products obtained from reactions of quinone imides.
The isolable o-benzoquinone imides are easily synthesized via PbIV oxidation of the corresponding 2-aminophenols. In a study published in 2001, Nicolaou explored the use of o-benzoquinone imides as Diels–Alder dienes, finding that they react through the N and O atoms, as desired.130 Importantly, their reactivity can be tuned by modifying the substituent on N. For example, an electron-withdrawing group (such as an acyl group) on the nitrogen should enhance the electrophilicity toward the enolate. Lectka chose to screen quinone imides derived from 2-amino-4,5-dichlorophenol131 (Fig. 6.5) as α-amino acid precursors for a variety of reasons—the starting material is inexpensive, it can be readily acylated with a virtually limitless variety of substituents, and is easily oxidized to the corresponding quinone imide. The chlorines in the 4- and 5- position are present to block undesired reactivity at the quinone ring, as well as to enhance the overall electrophilicity of the system.
Figure 6.5.
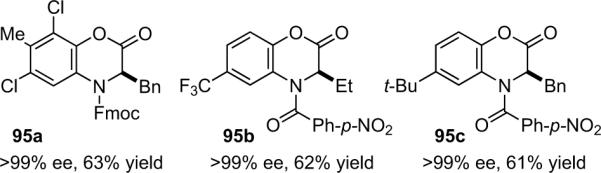
Benzoxazinone products.
The reaction of various combinations of acid chlorides and quinone imides, in the presence of 10 mol % BQd (7a) and Hünig's base in THF at −78 °C, forms the desired cycloadducts in fair yield and uniformly excellent ee (>99% ee in all cases, Fig. 6.5).132 The benzoxazinone cycloadducts are also readily converted into benzoxazines; a simple reduction with dimethyl sulfide–borane complex133 leads to the corresponding benzoxazine (Scheme 6.6) in good yield and with full retention of ee.
The efficient synthesis of non-natural α-amino acid derivatives has been a paramount goal of asymmetric catalysis for more than a generation. Many approaches to the synthesis of these compounds rely on metal-based catalysts, for example, asymmetric hydrogenation134 and Lewis acid catalyzed alkylations.135 The O'Donnell phase-transfer system is a good example of an `organocatalyzed' process.136 Another notable advance is represented by the asymmetric Strecker reaction that can be catalyzed by either metal-based or organic systems.137
Lectka has developed a complementary approach to the synthesis of α-amino acid derivatives that relies on an asymmetric cycloaddition of o-benzoquinone imides with chiral ketene enolates. The cycloadducts are derivatized in situ to provide the α-amino acid derivatives in high chemical yield, and very high enantioselectivity. As a testament to the flexibility of this method, a wide variety of R-substituents (derived from the acid chloride), N-acyl groups, and carboalkoxy groups can be incorporated into the optically enriched products (Figs. 6.6, 6.7, 6.8). The bicyclic products are activated esters and react smoothly with alcohols and amines to afford α-amino esters and amides (Fig. 6.6); this is accomplished by simply adding the desired nucleophile to the reaction pot once the cycloaddition is complete.
Figure 6.6.
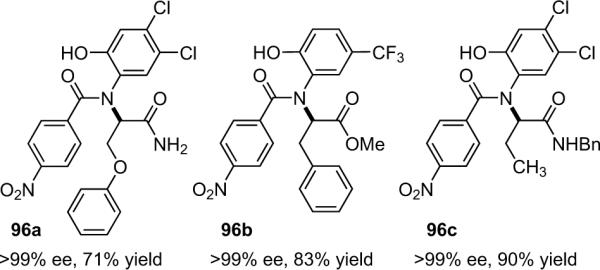
A selection of N-aryl α-amino acid derivatives.
Figure 6.7.
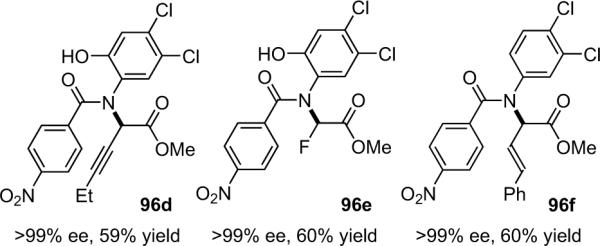
Highly biologically significant α-amino acid derivatives.
Figure 6.8.
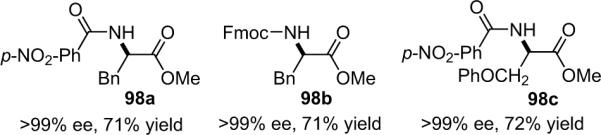
A selection of N-protected α-amino acid derivatives.
Among the amino acid derivative products, the most interesting are three biologically significant derivatives: β,γ-alkynyl amino acid (96d); α-fluoro amino acid (96e) and β,γ-alkenyl amino acid (96f, Fig. 6.7).138 These were produced in good yield and, once again, excellent ee. These three examples are of significance due not only to their high ee, but also to the difficulty of accessing them through other catalytic, asymmetric methods.139 Subsequent oxidation by CAN140 of the methyl esters affords α-amino acid derivatives, again without loss of ee (Fig. 6.8). The number of non-natural amino acid derivatives accessible by this method is considerable. In the case of N-Fmoc products, removal of the protecting group with piperidine followed by CAN oxidation leads to N-unprotected α-amino esters in good overall yield and with full retention of enantiomeric excess.
The yields for these cycloaddition reactions were good, but there was room for improvement. Reasoning that the sluggishness of the reaction may allow time for unwanted byproduct formation, the authors sought to further activate the quinone imides, through coordination with a Lewis acid.141 In this way, the reaction rate and subsequently the yield could be increased. o-Benzoquinone imides should be excellent candidates for Lewis acid activation,142 and they bear a striking resemblance to the imino esters employed in earlier bifunctional β-lactam forming reactions (see Section 4.2).
In screening metal triflates for their ability to increase the reaction rate and yield of the cycloaddition reaction, the authors found that, while both ZnII and InIII increased the yield, ScIII had the most pronounced effect. This bifunctional catalytic system (Scheme 6.7) improved the yield of every reaction, and, most importantly, did not degrade the enantioselectivity.141 Addition of a catalytic amount of scandium triflate to the reaction mixture provided up to a 42% increase in yield (Fig. 6.9).
Scheme 6.7.
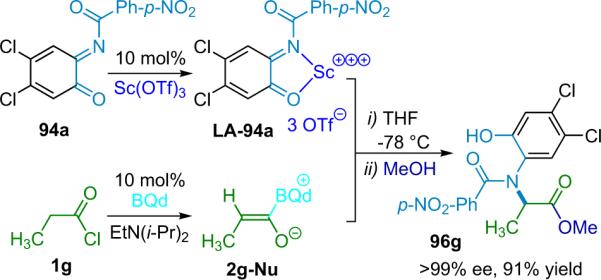
The bifunctional system to form α-amino acid derivatives.
Figure 6.9.
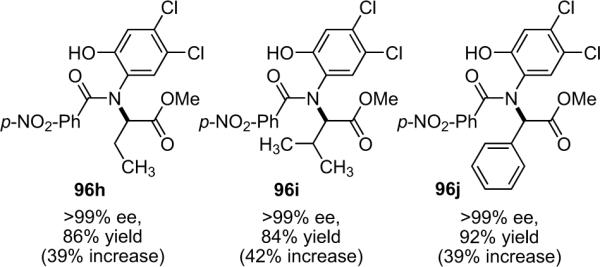
Examples of improved yield for the bifunctional system.
6.1.4. o-Quinone diimide reactions
Quinoxalinones (100), products of the Diels–Alder reaction between ketene enolates and o-benzoquinone diimides (99), are useful templates for drug development because of their structural relationship to benzodiazepines.143 These attractive targets exhibit a wide range of biological activity, including antidiabetic144 and antiviral effects,112 in particular against retroviruses such as HIV. In addition, they are partial agonists of the γ-aminobutyric acid (GABA)/benzodiazepine receptor complex,145 inhibitors of aldose reductase,146 and agonists of the AMPA and antiotensin II receptors.147 Related quinoxalines that are easily made from the respective quinoxalinones by a simple reduction also possess biological activity, for example, as inhibitors of cholesteryl ester transferase proteins.111
o-Benzoquinone diimides are easily prepared by PbIV oxidation of the respective 1,2-dianilides, as outlined by Adams and Way.148 Initially, the authors screened 4,5-dichloro diimide (99,R1=R2=Cl, Scheme 6.8) in a reaction with butyryl chloride, Hünig's base, and BQd at −78 °C in THF.149 The sluggish reaction formed several byproducts along with the desired cycloaddition product. Activation of the diimide by coordnation of a Lewis acid should render the system more electrophilic, thereby increasing the reaction rate; Lectka screened a number of metal triflates that were chosen based on his previous success with activation of imino esters.63b AlIII and InIII provided 64% and 65% yield, respectively, in the standard reaction (a large increase over the metal-free reaction), though ScIII and ZnII were far superior. For example, the initial rate of a standard reaction increased by over 2000% and cycloaddition product was isolated in 82% yield when 10 mol % Zn(OTf)2 was used as a cocatalyst. An IR experiment confirmed the notion that the Lewis acid operates through coordination with the diimide. Zn(OTf)2 was used as a cocatalyst in subsequent reactions, providing the bifunctional catalytic system depicted in Scheme 6.8.
Scheme 6.8.
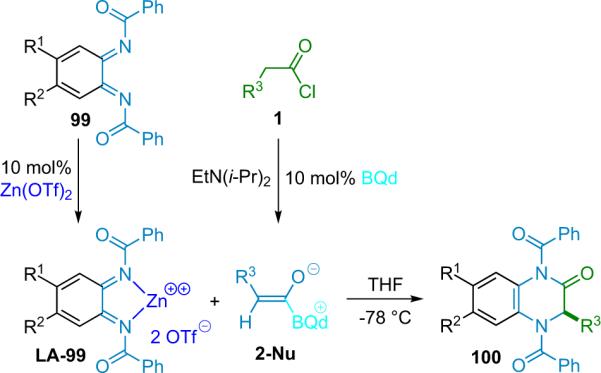
A bifunctional catalytic system to produce quinoxalinones.
This bifunctional catalytic methodology was thoroughly tested to determine its scope. A variety of substituted quinoxalinones were produced in good to excellent yield and all products were obtained in >99% ee (Fig. 6.10). In fact, the minor enantiomer was not detected by chiral phase HPLC in any chiral reaction, and for each reaction, only one regioisomer was obtained. The extreme selectivity consistently observed with this method was rationalized by the stepwise mechanism depicted in Scheme 6.9. The bifunctional reaction relies on quinone diimide activation by the Lewis acid (LA-99a) with concurrent nucleophilic activation of the ketene by BQd (2m-Nu). The regioselection comes from nucleophilic attack by the ketene enolate on more electrophilic nitrogen of the activated quinone diimide. Subsequent cyclizations of the stabilized intermediate provide the quinoxalinone product.
Figure 6.10.
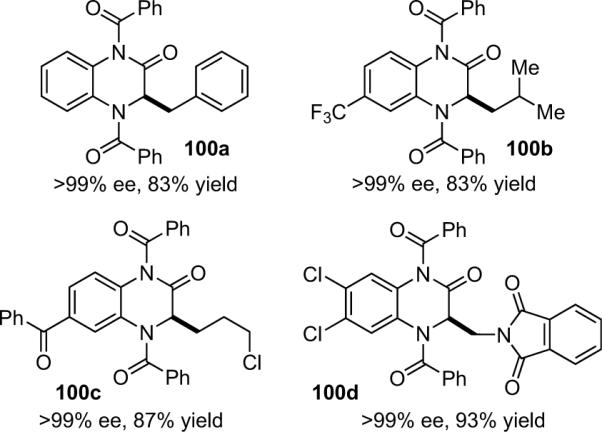
A sample of quinoxalinone products.
Scheme 6.9.
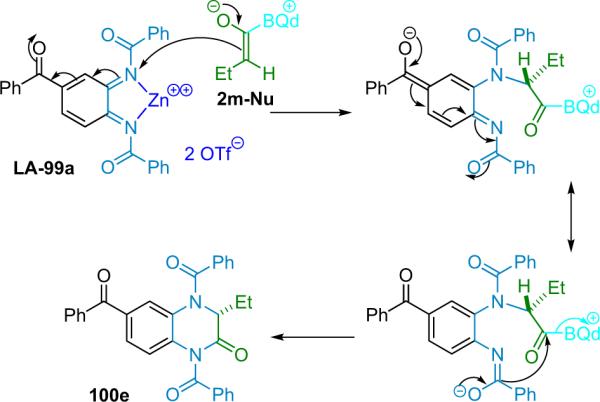
Proposed mechanism of regioselective cycloaddition.
The utility of this cycloaddition method was demonstrated through the synthesis of two drug targets. One compound, a quinoxalinone that exhibits strong activity against HIV (100f),112 was produced by the remarkably regioselective cycloaddition reaction between diimide 99b and 3-methylthiopropionyl chloride, mediated by cocatalysts BQ and Zn(OTf)2 (Scheme 6.10). This cycloaddition reaction gave the (R)-cycloadduct in >99% ee and moderate yield. Selective deprotection by TFA produces the target quinoxalinone 100f in 94% yield with full retention of ee. A second drug target, a quinoxaline that is known to inhibit cholesteryl ester transfer protein (101, Scheme 6.10),111 was produced by reduction of the cycloaddition product. Reduction by LiAlH4150 cleaves the benzoyl groups and simultaneously reduces the lactam carbonyl, affording the respective quinoxalines (101) in quantitative yield with full retention of ee.
Scheme 6.10.
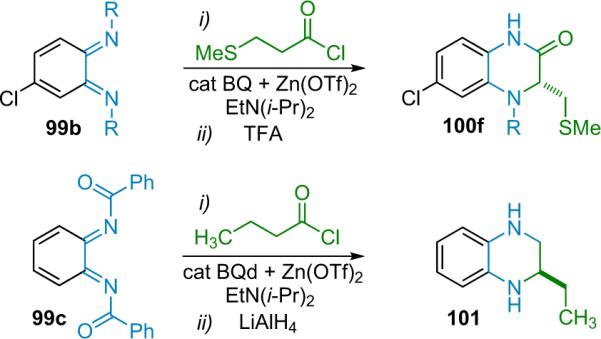
Drug target syntheses, R=CO2-i-Pr.
6.2. N-Thioacyl imine reactions
Nelson has successfully applied a bifunctional catalytic system using O-TMS quinine (6d) or quinidine (7d) and cocatalyst LiClO4 to the [4+2] cycloaddition reaction between N-thioacyl imines and ketenes.151 In this system, both the imine and the ketene are fomed in situ by deprotonation of the N-sulfinate mixed acetal and the acid chloride, respectively (Scheme 6.11). Nelson proposes that the Li+ acts to coordinate the transition state complex into a chair-like formation (103), enhancing the rate of reaction by drawing the reactive species together. A variety of cycloadducts (104) were formed, where R1=alkyl and R2=various aryl and alkyl groups, with consistently high ee (>98%) and dr (>97:3), and yields ranging from 51% to 76%.
Scheme 6.11.
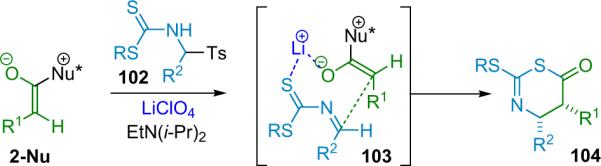
Nelson's bifunctional [4+2] cycloaddition reaction.
6.3. Formation of δ-lactones
The δ-lactone structure occurs in a wide range of natural products and other biologically active compounds. These subunits are found in antiviral and HIV protease inhibitors,152 anticancer compounds,153 and antibacterials.154 In addition, the majority of cholesterol lowering statin drugs like Zocor contain a β-hydroxy-δ-lactone structure, or its open form, for instance, Lipitor.155 Consequently, general catalytic methods to synthesize chiral δ-lactones are highly valuable. Only recently has catalytic asymmetric ketene chemistry been adapted to the formation of δ-lactones.
6.3.1. Conjugate addition/cyclization
Evans found that his Lewis acid catalyzed system for the [2+2] cycloaddition reaction between glyoxylate esters and silylketenes (see Scheme 3.7) was also amenable to δ-lactone formation.46 Using the same catalyst (107), trimethylsilylketene was reacted with unsaturated α-keto ester 105 to form δ-lactone 106 in high yield, ee, and dr (Scheme 6.12).
Scheme 6.12.
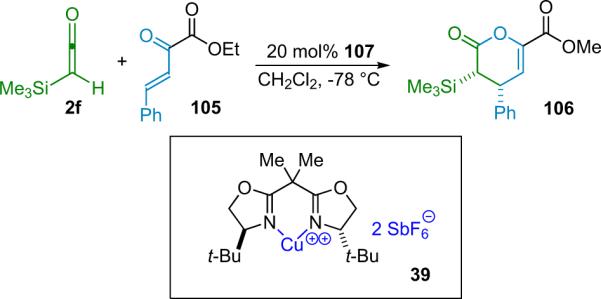
Evans's CuII catalyzed [4+2] cycloaddition reaction.
Ye et al. have recently used chiral N-heterocyclic carbene (NHC) catalysts to effect the reactions of disubstituted ketenes with a variety of electrophilic substrates (see Schemes 3.9, 3.13, and 4.10). The authors now report that δ-lactones (109) are selectively formed by NHC (44) catalysis of a disubstituted ketene cyclization reaction with α,β-unsaturated γ-ketoesters (108, Scheme 6.13).156 Interestingly, the trans-isomer is formed with high dr (16–40:1), but the fully epimerized cis-isomer can be obtained by deprotonation of the product by LDA and kinetically controlled reprotonation by aqueous HCl at −78 °C, usually in higher ee. There was only one regioisomer formed as well; this can be explained by nucleophilic attack by the catalyst-bound zwitterionic ketene enolate on the slightly more electrophilic carbon. The reaction works well for various aromatic and electron-withdrawn keto substitutents (R2, 108) as well as a variety of ketenes. Yields of the cycloaddition are moderate with good enantioselectivity, though excellent ee (95–99%) and dr (pure) were obtained for every product by recrystallization with consistently little or no loss of the desired enantiomer (50–70% overall yield).
Scheme 6.13.
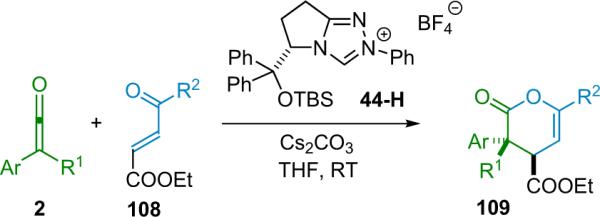
NHC catalyzed δ-lactone synthesis.
6.3.2. Ketene dienolates
Peters and Tiseni have developed a complimentary route to δ-lactones by utilizing ketene dienolates.157 Their dienolates are made in situ from β-methyl-α,β-unsaturated acid chlorides, purportedly through the unsaturated ketene (2n). Reaction with catalyst O-TMS-quinidine (TMS-Qd, 7d) forms the zwitterionic dienolate (2n-Nu), and the subsequent [4+2] cycloaddition reaction with Lewis acid activated trichloroacetaldehyde most likely takes place in the s-cis conformation of the dienolate (Scheme 6.14).157b The reaction works well with β-alkyl substitutents. Some β-(trialkylsilyl) substituted acid chlorides also work, improving the utility of the method, but these materials generally require a full equivalent of 7d to give satisfactory yield.
Scheme 6.14.
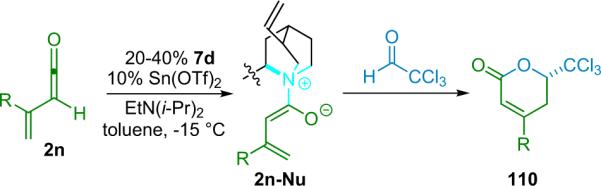
Activated aldehyde cycloaddition.
By modification of their system, the authors were able to extend the scope to include a variety of aryl aldehydes (25), as well as lower their catalyst loadings.157a The modified reaction uses a catalytic chiral vicinal amino alcohol (111) and excess Er(OTf)3 Lewis acid to effect the cycloaddition reaction (Scheme 6.15). In comparison to their previous system, the amino alcohol catalyzed reaction is much better in terms of materials loading and product scope, while raising the enantioselectivity (94% ee average). However, yields varied unpredictably—the 21 reported products were spread fairly evenly through the range of 23–91% yield.
Scheme 6.15.
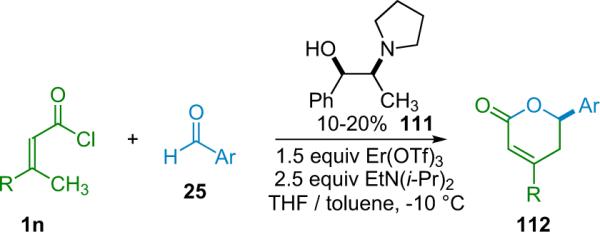
Amino alchohol catalyzed reaction.
Peters's proposed mechanism is similar to that proposed for the TMS-Qd catalyzed reaction, except that the authors do not assume that free ketene is an intermediate to the reaction. The major difference is that the amino alcohol binds to the ErIII metal in the presence of base, forming active catalyst LA-111 (Scheme 6.16). Subsequently, the ketene equivalent is inserted into the N–Er bond to make the active zwitterionic ketene dienolate (LA-2n-Nu). It is not clear whether the aldehyde binds to the ErIII before or after dienolate formation as shown, but the authors assume that all reactants are bound and organized by ErIII into a termolecular complex (113) prior to the C–C bond forming step.
Scheme 6.16.
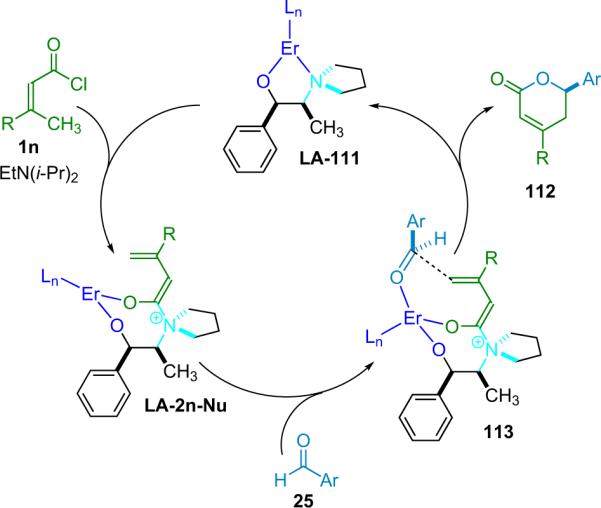
Proposed mechanism of δ-lactone formation.
The authors also demonstrate several derivativization schemes for their trichloromethyl δ-lactones (110).157b The trichloromethyl group can be reduced selectively by tetrabutyltin hydride, forming either 110b or 110c by controlling the stoichiometry and temperature (Scheme 6.17). These products can be derivatized in a number of ways; for example, basic hydrolysis of the monochloride 110c to the primary alcohol 10e proceeds without racemization. In addition, LiOH hydrolysis of the trichloride 110a at 60 °C produces the corresponding acid (110d) with only minor loss in optical purity. This reaction proceeds with inversion of stereochemistry; formation of an intermediate dichloroepoxide is implicated in the inverted hydrolysis.158
Scheme 6.17.
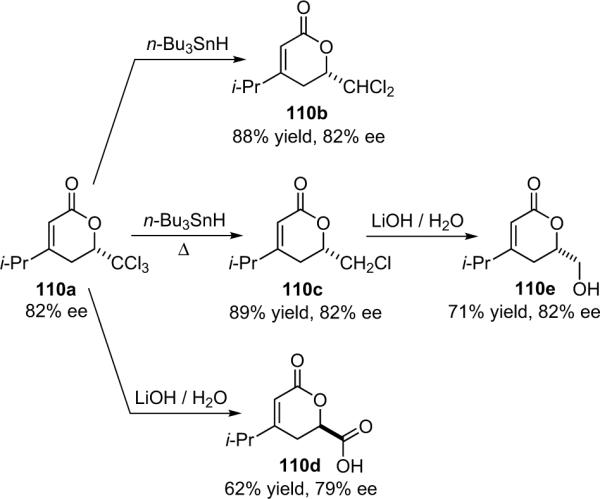
Simple δ-lactone derivatization.
7. Olefination of ketenes
Tang et al. have discovered a catalytic, asymmetric way to produce allenes from ketenes and ethyl diazoacetate.159 Their method does rely on a full equivalent of chiral phosphine, but they recover, reduce, and reuse 85% of the phosphine without significant effect on the reaction. In this chiral system, catalytic tetra(p-chlorophenyl)porphyrin iron chloride (116) activates the diazoacetate (114), forming an FeII carbene complex (Scheme 7.1). This activated complex transfers the carbene-type ligand to the chiral phosphine (115) to form the respective ylide, which undergoes a chiral Wittig substitution with the disubstituted ketene. The authors note that this mechanism is supported by the fact that the allene ester products (117) were consistently obtained in high ee (93–98%) when they run the reaction sequentially.
Scheme 7.1.
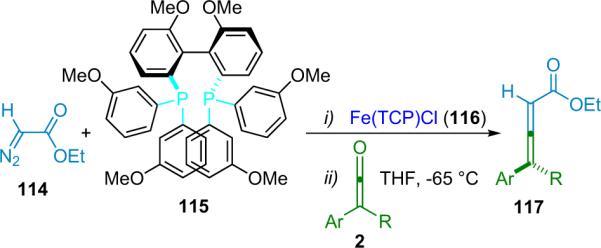
Synthesis of chiral allenes.
8. Conclusion
Ketenes and their catalytic enolate derivatives have an incredibly rich reactivity spectrum, and many of their products can be obtained with high enantioselectivity. In some cases, enantioselective reactions have been catalyzed by chiral Lewis acids, chiral nucleophiles including NHCs, cinchona alkaloid derivatives, and chiral DMAP derivatives, as well as various bifunctional combinations of organic and metallic catalysts. We have introduced several distinct reaction manifolds, including SN2 type substitution reactions (Section 5.4), [2+2] cycloaddition reactions (Sections 3 and 4), pseudo [4+2] pericyclic reaction patterns (Sections 5.1–5.3), and actual [4+2] cycloaddition reactions (Section 6), as well as several different types of bifunctional catalytic reaction activation by Lewis acid/base pairs. In almost all cases, ketene-derived products are synthetically and biologically useful, and usually difficult to synthesize by other means. In summary, harnessing the power of ketenes and ketene enolates has lead to a large (and growing) number of novel and practical catalytic, asymmetric reactions.
Acknowledgments
T.L. thanks the NIH (GM064559), Merck and Co., the Sloan, Dreyfus, and John Simon Guggenheim Memorial Foundations for support. D.H.P. thanks Johns Hopkins for a Zeltmann Fellowship.
Biographies

Daniel H. Paull received a B.S. from Rhodes College in 2003. He joined Professor Lectka's research group in 2004, where he investigated asymmetric fluorination reactions and obtained his Ph.D. in the Spring of 2009. He will be a postdoc in the laboratory of Stephen Martin at the University of Texas at Austin from September 2009.

Dr. Anthony Weatherwax was born in Los Angeles, CA and grew up in New York. He holds bachelor's degrees in anthropology (1993, SUNY Albany), art history (1998, University of Arizona), and chemistry (2001, University of Maryland). He joined the Lectka group in the Fall of 2001, received his Ph.D. in 2007, and since then he has been a postdoc in the labs of Andreas Pfaltz in Basel.

Thomas Lectka is a native of Detroit, MI. He obtained his B.A. from Oberlin College and his Ph.D. from Cornell University, where he worked in John McMurry's laboratory. After postdocs at Heidelberg with Rolf Gleiter and Harvard with Dave Evans, he began at Johns Hopkins University in 1994, where he was promoted to Professor in 2002. His research interests broadly span problems in synthetic methods and catalysis.
References and notes
- 1.Staudinger H. Chem. Ber. 1905;38:1735–1739. [Google Scholar]
- 2.For the history of ketenes and reviews, see: Tidwell TT. Ketenes. John Wiley & Sons; Hoboken, NJ: 2006. ; Tidwell TT. Eur. J. Org. Chem. 2006:563–576.; Tidwell TT. Angew. Chem., Int. Ed. 2005;44:5778–5785. doi: 10.1002/anie.200500098.; Temperley CM. Ketenes, their Cumulene Analogues, and their S, Se, and Te Analogues. In: Katritzky AR, Taylor RJK, editors. Comprehensive Organic Functional Group Transformations II. vol. 3. Elsevier; Amsterdam: 2005. pp. 573–603.; Miller R, Abaecherli C, Said A. Ketenes. 6th ed. Vol. A 15. Ullman's Encyclopedia of Industrial Chemistry; 2002. ; Orr RK, Calter MA. Tetrahedron. 2003;59:3545–3565.; Hyatt JA, Raynolds PW. Org. React. 1994;45:159–646.
- 3.For a more involved discussion and leading references, see Ref. 2a.
- 4.Moore HW, Wilbur DS. J. Org. Chem. 1980;45:4483–4491. [Google Scholar]
- 5.Kresze G, Runge W, Ruch E. Justus Liebigs Ann. Chem. 1972;756:112–127. [Google Scholar]
- 6.(a) Newman MS, Arkell A, Fukunaga T. J. Am. Chem. Soc. 1960;82:2498–2501. [Google Scholar]; (b) Olah GA, Wu A, Farooq O. Synthesis. 1989:568. [Google Scholar]
- 7.(a) Chiang Y, Kresge AJ, Popik VV. J. Am. Chem. Soc. 1999;121:5930–5932. [Google Scholar]; (b) Wagner BD, Arnold BR, Brown GS, Lusztyk J. J. Am. Chem. Soc. 1998;120:1827–1834. [Google Scholar]
- 8.Hegedus has found that trialkylammonium salts can interfere with the diastereoselectivity of the Staudinger reaction to produce β-lactams: Hegedus LS, Montgomery J, Narukawa Y, Snustad DC. J. Am. Chem. Soc. 1991;113:5784–5791.
- 9.BEMP: 2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazophosphorine.
- 10.(a) France S, Weatherwax A, Taggi AE, Lectka T. Acc. Chem. Res. 2004;37:592–600. doi: 10.1021/ar030055g. [DOI] [PubMed] [Google Scholar]; (b) Hafez AM, Taggi AE, Lectka T. Chem.—Eur. J. 2002;8:4114–4119. doi: 10.1002/1521-3765(20020916)8:18<4114::AID-CHEM4114>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]; (c) Hafez AM, Taggi AE, Dudding T, Lectka T. J. Am. Chem. Soc. 2001;123:10853–10859. doi: 10.1021/ja016556j. [DOI] [PubMed] [Google Scholar]; (d) Hafez AM, Taggi AE, Wack H, Drury WJ, III, Lectka T. Org. Lett. 2000;2:3963–3965. doi: 10.1021/ol006659r. [DOI] [PubMed] [Google Scholar]
- 11.Taggi AE, Hafez AM, Wack H, Young B, Drury WJ, III, Lectka T. J. Am. Chem. Soc. 2000;122:7831–7832. [Google Scholar]
- 12.(a) Hafez AM, Taggi AE, Wack H, Esterbrook J, Lectka T. Org. Lett. 2001;3:2049–2051. doi: 10.1021/ol0160147. [DOI] [PubMed] [Google Scholar]; (b) Taggi AE, Wack H, Hafez AM, France S. Org. Lett. 2002;4:627–629. doi: 10.1021/ol0172525. [DOI] [PubMed] [Google Scholar]
- 13.(a) Shah MH, France S, Lectka T. Synlett. 2003:1937–1939. [Google Scholar]; (b) Nelson A. Angew. Chem., Int. Ed. 1999;38:1583–1585. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1583::AID-ANIE1583>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]; (c) Tanabe Y, Yamamoto H, Yoshida Y, Miyawaki T, Utsumi N. Bull. Chem. Soc. Jpn. 1995;68:297–300. [Google Scholar]; (d) Sasson Y, Bilman N. J. Chem. Soc., Perkin Trans.2. 1989:2029–2033. Also see Ref. 12a. [Google Scholar]
- 14.For recent review, see: Tian S-K, Chen Y, Hang J, Tang L, McDaid P, Deng L. Acc. Chem. Res. 2004;37:621–631. doi: 10.1021/ar030048s.
- 15.Taggi AE, Hafez AM, Dudding T, Lectka T. Tetrahedron. 2002;58:8351–8356. [Google Scholar]
- 16.Pracejus H. Justus Liebigs Ann. Chem. 1960;634:9–22. [Google Scholar]
- 17.(a) Pracejus H, Kohl G. Justus Liebigs Ann. Chem. 1969;722:1–11. [Google Scholar]; (b) Pracejus H. Tetrahedron Lett. 1966:3809–3813. [Google Scholar]; (c) Pracejus H, Tille A. Chem. Ber. 1963;96:854–865. [Google Scholar]
- 18.Calmes M, Daunis J, Mai N. Tetrahedron. 1997;53:13719–13726. [Google Scholar]
- 19.Hodous BL, Ruble JC, Fu GC. J. Am. Chem. Soc. 1999;121:2637–2638. [Google Scholar]
- 20.Fu GC. Acc. Chem. Res. 2004;37:542–547. doi: 10.1021/ar030051b. [DOI] [PubMed] [Google Scholar]
- 21.Wiskur SL, Fu GC. J. Am. Chem. Soc. 2005;127:6176–6177. doi: 10.1021/ja0506152. [DOI] [PubMed] [Google Scholar]
- 22.Schaefer C, Fu GC. Angew. Chem., Int. Ed. 2005;44:4606–4608. doi: 10.1002/anie.200501434. [DOI] [PubMed] [Google Scholar]
- 23.Hodous BL, Fu GC. J. Am. Chem. Soc. 2002;124:10006–10007. doi: 10.1021/ja027466x. [DOI] [PubMed] [Google Scholar]
- 24.Pracejus H. Justus Liebigs Ann. Chem. 1960;634:23–29. [Google Scholar]
- 25.Dai X, Nakai T, Romero JAC, Fu GC. Angew. Chem., Int. Ed. 2007;46:4367–4369. doi: 10.1002/anie.200700697. [DOI] [PubMed] [Google Scholar]
- 26.The product of the reaction between benzyl alcohol and the isocyanate produced from phenyl isopropyl ketene, which was obtained in 92% yield and 95% ee, was also reported.
- 27.For a recent review and leading references, see: Yang HW, Romo D. Tetrahedron. 1999;55:6403–6434.
- 28.Staudinger H, Bereza S. Justus Liebigs Ann. Chem. 1911;380:243–247. [Google Scholar]
- 29.(a) Wynberg H, Staring EGJ. J. Org. Chem. 1985;50:1977–1979. [Google Scholar]; (b) Wynberg H, Staring EGJ. J. Am. Chem. Soc. 1982;104:166–168. [Google Scholar]
- 30.In their original publication, the (S)-enantiomer was assigned to the lactone product, but the authors subsequently demonstrated that inversion occurs during the basic hydrolysis, which proceeds through a dichloroepoxide intermediate with full inversion of stereochemistry upon opening. See: Wynberg H, Staring EGJ. J. Chem. Soc., Chem. Commun. 1984:1181–1182.
- 31.Tennyson R, Romo D. J. Org. Chem. 2000;65:7248–7252. doi: 10.1021/jo001010l. [DOI] [PubMed] [Google Scholar]
- 32.Zhu C, Shen X, Nelson SG. J. Am. Chem. Soc. 2004;126:5352–5353. doi: 10.1021/ja0492900. [DOI] [PubMed] [Google Scholar]
- 33.Calter MA, Tretyak OA, Flaschenriem C. Org. Lett. 2005;7:1809–1812. doi: 10.1021/ol050411q. [DOI] [PubMed] [Google Scholar]
- 34.Lin Y-M, Boucau J, Li Z, Casarotto V, Lin J, Nguyen AN, Ehrmantraut J. Org. Lett. 2007;9:567–570. doi: 10.1021/ol0626903. [DOI] [PubMed] [Google Scholar]
- 35.Tamai Y, Someya M, Fukumoto J, Miyano S. J. Chem. Soc., Perkin Trans. 1. 1994:1549–1550. [Google Scholar]
- 36.Tamai Y, Yoshiwara H, Someya M, Fukumoto J, Miyano S. J. Chem. Soc., Chem. Commum. 1994:2281–2282. [Google Scholar]
- 37.Dymock WB, Kocienski PJ, Pons J-M. Chem. Commun. 1996:1053–1054. [Google Scholar]
- 38.Zemribo R, Romo D. Tetrahedron Lett. 1995;36:4159–4162. [Google Scholar]
- 39.(a) Yang HW, Romo D. Tetrahedron Lett. 1998;39:2877–2880. [Google Scholar]; (b) Romo D, Harrison PHM, Jenkins SI, Riddoch RW, Park K, Yang HW, Zhao C, Wright GD. Bioorg. Med. Chem. 1998;6:1255–1272. doi: 10.1016/s0968-0896(98)00114-x. [DOI] [PubMed] [Google Scholar]
- 40.Nelson SG, Wan Z, Peelen TJ, Spencer KL. Tetrahedron Lett. 1999;40:6535–6539. [Google Scholar]
- 41.Nelson SG, Spencer KL. J. Org. Chem. 2000;65:1227–1230. doi: 10.1021/jo9913412. [DOI] [PubMed] [Google Scholar]
- 42.Nelson SG, Peelen TJ, Wan Z. J. Am. Chem. Soc. 1999;121:9742–9743. [Google Scholar]
- 43.Nelson SG, Wan Z. Org. Lett. 2000;2:1883–1886. doi: 10.1021/ol005968e. [DOI] [PubMed] [Google Scholar]
- 44.Nelson SG, Wan Z, Stan MA. J. Org. Chem. 2002;67:4680–4683. doi: 10.1021/jo025519n. [DOI] [PubMed] [Google Scholar]
- 45.Nelson SG, Spencer KL. Angew. Chem., Int. Ed. 2000;39:1323–1325. doi: 10.1002/(sici)1521-3773(20000403)39:7<1323::aid-anie1323>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 46.Evans DA, Janey JM. Org. Lett. 2001;3:2125–2128. doi: 10.1021/ol016096z. [DOI] [PubMed] [Google Scholar]
- 47.Gnanadesikan V, Corey EJ. Org. Lett. 2006;8:4943–4945. doi: 10.1021/ol061979h. [DOI] [PubMed] [Google Scholar]
- 48.Wilson JE, Fu GC. Angew. Chem., Int. Ed. 2004;43:6358–6360. doi: 10.1002/anie.200460698. [DOI] [PubMed] [Google Scholar]
- 49.He L, Lv H, Zhang Y-R, Ye S. J. Org. Chem. 2008;73:8101–8103. doi: 10.1021/jo801494f. [DOI] [PubMed] [Google Scholar]
- 50.(a) Calter MA, Liao W. J. Am. Chem. Soc. 2002;124:13127–13129. doi: 10.1021/ja027675h. [DOI] [PubMed] [Google Scholar]; (b) Calter MA, Liao W, Struss JA. J. Org. Chem. 2001;66:7500–7504. doi: 10.1021/jo0160367. [DOI] [PubMed] [Google Scholar]; (c) Calter MA, Guo X. J. Org. Chem. 1998;63:5308–5309. [Google Scholar]
- 51.Calter MA. J. Org. Chem. 1996;61:8006–8007. doi: 10.1021/jo961721c. [DOI] [PubMed] [Google Scholar]
- 52.Calter M, Guo X, Liao W. Org. Lett. 2001;3:1499–1501. doi: 10.1021/ol015814e. [DOI] [PubMed] [Google Scholar]
- 53.Lv H, Zhang Y-R, Huang X-L, Ye S. Adv. Synth. Catal. 2008;350:2715–2718. [Google Scholar]
- 54.Calter MA, Orr RK, Song W. Org. Lett. 2003;5:4745–4748. doi: 10.1021/ol0359517. [DOI] [PubMed] [Google Scholar]
- 55.Duffy RJ, Morris KA, Romo D. J. Am. Chem. Soc. 2005;127:16754–16755. doi: 10.1021/ja053478h. [DOI] [PubMed] [Google Scholar]
- 56.Purohit VC, Richardson RD, Smith JW, Romo D. J. Org. Chem. 2006;71:4549–4558. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]
- 57.Page MI, editor. The Chemistry of β-Lactams. Chapman Hall; New York, NY: 1992. [Google Scholar]
- 58.Staudinger H. Liebigs Ann. Chem. Vol. 356. 1907. pp. 51–123.; Palomo C, Aizpurua JM, Ganboa I, Oiarbide M. Eur. J. Org. Chem. 1999. pp. 3223–3235.; For recent article on the selectivity of the Staudinger reaction, see: Liang Y, Jiao L, Zhang S, Yu Z-X, Xu J. J. Am. Chem. Soc. Vol. 131. 2009. pp. 1542–1549.
- 59.(a) Gilman H, Speeter M. J. Am. Chem. Soc. 1943;65:2255–2256. [Google Scholar]; (b) Hart DJ, Ha D-C. Chem. Rev. 1989;89:1447–1465. [Google Scholar]; (c) Benaglia M, Cinquini M, Cozzi F. Eur. J. Org. Chem. 2000:563–572. [Google Scholar]
- 60.Toda F, Miyamoto H, Inoue M, Yasaka S, Matijasic I. J. Org. Chem. 2000;65:2728–2732. doi: 10.1021/jo991832m. [DOI] [PubMed] [Google Scholar]
- 61.Ishibashi H, Kameoka C, Kodama K, Ikeda M. Tetrahedron. 1996;52:489–502. [Google Scholar]
- 62.For a review on asymmetric ketene chemistry with recent information on β-lactam synthesis with auxiliaries, see Ref. 2f. Also see: Bose AK, Manhas MS, van der Veen JM, Bari SS, Wagle DR. Tetrahedron. 1992;48:4831–4844.; Evans DA, Sjogren EB. Tetrahedron Lett. 1985;26:3783–3786.
- 63.(a) Taggi AE, Hafez AM, Lectka T. Acc. Chem. Res. 2003;36:10–19. doi: 10.1021/ar020137p. [DOI] [PubMed] [Google Scholar]; (b) Ferraris D, Young B, Cox C, Dudding T, Drury WJ, III, Ryzhkov L, Taggi AE, Lectka T. J. Am. Chem. Soc. 2002;124:67–77. doi: 10.1021/ja016838j. [DOI] [PubMed] [Google Scholar]
- 64.Georg GI, editor. The Organic Chemistry of β-Lactams. VCH; New York, NY: 1993. and references therein. [Google Scholar]
- 65.Wack H, Drury WJ, III, Taggi AE, Ferraris D, Lectka T. Org. Lett. 1999;1:1985–1988. doi: 10.1021/ol9903234.; For examples of diastereoselective synthesis of trans-β-lactams, see: Abraham CJ, Paull DH, Dogo-Isonagie C, Lectka T. Synlett. 2009;10:1651–1654. doi: 10.1055/s-0029-1217348.; Weatherwax A, Abraham CJ, Lectka T. Org. Lett. 2005;7:3461–3463. doi: 10.1021/ol0511070.
- 66.France S, Guerin DJ, Miller SJ, Lectka T. Chem. Rev. 2003;103:2985–3012. doi: 10.1021/cr020061a. and references therein. [DOI] [PubMed] [Google Scholar]
- 67.Many of the pharmaceutically relevant β-lactams are monosubstituted in the 3-position of the azetidin-2-one. One could foresee this moiety arising from the reaction of monosubstituted ketenes with imines.
- 68.Taggi AE, Hafez AM, Wack H, Young B, Ferraris D, Lectka T. J. Am. Chem. Soc. 2002;124:6626–6635. doi: 10.1021/ja0258226. [DOI] [PubMed] [Google Scholar]
- 69.(a) Wack H, France S, Hafez AM, Drury WJ, III, Weatherwax A, Lectka T. J. Org. Chem. 2004;69:4531–4533. doi: 10.1021/jo049804d. [DOI] [PubMed] [Google Scholar]; (b) Hafez AM, Dudding T, Wagerle TR, Shah MH, Taggi AE, Lectka T. J. Org. Chem. 2003;68:5819–5825. doi: 10.1021/jo034150e. [DOI] [PubMed] [Google Scholar]
- 70.(a) Ogilvie WW, Yoakim C, Dô F, Haché B, Lagacé L, Naud J, O'Meara JA, Déziel R. Bioorg. Med. Chem. 1999;7:1521–1531. doi: 10.1016/s0968-0896(99)00094-2. [DOI] [PubMed] [Google Scholar]; (b) Han WT, Trehan AK, Wright JJK, Federici ME, Seiler SM, Meanwell NA. Bioorg. Med. Chem. 1995;3:1123–1143. doi: 10.1016/0968-0896(95)00101-l. [DOI] [PubMed] [Google Scholar]
- 71.(a) Ferraris D, Dudding T, Young B, Drury WJ, III, Lectka T. J. Org. Chem. 1999;64:2168–2169. [Google Scholar]; (b) Ferraris D, Young B, Cox C, Drury WJ, III, Dudding T, Lectka T. J. Org. Chem. 1998;63:6090–6091. doi: 10.1021/jo981079h. Also see Ref. 63b. [DOI] [PubMed] [Google Scholar]
- 72.Drury WJ, III, Ferraris D, Cox C, Young B, Lectka T. J. Am. Chem. Soc. 1998;120:11006–11007. [Google Scholar]; (b) Ferraris D, Young B, Dudding T, Drury WJ, III, Lectka T. Tetrahedron. 1999;55:8869–8882. [Google Scholar]
- 73.Ferraris D, Young B, Dudding T, Lectka T. J. Am. Chem. Soc. 1998;120:4548–4549. Also see Ref. 63b. [Google Scholar]
- 74.(a) France S, Wack H, Hafez AM, Taggi AE, Witsil DR, Lectka T. Org. Lett. 2002;4:1603–1605. doi: 10.1021/ol025805l. [DOI] [PubMed] [Google Scholar]; (b) France S, Shah MH, Weatherwax A, Wack H, Roth JP, Lectka T. J. Am. Chem. Soc. 2005;127:1206–1215. doi: 10.1021/ja044179f. [DOI] [PubMed] [Google Scholar]
- 75.Aggarwal VK, Mereu A, Tarver GJ, McCague R. J. Org. Chem. 1998;63:7183–7189. doi: 10.1021/jo980421n. [DOI] [PubMed] [Google Scholar]
- 76.Huang Y, Calter MA. Tetrahedron Lett. 2007;48:1657–1659. [Google Scholar]
- 77.(a) Yamamoto Y, Ito W. Tetrahedron. 1988;44:5415–5423. [Google Scholar]; (b) Juhl K, Hazell RG, Jørgensen KA. J. Chem. Soc., Perkin Trans. 1. 1999:2293–2297. [Google Scholar]
- 78.Hodous BL, Fu GC. J. Am. Chem. Soc. 2002;124:1578–1579. doi: 10.1021/ja012427r. [DOI] [PubMed] [Google Scholar]
- 79.Lee EC, Hodous BL, Bergin E, Shih C, Fu GC. J. Am. Chem. Soc. 2005;127:11586–11587. doi: 10.1021/ja052058p. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y-R, He L, Wu X, Shao P-L, Ye S. Org. Lett. 2008;10:277–280. doi: 10.1021/ol702759b. [DOI] [PubMed] [Google Scholar]
- 81.Duguet N, Campbell CD, Slawin AMZ, Smith AD. Org. Biomol. Chem. 2008;6:1108–1113. doi: 10.1039/b800857b. [DOI] [PubMed] [Google Scholar]
- 82.Berlin JM, Fu GC. Angew. Chem., Int. Ed. 2008;47:7048–7050. doi: 10.1002/anie.200802439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dochnahl M, Fu GC. Angew. Chem., Int. Ed. 2009;48:2391–2393. doi: 10.1002/anie.200805805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.(a) March J. Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 4th ed. John Wiley & Sons; New York, NY: 1992. [Google Scholar]; (b) Carey FA, Sundberg RJ. Advanced Organic Chemistry. 3rd ed. Plenum; New York, NY: 1990. [Google Scholar]
- 85.(a) House H. Modern Synthetic Reactions. 2nd ed. W.A. Benjamin; New York, NY: 1972. pp. 459–478. [Google Scholar]; (b) De Kimpe N, Verche R. The Chemistry of α-Haloketones, α-Haloaldehydes, and α-Haloimines. John Wiley & Sons; New York, NY: 1988. [Google Scholar]
- 86.France S, Weatherwax A, Lectka T. Eur. J. Org. Chem. 2005:475–479. [Google Scholar]
- 87.Paull DH, Scerba MT, Alden-Danforth E, Widger LR, Lectka T. J. Am. Chem. Soc. 2008;130:17260–17261. doi: 10.1021/ja807792c.; France S, Bernstein D, Weatherwax A, Lectka T. Org. Lett. 2005;7:3009–3012. doi: 10.1021/ol050980y.; France S, Wack H, Taggi AE, Hafez AM, Wagerle TR, Shah MH, Dusich CL, Lectka T. J. Am. Chem. Soc. 2004;126:4245–4255. doi: 10.1021/ja039046t.; Wack H, Taggi AE, Hafez AM, Drury WJ, III, Lectka T. J. Am. Chem. Soc. 2001;123:1531–1532. doi: 10.1021/ja005791j.; For a recent review see: Oestreich M. Angew. Chem., Int. Ed. 2005;44:2324–2327. doi: 10.1002/anie.200500478. Also see Ref. 86.
- 88.For examples, see: Shibata N, Kohno J, Takai K, Ishimaru T, Nakamura S, Toru T, Kanemasa S. Angew. Chem., Int. Ed. 2005;44:4204–4207. doi: 10.1002/anie.200501041.; Bertelsen S, Halland N, Bachmann S, Marigo M, Braunton A, Jørgensen KA. Chem. Commun. 2005:4821–4823. doi: 10.1039/b509366j.; Brochu MP, Brown SP, MacMillan DWC. J. Am. Chem. Soc. 2004;126:4108–4109. doi: 10.1021/ja049562z.; Duhamel P. Bull. Soc. Chim. Fr. 1996;133:457–459.; Durst T, Koh Kevin. Tetrahedron Lett. 1992;33:6799–6802.; Duhamel L, Angibaud P, Desmurs JR, Valnot JY. Synlett. 1991:807–808.; Evans DA, Ellman JA, Dorow RL. Tetrahedron Lett. 1987;28:1123–1126.
- 89.(a) Hintermann L, Togni Angew. Chem., Int. Ed. 2000;39:4359–4362. doi: 10.1002/1521-3773(20001201)39:23<4359::AID-ANIE4359>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Hintermann L, Togni A. Helv. Chim. Acta. 2000;83:2425–2453. [Google Scholar]
- 90.Walling C, Padwa A. J. Am. Chem. Soc. 1963;85:1597–1601. [Google Scholar]
- 91.Guy A, Lemaire M, Guette J-P. Synthesis. 1982;12:1018–1020. [Google Scholar]
- 92.(a) Ozeryanskii VA, Pozharskii AF, Vistorobskii NV. Russ. J. Org. Chem. 1997;33:251–256. [Google Scholar]; (b) Glowiak T, Majerz I, Malarski Z, Sobczyk L, Pozharskii AF, Ozeryanskii VA, grech E. J. Phys. Org. Chem. 1999;12:895–900. [Google Scholar]
- 93.D'Angeli F, Marchetti P, Bertolasi V. J. Org. Chem. 1995;60:4013–4016. [Google Scholar]
- 94.(a) Dogo-Isonagie C, Bekele T, France S, Wolfer J, Weatherwax A, Taggi AE, Paull DH, Dudding T, Lectka T. Eur. J. Org. Chem. 2007:1091–1100. doi: 10.1021/jo061522l. [DOI] [PubMed] [Google Scholar]; (b) Dogo-Isonagie C, Bekele T, France S, Wolfer J, Weatherwax A, Taggi AE, Lectka T. J. Org. Chem. 2006;71:8946–8949. doi: 10.1021/jo061522l. [DOI] [PubMed] [Google Scholar]
- 95.Seki M, Yamanaka T, Kondo K. J. Org. Chem. 2000;65:517–522. doi: 10.1021/jo991461+. [DOI] [PubMed] [Google Scholar]
- 96.Paquette LA, Hefferon GJ, Samodral R, Hanzawa Y. J. Org. Chem. 1983;48:1262–1266. [Google Scholar]
- 97.Molecular mechanics calculations confirm that there is a less than 0.5 kcal/mol difference between the diastereomeric acyl ammonium intermediates.
- 98.Recent reviews on asymmetric epoxidation: Aggarwal VK, Winn CL. Acc. Chem. Res. 2004;37:611–620. doi: 10.1021/ar030045f.; Yang D. Acc. Chem. Res. 2004;37:497–505. doi: 10.1021/ar030065h.; Sharpless KB. Angew. Chem., Int. Ed. 2002;41:2024–2032.; Bellucci G, Chiappe C, D'Andrea F. Tetrahedron: Asymmetry. 1995;6:221–230.
- 99.Nielsen LPC, Stevenson CP, Blackmond DG, Jacobsen EN. J. Am. Chem. Soc. 2004;126:1360–1362. doi: 10.1021/ja038590z. [DOI] [PubMed] [Google Scholar]
- 100.Takano S, Sugihara T, Kamikubo T, Ogasawara K. Heterocycles. 1991;32:1587–1591. [Google Scholar]
- 101.Lee EC, McCauley KM, Fu GC. Angew. Chem., Int. Ed. 2007;46:977–979. doi: 10.1002/anie.200604312. [DOI] [PubMed] [Google Scholar]
- 102.O'Hagan D, Harper DB. J. Fluorine Chem. 1999;100:127–133. [Google Scholar]
- 103.(a) Bégué, J-P, Bonnet-Delpon D. Biological Impacts of Fluorination: Pharmaceuticals Based on Natural Products. In: Tressaud A, editor. Fluorine and Health. Elsevier B.V.; Amsterdam: 2008. pp. 553–622. [Google Scholar]; (b) Bégué J-P, Bonnet-Delpon D. J. Fluorine Chem. 2006;127:992–1012. [Google Scholar]
- 104.For recent reviews of the pharmacology of fluorinated derivatives, see: Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/b610213c.; Thomas C. J. Curr. Top. Med. Chem. 2006;6:1529–1543. doi: 10.2174/156802606777951109.
- 105.Abraham CJ, Paull DH, Bekele T, Scerba MT, Dudding T, Lectka T. J. Am. Chem. Soc. 2008;130:17085–17094. doi: 10.1021/ja806818a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.For a recent review of isoquinoline alkaloid chemistry, see: Chrzanowska M, Rozwadowska MD. Chem. Rev. 2004;104:3341–3370. doi: 10.1021/cr030692k.
- 107.(a) White JD, Choi Y. Org. Lett. 2000;2:2373–2376. doi: 10.1021/ol0001463. [DOI] [PubMed] [Google Scholar]; (b) White JD, Choi Y. Helv. Chim. Acta. 2002;85:4306–4327. [Google Scholar]; (c) Evans DA, Wu J. J. Am. Chem. Soc. 2003;125:10162–10163. doi: 10.1021/ja0367602. [DOI] [PubMed] [Google Scholar]; (d) Ryu DH, Zhou G, Corey EJ. J. Am. Chem. Soc. 2004;126:4800–4802. doi: 10.1021/ja049323b. [DOI] [PubMed] [Google Scholar]; (e) Hu Q-Y, Zhou G, Corey EJ. J. Am. Chem. Soc. 2004;126:13708–13713. doi: 10.1021/ja046154m. [DOI] [PubMed] [Google Scholar]
- 108.(a) Paull DH, Wolfer J, Grebinski JW, Weatherwax A, Lectka T. Chimia. 2007;61:240–246. [Google Scholar]; (b) Alden-Danforth E, Scerba MT, Lectka T. Org. Lett. 2008;10:4951–4953. doi: 10.1021/ol802029e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wallace OB, Smith DW, Deshpande MS, Polson C, Felsenstein KM. Bioorg. Med. Chem. Lett. 2003;13:1203–1206. doi: 10.1016/s0960-894x(02)01058-2. [DOI] [PubMed] [Google Scholar]
- 110.(a) Croom KF, Goa KL. Drugs. 2003;63:2769–2802. doi: 10.2165/00003495-200363240-00008. [DOI] [PubMed] [Google Scholar]; (b) Bihel F, Kraus J-L. Org. Biomol. Chem. 2003;1:793–799. doi: 10.1039/b212064j. [DOI] [PubMed] [Google Scholar]; (c) Jarvest RL, Connor SC, Gorniak JG, Jennings LJ, Serafinowska HT, West A. Bioorg. Med. Chem. Lett. 1997;7:1733–1738. [Google Scholar]; (d) Sengupta SK, Trites DH, Madhavarao MS, Beltz WR. J. Med. Chem. 1979;22:797–802. doi: 10.1021/jm00193a009. [DOI] [PubMed] [Google Scholar]
- 111.Jones Z, Groneberg R, Drew M, Eary CT. 20,050,282,812. U.S. Patent. 2005
- 112.Rösner M, Billhardt-Troughton U-M, Kirsh R, Kleim J-P, Meichsner C, Riess G, Winkler I. 5,723,461. U.S. Patent. 1998
- 113.Patai S, editor. The Chemistry of the Quinonoid Compounds. Vol. 4. John Wiley & Sons; New York, NY: 1974. ; For a recent review see: Kharisov BI, Mendez-Rojas MA, Garnovskii AD, Ivakhnenko EP, Ortiz-Mendez U. J. Coord. Chem. 2002;55:745–770.
- 114.Some examples include: Weller DD, Stirchak EP. J. Org. Chem. 1983;48:4873–4879.; Sinclair IW, Proctor GR. J. Chem. Soc., Perkin Trans. 1. 1975:2485–2488.; Willstaetter R, Pfannenstiel A. Ber. Dtsch. Chem. Ges. 1904;37:4744–4746.; Minisci F, Citterio A, Vismara E, Fontana F, DeBernardinis S, Correale M. J. Org. Chem. 1989;54:728–731.
- 115.Magdziak D, Rodriguez AA, Van De Water RW, Pettus TRR. Org. Lett. 2002;4:285–288. doi: 10.1021/ol017068j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.(a) Ried W, Torok E. Liebigs Ann. Chem. 1965;687:187–190. [Google Scholar]; (b) Patai S, Rappoport Z, editors. The Chemistry of Quinonoid Compounds. Parts I and II. Vol. 2. John Wiley & Sons; New York, NY: 1988. [Google Scholar]
- 117.(a) Bryce-Smith D, Gilbert A. J. Chem. Soc., Chem. Commun. 1968:1319–1320. [Google Scholar]; (b) Liao C, Lin H, Lin J. J. Chin. Chem. Soc. 1980;27:87–95. [Google Scholar]
- 118.(a) Nair V, Kumar S. J. Chem. Soc., Perkin Trans. 1. 1996:443–447. [Google Scholar]; (b) Kaizer J, Speier G, Osz E, Giorgi M, Reglier M. Tetrahedron Lett. 2004;45:8011–8013. [Google Scholar]
- 119.Bekele T, Shah MH, Wolfer J, Abraham CJ, Weatherwax A, Lectka T. J. Am. Chem. Soc. 2006;128:1810–1811. doi: 10.1021/ja058077g. [DOI] [PubMed] [Google Scholar]
- 120.(a) Paull DH, Abraham CJ, Scerba MT, Alden-Danforth E, Lectka T. Acc. Chem. Res. 2008;41:655–663. doi: 10.1021/ar700261a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dudding T, Hafez AM, Taggi AE, Wagerle TR, Lectka T. Org. Lett. 2002;4:387–390. doi: 10.1021/ol017087t. [DOI] [PubMed] [Google Scholar]
- 121.Eguchi S, Suzuki T, Okawa T, Matsushita Y, Yashima E, Okamoto Y. J. Org. Chem. 1996;61:7316–7319. doi: 10.1021/jo9609283. [DOI] [PubMed] [Google Scholar]
- 122.Kar GK, Roy BC, Das Adhikari S, Ray JK, Brahma NK. Bioorg. Med. Chem. 1998;6:2397–2403. doi: 10.1016/s0968-0896(98)80015-1. [DOI] [PubMed] [Google Scholar]
- 123.Hungate RW, Chen JL, Starbuck KE, Vacca JP, McDaniel SL, Levin RB, Dorsey BD, Guare JP, Holloway MK, Whitter W, Darke PL, Zugay JA, Schleif WA, Emini EA, Quintero JC, Lin JH, Chen I-W, Anderson PS, Huff JR. Bioorg. Med. Chem. 1994;2:859–879. doi: 10.1016/s0968-0896(00)82037-4. [DOI] [PubMed] [Google Scholar]
- 124.Okayama T, Seki S, Ito H, Takeshima T, Hagiwara M, Morikawa T. Chem. Pharm. Bull. 1995;43:1683–1691. doi: 10.1248/cpb.43.1683. [DOI] [PubMed] [Google Scholar]
- 125.Park BK, Nakagawa M, Hirota A, Nakayama M. J. Antibiot. 1988;41:751–758. doi: 10.7164/antibiotics.41.751. [DOI] [PubMed] [Google Scholar]
- 126.Schultz AG, Shannon PJ. J. Org. Chem. 1985;50:4421–4425. [Google Scholar]
- 127.Hein SM, Gloer JB, Koster B, Malloch D. J. Nat. Prod. 2001;64:809–812. doi: 10.1021/np000617u. [DOI] [PubMed] [Google Scholar]
- 128.Dos Santos AA, Francke W. Tetrahedron: Asymmetry. 2006;17:2487–2490. [Google Scholar]
- 129.(a) Largeron M, Lockhart B, Pfeiffer B, Fleury M-B. J. Med. Chem. 1999;42:5043–5052. doi: 10.1021/jm991105j. [DOI] [PubMed] [Google Scholar]; (b) Largeron M, Fleury M-B. Tetrahedron Lett. 1998;39:8999–9002. [Google Scholar]
- 130.Nicolaou KC, Zhong Y, Baran PS, Sugita K. Angew. Chem., Int. Ed. 2001;40:2145–2149. [PubMed] [Google Scholar]
- 131.Heine HW, Williams DK, Rutherford JL, Ramphal J, Williams EA. Heterocycles. 1993;35:1126–1140. [Google Scholar]
- 132.Wolfer J, Bekele T, Abraham CJ, Dogo-Isonagie C, Lectka T. Angew. Chem., Int. Ed. 2006;45:7398–7400. doi: 10.1002/anie.200602801. [DOI] [PubMed] [Google Scholar]
- 133.Verma P, Singh S, Dikshit DK, Suprabhat R. Synthesis. 1988:68–70. [Google Scholar]
- 134.(a) Bhaduri S, Lahiri GK, Munshi P. J. Organomet. Chem. 2000;606:151–155. [Google Scholar]; (b) Gladiali S, Dore A, Fabbri D, Medici S, Pirri G, Pulacchini S. Eur. J. Org. Chem. 2000:2861–2865. [Google Scholar]; (c) Reetz MT, Mehler G. Tetrahedron Lett. 2003;44:4593–4596. [Google Scholar]
- 135.(a) Kobayashi S, Matsubara R, Nakamura Y, Kitagawa H, Sugiura M. J. Am. Chem. Soc. 2003;125:2507–2515. doi: 10.1021/ja0281840. [DOI] [PubMed] [Google Scholar]; (b) Matsubara R, Nakamura Y, Kobayashi S. Angew. Chem., Int. Ed. 2004;43:1679–1681. doi: 10.1002/anie.200353237. [DOI] [PubMed] [Google Scholar]
- 136.O'Donnell M. J. Acc. Chem. Res. 2004;37:506–517. doi: 10.1021/ar0300625. [DOI] [PubMed] [Google Scholar]
- 137.For examples, see: Groeger H. Chem. Rev. 2003;103:2795–2827. doi: 10.1021/cr020038p.; Huang J, Corey EJ. Org. Lett. 2004;6:5027–5029. doi: 10.1021/ol047698w.; Becker C, Hoben C, Schollmeyer D, Scherr G, Kunz H. Eur. J. Org. Chem. 2005:1497–1499.
- 138.Meffre P, LeGoffic F. Amino Acids. 1996;11:313–328. doi: 10.1007/BF00807939. [DOI] [PubMed] [Google Scholar]
- 139.(a) Huber DP, Stanek K, Togni A. Tetrahedron: Asymmetry. 2006;17:658–664. [Google Scholar]; (b) Mohar B, Baudoux J, Plaquevent J-C, Cahard D. Angew. Chem., Int. Ed. 2001;40:4214–4216. doi: 10.1002/1521-3773(20011119)40:22<4214::AID-ANIE4214>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 140.Kobayashi S, Kobayashi J, Ishiani H, Ueno M. Chem.—Eur. J. 2002;8:4185–4190. doi: 10.1002/1521-3765(20020916)8:18<4185::AID-CHEM4185>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 141.Paull DH, Alden-Danforth E, Wolfer J, Dogo-Isonagie C, Abraham CJ, Lectka T. J. Org. Chem. 2007;72:5380–5382. doi: 10.1021/jo070472x. [DOI] [PubMed] [Google Scholar]
- 142.Castellani CB, Gatti G, Millini R. Inorg. Chem. 1984;23:4004–4008. [Google Scholar]
- 143.For review and leading references, see: Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893–930. doi: 10.1021/cr020033s.
- 144.Gupta D, Ghosh NN, Chandra R. Bioorg. Med. Chem. Lett. 2005;15:1019–1022. doi: 10.1016/j.bmcl.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 145.Lee L, Murray WV, Rivero RA. J. Org. Chem. 1997;62:3874–3879. and references therein. [Google Scholar]
- 146.(a) Sarges R, Lyga JW. J. Heterocycl. Chem. 1988;25:1475–1479. [Google Scholar]; (b) Bunin BA, Ellman JA. J. Am. Chem. Soc. 1992;114:10997–10998. [Google Scholar]
- 147.Morales GA, Corbett JW, DeGrado WF. J. Org. Chem. 1998;63:1172–1177. [Google Scholar]
- 148.Adams R, Way JW. J. Am. Chem. Soc. 1954;76:2763–2769. [Google Scholar]
- 149.Abraham CJ, Paull DH, Scerba MT, Grebinski JW, Lectka T. J. Am. Chem. Soc. 2006;128:13370–13371. doi: 10.1021/ja065754d. [DOI] [PubMed] [Google Scholar]
- 150.Ikeda M, Ohno K, Uno T, Tamura Y. Tetrahedron Lett. 1980;21:3403–3406. [Google Scholar]
- 151.Xu X, Wang K, Nelson SG. J. Am. Chem. Soc. 2007;129:11690–11691. doi: 10.1021/ja074845n. [DOI] [PubMed] [Google Scholar]
- 152.Boyer FE, Prasad JVNV, Domagala JM, Ellsworth EL, Gajda C, Hagen SE, Markoski LJ, Tait BD, Lunney EA, Palovsky A, Ferguson D, Graham N, Holler T, Hupe D, Nouhan C, Tummino PJ, Urumov A, Zeikus E, Zeikus G, Gracheck SJ, Sanders JM, VanderRoest S, Brodfuehrer J, Iyer K, Sinz M, Gulnik SV, Erickson JW. J. Med. Chem. 2000;43:843–858. doi: 10.1021/jm990281p. [DOI] [PubMed] [Google Scholar]
- 153.(a) Shaw SJ, Sundermann KF, Burlingame MA, Myles DC, Freeze BS, Xian M, Brouard I, Smith AB., III J. Am. Chem. Soc. 2005;127:6532–6533. doi: 10.1021/ja051185i. [DOI] [PubMed] [Google Scholar]; (b) Lewy DS, Gauss C-M, Soenen DR, Boger DL. Curr. Med. Chem. 2002;9:2005–2032. doi: 10.2174/0929867023368809. [DOI] [PubMed] [Google Scholar]; (c) Fang X-P, Andersen JE, Chang C-J, McLaughlin JL, Fanwick PE. J. Nat. Prod. 1991;54:1034–1043. doi: 10.1021/np50076a017. [DOI] [PubMed] [Google Scholar]
- 154.Kato Y, Ogawa Y, Imada T, Iwasaki S, Shimazaki N, Kobayashi T, Komai T. J. Antibiot. 1991;44:66–75. doi: 10.7164/antibiotics.44.66. [DOI] [PubMed] [Google Scholar]
- 155.(a) Istvan ES, Deisenhofer J. Science. 2001;292:1160–1164. doi: 10.1126/science.1059344. [DOI] [PubMed] [Google Scholar]; (b) Endo A. J. Med. Chem. 1985;28:401–405. doi: 10.1021/jm00382a001. [DOI] [PubMed] [Google Scholar]
- 156.Zhang Y-R, Lv H, Zhou D, Ye S. Chem.—Eur. J. 2008;14:8473–8476. doi: 10.1002/chem.200801165. [DOI] [PubMed] [Google Scholar]
- 157.(a) Tiseni PS, Peters R. Org. Lett. 2008;10:2019–2022. doi: 10.1021/ol800742d. [DOI] [PubMed] [Google Scholar]; (b) Tiseni PS, Peters R. Angew. Chem., Int. Ed. 2007;46:5325–5328. doi: 10.1002/anie.200700859. [DOI] [PubMed] [Google Scholar]
- 158.The dichloroepoxide undergoes complete stereochemical inversion upon opening, see: Wynberg H, Staring EGJ. J. Chem. Soc., Chem. Commun. 1984:1181–1182.
- 159.Li C-Y, Wang X-B, Sun X-L, Tang Y, Zheng J-C, Xu Z-H, Zhou Y-G, Dai L-X. J. Am. Chem. Soc. 2007;129:1494–1495. doi: 10.1021/ja068642v. [DOI] [PubMed] [Google Scholar]