

Abstract
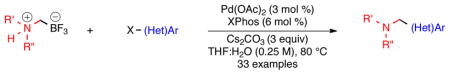
A reinvestigation into the chemical composition of potassium aminomethyltrifluoroborates is reported. These trifluoroborato salts have been reassigned as zwitterionic ammoniomethyltrifluoroborates. Minor adjustments to the previously disclosed reaction conditions are reported that permit a similar level of activity as nucleophiles in Suzuki–Miyaura cross-coupling reactions.
Introduction
Recently, we disclosed a series of reports outlining the synthesis1 of potassium aminomethyltrifluoroborates and their use as nucleophiles with aryl- and heteroaryl bromides2 and chlorides3 in Suzuki–Miyaura cross-coupling reactions. Decreased toxicity and simpler purification as compared to an analogous, specialized aminomethylstannane4 provided an impetus for exploiting the dissonant bond-forming connectivity conveyed through the agency of the aminomethyltrifluoroborates. Thus, performing an aminomethylation reaction via a Suzuki–Miyaura cross-coupling reaction between an aminomethyltrifluoroborate and an aryl electrophile is complementary to other commonly utilized routes to these materials, including alkylation of amines with benzylic halides, reductive amination of aromatic aldehydes, and processes initiated from aromatic nitriles. Aminomethylation of aromatic and heteroaromatic halides obviates the need to use lachrymal benzyl halides employed in the amine alkylation approach. Furthermore, the greater commercial availability5 of aryl- and heteroaryl chlorides as compared to the corresponding benzyl halides and aromatic nitriles and aldehydes employed6 in traditional routes allows inherently less expensive and more rapid access to starting materials and also provides greater structural diversity in targeted aminomethyl compounds.
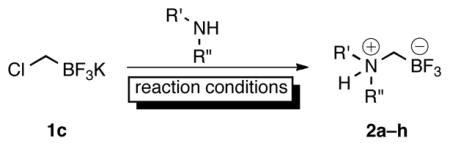
Because of their utility, we have continued to investigate the reported potassium aminomethyltrifluoroborates and their derivatives.7 During the course of these investigations and in particular through characterization by elemental analysis, we discovered that many of the samples assigned as potassium aminomethyltrifluoroborates 3 were instead composed principally of the internal salts 2 and varying amounts of KBr (Scheme 1). Previously, we believed that treatment with either KHCO3 or K2CO3 in acetone was sufficient to effect deprotonation, leading to the desired potassium salts, and this belief was supported by several observed changes in the 1H NMR spectra (Figure 1). In addition to the loss of the broad, exchangeable proton assigned to the protonated amine from the piperidine-derived preliminary internal salt, there was a coalescence of peaks, indicating an increase in fluxionality as well as a symmetrization of the axial and equatorial protons, as one might observe concomitant with deprotonation.
Scheme 1.

Previously Reported Synthesis of Potassium Aminomethyl Trifluoroborates
Figure 1.

Observed Changes in the 1H NMR Spectra of 2a after Exposure to Base.
Although their precise chemical identity and composition was now in question, aminomethyltrifluoroborates prepared in this manner had already proven to be versatile reagents with broad substrate scope and functional group compatibility. Herein, we disclose a reinvestigation of this chemistry and report the synthesis and isolation of ammoniomethyltrifluoroborates in addition to their use as nucleophilic partners in Suzuki–Miyaura cross-coupling reactions.
Results and Discussion
The first goal was to address whether the observations concerning the nature of the aminomethyltrifluoroborates were the result of a systemic misassignment, or represented a case-by-case event. Almost immediately difficulties were encountered in reproducing the previous procedures for the synthesis of the trifluoroborates;1,2 stirring 2a with either KHCO3 (20 min), or K2CO3 (30 min) in acetone did not lead to a replication of the reported spectra. Finally, after stirring the preliminary product with K3PO4 in acetone overnight, duplication of the observed changes in the 1H NMR spectra considered indicative of a deprotonation to the potassium aminomethyltrifluoroborate 3a (Figure 1) was observed, but these results were capricious and not reproducible. It was expected that with compounds presenting the same 1H NMR spectra as previously reported, an X-ray crystal structure of the potassium aminomethyltrifluoroborate could be acquired to confirm the structure, and the purity could be determined by elemental analysis. Although the composition of the internal salt 2a was confirmed by Xray analysis (see Figure S1), the structure of the presumed deprotonated potassium aminomethyl trifluoroborate salt 3a could not be. In agreement with the preliminary elemental analysis results, no potassium ions were detected8 in crystals formed from compounds having 1H NMR spectra matching those in Figure 1B. Further support of a systemic misassignment came from investigations with internal salt 2e. Although the same characteristics were exhibited in the 1H NMR of 2e when it was treated with base, elemental analysis of the resultant product thought to be 3e (Figure 2B) revealed that this sample could not be the potassium salt because it contained only 3.89% K instead of the expected 12.77% K. Although both analytical techniques convinced us that the deprotonation procedures were inconsistent at best and ineffective at worst, neither addressed the additional possibility of KBr or KI contamination in the final product.
Figure 2.

Observed Changes in the 1H NMR Spectra of 2e after Exposure to Base.
Previously, the aminomethyltrifluoroborates were prepared by alkylation of the desired amine with potassium iodomethyltrifluoroborate1 1a or potassium bromomethyltrifluoroborate2,3 1b under neat conditions for inexpensive amines or stoichiometric conditions in THF for the more valuable amines. The resulting crude ammoniomethyltrifluoroborates (2) were subjected to treatment with either KHCO3 or K2CO3, followed by filtration in hot acetone. To address the probable contamination with KI or KBr in addition to the precise composition of the aminomethyltrifluoroborates, a method was developed to synthesize many of the aminomethyltrifluoroborates from chloromethyltrifluoroborate 1c. Unexpectedly, in simply replacing bromochloromethane for dibromomethane in our optimized bromomethyltrifluoroborate synthesis (eq 1)9 the isolated yield of 1c reached a ceiling of a ~50%. We considered this insufficient as a replacement for such a robust starting material. Adapting Whiting’s10 procedure for the synthesis of pinacol (chloromethyl)boronate by quenching with aq KHF2 instead of ethereal pinacol resulted in a procedure that reliably delivered 70–77% yields of 1c on scales up to 5 g (eq 2).
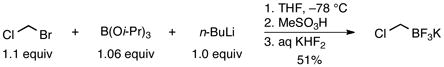 |
(1) |
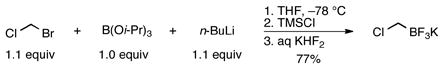 |
(2) |
The synthesis of the ammoniomethyltrifluoroborates was readily accomplished by reacting a variety of amines with 1c in a co-solvent mixture of THF and tert-butanol. In comparison to the conditions utilized for bromomethyltrifluoroborate (1b), increased reaction times were required (Table 1). A few examples (2b and 2h) resulted in increased yields by heating in acetone in a sealed tube. After the reactions were judged complete by 19F NMR, the solvent was removed in vacuo. The reaction mixture was subsequently suspended in hot acetone and filtered to remove the KCl byproduct. The internal salts were isolated substantially free from inorganic salt contamination, as KCl is less soluble in acetone than KBr. The procedure used to purify the isolated products (precipitation from hot acetone with diethyl ether) was similar to that commonly used for most potassium organotrifluoroborates. Most importantly, the majority of the ammoniomethyltrifluoroborates retained many of the favorable properties of potassium trifluoroborate salts as easy to manipulate air- and moisture-stable crystalline solids that have proved to be indefinitely bench-stable.
Table 1.
Synthesis of Ammoniomethyltrifluoroboratesa
 | |||||
|---|---|---|---|---|---|
| entry | nucleophile | reaction conditions | product | yield (%)b | |
| 1 | A | 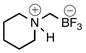 |
2a | 90c | |
| 2 | B | 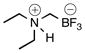 |
2b | 62c | |
| 3 | A | 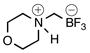 |
2c | 87 | |
| 4 | A | 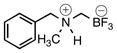 |
2d | 88 | |
| 5 | C | 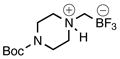 |
2e | 80 | |
| 6 |  |
A | 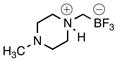 |
2f | 79 |
| 7 | 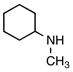 |
A | 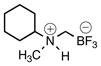 |
2g | 69 |
| 8 | B | 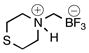 |
2h | 75 | |
5.00 mmol scale unless otherwise noted; Conditions: A, alkylamine (1.01–2.0 equiv), 3:1 THF:t-BuOH (1.0 M), 80 °C, 2–24 h; B, alkylamine (1.2–2.0 equiv), acetone (0.5 M), 80 °C, 16 h; C, alkylamine (1.01 equiv), 3:1 CPME:tert-amyl alcohol, 110 °C, 6.5 h;
Isolated yield;
10.0 mmol scale.
The behavior of the isolated internal salts in Suzuki–Miyaura cross-coupling reactions was next examined, particularly as to how they compared to the previously reported reaction conditions.2,3 Most of the original research samples were determined11 to be >90% pure by elemental analysis when reassigned as the internal salt with KBr contamination. Their previous structural misassignment, however, meant that a slightly larger excess (<25%) of the nucleophilic partner was used than originally reported.2 Aryl bromides could be cross-coupled with the ammoniomethyltrifluoroborate under almost identical reaction conditions, after adjusting for the effective molar excess of the trifluoroborate (i.e., 1.3 equiv trifluoroborate vs the reported 1.1 equiv, Table 2). Under these reaction conditions, we often observed <10% electrophile homocoupling. Isolated yields were decreased in the presence of 1.3 equiv of KBr (54%), not affected by 1.3 equiv of KCl (80%), and were not improved by the use of 4.0 equiv of Cs2CO3 (70%, see Table S1).
Table 2.
Cross-Coupling of Trifluoro(piperidin-1-ium-1- ylmethyl)borate with Aryl Bromidesa
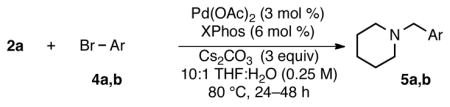
All reactions were carried out using 1.0 mmol of the aryl bromide and 1.3 mmol of the trifluoro(piperidin-1-ium-1-ylmethyl)borate;
Isolated yield.
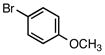
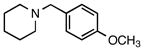
Aryl and heteroaryl chlorides also behaved similarly under the previously reported cross-coupling reaction conditions3 when a few minor modifications were implemented. After adjusting for the effective molar excess of the trifluoroborate (i.e., 1.2 equiv of trifluoroborate vs the reported 1.01 equiv), changing the solvent ratio to 4: 1 THF: H2O, and increasing the reaction time to 45 h, aryl and heteroaryl chlorides remained competent cross-coupling partners for the ammoniomethyltrifluoroborates (Table 3).
Table 3.
Cross-Coupling of Trifluoro(piperidin-1-ium-1- ylmethyl)borate with Aryl Chloridesa
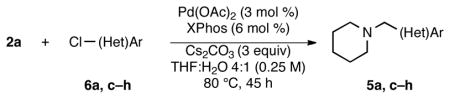 | ||||
|---|---|---|---|---|
| entry | aryl chloride | product | yield (lit3, %)b | yield (%)b |
| 1 |
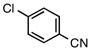 6a |
5a | 92c | 81 |
| 2 |
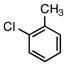 6c |
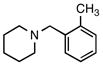 5c |
63 | 73 |
| 3 |
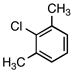 6d |
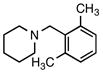 5d |
66 | 52 |
| 4 |
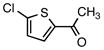 6e |
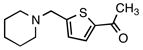 5e |
93 | 83 |
| 5 |
6f |
5f |
78 | 70 |
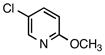
| 6 |
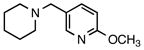
 6g |
 5g |
87 | 96 |
| 7 |
 6h |
5h |
n/a | 68 |
All reactions were carried out using 1.0 mmol of the aryl chloride and 1.2 mmol of the trifluoro(piperidin-1-ium-1-ylmethyl)borate;
Isolated yield;
10:1 CPME:H2O (0.25 M) was used as the solvent, 95 °C.
The reaction conditions remained general across electron-deficient (Table 3, entry 1) and hindered (Table 3, entries 2 and 3) aryl chlorides, providing moderate to good yields of the desired products. Heteroaryl chlorides were tolerated, including those with complementary functional groups, such as aldehydes and ketones (Table 3, entries 4 and 5). 3-Chloropyridines participated in good to excellent yields (Table 3, entries 6 and 7), whereas coupling with heterocycles chlorinated adjacent to nitrogen, such as 2-chloropyridine and 2-chloropyrimidine, remained elusive. Ethanol12 or co-solvent mixtures, including CH3CN, tert-butanol, tert-amyl alcohol, and n-butanol failed to resolve the challenges of these cross-coupling partners.
To illustrate the scope of the ammoniomethyltrifluoroborate cross-coupling partner, the breadth of available ammoniomethyltrifluoroborates (Table 1) was cross-coupled with both electron-rich 4-chloroanisole (Table 4) and 3-chloropyridine (Table 5). Most of the ammoniomethyl derivatives participated in the cross-coupling with 4-chloroanisole in good yields, whereas 3-chloropyridine resulted in more moderate yields.
Table 4.
Cross-Coupling of Various N,N-Dialkylammoniomethyltrifluoroborates with 4-Chloroanisolea
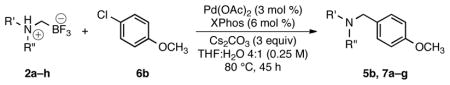 | ||||
|---|---|---|---|---|
| entry | nucleophile | product | yield (lit3, %)b | yield (%)b |
| 1 | 2a | 5b | 94 | 90 |
| 2 | 2b |
 7a |
84 | 63 |
| 3 | 2c |
 7b |
75 | 85 |
| 4 | 2d |
7c |
95 | 87 |
| 5 | 2e |
7d |
74 | 98 |
| 6 | 2f |
7e |
68 | 76 |
| 7 | 2g |
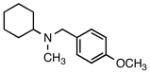 7f |
75 | 81 |
| 8 | 2h |
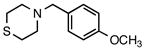 7g |
n/a | 60c |
All reactions were carried out using 1.0 mmol of 4-chloroanisole and 1.2 mmol of the ammoniomethyltrifluoroborate;
Isolated yield;
Average of 3 trials.
Table 5.
Cross-Coupling of Various N,N-Dialkylammoniomethyltrifluoroborates with 3-Chloropyridinea
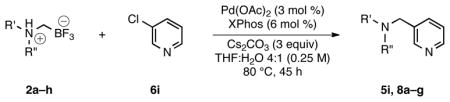 | ||||
|---|---|---|---|---|
| entry | nucleophile | product | yield (lit3, %)b | yield (%)b |
| 1 | 2a |
5i |
77 | 61 |
| 2 | 2b |
8a |
n/a | 30 |
| 3 | 2c |
8b |
n/a | 64 |
| 4 | 2d |
8c |
n/a | 83 |
| 5 | 2e |
8d |
n/a | quant |
| 6 | 2f |
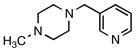 8e |
n/a | --c |
| 7 | 2g |
 8f |
n/a | 75 |
| 8 | 2h |
8g |
n/a | 38 |
All reactions were carried out using 1.0 mmol of 3-chloropyridine and 1.2 mmol of the ammoniomethyltrifluoroborate;
Isolated yield;
Unable to isolate any desired product.
Conclusion
In summary, the products generated after alkylation of amine nucleophiles with chloromethyltrifluoroborate (1c) have been identified as ammoniomethyltrifluoroborates, and it has been confirmed that treatment with base is insufficient to transform them to the corresponding potassium aminomethyltrifluoroborates. The described ammoniomethyltrifluoroborates retained many of the favorable characteristics associated with materials previously assigned as potassium trifluoroborate salts. Furthermore, it has been established that minor changes to the previously reported conditions are all that are necessary to achieve a cross-coupling of the isolated and pure ammoniomethyltrifluoroborates with both aryl- and heteroaryl bromides and chlorides. Finally, it should be emphasized that the previously reported1–3 deprotonation procedure with potassium carbonate is required and sufficient to provide full conversion to the potassium trifluoroborates when a less basic nitrogen has been incorporated within the organoboron substructure (e.g., with pyridyl- and quinolinyltrifluoroborates).
Experimental Section
General
Tetrahydrofuran (THF) was distilled from sodium/benzophenone prior to use. Acetone, diethyl ether (Et2O), and tert-butyl alcohol (t-BuOH), Pd(OAc)2, XPhos (2-dicyclohexylphosphino-2′4′6′-triisopropylbiphenyl), and Cs2CO3 were used as received. H2O was sparged with nitrogen for at least 20 min prior to use. Standard benchtop techniques were employed for handling air-sensitive reagents.
Amines were fractionally distilled under nitrogen from potassium hydroxide onto activated molecular sieves. All aryl bromides and chlorides were used as received.
Melting points (°C) are uncorrected. NMR spectra were recorded on a 500 or 400 MHz spectrometer. 1H NMR spectra were referenced using residual undeuterated solvent as an internal reference (δ 7.26 for CDCl3; δ 2.50 for DMSO-d6; δ 2.05 for acetone-d6; δ 1.94 for CD3CN). 13C NMR spectra were referenced to either the δ 77.0 resonance of CDCl3, the δ 39.5 resonance of DMSO-d6, the δ 29.84 resonance of acetone-d6, or the δ 1.32 resonance of CD3CN. 19F NMR chemical shifts were referenced to external CFCl3 (0.0 ppm). 11B NMR spectra were obtained on a spectrometer equipped with the appropriate decoupling accessories. All 11B NMR chemical shifts were referenced to external BF3·OEt2 (0.0 ppm) with a negative sign indicating an upfield shift. Data are presented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, b = broad), coupling constant J (Hz) and integration. Infrared spectra were recorded on a FT-IR instrument with a Horizontal Attenuated Total Reflectance (HATR) device. Analytical thin-layer chromatography (TLC) was performed on TLC silica gel plates (0.25 mm) precoated with a fluorescent indicator. Standard flash chromatography procedures were followed using 32–63 μm silica gel. Visualization was effected with ultraviolet light and KMnO4. Reactions conducted in microwave vials were heated conventionally.
Preparation of Potassium Chloromethyltrifluoroborate (1c)
BrCH2Cl (3.0 mL, 1.991 g/mL, 44.0 mmol) and B(O-iPr)3 (8.25 mL, 0.912 g/mL, 40.0 mmol) were placed in an oven-dried 250-mL three-neck flask equipped with a stirrer bar and internal thermometer under N2. Anhydrous THF (39 mL) was added from a syringe. The reaction mixture was cooled to an internal temperature of −50 °C. n-BuLi (17.6 mL, 2.5 M in hexanes, 44.0 mmol) was added to an oven-dried 50-mL pear-shaped flask and cooled in a Dry Ice/acetone bath. The pre-cooled n-BuLi was added dropwise at a rate of 1 drop/s, maintaining an internal temperature below −50 °C. After the addition of n-BuLi was complete, the reaction mixture was allowed to stir at −50 °C for 30 min before TMSCl (6.1 mL, 0.856 g/mL, 48 mmol) was added dropwise via syringe. After the reaction mixture was allowed to stir for 10 min, the cooling bath was removed and the reaction mixture was allowed to stir at rt for 24 h. Then, the flask was cooled in an ice water bath and sat. aq KHF2 (36 mL, ~4.5 M, 160 mmol) was added dropwise. The reaction mixture was stirred for 30 min (and judged complete by 11B NMR) before being concentrated in vacuo. Residual water was removed by azeotroping with toluene before drying in vacuo overnight. The dry, crude mixture was purified by Soxhlet extraction with 250 mL HPLC grade acetone for 10 h. The acetone extracts were concentrated in vacuo to a volume of 50 mL, and Et2O (5 mL) was added to precipitate the trifluoroborate product. Additional Et2O (200 mL) was added to assist filtration. Filtration gave a 77% yield of 1c (4.80 g, 30.7 mmol) as a white powder: mp 180–184 °C; νmax(HATR)/cm−1 1418, 1246, 1140, 1112, 1068, 994, 966, 774, 737, 684; 1H NMR (500 MHz, acetoned6) δ 2.43 (bs, 2H); (DMSO-d6) δ 2.31 (bs, 2H); (CD3CN) δ 2.44 (bs, 2H); 13C NMR (125 MHz, acetone-d6) δ no peaks observed; 19F NMR (470 MHz, acetone-d6) δ −146.8 (q, J = 51.0 Hz); (DMSO-d6) δ −143.3 (q, J = 50.4 Hz); (CD3CN) δ −146.4 (q, J = 51.0 Hz); 11B NMR (128 MHz, acetone-d6) δ 1.9 (q, J = 51.4 Hz); (DMSO-d6) δ 2.0–0.2 (m); (CD3CN) δ 1.5 (q, J = 51.2 Hz); Anal. Calc. CH2BClF3K: C, 7.68; H, 1.29; N: 0.0; found: C, 7.93; H, 1.02; N, <0.02.
General Experimental Procedure for the Preparation of Ammoniomethyl Trifluoroborates. Reaction Conditions A. Preparation of Trifluoro(piperidin-1-ium-1-ylmethyl)borate (2a)
An oven-dried 10–20-mL microwave vial equipped with a stirrer bar was charged with 1c (1.56 g, 10.0 mmol) and sealed with a cap lined with a disposable PTFE septum. The vial was then evacuated under vacuum and purged with N2 (3×). Anhydrous THF (5.5 mL), t-BuOH (2.5 mL), and piperidine (2.0 mL, 8.62 g/mL, 20.0 mmol) were added via syringes (solid amines were added with 1c). The reaction mixture was stirred and heated to 80 °C for 2 h and judged complete by 19F NMR. At this point the reaction mixture was transferred to a 100-mL round-bottom flask, and the volatiles were removed in vacuo. The crude solid was dried under high vacuum overnight before being dissolved in a solution of hot HPLC acetone and filtered to remove KCl. The filtrate was concentrated in vacuo, dissolved in a minimal amount of hot acetone (20 mL), and precipitated by the dropwise addition of Et2O (5 mL). Additional Et2O (150 mL) was added to facilitate filtering. Filtration and drying overnight in vacuo over P2O5 afforded 2a (1.51 g, 90%, 9.04 mmol) as a white powder: mp 147–150 °C; ν(HATR)/cm−1 3405 (bs, R3NH+), 3176, 2950, 1458, 1307, 1058, 980, 956; 1H NMR (500 MHz, CD3CN) δ 6.04 (s, 1H), 3.47 (d, J = 12.4 Hz, 2H), 2.80 (t, J = 12.1 Hz, 2H), 2.07 (s, 2H), 1.81 (s, 2H), 1.71 (s, 3H), 1.41 (d, J = 10.9 Hz, 1H); 13C NMR (126 MHz, CD3CN) δ 56.5, 23.9, 22.3; 19F NMR (471 MHz, CD3CN) δ −141.9 (q, J = 49.7 Hz); 11B NMR (128 MHz, CD3CN) δ 2.3 (q, J = 51.4 Hz); Anal. Calcd. for C6H13BF3N: C, 43.16; H, 7.85; N, 8.39; found: C, 42.95; H, 7.84; N, 8.39.
Reaction Conditions B. Preparation of [(Diethylammonio)methyl]trifluoroborate (2b)
An ovendried 10–20-mL microwave vial equipped with a stirrer bar was charged with 1c (1.56 g, 10.0 mmol), and sealed with a cap lined with a disposable PTFE septum. The vial was then evacuated under vacuum then purged with N2 (3×). Acetone (20 mL) and Et2NH (2.0 mL, 0.707 g/mL, 20.0 mmol) were added via syringes. The reaction mixture was heated to 80 °C for 24 h and judged complete by 19F NMR. At this point the reaction mixture was transferred to a 100-mL round-bottom flask, and the volatiles were removed in vacuo. The crude solid was dried under high vacuum overnight before being dissolved in a solution of hot HPLC acetone and filtered to remove KCl. The filtrate was concentrated in vacuo, dissolved in a minimal amount of hot acetone (20 mL), and precipitated by the dropwise addition of Et2O (100 mL). Filtration and drying overnight in vacuo over P2O5 afforded 2b (0.895 g, 57%, 5.77 mmol) as an off-white powder: mp 108–111 °C; ν(HATR)/cm−1 3592 (bs, R3NH+), 3188, 1470, 1019, 988; 1H NMR (500 MHz, CD3CN) δ 6.25–5.87 (m, 1H), 3.23–3.02 (m, 4H), 2.08 (s, 2H), 1.23 (q, J = 7.5 Hz, 6H); 13C NMR (126 MHz, CD3CN) δ 50.4, 9.3; 19F NMR (471 MHz, CD3CN) δ −142.4 (q, J = 49.9 Hz); 11B NMR (128 MHz, CD3CN) δ 2.0 (q, J = 51.3 Hz); Anal. Calc. for C5H13BF3N: C, 38.75; H, 8.46; N, 9.04; found: C, 38.14 (low); H, 8.34 (low); N, 8.79 (low); HRMS (ESI-TOF): calcd for C5H12BF3N− [M–H]−: 154.1015; found: 154.1016.
Trifluoro(morpholino-4-iummethyl)borate (2c)
According to general reaction conditions A with morpholine (0.87 mL, 0.996 g/mL, 10.0 mmol) and 1c (0.782 g, 5.0 mmol) for 1.5 h, 2c was obtained (0.739 g, 87%, 4.37 mmol) as a pale yellow powder: mp 170–172 °C; ν(HATR)/cm−1 3537, 3112, 1638, 1442, 1262, 1122, 1079, 1054, 1028, 960; 1H NMR (500 MHz, CD3CN) δ 6.57 (s, 1H), 3.93 (d, J = 13.0 Hz, 2H), 3.73 (dd, J = 18.1, 6.7 Hz, 2H), 3.43 (d, J = 13.5 Hz, 2H), 3.06–2.92 (m, 2H), 2.15 (s, 2H); 13C NMR (126 MHz, CD3CN) δ 64.7, 55.3; 19F NMR (471 MHz, CD3CN) δ −141.8 (q, J = 50.2 Hz); 11B NMR (128 MHz, CD3CN) δ 2.9–1.0 (m); Anal. Calc. for C5H11BF3NO: C, 35.54; H, 6.56; N, 8.29; found: C, 34.44 (low); H, 6.74 (high); N, 7.92 (low); HRMS (ESI-TOF): calcd for C5H10BF3NO− [M–H]−: 168.0808; found: 168.0809.
{[Benzyl(methyl)ammonio]methyl}trifluoroborate (2d)
According to general reaction conditions A with N-benzylmethylamine (0.65 mL, 0.939 g/mL, 5.05 mmol) and 1c (0.782 g, 5.0 mmol) for 16 h, 2d was obtained (0.895 g, 88%, 4.40 mmol) as an off-white powder: mp 109–112 °C; ν(HATR)/cm−1 3188, 1458, 1318, 1045, 1003, 934, 780, 748, 700; 1H NMR (500 MHz, CD3CN) δ 7.47 (s, 5H), 6.56 (bs, 1H), 4.30 (d, J = 13.0 Hz, 1H), 4.08 (d, J = 13.0 Hz, 1H), 2.71 (s, 3H), 2.17 (bs, 1H), 2.03 (bs, 1H); 13C NMR (126 MHz, CD3CN) δ 132.0, 131.5, 130.7, 130.0, 62.3, 43.3; 19F NMR (471 MHz, CD3CN) δ −141.94 (q, J = 47.4 Hz); 11B NMR (128 MHz, CD3CN) δ 2.0 (q, J = 50.7 Hz); Anal. Calc. for C9H13BF3N: C, 53.25; H, 6.45; N, 6.90; found: C, 52.05 (low); H, 5.98 (low); N, 6.68; HRMS (ESI-TOF): calcd for C9H13BF3N− [M–H]−: 202.1015; found: 202.1018.
{[4-(tert-Butoxycarbonyl)piperazin-1-ium-1-yl]methyl}trifluoroborate (2e)
According to general reaction conditions A with tert-butyl piperazine-1-carboxylate (0.940 g, 5.05 mmol) and 1c (0.782 g, 5.0 mmol) for 6.5 h in 3:1 CPME:tert-amyl alcohol, 2e was obtained (1.07 g, 80%, 3.99 mmol) as an off-white powder: mp 185 °C (dec); ν(HATR)/cm−1 3177, 1697, 1646 (C=O), 1421, 1284, 1170, 1143, 1034, 959; 1H NMR (360 MHz, acetone-d6) δ 7.43 (s, 1H), 4.17 (d, J = 14.6 Hz, 2H), 3.65 (d, J = 13.1 Hz, 2H), 3.47–3.27 (m, 2H), 3.05 (td, J = 12.6, 3.5 Hz, 2H), 2.20 (s, 2H), 1.46 (s, 9H); 13C NMR (126 MHz, acetone-d6) δ 154.6, 80.8, 55.0, 41.4, 28.4; 19F NMR (339 MHz, acetone-d6) δ −142.1 (q, J = 43.7 Hz); 11B NMR (128 MHz, acetone-d6) δ 2.2 (q, J = 49.5 Hz); Anal. Calc. for C10H20BF3N2O2: C, 44.80; H, 7.52; N, 10.45; found: C, 44.52; H, 7.52; N, 10.40.
Trifluoro[(4-methylpiperazin-1-ium-1-yl)methyl]borate (2f)
According to general reaction conditions A with 1-methylpiperazine (0.61 mL, 0.903 g/mL, 5.50 mmol) and 1c (0.782 g, 5.0 mmol) for 4 h, 2f was obtained (0.722 g, 79%, 3.96 mmol) as a white powder: mp 73–78 °C; ν(HATR)/cm−1 3601, 3168, 2808, 1455, 1068, 1040, 966; 1H NMR (500 MHz, CD3CN) δ 6.16 (s, 1H), 3.44 (d, J = 12.3 Hz, 2H), 2.96 (t, J = 11.0 Hz, 2H), 2.82 (d, J = 12.5 Hz, 2H), 2.31 (t, J = 11.4 Hz, 2H), 2.25 (d, J = 4.0 Hz, 3H), 2.11 (s, 2H); 13C NMR (126 MHz, CD3CN) δ 55.4, 52.6, 45.6; 19F NMR (471 MHz, CD3CN) δ −141.9 (q, J = 47.7 Hz); 11B NMR (128 MHz, CD3CN) δ 2.0 (q, J = 50.7 Hz); Anal. Calc. for C6H14BF3N2: C, 39.60; H, 7.75; N, 15.39; found: C, 38.05 (low); H, 7.66 (low); N, 14.44 (low); HRMS (ESI-TOF): calcd for C6H13BF3N2 − [M–H]−: 181.1124; found: 181.1120.
{[Cyclohexyl(methyl)ammonio]methyl}trifluoroborate (2g)
According to general reaction conditions A with N-methylcyclohexylamine (0.72 mL, 0.868 g/mL, 5.50 mmol) and 1c (0.782 g, 5.0 mmol) for 24 h, 2g was obtained (0.671 g, 69%, 3.44) as a white powder: mp 105–108 °C; ν(HATR)/cm−1 3182, 2942, 2863 1293, 1061, 1024, 985, 944; 1H NMR (500 MHz, DMSO-d6) δ 8.38–7.84 (m, 1H), 2.95 (s, 1H), 2.57 (s, 3H), 2.07–1.50 (m, 7H), 1.37–0.99 (m, 5H); 13C NMR (126 MHz, DMSO-d6) δ 64.5, 38.8, 25.9, 25.7, 24.8, 24.5; 19F NMR (471 MHz, DMSO-d6) δ −138.2; 11B NMR (128 MHz, DMSO-d6) δ 1.9; Anal. Calc. for C8H17BF3N: C, 49.27; H, 8.79; N, 7.18; found: C, 48.04 (low); H, 8.04 (low); N, 6.84; HRMS (ESI-TOF): calcd for C8H16BF3N− [M–H]−: 194.1330; found: 194.1328.
Trifluoro(thiomorpholino-4-iummethyl)borate (2h)
According to general reaction conditions B and reaction with thiomorpholine (0.57 mL, 1.026 g/mL, 6.0 mmol) and 1c (0.782 g, 5.0 mmol) for 16 h, 2h was obtained (0.693 g, 75%, 3.74 mmol) as an off-white powder after trituration with cold acetone: mp 188–189 °C; ν(HATR)/cm−1 3178, 1736, 1408, 1075, 1028, 967, 952, 925; 1H NMR (500 MHz, CD3CN) δ 6.26 (s, 1H), 3.72 (d, J = 12.0, 2H), 3.2–2.92 (m, 4H), 2.77 (d, J = 14.0, 2H), 2.14 (s, 2H);13C NMR (126 MHz, CD3CN) δ 57.0, 25.4; 19F NMR (471 MHz, CD3CN) δ −141.6 (q, J = 49.3 Hz); 11B NMR (128 MHz, CD3CN) δ 1.6 (q, J = 51.1 Hz); Anal. Calc. for C5H11BF3NS: C, 32.46; H, 5.99; N, 7.57; S, 17.33 found: C, 32.39; H, 5.86; N, 7.36; S, 17.14.
General Experimental Procedure for the Suzuki–Miyaura Cross-Coupling Reactions of Aryl Bromides. Preparation of 4-(Piperidin-1-ylmethyl)benzonitrile (5a)
A 2–5 mL microwave vial equipped with a stirrer bar was charged with 2a (0.217 g, 1.3 mmol), Cs2CO3 (0.977 g, 3.0 mmol), 4a (0.182 g, 1.0 mmol), Pd(OAc)2 (0.067 g, 0.03 mmol), and XPhos13 (0.029 g, 0.06 mmol), then sealed with a cap lined with a disposable PTFE septum. The vial was then evacuated under vacuum and purged with N2 (3×). Anhydrous THF (3.63 mL), and H2O (0.36 mL) were added by syringe (aryl bromides that were liquids at room temperature were added by syringe) and the reaction mixture was stirred and heated at 80 °C for 24 h, then cooled to rt and diluted with H2O (1 mL). The reaction mixture was extracted with EtOAc (3 × 3 mL). The combined organics were dried (MgSO4), filtered through Celite, and concentrated in vacuo. Purification by flash column chromatography, eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N afforded 5a (0.156 g, 78%, 0.78 mmol) as a yellow oil: Rf = 0.08 (silica gel, hexanes:EtOAc 7:1 with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.3
1-(4-Methoxybenzyl)piperidine (5b)
According to the general procedure with 4b (0.187 g, 1.0 mmol) for 44 h, 5b was obtained in 68% yield (0.140 g, 0.68 mmol) as a yellow oil after silica gel column chromatography (eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2 % Et3N): Rf = 0.05 (7:1 hexanes:EtOAc with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.2
General Experimental Procedure for the Suzuki–Miyaura Cross-Coupling Reactions of Aryl-and Heteroaryl Chlorides. Preparation of 4-(Piperidin-1-ylmethyl)benzonitrile (5a)
A 2–5 mL microwave vial equipped with a stirrer bar was charged with 2a (0.200 g, 1.2 mmol), Cs2CO3 (0.977 g, 3.0 mmol), 6a (0.138 g, 1.0 mmol), Pd(OAc)2 (0.067 g, 0.03 mmol), and XPhos (0.029 g, 0.06 mmol), then sealed with a cap lined with a disposable PTFE septum. The vial was then evacuated under vacuum and purged with N2 (3×). Anhydrous THF (3.2 mL) and H2O (0.80 mL) were added by syringe (aryland heteroaryl chlorides that were liquids at room temperature were added by syringe) and the reaction mixture was stirred and heated at 80 °C for 45 h, then cooled to rt and diluted with H2O (1 mL). The reaction mixture was extracted with EtOAc (3 × 3 mL). The combined organics were dried (MgSO4), filtered through Celite, and concentrated in vacuo. Purification by flash column chromatography, eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 4:1 hexanes:EtOAc with 0.2% Et3N afforded 5a (0.163 g, 81%) as a yellow oil: Rf = 0.06 (silica gel, hexanes:EtOAc 7:1 with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.3
1-(2-Methylbenzyl)piperidine (5c)
According to the general procedure with 6c (0.126 g, 1.0 mmol), 5c was obtained in 73% yield (0.138 g, 0.73 mmol) as a pale yellow oil after silica gel column chromatography (eluting with 9:1 hexanes:EtOAc with 0.2% Et3N to 7:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.2 (silica gel, 7:1 hexanes:EtOAc with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.3
1-(2,6-Dimethylbenzyl)piperidine (5d)
According to the general procedure with 6d (0.145 g, 1.0 mmol), 5d was obtained in 53% yield (0.107 g, 0.53 mmol) as a yellow solid after silica gel column chromatography (eluting with 99:1 hexanes:EtOAc to 9:1 hexanes:EtOAc): Rf = 0.5 (silica gel, 9:1 hexanes:EtOAc); mp 25–26 °C; 1H and 13C NMR spectra are comparable to those reported in the literature.3
1-[5-(Piperidin-1-ylmethyl)thiophen-2-yl]ethanone (5e)
According to the general procedure with 6e (0.161 g, 1.0 mmol), 5e was obtained in 83% yield (0.184 g, 0.82 mmol) as a yellow solid after silica gel column chromatography (eluting with 9:1 CHCl3:EtOAc to 100% EtOAc): Rf = 0.08 (silica gel, 8:2 CHCl3:EtOAc); mp 50–52 °C; NMR spectra are comparable to those reported in the literature.3
5-(Piperidin-1-ylmethyl)thiophene-2-carbaldehyde (5f)
According to the general procedure with 6f (0.147 g, 1.0 mmol), 5f was obtained in 70% yield (0.147 g, 0.70 mmol) as an orange oil after silica gel column chromatography (eluting with 9:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 1% Et3N): Rf = 0.2 (silica gel, 8:2 CHCl3:EtOAc); 1H and 13C NMR spectra are comparable to those reported in the literature.3
2-Methoxy-5-(piperidin-1-ylmethyl)pyridine (5g)
According to the general procedure with 6g (0.144 g, 1.0 mmol), 5g was obtained in 96% yield (0.197 g, 0.96 mmol) as a yellow oil after silica gel column chromatography (eluting with 8:2 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.1 (silica gel, 8:2 CHCl3:EtOAc); 1H and 13C NMR spectra are comparable to those reported in the literature.3
2-Fluoro-5-(piperidin-1-ylmethyl)pyridine (5h)
According to the general procedure with 6h (0.132 g, 1.0 mmol), 5h was obtained in 68% yield (0.132 g, 0.68 mmol) as a yellow oil after silica gel column chromatography (eluting with 8:2 CHCl3:EtOAc to 99:1 EtOAc:Et3N): Rf = 0.04 (silica gel, 8:2 CHCl3:EtOAc); ν(HATR)/cm−1 2937, 1598, 1483, 1245, 1114, 908, 858, 832, 755, 733; 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.78 (t, J = 8.1 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 3.44 (s, 2H), 2.36 (bs, 4H), 1.61–1.49 (m, 4H), 1.48–1.38 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 163.1 (d, J = 237.9 Hz), 147.8 (d, J = 14.5 Hz), 142.1 (d, J = 7.9 Hz), 132.0 (d, J = 4.4 Hz), 109.2 (d, J = 37.4 Hz) 60.1, 54.5, 26.0, 24.4; 19F NMR (471 MHz, CDCl3) δ −70.8; HRMS (ESI-TOF): calcd for C11H16FN2 + [M+H]+: 195.1298; found: 195.1293.
1-(4-Methoxybenzyl)piperidine (5b)
According to the general procedure with 6b (0.142 g, 1.0 mmol), 5b was obtained in 90% yield (0.185 g, 0.90 mmol) as a yellow oil after silica gel column chromatography (eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.05 (silica gel, 7:1 hexanes:EtOAc with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.2
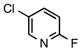
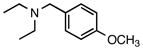
N-Ethyl-N-(4-methoxybenzyl)ethanamine (7a)
According to the general procedure with 2b (0.186 g, 1.2 mmol) and 6b (0.142 g, 1.0 mmol), 7a was obtained in 63% yield (0.122 g, 0.63 mmol) as a yellow oil after silica gel column chromatography (eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.08 (silica gel, hexanes:EtOAc 7:1 with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.3
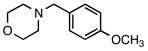
4-(4-Methoxybenzyl)morpholine (7b)
According to the general procedure with 2c (0.203 g, 1.2 mmol) and 6b (0.142 g, 1.0 mmol), 7b was obtained in 85% yield (0.176 g, 0.85 mmol) as a yellow oil after silica gel column chromatography (eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.05 (silica gel, 7:1 hexanes:EtOAc with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.2
N-Benzyl-1-(4-methoxyphenyl)-N-methylmethanamine (7c)
According to the general procedure with 2d (0.244 g, 1.2 mmol) and 6b (0.142 g, 1.0 mmol), 7c was obtained in 87% yield (0.211 g, 0.87 mmol) as a clear, colorless oil after silica gel column chromatography (eluting with 9:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.4 (silica gel, 7:1 hexanes:EtOAc with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.2
tert-Butyl-4-(4-methoxybenzyl)piperazine-1-carboxylate (7d)
According to the general procedure and reaction with 2e (0.322 g, 1.2 mmol) and 6b (0.142 g, 1.0 mmol), 7d was obtained in 98% yield (0.302 g, 0.98 mmol) as a white solid after silica gel column chromatography (eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.09 (silica gel, hexanes:EtOAc 7:1 with 0.2% Et3N); mp 55–57 °C; 1H and 13C NMR spectra are comparable to those reported in the literature.2
1-(4-Methoxybenzyl)-4-methylpiperazine (7e)
According to the general procedure and reaction with 2f (0.218g, 1.2 mmol) and 6b (0.142 g, 1.0 mmol) 7e was obtained in 76% yield (0.168 g, 0.76 mmol) as a dark yellow oil after silica gel column chromatography (eluting with 8:2 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.4 (silica gel, EtOAc:Et3N 99:1); 1H and 13C NMR spectra are comparable to those reported in the literature.3
N-(4-Methoxybenzyl)-N-methylcyclohexanamine (7f)
According to the general procedure with 2g (0.234 g, 1.2 mmol) and 6b (0.142 g, 1.0 mmol), 7f was obtained in 81% yield (0.189 g, 0.81 mmol) as a clear orange oil after silica gel column chromatography (eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.08 (silica gel, hexanes:EtOAc 7:1 with 0.2% Et3N); 1H and 13C NMR spectra are comparable to those reported in the literature.3
4-(4-Methoxybenzyl)thiomorpholine (7g)
According to the general procedure with 2h (0.222g, 1.2 mmol) and 6b (0.142 g, 1.0 mmol), 7g was obtained in 72% yield (0.160 g, 0.72 mmol) as a yellow oil after silica gel column chromatography (eluting with 7:1 hexanes:EtOAc with 0.2% Et3N to 1:1 hexanes:EtOAc with 0.2% Et3N): Rf = 0.4 (silica gel, hexanes:EtOAc 7:1 with 0.2% Et3N); ν(HATR)/cm−1 2908, 2803, 1611, 1511, 1242, 1035, 957, 823; 1H NMR (500 MHz, CDCl3) δ 7.21 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 3.80 (s, 3H), 3.45 (s, 2H), 2.74–2.61 (m, 8H); 13C NMR (126 MHz, CDCl3) δ 158.8, 130.3, 130.1, 113.7, 63.2, 55.3, 54.9, 28.1; HRMS (ESI-TOF): calcd for C12H18NOS+ [M+H]+: 224.1109; found: 224.1103.
3-(Piperidin-1-ylmethyl)pyridine (5i)
According to the general procedure with 6i (0.114 g, 1.0 mmol), 5i was obtained in 61% yield (0.107 g, 0.61 mmol) as an orange oil after silica gel column chromatography (eluting with 9:1 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.4 (silica gel, EtOAc:Et3N 99:1); 1H and 13C NMR spectra are comparable to those reported in the literature.2
N-Ethyl-N-(pyridin-3-ylmethyl)ethanamine (8a)
According to the general procedure with 2b (0.186 g, 1.2 mmol) and 6i (0.114 g, 1.0 mmol), 8a was obtained in 30% yield (0.050 g, 0.30 mmol) as an orange oil after silica gel column chromatography (eluting with 9:1 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.4 (silica gel, EtOAc:Et3N 99:1); ν(HATR)/cm−1 3029, 2966, 2935, 2806, 1576, 1424, 1201, 714; 1H NMR (500 MHz, CDCl3) δ 8.54 (d, J = 1.5 Hz, 1H), 8.48 (dd, J = 4.7, 1.3 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.24 (dd, J = 7.7, 4.8 Hz, 1H), 3.57 (s, 2H), 2.52 (q, J = 7.1 Hz, 4H), 1.04 (t, J = 7.1 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 150.4, 148.4, 136.6, 135.5, 123.4, 55.0, 46.9, 11.9; HRMS (ESI-TOF): calcd for C10H17N2 + [M+H]+: 165.1392; found: 165.1389.
4-(Pyridin-3-ylmethyl)morpholine (8b)
According to the general procedure with 2c (0.203 g, 1.2 mmol) and 6i (0.114 g, 1.0 mmol), 8b was obtained in 64% yield (0.113 g, 0.63 mmol) as an orange oil after silica gel column chromatography (eluting with 9:1 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.06 (silica gel, CHCl3:EtOAc 8:2); ν(HATR)/cm−1 3416, 2809, 1115, 1007, 865, 715; 1H NMR (500 MHz, CD3CN) δ 8.54–8.39 (m, 2H), 7.69 (s, 1H), 7.30 (s, 1H), 3.61 (s, 4H), 3.49 (s, 2H), 2.39 (s, 4H).; 13C NMR (126 MHz, CDCl3) δ 150.7, 148.9, 136.9, 133.4, 123.5, 67.1, 60.7, 53.7; HRMS (ESI-TOF): calcd for C10H15N2O+ [M+H]+: 179.1184; found: 179.1183.
N-Benzyl-N-methyl-1-(pyridin-3-yl)methanamine (8c)
According to the general procedure with 2d (0.244 g, 1.2 mmol) and 6i (0.114 g, 1.0 mmol), 8c was obtained in 83% yield (0.178 g, 0.83 mmol) as a pale, yellow oil after silica gel column chromatography (eluting with 9:1 CHCl3:EtOAc to 100% EtOAc): Rf = 0.2 (silica gel, CHCl3:EtOAc 8:2); ν(HATR)/cm−1 3028, 2788, 1576, 1453, 1425, 1367, 1023, 788, 740, 714, 699; 1H NMR (500 MHz, CDCl3) δ 8.57 (s, 1H), 8.49 (dd, J = 4.7, 1.4 Hz, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.40–7.28 (m, 4H), 7.28–7.20 (m, 2H), 3.53 (s, 2H), 3.50 (s, 2H), 2.18 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 150.4, 148.6, 139.0, 136.5, 134.8, 128.9, 128.4, 127.2, 123.4, 62.0, 58.9, 42.2; HRMS (ESI-TOF): calcd for C14H17N2 + [M+H]+: 213.1392; found: 213.1386.
tert-Butyl-4-(pyridin-3-ylmethyl)piperazine-1-carboxylate (8d)
According to the general procedure with 2e (0.322 g, 1.2 mmol) and 6i (0.114 g, 1.0 mmol, 8d was obtained in 99% yield (0.274 g, 0.99 mmol) as a yellow solid after silica gel column chromatography (eluting with 8:2 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.1 (silica gel, CHCl3:EtOAc 8:2); mp 93 °C (dec); ν(HATR)/cm−1 2975, 2950, 2809, 1679 (C=O), 1426, 1240, 1163, 1129, 998; 1H NMR (500 MHz, CDCl3) δ 8.53 (s, 1H), 8.52–8.48 (m, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.27–7.23 (m, 1H), 3.51 (s, 2H), 3.45–3.38 (m, 4H), 2.42–2.34 (m, 4H), 1.44 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 154.8, 150.6, 148.9, 136.7, 133.5, 123.4, 79.7, 60.3, 52.9, 44.2, 43.3, 28.5; HRMS (ESI-TOF): calcd for C15H24N3O2 + [M+H]+: 278.1869; found: 278.1860.
N-Methyl-N-(pyridin-3-ylmethyl)cyclohexanamine (8f)
According to the general procedure with 2g (0.234 g, 1.2 mmol) and 6i (0.114 g, 1.0 mmol), 8f was obtained in 75% yield (0.154 g, 0.75 mmol) as a clear, yellow oil after silica gel column chromatography (eluting with 9:1 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.2 (silica gel, CHCl3:EtOAc 8:2); ν(HATR)/cm−1 3030, 2928, 2853, 1450, 1426, 1026, 793, 714; 1H NMR (500 MHz, CDCl3) δ 8.52 (s, 1H), 8.48 (d, J = 4.7 Hz, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.23 (dd, J = 7.7, 4.9 Hz, 1H), 3.57 (s, 2H), 2.48–2.37 (m, 1H), 2.18 (s, 3H), 1.91–1.76 (m, 4H), 1.68–1.58 (m, 1H), 1.37–1.16 (m, 4H), 1.17–1.05 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 150.4, 148.4, 136.5, 136.0, 123.4, 62.8, 55.3, 37.7, 28.8, 26.5, 26.1; HRMS (ESI-TOF): calcd for C13H21N2 + [M+H]+: 205.1705; found: 205.1706.
4-(Pyridin-3-ylmethyl)thiomorpholine (8g)
According to the general procedure with 2h (0.222 g, 1.2 mmol) and 6i (0.114 g, 1.0 mmol), 8g was obtained in 39% yield (0.075 g, 0.39 mmol) as a dark orange oil after silica gel column chromatography (eluting with 8:2 CHCl3:EtOAc to 98:2 EtOAc:Et3N): Rf = 0.10 (silica gel, CHCl3:EtOAc 8:2); ν(HATR)/cm−1 3025, 2925, 2806, 1457, 1424, 1286, 1101, 1006, 955, 801, 708; 1H NMR (500 MHz, CDCl3) δ 8.50 (s, 1H), 8.47 (d, J = 3.7 Hz, 1H), 7.61 (d, J = 7.7 Hz, 1H), 7.22 (dd, J = 7.6 Hz, 4.9, 1H), 3.49 (s, 2H), 2.72–2.59 (m, 8H); 13C NMR (126 MHz, CDCl3) δ 150.5, 148.8, 136.7, 133.7, 123.5, 61.0, 55.0, 28.1; HRMS (ESI-TOF): calcd for C10H15N2S+ [M+H]+: 195.0956; found: 195.0959.
Supplementary Material
Acknowledgments
This research was supported by the NIH (R01 GM-081376), and Sigma-Aldrich. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data, and Dr. Patrick Carroll (University of Pennsylvania) is acknowledged for obtaining X-ray data.
Footnotes
Supporting Information Available: Full experimental details and NMR spectra for all compounds and X-ray crystal structure data for compound 2a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Molander GA, Ham J. Org Lett. 2006;8:2031–2034. doi: 10.1021/ol060375a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molander GA, Sandrock DL. Org Lett. 2007;9:1597–1600. doi: 10.1021/ol070543e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molander GA, Gormisky PE, Sandrock DL. J Org Chem. 2008;73:2052–2057. doi: 10.1021/jo800183q. [DOI] [PubMed] [Google Scholar]
- 4.Jensen MS, Yang C, Hsiao Y, Rivera N, Wells KM, Chung JYL, Yasuda N, Hughes DL, Reider PJ. Org Lett. 2000;2:1081–1084. doi: 10.1021/ol005641d. [DOI] [PubMed] [Google Scholar]
- 5.Compare 2,779,133 commercially available aryl- and heteroaryl chlorides versus 397,551 commercially available aryl- and heteroaryl nitriles and aldehydes. SciFinder, version 2010, Chemical Abstracts Service: Columbus, OH, 2010. (as of December 28, 2010).
- 6.Schaumann E. In: Science of Synthesis: Houben-Weyl Methods of Molecular Transformations. Enders D, editor. Vol. 40. Thieme; New York: 2008. pp. 7–411. [Google Scholar]
- 7.Molander GA, Hiebel MA. Org Lett. 2010;12:4876–4879. doi: 10.1021/ol102039c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.After this observation was made, the collected data was not refined to a final structure.
- 9.Molander GA, Canturk B. Org Lett. 2008;10:2135–2138. doi: 10.1021/ol800532p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whiting A. Tetrahedron Lett. 1991;32:1503–1506. [Google Scholar]
- 11.Sandrock DL. PhD Dissertation. University of Pennsylvania; Philadelphia, PA: 2010. Suzuki–Miyaura Cross-Coupling Reactions and Method Development of Potassium Organotrifluoroborates. [Google Scholar]
- 12.Molander GA, Canturk B, Kennedy LE. J Org Chem. 2009;74:973–980. doi: 10.1021/jo802590b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.XPhos = 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.