

Abstract
Stromal fibroblasts regulate epithelial cell behavior through direct and indirect cell–cell interactions. To clarify the role of TGF-β signaling in stromal fibroblasts during mammary development and tumorigenesis, we conditionally knocked out the TGF-β type II receptor gene in mouse mammary fibroblasts (Tgfbr2fspKO). Tgfbr2fspKO mice exhibit defective mammary ductal development, characterized in part by increased ductal epithelial cell turnover associated with an increase in stromal fibroblast abundance. Tgfbr2fspKO mammary fibroblasts transplanted with mammary carcinoma cells promote growth and invasion, which is associated with increased activating phosphorylation of the receptors: erbB1, erbB2, RON, and c-Met. Furthermore, the increased receptor phosphorylation correlates with increased secretion of the cognate ligands by Tgfbr2fspKO fibroblasts. Treatment of tumor cells with fibroblast-conditioned medium leads to increased tumor cell proliferation and motility, which are blocked by addition of pharmacologic inhibitors of TGF-α signaling or neutralizing antibodies to macrophage-stimulating protein (MSP), HGF, or c-Met. These studies characterize a significant role for stromal TGF-β signaling in mammary tissue homeostasis and mammary tumor progression via regulation of TGF-α, MSP, and HGF signaling pathways.
Keywords: TGF-β, TGF-β receptor type II, mammary gland, tumorβstromal interactions, tumor progression, subrenal grafting
Introduction
Development and maintenance of normal epithelial tissue requires a continuous exchange of signals within the surrounding stromal environment. Tissue fibroblasts comprise a major cell type in stroma, and their function in regulating blood vessel formation, extracellular matrix deposition, protease activity, and growth factor expression determines the differentiation states of many tissues including the mammary epithelium (Cunha and Hom, 1996; Silberstein, 2001; Simian et al., 2001; Haslam and Woodward, 2003). Abnormal changes in the stromal environment can cause profound reactions in the mammary epithelium, contributing to transformation and malignancy (Wiseman and Werb, 2002; Kuperwasser et al., 2004).
The impact of the stromal environment has been well demonstrated in the mammary gland. In early studies of breast cancer, it was shown that grafting of embryonic mammary mesenchymal tissue to adult mammary gland promoted tumor growth (Sakakura et al., 1981). In vitro and in vivo studies showed that the presence of tumor-derived fibroblasts accelerated mammary tumor growth and invasion in vivo, while fibroblasts from normal mammary tissue retarded tumor cell activity (Camps et al., 1990; Shekhar et al., 2001; Sadlonova et al., 2004). These studies indicate that phenotypic changes in stromal fibroblasts significantly influence epithelial cell fate. Similar tumor-promoting effects of fibroblasts have been reported with malignant human and mouse prostate cells, melanoma, lymphoma, and bladder cancer cells (Leonard et al., 1987; Camps et al., 1990; Tuxhorn et al., 2002a), indicating that stromal changes affect multiple tissues. In cancer patients, poor prognosis is often associated with desmoplasia, a pro-tumorigenic phenotype characterized by an abundance of fibroblasts, remodeling of the ECM and induction of tumor angiogenesis (Wiseman and Werb, 2002; De Wever and Mareel, 2003). Moreover, in breast cancer patients, upregulated secretion of motility factors and loss of heterozygosity of putative tumor suppressor genes in stromal cells have frequently been detected, indicating that genetic changes in the stroma may precede genotypic changes in the mammary epithelium (Schor et al., 1994; Moinfar et al., 2000). As such studies indicate the significant contributions of stromal cells on tumor cell behavior, it would be of great interest to understand the molecular mechanisms governing stromal–epithelial interactions.
A number of studies have investigated the molecular mechanisms governing stromal–epithelial interactions and TGF-β signaling has emerged as one key regulator of these interactions. TGF-β signaling exerts multiple effects in both stromal and epithelial cells including inhibiting cell proliferation, promoting motility and regulating differentiation. All these cellular functions are achieved by binding of the TGF-β ligand to its cell surface type II receptor, which leads to recruitment and activation of its type I receptor and subsequent downstream signaling in the cytoplasm of multiple pathways including SMAD, MAPK and Rho pathways (Daniel et al., 2001; Pollard, 2001). In vitro, TGF-β activates fibroblasts to a myofibroblast state, inducing production of growth factors, angiogenic factors, extracellular matrix (ECM) proteins and proteases (Derynck et al., 2001; Ihn, 2002; De Wever and Mareel, 2003), a phenotype observed during desmoplasia. Overexpression in transgenic mice of a kinase-deficient TGF-β type II receptor (T RII) in the mammary stroma results in mammary epithelial hyperplasia and increased hepatocyte growth factor (HGF) expression (Joseph et al., 1999), while increased TGF-β expression and activity often correlates with the occurrence of desmoplasia (Hagedorn et al., 2001; Lei et al., 2002; Tuxhorn et al., 2002b). As previous studies have shown a connection between TGF-β signaling to increased desmoplasia and tumor malignancy, the TGF-β signaling pathway in stromal fibroblasts has been a suggested target in cancer therapies. However, the functions and mechanisms of stromal-derived TGF-β signaling have remained poorly defined. Recent studies in our laboratory investigated the effects of stromal–epithelial interactions in the prostate and forestomach by conditionally inactivating expression of the T RII in fibroblasts through deletion of exon 2 of the Tgfbr2 gene. Crossing transgenic mice expressing Cre under the control of the fibroblast-specific protein 1 (FSP1 or S100A4) promoter with mice homozygous for Floxed Tgfbr2 (Tgfbr2flox/flox) generated progeny deficient in stromal Tβ RII expression (Tgfbr2fspKO). While Tgfbr2fspKO mice did not exhibit phenotypic changes in lung, liver, and kidney tissues, they consistently developed squamous cell carcinoma of the forestomach and prostatic intraepithelial neoplasia (Chytil et al., 2002; Bhowmick et al., 2004). These observations indicate that suppression of fibroblastic TGF-β signaling may impair normal epithelial cell fate of specific tissue types.
As the role for TGF-β signaling in fibroblasts on mammary development and tumor progression remained unclear, we characterized the in vivo and in vitro effects of Tβ RII-deficient fibroblasts on the mammary epithelium. Female Tgfbr2fspKO mice exhibit severe defects in mammary gland development, including increased ductal epithelial cell turnover and increased fibroblast abundance. Mammary carcinoma cells co-implanted with Tgfbr2fspKO fibroblasts in vivo exhibited increased tumor growth and invasion associated with increased tumor cell survival, proliferation, and tumor angiogenesis. Furthermore, we demonstrate that loss of TGF-β signaling in fibroblasts results in increased TGF-α, macrophage-stimulating protein (MSP), and HGF production, activating pathways that contribute to increased malignant tumor cell proliferation and motility in vivo and in vitro. Our studies provide strong evidence that inhibiting TGF-β signaling in stromal fibroblasts may serve to alter stromal–epithelial paracrine signaling interactions in mammary tissues and enhance mammary tumor progression. Therefore, it is important to thoroughly understand the function of TGF-β signaling in tumor progression and its role in stromal–epithelial interactions in order to design new drugs with improved effectiveness and specificity.
Results
FSP1 promoter activity is exclusive to fibroblasts in the mammary gland
The specificity of the FSP1 promoter in mouse mammary tissues was determined by examining expression patterns of FSP1 activity in transgenic reporter mice expressing green fluorescent protein (GFP) under the control of the FSP1 promoter (FSP.GFP). Mammary gland development in FSP.GFP mice was normal and comparable to wild-type (WT) mice. GFP-expressing cells were detected throughout the stromal compartment, but were not detected in myoepithelial cells, epithelial cells, or adipocytes (Figure 1a). Similar expression patterns driven by FSP1 promoter activity were also detected in mammary glands of ROSA26A Cre reporter mice that showed lacZ staining specifically in FSP1-expressing cells (Figure 1b), indicating that FSP1 promoter activity was specific to fibroblasts in the mammary gland. A population of fibroblastic cells that did not express GFP or lacZ was detected (Figure 1a and b), indicating that the FSP1 promoter was not active in all stromal cells and indicating heterogeneity of fibroblastic cell types in the gland. PCR analyses of stromal regions micro-dissected from mammary tissue and cultured fibroblasts from Tgfbr2fspKO and Tgfbr2flox/flox mice showed that excision of exon 2 of the Tgfbr2 gene occurred in stromal cells in the mammary gland of Tgfbr2fspKO mice (Figure 1c and d) and was absent in the mammary gland of Tgfbr2flox/flox mice. To determine whether excision of exon 2 of Tgfbr2 was specific to stromal cells, mammary epithelial tissue was also microdissected from Tgfbr2fspKO mice and examined for Tgfbr2 allelic recombination by PCR. Tgfbr2fspKO mammary epithelial cells were also extracted and placed in primary culture. As shown in Figure 1e and f, Tgfbr2fspKO mammary epithelial tissue and cultured cells do not show recombination of the Tgfbr2 allele, in comparison to stromal tissue and cultured stromal cells of Tgfbr2fspKO. These results indicate that deletion of exon 2 of Tgfbr2 was specific to stromal cells.
Figure 1.

FSP1 promoter activity is exclusive to fibroblasts in the mammary gland. (a) Analysis of GFP expression in the stromal cells of mammary tissue from FSP.GFP transgenic reporter mice as visualized under fluorescence microscopy. GFP-expressing cells are indicated by arrowheads. Non-GFP-expressing stromal cells are indicated by arrows. (b) Histological analysis of lacZ staining in stromal cells from mammary tissue sections of ROSA26A Cre reporter mice as indicated by arrowheads. Non-lacZ-stained stromal cells are indicated by arrows. Scale bars represent 40 μm. (c) FSP.Cre-mediated recombination in fibroblasts was determined by PCR analyses of DNA extracted from the mammary stromal tissue of Tgfbr2fspKO and control mice. Stromal tissue was micro-dissected by laser capture from mammary tissue sections as indicated by arrowheads. Left and right panels indicate before and after laser capture microdissection, respectively. DNA, extracted for PCR analysis, was used to address recombination of the Tgfbr2 allele in the mammary stroma of Tgfbr2fspKO mice, as indicated by arrowheads. The positive control was generated using DNA extracted from PyVmT cells containing floxed Tgfbr2 and expressing Cre under the control of the MMTV promoter. (d) Recombination of Tgfbr2 alleles in cultured mammary fibroblasts was verified by PCR. (e) Specificity of FSP.Cre-mediated recombination was determined by PCR analyses of DNA extracted from mammary stromal (S) and epithelial (E) tissue of Tgfbr2fspKO mice. Epithelial tissue was micro-dissected by laser capture from mammary tissue sections as indicated by arrowheads and subjected to PCR analyses for recombination of the Tgfbr2 allele. Left and right panels indicate before and after laser capture microdissection, respectively. The positive control was generated using DNA extracted from PyVmT cells containing floxed Tgfbr2 and expressing Cre under the control of the MMTV promoter. (f) Specificity of recombination of Tgfbr2 alleles was confirmed by PCR analyses on DNA extracted from cultured mammary fibroblasts and epithelial cells. The positive control was generated using DNA extracted from PyVmT cells containing floxed Tgfbr2 and expressing Cre under the control of the MMTV promoter. The negative control was generated using DNA extracted from PyVmT cells containing floxed Tgfbr2 and not expressing Cre.
Cre-mediated TβRII homozygous deletion in fibroblasts inhibits mammary ductal morphogenesis and terminal end bud formation
Whole-mount analyses of mammary gland tissue from female Tgfbr2fspKO mice at 6 weeks of age showed markedly reduced ductal elongation and branching associated with small terminal end buds (Figure 2a). In addition, 30% of homozygous knockout female mice examined exhibited mammary gland tissue devoid of mature ducts and terminal end buds, with a significant reduction of fat pad formation (data not shown). Mammary development in Tgfbr2flox/ flox mice appeared normal and comparable to WT mice. By crossing Tgfbr2fspKO with FSP.GFP mice to generate Tgfbr2fspKO progeny expressing GFP (Tgfbr2fspKO.GFP), we observed an increase in the number of fibroblasts expressing FSP.GFP in the mammary tissue of female Tgfbr2fspKO.GFP mice (Figure 2b). These results suggest that Tgfbr2 deletions blocked autocrine TGF-β signaling, resulting in increased fibroblast proliferation.
Figure 2.

Tgfbr2fspKO mice exhibit severe defects in mammary gland development. (a) Whole mounts of mammary gland tissue isolated from female Tgfbr2fspKO and Tgfbr2flox/flox mice at 6 weeks of age, showing differences in ductal development at low and high magnifications. In the Tgfbr2flox/flox, ducts have extended well beyond the central lymph node (*), but this is not the case in Tgfbr2fspKO mammary glands. Arrowheads indicate terminal end buds. (b) A significant increase in the number of fibroblasts was detected in the mammary glands of Tgfbr2fspKO.GFP mice (n=15), compared to Tgfbr2flox/flox mice (n=15), which was determined by quantification of FSP.GFP-expressing cells. Red oval indicates mean. (c) Quantitation of Ki67 immunostaining of mammary gland tissue from mice at 6 weeks of age indicates an increase in cell proliferation in the ductal epithelium (n=20) of Tgfbr2fspKO mice compared to Tgfbr2flox/flox (n=20). A decrease in cell proliferation was detected in the terminal end bud cells of Tgfbr2fspKO mice (n=13) compared to Tgfbr2flox/flox mice (n=16). Red oval indicates the mean. Statistical significance was determined two-tailed Student’s t-test. (d) Quantitation of cleaved caspase 3 immunostaining of mammary ducts and terminal ends of mammary gland tissue isolated from mice at 6 weeks of age indicate increase in ductal epithelial apoptosis in Tgfbr2fspKO mice (n=8) compared to Tgfbr2flox/flox mice (n=6). No significant change in apoptosis was detected in terminal end bud cells in Tgfbr2fspKO mice (n=16) compared to Tgfbr2flox/flox mice (n=16). Red oval indicates the mean. Statistical significance was determined by two-tailed Student’s t-test.
To further examine the effects of stromal-specific Tgfbr2 deletions on mammary development, we analysed the ductal epithelium for changes in cell proliferation and apoptosis. By immunostaining for the cell proliferation marker Ki67, we observed a statistically significant 1.8-fold increase in proliferation in the ductal epithelial cells in Tgfbr2fspKO mammary glands, while Ki67 staining was significantly decreased (1.4-fold) in the terminal end buds (Figure 2c). Analyses of apoptosis by caspase 3 immunostaining revealed a corresponding 2.5-fold increase in apoptosis in ductal epithelium with knockout of Tgfbr2 in fibroblasts (Figure 2d). There was no significant difference in caspase 3 staining in the terminal end buds between the knockout and control mice. These results suggested that loss of TGF-β signaling in the stroma altered paracrine signaling to the mammary epithelium, leading to increased cell turnover of the ductal epithelium and decreased proliferation of terminal end bud epithelium. It was not possible to carry out studies of the effect of Tgfbr2 deletion on mammary tumor formation in vivo because the Tgfbr2fspKO die at about 8 weeks of age, likely due to the forestomach carcinomas (24). Therefore, we characterized Tgfbr2fspKO mammary fibroblast in culture and examined their effect growth and behavior on mammary carcinoma cell line xenograft models.
Homozygous Tgfbr2 deletion in fibroblasts inhibits responsiveness to TGF-β
Fibroblast cell lines were generated by spontaneous immortalization of primary mammary fibroblasts and clonal populations of fibroblasts were obtained that were 100% recombined. By Southern blot analyses, recombination efficiencies of the floxed Tgfbr2 alleles in primary and immortalized (heterogeneous) Tgfbr2fspKO fibroblasts ranged from approximately 20 to 50%, whereas cloned immortalized cells exhibited 100% recombination (Figure 3a).
Figure 3.

Tgfbr2fspKO fibroblasts exhibit defective responses to TGF-β signaling. (a) Southern blot analysis of mammary fibroblast DNA showing recombination efficiencies of Tgfbr2 floxed alleles of DNA samples from primary fibroblasts (lanes 1–3), immortalized heterogenous (lanes 4–5), and immortalized clonal fibroblasts (lanes 6–7). One cell line representative of three Tgfbr2flox/flox and of three Tgfbr2fspKO clones examined is shown here. Numbered lanes are as follows: (1) Tgfbr2flox/flox, (2) Tgfbr2fspKO, (3) TgfbrfspKO, (4) Tgfbr2flox/flox, (5) Tgfbr2fspKO, (6) Tgfbr2flox/flox, (7) Tgfbr2fspKO, (8) control for Tgfbr2flox/flox, (9) control for Tgfbr2fspKO. Control DNA samples were isolated from MMTV.PyVmT transgenic mice expressing MMTV-Cre with Floxed Tgfbr2. Recombination efficiency was determined by phosphorimager analyses. (b) Primary or immortalized mammary fibroblasts from Tgfbr2flox/flox and Tgfbr2fspKO mice were treated with TGF-β and subjected to 3[H]thymidine incorporation. One representative of three Tgfbr2flox/flox and three Tgfbr2fspKO clones is shown here. (c) Primary or (d, e) immortalized fibroblasts from Tgfbr2flox/flox and Tgfbr2fspKO mice were treated with TGF-β, and counted by a hemocytometer at the indicated time-points.
We next compared TGF-β responsiveness between Tgfbr2fspKO and Tgfbr2flox/flox fibroblasts by [3H]thymidine incorporation. As shown in Figure 3b, Tgfbr2flox/flox fibroblasts respond to TGF-β treatment with a 50% reduction in [3H]thymidine incorporation, whereas Tgfbr2fspKO cells treated with TGF-β exhibited levels of [3H]thymidine incorporation comparable to untreated cells. Similar results were obtained by quantitation of cell numbers by a hemocytometer every 24 h (Figure 3c–e). These studies indicated that while rates of proliferation remained similar between untreated Tgfbr2fspKO cells and Tgfbr2flox/flox cells, cell proliferation was not inhibited in Tgfbr2fspKO cells treated with TGF-β. Primary and immortalized fibroblasts gave similar results. Responsiveness to TGF-β was also examined by Western blot analyses of TGF-β-inducible markers and pathways, including smooth muscle actin, fibronectin, and SMAD2 phosphorylation.
Tgfbr2fspKO fibroblasts promote mammary carcinoma growth and invasion in vivo
To address the role of TGF-β signaling by stromal cells on mammary tumor progression, we utilized mammary carcinoma cells, which were derived from transgenic mice overexpressing polyomavirus middle T antigen (PyVmT) under the control of the MMTV promoter. PyVmT tumor formation is rapid, and histologically analogous to human breast cancers, making it a good model in which to study mammary tumor progression (Guy et al., 1992; Cardiff, 2001a; Cardiff et al., 2001b; Maglione et al., 2001). Moreover, PyVmT mammary carcinoma cells have been shown to mimic the clinical behavior of human breast cancer cells including invasion and metastasis into multiple tissues (Guy et al., 1992; Lin et al., 2003). To prevent the potential interference of endogenous host fibroblasts, these PyVmT cells were implanted with either Tgfbr2fspKO or Tgfbr2flox/flox fibroblasts under the renal kidney capsules of nude mice, a site shown to be largely devoid of fibroblasts (Cunha et al., 2000). In the absence of fibroblasts, grafting of PyVmT tumor cells under the renal kidney capsule resulted in limited and insignificant growth and invasion, indicating that PyVmT tumor growth remained dependent on stromal interactions (data not shown). When PyVmT mammary tumor cells were implanted with primary and immortalized Tgfbr2fspKO fibroblasts, an approximate twofold increase in tumor mass was observed compared to PyVmT grafted with Tgfbr2flox/flox fibroblasts (Table 1). By H&E staining, we detected an increase in tumor invasion into the kidney parenchyma of PyVmT cells grafted with Tgfbr2fspKO fibroblasts (Figure 4a). To determine whether these effects were restricted specifically to the PyVmT cell line, the 4T1 mouse mammary carcinoma cell line (Aslakson and Miller, 1992) was also grafted with Tgfbr2fspKO or Tgfbr2flox/flox fibroblasts under the renal capsule. An approximately twofold increase in tumor mass of 4T1 cells grafted with Tgfbr2fspKO cells (data not shown) was observed, indicating that the effects of Tgfbr2fspKO stromal cells on mammary tumor growth were not restricted to the specific genetic background of PyVmT cells, but may represent a more global regulatory mechanism on tumor growth. Taken together, these data suggest that, in the absence of TGF-β signaling, stromal fibroblasts interact with tumor cells to enhance growth and invasion.
Table 1. Effect of loss of fibroblast TGF-β p signaling on growth of PyVmT subrenal capsule grafts.
PyVmT carcinoma cells were grafted with primary, heterogeneous immortalized, or clonal immortalized Tgfbr2 flox/flox or Tgfbr2fspKO fibroblasts in the subrenal capsule of nude mice and harvested 30 days later. Kidney tissue containing tumor masses were weighed. Statistical significance was determined by two-tailed Student's t-test
| Type of fibroblasts grafted with PyVmT | PyVmT:Tgfbr2flox/flox (mean tumor/kidney mass+s.d. (g)) | PyVmT:Tgfbr2flspKO (mean tumor/kidney mass+s.d. (g)) | Mean weight ratio PyVmT:Tgfbr2fspKO/PyVmT:Tgfbr2flox/fl°x | Student’s t-test P-value | n |
|---|---|---|---|---|---|
| Primary | 0.42+0.16 | 0.76+0.42 | 1.83 | 0.000015 | 10 |
| Immortalized (heterogeneous) | 0.46+0.1 | 1.06+0.8 | 2.16 | 0.013 | 8 |
| Immortalized (clonal) | 0.4+0.094 | 0.77+0.15 | 1.76 | 0.024 | 6 |
Figure 4.
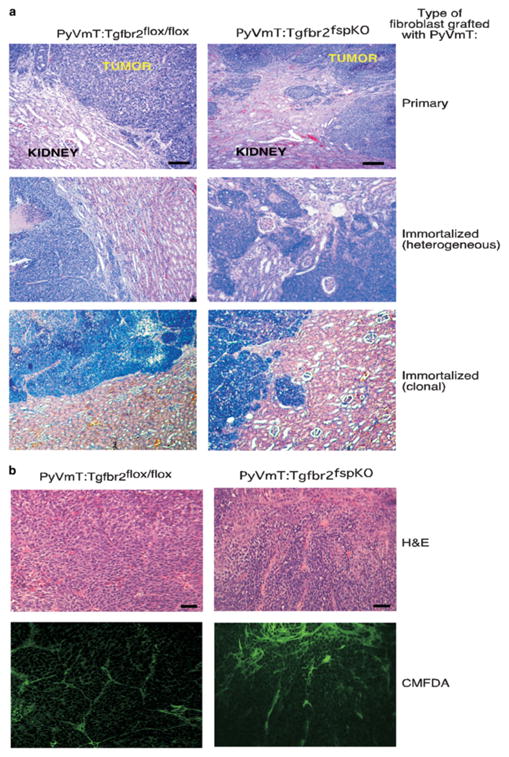
Subrenal capsule grafting of PyVmT tumor cells with Tgfbr2fspKO fibroblasts result in increased tumor growth and invasion in vivo. (a) H&E staining of tumor tissue isolated 30 days after grafting showing tumor invasion into the kidney parenchyma. Scale bars represent 40 μm. (b) Exogenous fibroblasts, not endogenous fibroblasts, are primary contributors to tumor growth. Tgfbr2flox/flox and Tgfbr2fspKO cells were labeled with CMFDA flourochrome and grafted with PyVmT. Tumors were harvested 30 days after grafting, formalin fixed and stained by H&E. Fibroblasts were visualized by fluorescent microscopy. To control for background fluorescence, sections, sections containing labeled fibroblasts were compared to tumor sections containing unlabeled fibroblasts; the latter showed no fluorescence. Scale bars represent 40 μm.
Previous studies have indicated the possibility that myofibroblasts can originate from multiple cell types including bone marrow-derived cells, smooth muscle pericytes, and myoepithelial cells, which contribute to tumor progression (Ronnov-Jessen et al., 1995). Therefore, we investigated the possibility that host cells contributed to the formation of the stromal compartment during tumor formation. Tgfbr2fspKO or Tgfbr2flox/flox fibroblasts were labeled with a flourochrome dye (CMFDA), which permits visualization of these cells by fluorescence microscopy, and were grafted with PyVmT tumor cells under the renal capsule. Tumors harvested 30 days post-implantation were analysed by histology and fluorescence microscopy. As shown in Figure 4b, the majority of the cells in the stromal compartment were composed of CFMDA-labeled fibroblasts, indicating that exogenous fibroblasts most likely contributed to tumor growth.
To further determine the effects of Tgfbr2fspKO fibroblasts on PyVmT tumor growth, we performed histological analyses to measure tumor cell proliferation, apoptosis, and tumor angiogenesis. Immunostaining of tumor tissue with Ki67 indicated a 1.8-fold increase in cell proliferation of PyVmT cells grafted with Tgfbr2fspKO fibroblasts, while immunostaining for TUNEL-positive nuclei revealed a 1.5-fold decrease in apoptosis (Figure 5a and b). By immunostaining for the endothelial cell marker CD31, we measured a 1.6-fold increase in blood vessel density in PyVmT cells grafted with Tgfbr2fspKO fibroblasts (Figure 5c). The data indicate enhancement of PyVmT tumor cell proliferation and survival, as well as increased tumor angiogenesis in the presence of Tgfbr2fspKO fibroblasts.
Figure 5.

PyVmT grafted with Tgfbr2fspKO cells exhibit increased tumor angiogenesis, increased proliferation and decreased apoptosis. Tumor tissue was harvested 30 days post grafting, formalin fixed and subjected in immunohistochemical staining for (a) Ki67 (b) apoptotic nuclei by TUNEL and (c). CD31 expression as indicated by arrowheads. Quantitation of CD31 immunostaining was determined by measuring pixel density using Scion Image Software of five fields of two sections per sample (n=18). Quantitation of Ki67 and TUNEL immunostaining were determined by counting number of peroxidase stained nuclei over the total number of hematoxylin-stained nuclei in five fields of two sections per sample (n=18). Scale bars represent 40 μm. Quantitation of PyVmT cells grafted with immortalized heterogeneous fibroblasts is depicted here. Statistical significance was determined by Poisson regression.
Tgfbr2fspKO fibroblasts increase receptor phosphorylation of erbB, RON and c-Met receptor kinases in tumor cells
As an increase in tumor growth suggested possible changes in paracrine signaling between Tgfbr2fspKO fibroblasts and tumor cells, we examined for changes in biochemical signaling pathways in PyVmT tumor cells. The erbB family has been shown to play a prominent role in mammary tumor progression, and studies have reported a potential role for the c-Met receptor kinases in stromal–epithelial interactions (Joseph et al., 1999; Pollard, 2001; Wiseman and Werb, 2002; Gorska et al., 2003; Roskoski, 2004; Wang et al., 2004). As shown in Figure 6a, PyVmT tumor cells grafted with Tgfbr2fspKO fibroblasts exhibit an increase (relative to those grafted with Tgfbr2flox/flox fibroblasts) in phosphorylation levels of erbB2 and erbB1, co-receptors for members of the EGF receptor family, as well as c-Met protein and RON, receptors for the members of the HGF ligand superfamily. The data suggest that Tgfbr2fspKO fibroblasts promote tumor growth by upregulating activity of growth factor receptors. We then determined whether the enhanced receptor kinase activity was due to increased ligand secretion by fibroblasts. Fibroblast-conditioned medium was assayed for expression of TGF-β, ligand for the erbB receptor family, as well as HGF and MSP, ligands for c-Met and Ron receptors, respectively. By ELISA assay (Figure 6b–d), we found that Tgfbr2fspKO fibroblasts express higher levels of TGF-α, MSP, and HGF than Tgfbr2flox/flox fibroblasts. Interestingly, we did not detect any differences in levels of secretion of TGF-β 1 between Tgfbr2fspKO and Tgfbr2flox/flox fibroblasts (data not shown). These results indicate that suppression of TGF-β signaling in fibroblasts increased expression of multiple growth factors to alter tumor receptor signaling.
Figure 6.

Tgfbr2fspKO fibroblasts increased receptor tyrosine kinase phosphorylation in PyVmT tumors. Cell lysates prepared from 30-day grafts containing PyVmT cells and fibroblasts were subjected to Western blot analysis for: (a) phosphorylation of the indicated cell surface receptors; (b) levels of soluble TGF-α, (c) MSP, and (d). HGF in conditioned media from primary and immortalized Tgfbr2flox/flox and Tgfbr2fspKO fibroblasts were determined by ELISA. Statistical significance was determined by two-tailed Student’s t-test.
TGF-β signaling in fibroblasts mediates TGF-α, MSP, and HGF signaling to regulate mammary tumor cell proliferation and migration
To determine the relative contribution of EGF, MSP, and HGF receptor signaling in regulating tumor cell activity, we examined the effects of fibroblast-conditioned medium on PyVmT cell proliferation by 3[H]thymidine incorporation and motility by wounding assay. As shown in Figure 7, PyVmT cells cultured with conditioned medium from Tgfbr2fspKO fibroblasts led to a significant increase in cell proliferation and motility, compared to treatment with conditioned medium from Tgfbr2flox/flox fibroblasts. A significant decrease in PyVmT cell proliferation and motility was observed upon addition of the EGF receptor tyrosine kinase inhibitor gefitinib (IRESSA, ZD1839) (Wakeling et al., 2002) to the conditioned medium from Tgfbr2fspKO fibroblasts as well as from Tgfbr2flox/flox fibroblasts. Further, upon addition of MSP, HGF or c-Met receptor neutralizing antibodies to fibroblast-conditioned medium, we detected a significant decrease in PyVmT cell proliferation and motility induced by conditioned medium from Tgfbr2fspKO fibroblasts, as well a modest decrease in proliferation and motility induced by conditioned medium from Tgfbr2flox/flox fibroblasts. Flow cytometry analysis of propidium iodide-stained PyVmT cells indicated that gefitinib did not affect cellular apoptosis, but inhibited PyVmT cell proliferation induced by fibroblast-conditioned medium by inhibiting the cell cycle (data not shown). These data are consistent with the 3[H]thymidine incorporation experiments, indicating a role for EGFR signaling in tumor cell proliferation. These results indicate that TGF-β signaling in fibroblasts mediates tumor cell growth and motility via a network of multiple signaling pathways, including HGF, MSP, and EGF-related ligands.
Figure 7.

Elevated levels of TGF-α, MSP, and HGF secretion by Tgfbr2fspKO fibroblasts contribute to PyVmT proliferation and migration. (a) PyVmT cells were treated with fibroblast-conditioned medium from Tgfbr2flox/flox and Tgfbr2fspKO clonal fibroblasts in the presence or absence of gefitinib, or neutralizing antibodies to HGF, c-Met, or MSP for 24 h, and proliferation was measured by [3H]thymidine incorporation. Statistical comparison of proliferation between treatment of PyVmT tumor cells with conditioned medium from Tgfbr2flox/flox and from Tgfbr2fspKO clonal fibroblasts was determined by two-tailed Student’s t-test. P-value significance is indicated by *. (b) PyVmT cells were wounded, and then treated with fibroblast-conditioned medium from Tgfbr2flox/flox and Tgfbr2fspKO clonal fibroblasts in the presence or absence of gefitinib, or neutralizing antibodies to HGF, c-Met, or MSP. Migration was assessed at 12 h by wound closure using Scion Image software. Results of conditioned medium experiments from one Tgfbr2flox/flox and one Tgfbr2fspKO cell line are depicted here and are representative of three Tgfbr2flox/flox and three Tgfbr2fspKO cell lines examined. Statistical comparison of migration between treatment of PyVmT with conditioned medium from Tgfbr2flox/flox and Tgfbr2fspKO clonal fibroblasts was determined by two-tailed Student’s t-test. P-value significance is indicated by *.
Discussion
Epithelial cells communicate with the stromal microenvironment through complex interacting signals among growth factors, growth factor receptors, extracellular matrix proteins, and proteases. Genetic alterations in stromal cells have been shown to contribute to changes in epithelial cell fate (Mueller and Fusenig, 2002; Wiseman and Werb, 2002; De Wever and Mareel, 2003) in part by altering paracrine signaling interactions between the stromal cells and epithelium. There is mounting evidence that TGF-β signaling in fibroblasts can regulate tumor growth in mouse models, as well as in human tissues (Wiseman and Werb, 2002; De Wever and Mareel, 2003; Kuperwasser et al., 2004); however, the effects and molecular mechanisms of TGF-β signaling in fibroblasts on mammary tumor progression have remained undefined. In this report, we demonstrate that suppression of the TGF-β response in stromal fibroblasts alter stromal–epithelial interactions via the TGF-α, MSP, and HGF signaling networks to disrupt normal mammary development and enhance tumor growth and cell motility. Increased expression of TGF-β and MSP by fibroblasts in response to loss of TGF-β signaling has not been reported previously, and the finding of increased HGF expression confirms the data from a recent report on stromal–epithelial interactions in the prostate and forestomach (Bhowmick et al., 2004). The data indicate that increased expression of multiple growth factors by the Tgfbr2fspKO fibroblasts is responsible for the striking effects these cells have on tumor progression. Further, these findings suggest that stromal TGF-β signaling possess dual roles in normal mammary development and tumor progression similar to the dual role found in epithelial TGF-β signaling (Akhurst and Derynck, 2001; Derynck et al., 2001; Akhurst, 2002; Tang et al., 2003).
In contrast to the mammary tumor growth-promoting effects of Tgfbr2fspKO fibroblasts, normal mammary ductal development was severely impaired in female Tgfbr2fspKO/fspKO mice. We did detect a significant reduction in adipocytes, an increase in the number of fibroblasts, and an increased ductal epithelial cell turnover, suggesting that loss of T RII in fibroblasts impaired normal activity of multiple cell types, along with altering paracrine signaling interactions between the stroma and epithelium. It is possible that a part of the mammary gland phenotype, such as reduced ductal elongation, is due to nutritional impairment secondary to the forestomach carcinomas. However, Tgfbr2fspKO mice maintaining the same size and weight of non-knockout littermates still demonstrated the mammary gland phenotype described. As shown in Figure 3, Tgfbr2fspKO fibroblasts do not respond to TGF-β inhibition of cell proliferation; therefore, it is not surprising that deficiency of T RII would result in increased growth of fibroblasts. As a subpopulation of mammary stromal cells expressed an FSP1 promoter and therefore Cre protein in TgfbrfspKO mice, it likely that this subset of stromal cells would be responsible for the increased numbers of fibroblasts. The increased levels of growth factor secretion by Tgfbr2fspKO fibroblasts are one likely mechanism responsible for increased ductal epithelial cell turnover, and previous studies have documented opposing regulatory mechanisms between TGF-β and HGF in the mammary epithelium (Pollard, 2001; Liu, 2004). It is also possible that Tgfbr2fspKO fibroblasts may express growth inhibitory factors responsible for retarded ductal outgrowth; expression profiling between Tgfbr2flox/flox andTgfbr2fspKO fibroblasts is currently underway to further identify genetic and phenotypic due to loss of TGF-β signaling in fibroblasts.
It is interesting to note that the composition of the stroma was severely affected in Tgfbr2fspKO mice. Normally, the mammary stroma is composed predominantly of adipocytes and fibroblasts (Cunha and Hom, 1996). The inverse proportion of adipocytes and fibroblasts in Tgfbr2fspKO mice suggests that TGF-β signaling may play a role in the differentiation of the mesenchyme during embryonic development. As FSP1 promoter activity has been shown to be active as early during embryonic day 8 in mesenchymal tissues (Strutz et al., 1995), it is possible that inhibition of TGF-β signaling in early mesenchymal tissues would affect differentiation of the fat pad precursor and mammary mesenchyme. These possibilities would be consistent with previous studies showing that TGF-β signaling regulates adipocyte and fibroblast differentiation in vitro. It is likely that the decreased numbers of adipocytes would also affect mammary gland development as adipocytes have been shown to secrete numerous growth factors and store steroid hormones influencing the mammary epithelium (Gregoire, 2001). Moreover, studies have shown that adipocyte tissue regulates mammary ductal outgrowth in vivo (Couldrey et al., 2002) and alveolar morphogenesis in vitro (Zangani et al., 1999). The decreased numbers of adipocytes would likely affect paracrine signaling interactions among adipocytes, fibroblasts, and the mammary epithelium. Thus, the combined effects of stemming from increased fibroblast growth and decreased adipocyte formation would severely retard epithelial ductal outgrowth and morphogenesis. While the mechanisms of fibroblast–adipocyte and adipocyte–epithelial interactions remain largely unclear, it would be of great interest in the future to further study these cellular interactions to further elucidate the mechanisms of mesenchymal differentiation during development, in particular the role of TGF-β signaling plays in the mesenchyme during mammary gland development.
Studies have shown cooperation between TGF-β and H-Ras as well as TGF-β and chemical carcinogens in the induction of mammary and hepatocellular carcinomas (Bottinger et al., 1997; Kanzler, 2001; Tang et al., 2003). Similarly, upon grafting Tgfbr2fspKO fibroblasts with PyVmT cells, it is possible that the increased growth factor secretion from Tgfbr2fspKO fibroblasts synergizes with the PyVmT, which transforms mammary epithelium through a number of pathways including: shc, src, PI-3 kinase, erbB, and cyclin D1 pathways (Guy et al., 1992; Webster et al., 1998; Lin et al., 2003), to enhance tumor growth and invasion. Tumor growth may also have been enhanced by increased blood vessel density due to the elevated levels of HGF and TGF-β, both of which possess potent angiogenic properties (Grotendorst et al., 1989; Zhang et al., 2003). Moreover, preliminary studies on the effects of Tgfbr2fspKO fibroblasts on mammary metastasis indicate a possible increase in tumor cell intravasation and lung metastases. These results suggest that loss of TGF-β signaling in fibroblasts promotes tumor progression via multiple signaling mechanisms.
As such, our studies demonstrate that the increase in mammary tumor growth and invasion by Tgfbr2fspKO fibroblasts is mediated by TGF-α, MSP, and HGF signaling pathways. Increases in protein levels of TGF-α, MSP, and HGF secreted by Tgfbr2fspKO fibroblasts correlated with increased phosphorylation of their respective receptor activity in tumor cells in vivo. To further elucidate the relative contributions of TGF-α-, MSP-, and Ron-mediated signaling pathways, in vitro studies involving treatment of PyVmT tumor cells with fibroblast-conditioned medium were performed. In such studies, we demonstrate that the elevated levels of growth factor secretion by Tgfbr2fspKO fibroblasts enhance tumor cell proliferation and migration; such contributions are abolished by addition of EGFR tyrosine kinase inhibitor or neutralizing antibodies to MSP and HGF and its c-Met receptor to the conditioned medium. While the relevance of erbB signaling in cancer has long been established (Graf and Beug, 1983; Maguire and Greene, 1989), these present studies characterize mechanisms of erbB signaling in the context of stromal–tumor interactions which have previously been unclear. The potent inhibition of PyVmT cell proliferation and migration caused by treatment with the EGFR inhibitor gefitinib indicates that an increase in erbB signaling significantly contributes to tumor growth and motility induced by Tgfbr2fspKO fibroblasts. As the PyVmT antigen has been shown to upregulate erbB receptor expression (Webster et al., 1998), the elevated levels of TGF-αsupplied by Tgfbr2fspKO likely enhance tumor growth and invasion through increased erbB signaling. Since TGF-αcan activate erbB2 and the EGF receptor tyrosine kinase inhibitor gefitinib has been shown to also block phosphorylation of erbB2 (Moulder et al., 2001), the effect of the small-molecule inhibitor could reflect inactivation of both erbB1 and erbB2. We also detected similar findings with HGF as addition of neutralizing antibodies to HGF and its receptor c-Met reduces tumor cell proliferation and migration induced by conditioned medium from Tgfb2fspKO fibroblasts.
These results are consistent with previous reports in which HGF-c-Met activity is upregulated in tissues where TGF-β activity is downregulated, most notably during mammary gland development where stromal tissues secrete HGF to regulate mammary ductal outgrowth (Pepper et al., 1995), and in mammary hyperplasia (Joseph et al., 1999). Our studies thus indicate that loss of TGF-β in fibroblast signaling leads to increased HGF-c-Met signaling via increased ligand expression to mediate mammary tumor cell growth and invasion. In a related manner, expression of another member of the HGF ligand family, MSP, is mediated by TGF-β signaling in fibroblasts. While the MSP has been shown to regulate macrophage activity during inflammatory events (Leonard and Skeel, 1979; Skeel and Leonard, 1994), much less is known about the signaling mechanisms of MSP and RON in tumor progression. Through in vivo studies and conditioned medium experiments, we demonstrate that loss of TGF-β signaling in fibroblasts results in increased RON signaling in mammary tumor cells contributing to tumor cell growth and motility. Crosstalk between RON and c-Met has been reported in ovarian cancer cell lines (Wang et al., 2002) in which heterodimerization of RON and c-Met may occur through ligand binding of either HGF or MSP. It is possible therefore that suppression of TGF-β signaling leads to synergy between HGF and MSP signaling in tumor cells and that neutralizing antibody binding of c-Met may also inhibit MSP signaling. Studies are underway to further elucidate the relevance of c-Met and RON co-signaling in TGF-β-mediated stromal–tumor cell interactions. These results support the possibility that suppression of TGF-β signaling in fibroblasts enhances tumor progression through synergism with multiple signaling mechanisms.
The strikingly different effects of Tgfbr2fspKO fibroblasts on normal and transformed mammary epithelium suggest complex dual roles of fibroblastic TGF-β signaling in regulating mammary epithelial cell behavior as both growth promoting and growth suppressive. Disruption of mammary gland development in Tgfbr2fspKO mice suggests that normal TGF-β signaling in fibroblasts functions to promote normal epithelial outgrowth via strict regulation of paracrine-signaling interactions between the stroma and epithelium. Our data suggest that suppression of TGF-β signaling in fibroblasts lead to higher levels of growth factors which would contribute to de-regulation of growth and apoptotic regulatory signals to the mammary epithelium, leading to retarded epithelial growth. Moreover, these findings suggest that while suppression of TGF-β signaling in fibroblasts lead to increased levels of growth factors, these changes are not sufficient to transform normal mammary epithelium to invasive carcinoma, as was detected in the forestomach of Tgfbr2fspKO mice (Bhowmick et al., 2004). Instead, suppression of TGF-β signaling in fibroblasts would require synergism with additional mutations, such as alterations in the growth and apoptotic pathways as detected in PyVmT tumor cells to promote mammary tumor growth. As autocrine TGF-β signaling in the epithelium during mammary tumor progression has been shown to be both tumor suppressive and tumor promoting (Akhurst and Derynck, 2001; Derynck et al., 2001), our findings suggest similar roles of growth suppression and promotion of fibroblastic TGF-β signaling in mammary tumor progression. Taken together, the effects of targeting TGF-β signaling in fibroblasts may not only be tissue specific but may depend on the transformative state of the epithelium.
In summary, our studies characterize mechanisms of fibroblastic TGF-β signaling in mammary tumor growth and invasion. We are currently investigating other possible mechanisms affected by changes in the genetic profile of Tgfbr2fspKO fibroblasts, including possible changes on extracellular matrix deposition and protease activity. It is of current interest to investigate the use of TGF-β inhibitors to block cancer progression in the field of molecular therapeutics (Wiseman and Werb, 2002; Siegel and Massague, 2003; Sun, 2004). Our studies demonstrate the complexities of the TGF-β signaling network in regulating stromal–epithelial interactions. Further understanding of the complex, multiple effects of TGF-β in both the stroma and tumor cells will facilitate the development and appropriate use of cancer therapies targeting the TGF-β pathways.
Materials and Methods
Mouse strains and maintenance
Female nude (nu/nu) mice were obtained from Harlan Mutant Mouse Research Center (North Carolina). FSP.GFP mice of BalbC background have been described (Iwano et al., 2002). Rosa26 lacZ reporter mice carry lacZ genes that were conditionally silenced by spliced donor sequences flanked by lox P sites (Soriano, 1999). Rosa26 Cre reporter mice were generated by crossing Rosa26 lacZ reporter mice (C57/Bl6 background) with FSP.Cre mice (CF57/Bl6 background). Animals positive for the lacZ transgene and for the Cre gene were identified by PCR analysis of genomic DNA from tail biopsy. The following primers were used to distinguish between WT and silenced lacZ genes: 5′-AAAGTCGCTCTGAGTTGGTTAT-3′, 5′-GCGAAGAGTTGTCCTCAACC-3′ and 5′-GGAGCGGGAGAAATGGATATG-3′ (Soriano, 1999). The following primers were used to identify the Cre gene: 5′-CCGGTTATTCAACTTGCACC-3′ and 5′-CTGCATTACCGGTCGATGCAAC′-3′. The Tgfbr2fspKO mice were generated by crossing FSP.Cre mice (CF57Bl/6) with Tgfbr2flox/flox mice (C57BL/6). Animals positive for the Cre gene and homozygous for the Floxed Tgfbr2 gene were identified by PCR analysis of genomic DNA from tail biopsy. The following primers were used to identify Floxed Tgfbr2: 5′-TAAACAAGGTCCGGAGCCCA-3′ and 5′-ACTTCTGCAAGAGGTCCCCT-3′, while the presence of the Cre gene was identified using primers described above. The Tgfbr2fspKO.GFP mice (C57BL/ 6) were generated by initially crossing Tgfbr2flox/flox mice (C57BL/6) with FSP.GFP mice (Balb/C) for three generations. Animals homozygous for Floxed Tgfbr2 gene and positive for GFP were then crossed with animals homozygous for the Tgfbr2 gene and positive for the Cre gene to generate Tgfbr2fspKO.GFP progeny. Tgfbr2fspKO.GFP mice were identified by PCR analyses for the presence of homozygous Floxed Tgfbr2 genes (primers described above), Cre genes (primers described above), and GFP genes. The following primers were used to identify GFP: 5′-GTGATTTGGGTCATGCTCAG-3′ and 5′-GAAACAGCTCCTCGCCCTTGC-3′ (Iwano et al., 2002). These animals were maintained in accordance with AAALC and Vanderbilt University guidelines.
Laser capture microdissection and PCR analyses
Cre-mediated recombination in mammary stromal tissue was assessed by laser capture dissection of mammary stromal tissue and PCR analyses of extracted DNA. Briefly, 10 m sections were prepared from paraffin-embedded mammary tissues from Tgfbr2fspKO or Tgfbr2flox/ flox mice, deparaffinized in xylenes and alcohols, and then stained in eosin. Stromal tissue was then micro-dissected by laser using the PixCell®IIe Capture Microdissection system (LCM) and DNA was extracted by PicoPure DNA extraction kit (Arcturus, Mountain View, CA, USA) according to the manufacturer’s protocol. Cre-mediated recombination of exon 2 of the Tgfbr2 gene was assessed by PCR using the following primers: 5′-AGGGATGAATGGGCTTGCTT-3′ and 5′-CTCACCTCAGAGCCTGATTA-3′.
Fibroblast and tumor cell culture
Primary fibroblasts were isolated from the thoracic and inguinal mammary glands from 6-week-old Tgfbr2flox/flox and Tgfbr2fspKO mice using a modified protocol as described (Bitzer et al., 2000). Fibroblasts were cultured onto 10 mm dishes coated with rat tail collagen type I (BD Biosciences, Palo Alto, CA, USA) in DMEM/F12 medium containing 5% adult bovine serum (ABS), for a minimum of three passages prior to in vitro and in vivo studies. PyVmT cells were isolated from PyVmT transgenic mice on an FVB genetic background as described (Medina and Kittrell, 2000). Briefly, mammary tumors from MMTV-PyVmT mice were digested in collagenase and hyalorinidase and cultured on plastic in DMEM media containing 2% ABS with antibiotics. Cultured cells were differentially trypsinized to exclude stromal cells and cultured in DMEM/2% ABS to exclude the survival of stromal cells. The identity and purity of PyVmT tumor cells was confirmed by immunofluoresence staining for PyVmT expression.
Generation of immortalized fibroblasts
Floxed T RII and Tgfbr2fspKO fibroblasts were immortalized as described (Tolstonog et al., 2001). Primary Tgfbr2fspKO and Tgfbr2flox/flox fibroblasts were maintained in DMEM/F12/5% ABS on collagen-coated plates at complete confluency for an extended period of time before subpassage. This procedure was repeated until the cells underwent senescence; the cells were maintained through subsequent blast crises and were allowed to double over time. Immortalization of fibroblasts was determined by their ability to overcome cell–cell contact inhibition of proliferation, attachment and survival on plastic, and by their ability to survive in low-serum or serum-free conditions. A heterogeneous population of immortalized cells was analysed by Western blot analysis for expression of smooth muscle actin and vimentin and assayed for TGF-β responsiveness, as described below. Clonal populations of fibroblasts were obtained by FACS sorting of individual cells into 96-well plates. Tgfbr2flox/flox and Tgfbr2fspKO fibroblasts were distinguished by PCR to identify the presence of: Floxed Tgfbr2 (primers described above), Cre (primers described above), and to identify recombination of the Floxed Tgfbr2 alleles.
Southern blot analysis
Recombination of the Tgfbr2 allele was assessed by Southern blot analysis in cultured mammary fibroblasts isolated from Tgfbr2flox/flox and Tgfbr2fspKO mice according to the methods described by Chytil et al. (2002).
Cell proliferation
Fibroblast cell proliferation was measured by [3H]thymidine incorporation and by manual cell counting. To assay 3[H]thymidine incorporation, fibroblasts (20 000) were plated per well in 24-well plates (Nalge NUNC International, Rochester, NY, USA), starved for 24 h in growth-factor-deprived media, then incubated in DMEM/F12/5% ABS in the presence or absence of 5 ng/ml TGF-β 1 (R&D systems), treated with 1 μCi [3H]dTh for 24 h, and then assayed for 3[H]thymidine incorporation using a beta scintillation counter. To manually count cells, fibroblasts were plated and treated with TGF-β as described above, trypsinized, then counted by a hemocytometer at 0, 24 and 48 h. Measurements represent the mean number of cells per field by a hemocytometer.
Assessment of tumor progression
Grafting of collagen-embedded cells was performed according to the methods of Hayward et al. (1998). Briefly, 1 × 105 PyVmT cells were re-suspended together with 2.5 × 105 Tgfbr2fspKO or Tgfbr2flox/flox fibroblasts in 50 μl of collagen for one graft. The collagen-embedded cells were cultured in DMEM/F12 5% ABS for 24 h and then implanted under the renal capsule layer of the kidneys in female nude mice, 8–10 weeks of age. Tumor tissues were collected 30 days post-implantation, and weighed. Statistical significance was assessed by two-tailed, paired Student’s t-test.
Histology
Mammary tissue from Tgfbr2flox/flox and Tgfbr2fspKO mice were subjected to whole-mount staining according to the methods by Medina and Kittrell (2000). Briefly, mammary gland tissue was fixed in 4% paraformaldehyde at 4°C overnight and dehydrated in 70, 95, and 100% ethanol for 1 h each at 4°C. The tissue was then subjected to treatment in acetone, 100 and 95% ethanol, stained with hematoxylin for 3 h, and then dehydrated in 70, 90, and 100% ethanol, and 100% xylene.
To visualize β-galactosidase activity in Rosa26 Cre reporter mice, mammary tissue from tissue was fixed in 0.2% glutaraldehyde for 1 h at 4°C, permeabilized in PBS buffer containing 2 mm MgCl2, 0.01% sodium deoxycholate, and 0.02% NP-40 for 45 min, and then stained with PBS buffer containing 5 mm potassium ferricyanide and 5 mm potassium ferrocyanide, and 5 mg/ml X-gal for 12 h at 30°C. The treated tissue was further fixed in 4% paraformaldehyde for 1 h, dehydrated in 70% ethanol, embedded in paraffin, and sectioned to 5 μm thickness. Sections were counterstained with neutral Red.
To visualize GFP expression in FSP. GFP mice, mammary gland tissue was extracted, fixed in 4% paraformaldehyde 4°C overnight, and dehydrated in 70, 95, and 100% ethanol for 1 h each at 4°C. The tissue was then paraffin embedded, sectioned to 5 m thickness and counterstained with Hoescht dye. GFP expression was visualized by immunofluoresence microscopy.
Immunohistochemistry
Paraformaldehyde-fixed mammary tissues and tumor tissues were immunoperoxidase stained for cleaved caspase 3 (Cell Signaling Technologies), Ki67 (Pharmingen), and CD31 expression (Pharmingen) by the Vanderbilt Histology Core Facility. Apoptosis in tumor sections was analysed by TUNEL (Intergen) and visualized by peroxidase staining (Vectastain Elite kit, Vector Laboratories). Sections were counterstained with hematoxylin. Blood vessel density was calculated by determining the relative area of CD31-positive staining in at least five fields at 20 magnification, while proliferative and apoptotic indices were calculated by determining the relative area of positive stained cells to total number of cells in at least five fields at 20 magnification using Scion Image software. Statistical significance was determined by Poisson Regression by the Vanderbilt Biostatistics Core Facility.
Western blot analyses
Grafts containing PyVmT and fibroblasts were lysed in RIPA buffer supplemented with a protease inhibitor cocktail containing leupeptin, aprotinin, and pepstatin (Sigma, St Louis, MS, USA) and sodium orthovanadate (Sigma). In all, 50 g of protein were fractionated on 8–12% SDS–PAGE gels. The proteins were transferred to nitrocellulose membranes and then probed with antibodies (1 : 1000) to the phosphorylated and unphosphorylated forms of c-Met (Cell Signaling Technologies, Beverly, MA, USA), erbB1 (Cell Signaling Technologies), erbB2 (Cell Signaling Technologies), and RON (Santa Cruz Biotechnologies). Specific immunoreaction was detected using IgG antibodies conjugated to horseradish peroxidase (Amersham Pharmacia Biotechnology, Arlington Heights, IL, USA) and ECL Plus chemiluminesence (Amersham Pharmacia Biotechnology). Data are a representation of three independent samples per cell type.
Flourochrome labeling of cells
To label fibroblasts with the green flourochome CMFDA (Molecular Probes, Eugene, OR, USA), 250 000 fibroblasts were plated in a 6 mm dish for 24 h, washed with PBS, and then labeled with 3 m CMFDA for 45 min in PBS at 37°C. Excess dye was removed by extensive washing with PBS and fibroblasts were re-incubated with complete medium for a minimum for 4 h before recombining with PyVmT cells for subrenal graft assays.
Determination of TGF-α, MSP, and HGF levels
Immortalized and primary cultures of fibroblasts from Tgfbr2flox/flox and Tgfbr2fspKO mammary glands were grown in 10 mm plates in DMEM/F12 media containing 5% ABS and antibiotics. At 80% confluency, the cells were incubated in 4 ml per plate of DMEM/F12 media containing 0.1% ABS for 24 h. The conditioned medium was concentrated in YM80 Centricon (Amicon Millipore, Billerica, MA, USA) filtration units and assayed for HGF, MSP, and TGF-α by ELISA (R&D systems). Statistical significance was determined by two-tailed Student’s t-test.
Assessment of PyVmT-cell proliferation
PyVmT (20 000 cells) were cultured in 24-well plates and treated with fibroblast-conditioned medium from Tgfbr2flox/flox and Tgfbr2fspKO clonal fibroblasts in the presence or absence of 100 nm gefitinib or 500 ng/ml of neutralizing antibodies to HGF, c-Met, or MSP (R&D systems) for 24 h. Cells were then treated with 1 μCi of 3[H]thymidine and incubated for another 24 h before assaying for 3[H]thymidine incorporation. Experiments were repeated in triplicate using three different Tgfbr2flox/flox and Tgfbr2fspKO clonal fibroblast cell lines. Results of conditioned medium from one Tgfbr2flox/flox and one Tgfbr2fspKO cell line are depicted here and are representative of all cell lines examined. Statistical significance was determined by two-tailed Student’s t-test.
Assessment of PyVmT-cell migration
PyVmT (20 000 cells) were cultured in 24-well plates and treated with fibroblast-conditioned medium from Tgfbr2flox/flox and Tgfbr2fspKO clonal fibroblasts in the presence or absence of 100 nm gefitinib (kindly provided by Plan Wakeling, Astrazeneca Pharmaceuticals) and 500 ng/ ml of neutralizing antibodies to HGF, c-Met, and MSP (R&D systems) for 12 h. A vertical wound was made to the cells with a sterile pipet tip and phase-contrast images were taken of each sample at 0 and 12 h. Wound closure was assessed using Scion Image Software. Experiments were repeated in triplicate using three different Tgfbr2flox/flox and Tgfbr2fspKO clonal fibroblast cell lines. Results of conditioned medium from one Tgfbr2flox/flox and one Tgfbr2fspKO cell line are depicted here and are representative of all cell lines examined. Statistical significance was determined by two-tailed Student’s t-test.
Acknowledgments
This work was supported by grants number CA102162 and CA85492 (to HLM) from the National Cancer Institute DHHS, and by the TJ Martell Foundation. The following Shared Resources of the Vanderbilt-Ingram Cancer Center provided outstanding assistance and are supported by grant number P30 CA68485: Statistics and Human Acquisition and Pathology Cores. The Mouse Pathology Core is supported by the NIH grant AR41943. We thank Bonnie LaFleur for biostatistical assistance, Dana Brantley-Sieders, and Jin Chen for critical reading of this manuscript.
Abbreviations
- TβRII
TGF-β type II receptor
- PyVmT
polyomavirus middle T antigen
- MSP
macrophage-stimulating protein
- FSP1
fibroblast-specific protein 1
- GFP
green fluorescent protein
- ABS
adult bovine serum
References
- 1.Akhurst RJ. J Clin Invest. 2002;109:1533–1536. doi: 10.1172/JCI15970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akhurst RJ, Derynck R. Trends Cell Biol. 2001;11:S44–S50. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 3.Aslakson C, Miller FR. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- 4.Bhowmick N, Chytiil A, Plieth D, Gorska A, Dumont N, Shappel S, Washington M, Neilson E, Moses H. Science. 2004;303:847–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 5.Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M, Bottinger EP. Genes Dev. 2000;2:187–197. [PMC free article] [PubMed] [Google Scholar]
- 6.Bottinger EP, Jakubczak JL, Haines DC, Bagnall K, Wakefield LM. Cancer Res. 1997;57:5564–5570. [PubMed] [Google Scholar]
- 7.Camps J, Chang S, Hsu T, Freeman M, hong S, Zhau H, von Eschenbach A, Chung L. Proc Natl Acad Sci USA. 1990;87:75–79. doi: 10.1073/pnas.87.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardiff R. Microsc Res Tech. 2001a;52:224–230. doi: 10.1002/1097-0029(20010115)52:2<224::AID-JEMT1007>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 9.Cardiff R, Wagner U, Henninghausen L. Vet Pathol. 2001b;38:357–358. doi: 10.1354/vp.38-4-357. [DOI] [PubMed] [Google Scholar]
- 10.Chytil A, Magnuson MA, Wright CVE, Moses HL. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 11.Couldrey C, Moitra J, Vinson C, Anver M, Nagashima K, Green J. Dev Dyn. 2002;223:459–468. doi: 10.1002/dvdy.10065. [DOI] [PubMed] [Google Scholar]
- 12.Cunha G, Hom Y. J Mamm Gland Biol. 1996;1:21–34. doi: 10.1007/BF02096300. [DOI] [PubMed] [Google Scholar]
- 13.Cunha G, Hom YK, Young P, Brody J. In: Methods in Mammary Gland Biology. Ip MM, editor. Kluwer Academic/Plenum Publishers; New York: 2000. [Google Scholar]
- 14.Daniel CW, Robinson S, Silberstein GB. Bioactive Components of Human Milk. Plenum Publishers; New York: 2001. pp. 61–69. [Google Scholar]
- 15.De Wever O, Mareel M. J Pathol. 2003;4:429–447. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- 16.Derynck R, Akhurst RJ, Balmain A. Nature. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 17.Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL. Am J Pathol. 2003;163:1539–1549. doi: 10.1016/s0002-9440(10)63510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graf T, Beug H. Cell. 1983;34:7–9. doi: 10.1016/0092-8674(83)90130-7. [DOI] [PubMed] [Google Scholar]
- 19.Gregoire FM. Exp Biol Med. 2001;226:997–1002. doi: 10.1177/153537020122601106. [DOI] [PubMed] [Google Scholar]
- 20.Grotendorst G, Soma Y, Takehara K, Charette M. Cell Physiol. 1989;139:617–623. doi: 10.1002/jcp.1041390323. [DOI] [PubMed] [Google Scholar]
- 21.Guy C, Cardiff R, Muller W. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagedorn H, Bachmeir B, Nerlich A. Int J Oncol. 2001;18:669–681. doi: 10.3892/ijo.18.4.669. [DOI] [PubMed] [Google Scholar]
- 23.Haslam S, Woodward T. Breast Cancer Res. 2003;5:208–215. doi: 10.1186/bcr615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayward S, Haughney PC, Rosen MA, Greulich KM, Weier HU, Dahiya R, Cunha GR. Differentiation. 1998;63:131–140. doi: 10.1046/j.1432-0436.1998.6330131.x. [DOI] [PubMed] [Google Scholar]
- 25.Ihn H. Curr Opin Rheum. 2002;14:681–685. doi: 10.1097/00002281-200211000-00009. [DOI] [PubMed] [Google Scholar]
- 26.Iwano M, Plieth D, Danoff T, Xue C, Okada H, Neilson E. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joseph H, Gorska AE, Sohn P, Moses HL, Serra R. Mol Biol Cell. 1999;10:1221–1234. doi: 10.1091/mbc.10.4.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanzler S. Oncogene. 2001;16:5015–5024. doi: 10.1038/sj.onc.1204544. [DOI] [PubMed] [Google Scholar]
- 29.Kuperwasser C, Chavarria T, Wu G, Gray JW, Carey L, Richardson A, Weinberg RA. Proc Natl Acad Sci USA. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei X, Bandyopadhyay A, Le T, Sun L. Oncogene. 2002;21:7514–7523. doi: 10.1038/sj.onc.1205966. [DOI] [PubMed] [Google Scholar]
- 31.Leonard E, Skeel AH. Adv Exp Med Biol. 1979;121B:181–194. doi: 10.1007/978-1-4684-8914-9_16. [DOI] [PubMed] [Google Scholar]
- 32.Leonard J, Johnson D, Felsen R, Tanney LE, Royston I, Dillman R. Cancer Res. 1987;47:2899–2902. [PubMed] [Google Scholar]
- 33.Lin E, Jones J, Zhu L, Whitney K, Muller W, Pollard J. Am J Pathol. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y. Am J Physiol Renal Physiol. 2004;287:F7–F16. doi: 10.1152/ajprenal.00451.2003. [DOI] [PubMed] [Google Scholar]
- 35.Maglione J, Moghanak D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
- 36.Maguire HJ, Greene MI. Semin Oncol. 1989;16:148–155. [PubMed] [Google Scholar]
- 37.Medina D, Kittrell F. In: Methods in Mammary Gland Biology and Breast Cancer Research. Ip MM, Asch BB, editors. Kluwer Academic/Plenum Publishers; New York: 2000. pp. 137–147. [Google Scholar]
- 38.Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Cancer Res. 2000;60:2562–2566. [PubMed] [Google Scholar]
- 39.Moulder S, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Cancer Res. 2001;61:8887–8895. [PubMed] [Google Scholar]
- 40.Mueller M, Fusenig N. Differentiation. 2002;70:486–497. doi: 10.1046/j.1432-0436.2002.700903.x. [DOI] [PubMed] [Google Scholar]
- 41.Pepper M, Soriano JV, Menoud PA, Sappino AP, Orci L, Montesano R. Exp Cell Res. 1995;219:204–210. doi: 10.1006/excr.1995.1220. [DOI] [PubMed] [Google Scholar]
- 42.Pollard J. Breast Cancer Res. 2001;3:230–237. doi: 10.1186/bcr301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ronnov-Jessen L, Petersen O, Koteliansky V, Bissell M. J Clin Invest. 1995;95:859–873. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roskoski R., Jr Biochem Biophys Res Commun. 2004;319:1–11. doi: 10.1016/j.bbrc.2004.04.150. [DOI] [PubMed] [Google Scholar]
- 45.Sadlonova A, Novak Z, Johnson MR, Bowe DB, Gault SR, Page GP, Thottassery JV, Welch DR, Frost AR. Breast Cancer Res. 2004;7:R46–R59. doi: 10.1186/bcr949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakakura T, Sakagami Y, Nishizuka Y. J Natl Cancer Inst. 1981;66:953–959. [PubMed] [Google Scholar]
- 47.Schor AM, Rushton G, Ferguson JE, Howell A, Redford J, Schor SL. Int J Cancer. 1994;59:25–32. doi: 10.1002/ijc.2910590107. [DOI] [PubMed] [Google Scholar]
- 48.Shekhar MP, Werdell J, Santner SJ, Pauley RJ, Tait L. Cancer Res. 2001;61:1320–1326. [PubMed] [Google Scholar]
- 49.Siegel P, Massague J. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 50.Silberstein G. Breast Cancer Res. 2001;3:218–223. doi: 10.1186/bcr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simian M, Hirai Y, Navre M, Werb Z, Lochter A, Bissel MJ. Development. 2001;128:3117–3131. doi: 10.1242/dev.128.16.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skeel A, Leonard EJ. J Immunol. 1994;152:4618–4623. [PubMed] [Google Scholar]
- 53.Soriano P. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 54.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun L. Front Biosci. 2004;1:1925–1935. doi: 10.2741/1382. [DOI] [PubMed] [Google Scholar]
- 56.Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver M, Wakefield LM. J Clin Invest. 2003;112:1116–1124. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tolstonog GV, Shoeman RL, Traub U, Traub P. DNA Cell Biol. 2001;20:509–529. doi: 10.1089/104454901317094945. [DOI] [PubMed] [Google Scholar]
- 58.Tuxhorn J, Ayala G, Smith M, Smith V, Dang T, Rowley D. Clin Cancer Res. 2002a;8:2912–2923. [PubMed] [Google Scholar]
- 59.Tuxhorn J, McAlhany S, Yang F, Dang T, Rowley D. Cancer Res. 2002b;62:6021–6025. [PubMed] [Google Scholar]
- 60.Wakeling A, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. Cancer Res. 2002;62:5749–5754. [PubMed] [Google Scholar]
- 61.Wang D, Shen Q, Chen YQ, Wang MH. Oncogene. 2004;23:1668–1680. doi: 10.1038/sj.onc.1207282. [DOI] [PubMed] [Google Scholar]
- 62.Wang MH, Zhou YQ, Chen YQ. Scand J Immunol. 2002;56:545–553. doi: 10.1046/j.1365-3083.2002.01177.x. [DOI] [PubMed] [Google Scholar]
- 63.Webster M, Hutchinson JN, Rauh MJ, Muthuswamy SK, Anton A, Tortorice CG, Cardiff RD, Graham FL, Hassell JA, Muller WJ. Mol Cell Biol. 1998;18:2344–2359. doi: 10.1128/mcb.18.4.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiseman BS, Werb Z. Science. 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zangani D, Darcy KM, Shoemaker S, Ip MM. Exp Cell Res. 1999;247:399–409. doi: 10.1006/excr.1998.4373. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y, Su Y, Volpert OV, Vande Woude GF. Proc Natl Acad Sci USA. 2003;100:12718–12723. doi: 10.1073/pnas.2135113100. [DOI] [PMC free article] [PubMed] [Google Scholar]