

Abstract
In this study, various 3′R,4′R-disubstituted-2′,2′-dimethydihydropyrano[2,3-f]chromone (DSP) derivatives were discovered as potent chemosensitizers in the treatment of multidrug resistant cancer cells. Twenty-four DSP analogs (5–28) were synthesized and evaluated against a multi-drug resistant (MDR) cell line (KB-Vin) with and without vincristine (VCR). All DSP analogs exhibited low intrinsic cytotoxicity. However, in combination treatment, most DSPs reversed resistance to VCR and lowered the GI50 value of VCR by 12–349-fold. At a concentration of 1μg/mL, three compounds, 11, 14 and 21, fully reversed resistance to VCR in KB-Vin cancer cells, a twofold increase compared to verapamil, a first generation chemosensitizer. Detailed structure-activity relationship (SAR) conclusions were established based on 3′ and 4′ substitutions. Moreover, a preliminary mechanism study indicated that the chemosensitizing activity of DSP analogs results from inhibition of P-glycoprotein (P-gp) over-expressed in MDR cancer cells.1
Introduction
Resistance of cancer cells to anticancer drugs remains one of the major obstacles in achieving an effective treatment for cancer. Three primary mechanisms of anti-cancer drug resistance have been identified: decreased uptake of water-soluble drugs; various changes in cells that affect the capacity of cytotoxic drugs to kill cells; and the most commonly encountered, increased energy-dependent efflux of hydrophobic cytotoxic drugs by one of a family of energy-dependent transporters, such as P-gp (also known as MDR1).1 One approach used in clinical research to reverse MDR is the development of chemosensitizers, especially drugs that target P-gp and inhibit its activity. Verapamil and cyclosporine A (Figure 1) are examples of first generation chemosensitizers that inhibit the activity of P-gp and were evaluated clinically as adjuvants for chemotherapy. Both compounds are able to increase the intracellular concentration of cytotoxic agents, such as VCR, cyclophosphamide, dexamethasone, and doxorubicin, in MDR cells.2, 3 Moreover, both verapamil and cyclosporine A are widely used medications, verapamil as a calcium channel blocker to treat hypertension and cyclosporine A as an immunosuppressant to reduce the activity of the patient's immune system in post-allogeneic organ transplant. However, clinical studies showed that both compounds were generally toxic at the doses required to attenuate P-gp function.4 Since then, several potent and selective next generation inhibitors have been developed and investigated, but none of them have yet shown significant clinical benefit. Inhibition of P-gp by chemosensitizers may also be problematic because drug distribution and elimination can be affected by these agents. 4, 5 However, it is still possible that the ideal chemotype for a chemosensitizer has yet to be identified and developed.
Figure 1.

It has been reported that the naturally existing pyranocoumarin (±)-praeruptorin A (PA) and its khellactone analogs with 3′,4′-modifications (Figure 2) can reverse P-gp-mediated MDR.6, 7 (±)-3′-O-4′-O-bis(3,4-Dimethoxycinnamoyl)-cis-khellactone (DMDCK, 3) showed the highest potency among these analogs. At a concentration of 4 μM, 3 was able to increase the activity of the chemotherapy drug vinblastine over 110-fold.7 However, even at such a high concentration, 3 was unable to completely reverse vinblastine resistance in the liver cancer cell system used. 7 In addition, it was also reported that the side chains at the 3′ and 4′ positions of the khellactone system play an important role in maintaining high activity. Unfortunately, only a few khellactone analogs with 3′,4′ modifications have been reported to show potent chemosensitizing activity, so detailed SAR information is unavailable. Therefore, continuing research to develop novel active analogs is merited, and forms the basis for the work on the DSP series reported herein.
Figure 2.

Structures of (+/−) Praeruption A, DCK, MMDCK, DMDCK and DSP
Design
Even before the structure of P-gp protein was resolved,8 a general pharmacophore model for P-gp binding and inhibition was proposed based on verapamil.9 The pharmacophore consists of a butterfly-shaped molecule with two hydrophobic motifs (aromatic ring system) connected with a linker that contains hydrogen bond (HB) acceptors and/or donors. DMDCK (3) was subsequently shown to fit the pharmacophore model and could be readily docked into verapamil's hypothetical binding site on the protein subunit of P-gp.7, 9
DSPs (Figure 2) share certain structural similarities to DMDCK (3), which prompted us to design and synthesize a series of 3′,4′-disubstituted-pyranochromone derivatives as new chemosensitizing agents. It is likely that the pyranochromone moiety has similar HB properties to those of khellactone in DMDCK. Incorporation of two side chains at the 3′ and 4′ positions with substitutions containing aromatic moieties could mimic the hydrophobic moieties in verapamil and DMDCK (3). Based on this rationale, a series of 3′,4′-disubstituted-pyranochromone derivatives with varied 3′ and 4′ side chains were designed, synthesized, and screened against the MDR human esophageal cancer cell line (KB-Vin) with or without combination of the anticancer drug VCR. This approach allowed us to evaluate the intrinsic cytotoxicity of DSP analogs, as well as their ability to reverse MDR.
Chemistry
Compound 4, 3′R,4′R-2′,2′-dimethyl-dihydropyranochromone, is the core structure of all target compounds and a key intermediate in all synthetic pathways. The synthesis of 4 has been reported previously.10,11 Side chains with varied chain lengths and aromatic or non-aromatic functional groups at the chain terminus were introduced at the 3′ and 4′ positions of the core scaffold (Scheme 1).
Scheme 1.

Synthesis of 3′,4′-dibenzoyl-pyranodihydrochromone derivatives (5 - 28). Reagents and Condition: (i) diverse benzoyl chlorides, DMAP, anhydrous CH2Cl2, rt ;(ii) phenylacetyl chlorides or 3-phenylpropanoyl chloride, DMAP, anhydrous CH2Cl2, rt; (iii) trans-cinnamoyl chlorides, DMAP, CH2Cl2, rt; (iv) cyclohexanecarbonyl chloride, DMAP, anhydrous CH2Cl2, rt; (v) benzyl bromide, NaH/DMF, N2, 0°C; (vi) diverse dialkylcarbamyl chlorides or cyclocarbamyl chlorides, NaH, rt.
Compound 4 stirred with substituted benzoyl chloride or cyclohexyl carbonyl chloride in the presence of DMAP at room temperature to afford 5–16 and 23. Reaction of phenylacetyl chlorides or 3-phenylpropanoyl chloride with 4 gave 17–19, while reaction of 4 with trans-cinnamoyl chlorides afforded 20–21.
3′,4′-Dibenzyloxy substituted DSP (22) was generated by reacting 4 with benzyl bromide in the presence of NaH in DMF under N2 atmosphere (Scheme 1). 12
The synthesis of carbamates 24–28 was accomplished by treatment of 4 with N,N-dialkylcarbamylchloride or pyrrolidine and piperidine carbonyl chloride, respectively, in anhydrous THF in the presence of NaH (Scheme 1). 13
Results and Discussion
Chemosensitization Activity In Vitro
VCR is a potent mitotic inhibitor, and is widely used in combination chemotherapy of acute leukemias and some solid tumors. The GI50 value of VCR against the KB (drug-sensitive) cell line was 0.009 μg/mL (Table 1). Against the KB-Vin cell line, a multi-drug resistant cancer cell line, the cytotoxic effect of VCR decreases dramatically, over 270-fold, and its GI50 value is 2.44 μg/mL (Table 1). In order to determine the ability of DSPs to reverse MDR in KB-Vin cells, the cytotoxic effect of VCR was evaluated with or without the addition of each newly synthesized DSP analog.
Table 1.
Intrinsic cytotoxicity of indicated compounds and cytotoxicity of DSP + VCR formula
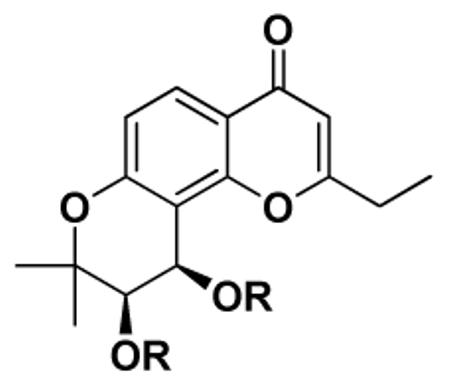 | |||||
|---|---|---|---|---|---|
| Compound used in combination |
R | GI50 of single drug formula (μg/mL) a |
GI50 of VCR + 1μg/mL DSP (or verapamil) (μg/mL) b |
Cytotoxicity fold increase of VCR c |
Molar Concentration (μM) d |
| 5 |
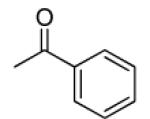
|
8.79 | >0.2 | <12.2 | 2.0 |
| 6 |
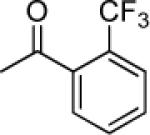
|
> 31.5 | >0.2 | < 12.2 | 1.58 |
| 7 |
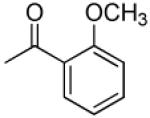
|
>31.5 | 0.087 | 28.1 | 1.79 |
| 8 |
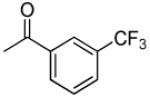
|
> 31.5 | >0.2 | < 12.2 | 1.58 |
| 9 |
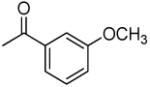
|
> 35.8 | 0.09 | 27.1 | 1.79 |
| 10 |
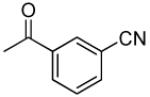
|
> 2.5 | 0.0152 | 160.5 | 1.82 |
| 11 |
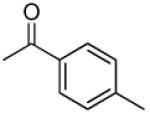
|
20.6 | 0.007 | 348.6 | 1.90 |
| 12 |
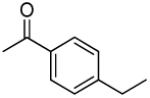
|
> 13.6 | 0.12 | 20.3 | 1.81 |
| 13 |

|
>31.5 | 0.15 | 16.3 | 1.58 |
| 14 |
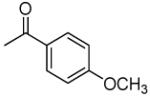
|
9.0 | 0.0095 | 256.8 | 1.79 |
| 15 |
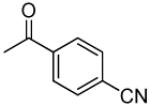
|
>5.5 | 0.013 | 187.7 | 1.82 |
| 16 |
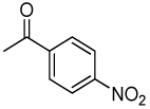
|
>34.0 | 0.012 | 203.3 | 1.70 |
| 17 |
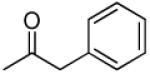
|
8.33 | 0.03 | 81.3 | 1.90 |
| 18 |
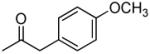
|
13.3 | 0.071 | 34.4 | 1.71 |
| 19 |
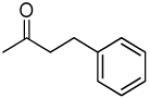
|
10.1 | 0.023 | 106.1 | 1.81 |
| 20 |
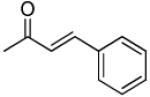
|
9.44 | 0.011 | 221.8 | 1.82 |
| 21 |
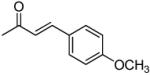
|
>36.4 | 0.0085 | 287.1 | 1.64 |
| 22 |
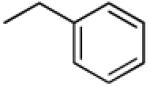
|
>15.7 | 0.060 | 40.7 | 2.13 |
| 23 |
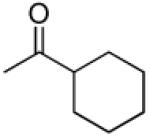
|
>20 | >0.2 | < 12.2 | 1.96 |
| 24 |
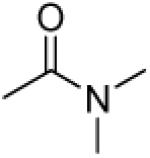
|
>20 | >0.2 | < 12.2 | 2.31 |
| 25 |
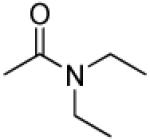
|
>20 | 0.021 | 116.2 | 2.05 |
| 26 |
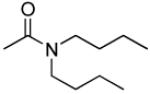
|
>20 | >0.2 | < 12.2 | 1.67 |
| 27 |
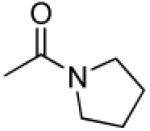
|
>20 | 0.098 | 24.9 | 2.06 |
| 28 |
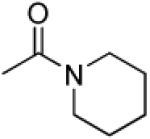
|
>20 | 0.11 | 22.2 | 1.95 |
| verapamil | >31.5 | 0.017 | 141.2 | 2.0 | |
| VCR | 2.44 | --- | --- | ||
|
VCR (in KB cell line) |
0.009 | ||||
Cytotoxicity of single compound as GI50 values for KB-Vin cell line or as indicated, the concentration that caused 50% reduction in absorbance at 562 nm relative to untreated cells using the sulforhodamine B assay.
Mean GI50 value of VCR + indicated compounds (1 μg/mL), from 2 or more independent tests (SD listed in supporting information).
Cytotoxicity fold increase = 2.44 (GI50 of VCR in KB-Vin) / GI50 of VCR+DSP formula
Molar concentration of DSP and verapamil that were used in the combination formula. Except for compounds 22, 24, 25 and 27, all other DSPs were added at the same or lower molar concentration as verapamil in this assay.
In order to exclude the possibility that cytotoxicity of the combination treatment with VCR and DSP might result from the cytotoxic effect of DSPs, all newly synthesized DSPs were also screened individually against KB-Vin cells, and the results are shown in Table 1. In general, all DSP analogs exhibited low intrinsic cytotoxic activity, similar to that of the prototype first generation P-gp inhibitor verapamil.
The chemosensitizing activity of DSPs was then evaluated by measuring the cytotoxic effect of VCR in the presence of DSP analogs at 1 μg/mL, using verapamil as the positive control. As seen from the data in Table 1, most of the DSP analogs lowered the GI50 value of VCR, and thus, reversed VCR resistance in the range of 12–349-fold. In comparison, verapamil increased VCR sensitivity by 141-fold.
Compounds 5–16 are pyranochromones with benzoyloxy groups at the 3′ and 4′ positions. Different functional groups were selectively introduced on the o-, m- or p-position of the benzoyl rings. As shown in Table 1, except for 10, non-substituted, o-, and m-substituted benzoyloxy-DSPs (5–9) exhibited lower chemosensitizing activity than corresponding p-substituted compounds. Compounds 5, 6, and 8 failed to decrease the GI50 of VCR below 0.2 μg/mL, while o- and m-methoxy-substituted benzoyloxy-DSPs (7 and 9) exhibited only moderate chemosensitizing activity by increasing the potency of VCR around 28-fold. Compound 10, with an m-CN substituent, exhibited better chemosensitizing activity (160-fold).
DSP analogs with p-substituted benzoyloxy rings (11–16) showed substituent-dependent activity. Compounds 11 and 14 with methyl and methoxy substituents, respectively, showed the most promising chemosensitizing activity. GI50 values of the combinations of 11 and 14 with VCR were 0.007 and 0.0095 μg/mL, respectively, corresponding to 349- and 257-fold increased potency relative to VCR alone. The improved potency was greater than that with verapamil. Interestingly, 12, with an ethyl group, had a substantially lower effect (20.3-fold VCR increase), and 13, substituted with a trifluoromethyl group, improved VCR activity by only 16.3-fold. Compounds 15 and 16, with electron-withdrawing groups at the p-position, showed enhancements of approximately 200-fold, which was better than that of verapamil. These results suggested that p-substituents are generally preferred over o- or m-substituents for chemosensitizing activity. Substituents with extended conjugation, e.g., CN or NO2 groups, and smaller less sterically hindered groups may benefit the efficacy of 3′,4′-disubstituted DSPs as new potent chemosensitizers.
Compounds 17–21 were designed, synthesized, and evaluated in order to understand the effects of adding a linker between the ester carbonyl group and the aromatic substituents in the side chains. Introducing a saturated linker slightly increased the chemosensitizing ability, with improved GI50 values of the VCR-DSP combination from over 0.2 μg/mL for 5 (−O2CC6H5), 0.03 μg/mL for 17 (−O2CCH2C6H5), and 0.023 μg/mL for 19 (−O2CCH2CH2C6H5). Unexpectedly, replacing the phenyl group of 17 with a p-methoxyphenyl in 18 resulted in a 2.4-fold reduction in chemosensitizing efficacy, and 7.5 times reduction compared with 14, which also contains a p-methoxyphenyl group, but linked directly to the ester carbonyl moiety. Compounds 20 and 21 have a conjugated double bond between the ester carbonyl group and the phenyl ring. Interestingly, combination of either compound with VCR led to significantly enhanced activity. Compound 20, with an unsaturated vinyl group, showed twofold increased efficacy compared to 19, with a saturated ethylene group. Compound 21, with p-methoxycinnamoyloxy 3′,4′ side chains, was able to fully restore the cytotoxcitiy of VCR. The GI50 value of the VCR-21 combination was 0.0085 μg/mL, which was comparable to the values with 11 and 14, Thus, cinnamic and p-methoxycinnamic esters linked to the core dihydropyranochromone structure favored MDR reversal activity.
In order to investigate the impact of the carbonyl group in the active analogs, 22 was synthesized. In this compound, the carbonyl moieties in the 3′ and 4′ side chains have been replaced with a methylene group. With an ether rather than ester side chain, 22 will likely be chemically and metabolically more stable than its analog 5. Interestingly, 22 showed better capability to reverse MDR than 5. This finding shows that the carbonyl group may not be essential for activity. Further SAR studies are needed to confirm this hypothesis.
To further investigate the importance of the 3′,4′-side chains, compounds 23–28, with various 3′,4′-carbamate groups, were designed and synthesized. Most of these compounds, which do not have an aromatic substituent in the side chains, showed either no or marginal chemosensitizing activity. The exception was 25 (N,N-diethylcarbamoyloxy), which showed ten times better efficacy than its counterparts 24 (N,N-dimethylcarbamoyloxy) and 26 (N,N-dibutylarbamoyloxy). 25 also presented 5-fold more potency than its cyclic analogs 27 and 28, suggesting that the orientation of the N-alkyl substitutions at 3′ and 4′ positions affects the interactions of the drug molecules and the binding pocket in the target protein, for example, the different orientation between chain alkyl groups (25) and cyclic groups (27, 28), which, in turn, affects the activity.
In summary, three compounds, 11, 14 and 21, were found to be the most potent chemosensitizers of VCR among 24 newly synthesized compounds. At a non-toxic concentration, they are able to fully restore the cytotoxicity of VCR against KB-Vin, so that the MDR cell line was as sensitive as the parent KB cell line. They were also over two times better than the positive control verapamil (Table 1). While most of the active compounds contain 3′,4′-ester side chains ending in an aromatic ring, 25, which contains two carbamate side chains and no aromatic system, also exhibited considerable potency. The dicarbamate 25 should also exhibit increased polarity and different physicochemical properties, which may benefit further development of new analogs with interesting biological activity.
The SAR study on 3′,4′ side chains suggested the following conclusions. 1. A terminal aromatic ring is important but not necessary to maintain high chemosensitizing capability. 2. Substituents on the aromatic rings can significant impact the activity. Moieties that have increased electron density, extended conjugation systems, or HB effects can benefit efficacy. 3. The characteristic of the linker between the core structure and the aromatic ring system can also affect the chemosensitizing potential. Extending the conjugation system by adding double bond(s) benefits the activity.
Previous modification studies of PA and DCK (1) revealed that aromatic rings in the 3′ and 4′ side chains of the khellactone benefit the chemosensitizing activity, and in addition, methoxy groups substituted on cinnamoyl side chains could impact the activity.7 Those results are mostly, but not fully, consistent with those reported here for pyranochromone-based DSP active analogs. For example, we found that 25, without an aromatic ring in the 3′,4′-side chains displayed good chemosensitizing ability, and thus, an aromatic ring is not essential for activity. In addition, our studies showed both positive (14 vs 5, 21 vs 20) and negative (18 vs 17) effects for the addition of p-methoxy group on a phenyl ring. Fong et al. also reported that p-methoxycinnamoyl-khellactone (MMDCK, 2) exhibited decreased potency compared with DCK (1).7
Due to the different cell lines and MDR substrates, it is difficult to directly compare the activities between the prior khellactone compounds and the DSP analogs reported here. However, in both HepG2/Dox and KB-V1 cell lines, the best reported khellactone compound, DMDCK (3) at 4 μM, could not completely restore the cytotoxicity of vinblastine, an alkaloid analog of VCR. In contrast, DSP analogs 11, 14 and 21 completely restored the cytotoxic effect of VCR suggesting they are more efficacious and more potent than DMDCK (3).
Effect on Cellular P-gp Activity
The acquired VCR-resistance in the KB-Vin cell line is due to enhanced drug efflux via P-gp over-expression.14 Our research results demonstrate that addition of DSPs could restore VCR's cytotoxity againt KB-Vin, suggesting that DSP analogs might function like verapamil as P-gp inhibitors. To test this hypothesis, the effect of DSP analogs on efflux of the P-gp substrate calcein-AM was investigated using the established and widely used assay for cellular P-gp function.15 Inhibition of P-gp leads to the accumulation of calcein-AM in the cytoplasm and concomitant degradation by esterase action to give a fluorescent derivative that can be conveniently monitored as a quantitative measure of P-gp inhibition. Four DSP analogs (8, 13–15) were selected and their ability to facilitate calcein-AM influx was examined and compared to that of verapamil as the positive inhibitor control. The results are shown in Figure 3. Compounds 15 and 14, which showed improved activity versus verapamil in the chemosensitization assay (Table 1), also exerted more potent inhibition of cellular P-gp based on the functional assay results. Compound 13, which showed weaker chemosensitizing activity versus verapamil (16- and 142- fold respectively, Table 1), was correspondingly less active as a P-gp inhibitor. Compound 8 exerted no effect on calcein-AM efflux, which is consistent with its lack of chemosensitization activity. These results demonstrate that the rank-order of chemosensitizing efficacy of DSP analogs is consistent with their relative inhibitory activity toward cellular P-gp. Thus, DSP analogs most likely act as pump inhibitors in the KB-MDR cell system.
Figure 3.

Conclusions
Novel DSP analogs with significant chemosensitizing activity in a MDR human cancer cell line were discovered. An SAR study on the 3′ and 4′ side chains of pyranochromone ring was conducted with the following conclusions. p-Substitution on the aromatic ring in the side chain is better than o- and m-substitution. Small substitutions that can increase electron density or provide hydrogen-bonding capacity at the putative interaction domain on P-gp benefit the activity. The length and identity of the linker is also important and a conjugated double-bond is favorable. Compared with verapamil, a prototype first generation chemosensitizer, nine DSPs exhibited improved or comparable chemosensitizing potential. Compounds 11, 14, and 21 fully overcame VCR resistance in KB-Vin cancer cells at a concentration of 1 μg/mL (1.64–1.90 μM range). Compared to recent work on khellactone-based chemosensitizers, our study provides a more comprehensive SAR and discovered DSP analogs with more promising potency at lower concentration. Preliminary work indicates that the chemosensitizing activity of DSP analogs results from P-gp inhibition. Detailed mechanistic studies are currently ongoing.
Experimental Section
Chemistry
Melting points were measured with a Fisher Johns melting apparatus without correction. The proton nuclear magnetic resonance (1H NMR) spectra were measured on a 300 MHz Varian Gemini 2000 spectrometer and a Varian Inova 400 MHz spectrometer using TMS as internal standard. The solvent used was CDCl3 unless indicated. Microwave reactions were performed with a Biotage initiator EXP US. Mass spectra were measured on a Shimazu LCMS-2010 (ESI-MS). Optical rotation was measured with a Jasco Dip-2000 digital polarimeter at 20 °C at the sodium D line. Thin-layer chromatography (TLC) was performed on PLC silica gel 60 F254 plate (20 × 20, Merck). Biotage Flash and Isco companion systems were used for medium-pressure column chromatography. Silica 40 μM columns from Grace Inc. were used for column chromatography. All final compounds are >95% pure on the basis of HPLC.
Synthesis of compounds 5-21 and 23
A mixture of compound 4 (1 equiv), substituted benzoyl cholorde, cyclohenxal chloride, phenylacetyl chlorides, 3-phenylpropanoyl chloride, or trans-cinnamoyl chlorides (3 equiv), and DMAP (4 equiv) were stirred in CH2Cl2 for 1-2 h at rt, monitored by TLC. At completion, the mixture was diluted with CH2Cl2 and washed with water and brine. The solvent was then removed under reduced pressure and the residue was purified by PTLC with hexane:EtOAc = 3:2 to afford the appropriate compounds (5–21 and 23).
3′R,4′R-Di-benzoyloxy-2′,2′-dimethyl-2-ethyldihydropyrano[2,3-f]chromone (5)
85% yield (starting from 30 mg of 4); white solid; mp 63-65 °C; 1NMR δ 8.15 (1H, d, J = 8.8 Hz, H-5), 8.10-8.13 (1H, m, benzoyl-3′,4′), 7.87-7.90, 7.46-7.61, 7.32-7.46 (each 3H, m, bezoyl-3′,4′), 7.01 (1H, d, J = 4.8 Hz, H-4′), 6.99 (1H, d, J = 8.8 Hz, H-6), 6.08 (1H, s, H-3), 5.66 (1H, d, J = 4.8 Hz, H-3′), 2.34 (2H, q, J = 8.0 Hz, CH2CH3-2), 1.69, 1.54 (each 3H, s, CH3-2′), 1.02 (3H, t, J = 8.0 Hz, CH2CH3-2); [α]D −82.9 ° (c = 0.0035, CHCl3); HRMS for (M+ + H): calcd m/z 499.1757, found: 499.1735.
3′R,4′R-Di-(2-trifluoromethylbenzoyloxy)-2′,2′-dimethyl-2-ethyldihydropyrano[2,3-f] chromone (6)
88% yield (starting from 30 mg of 4); white solid; mp = 104-106 °C; 1NMR δ 7.95 (1H, d, J = 8.8 Hz, H-5), 7.50-7.80 (8H, m, benzoyl-3′,4′), 6.65 (1H, d, J = 8.8 Hz, H-6), 6.30 (1H, s, H-3), 5.85 (1H, d, J = 4.8 Hz, H-4′), 5.50 (1H, d, J = 4.8 Hz, H-3′), 2.34 (2H, q, J = 8.0 Hz, CH2CH3-2), 1.69, 1.54 (each 3H, s, CH3-2′), 1.02 (3H, t, J = 8.0 Hz, CH2CH3-2). [α]D −34.7 ° (c = 0.0019, CHCl3); HRMS for (M+ + H): calcd m/z 635.1504, found: 635.1493.
3′R,4′R-Di-(2-methoxybenzoyloxy)-2′,2′-dimethyl-2-ethyldihydropyrano[2,3-f]chromone (7)
80% yield (starting from 30 mg of 4); white solid; mp = 104-106 °C; 1NMR δ 8.18, 7.81, 7.63, 7.07 (each 1H, d, J = 7.6 Hz, methoxybenzoyl-3′,4′), 8.11 (1H, d, J = 8.8 Hz, H-5), 7.44, 7,42, 6.93, 6.90 (each 1H, t, J = 7.6 Hz, methoxybenzoyl-3′,4′), 7.00 (1H, d, J = 4.8 Hz, H-4′), 6.94 (1H, d, J = 9.0, H-5), 6.09 (1H, s, H-3), 5.62 (1H, d, J = 4.8 Hz, H-3′), 3.77, 3.70 (each 3H, s, methoxybenzoyl-OCH3-3′,4′), 2.48 (2H, q, J = 7.5 Hz, CH2CH3-2), 1.63, 1.56 (each 3H, s, CH3-2′), 1.14 (3H, t, J = 7.5 Hz, CH2CH3-2); [α]D −40.0 ° (c = 0.0007, CHCl3); HRMS for (M+ + H): calcd m/z 559.1963, found: 559.1937.
3′R,4′R-Di-(3-trifluoromethylbenzoyloxy)-2′,2′-dimethyl-2-ethyldihydropyrano[2,3-f] chromone (8)
80% yield (starting from 100 mg of 4); white solid; mp = 124-126 °C; 1NMR δ 8.15 (1H, d, J = 8.8 Hz, H-5), 8.02-8.07 (4H, m, benzoyl-3′,4′), 7.77-7.82 (2H, m, benzoyl-3′,4′), 7.49-7.55 (2H, m, benzoyl-3′,4′), 6.99 (1H, d, J = 4.2 Hz, H-4′), 6.98 (1H, d, J = 8.8 Hz, H-6), 6.06 (1H, s, H-3), 5.67 (1H, d, J = 4.2 Hz, H-3′), 2.33 (2H, q, J = 7.6 Hz, CH2CH3-2), 1.68, 1.54 (each 3H, s, CH3-2′), 1.02 (3H, t, J = 7.6 Hz, CH2CH3-2); [α]D −74.6 ° (c = 0.0013, CHCl3); HRMS for (M+ + H): calcd m/z 635.1504, found: 635.1514.
3′R,4′R-Di-(3-methoxybenzoyloxy)-2′,2′-dimethyl-2-ethyldihydropyrano[2,3-f]chromone (9)
80% yield (starting from 30 mg of 4); white solid; mp = 90-92 °C; MS-ESI + (m/z, %) 559 (M+ + 1, 100); 1NMR δ 8.09 (1H, d, J = 8.4 Hz, H-5), 7.71, 7.68 (each 1H, d, J = 7.6 Hz, benzoyl-3′,4′), 7.61 (2H, s, benzoyl-3′,4′), 7.38 (2H, t, J = 8.4, 7.6 Hz, benzoyl-3′,4′), 7.15 (2H, J = 8.4 Hz, benzoyl-3′,4′), 6.93 (1H, d, J = 8.4 Hz, H-6), 6.17 (1H, s, H-3), 5.53 (1H, d, J = 4.8 Hz, H-4′), 5.41 (1H, d, J = 4.8 Hz, H-3′), 3.87, 3.85 (each 3H, s, 3-methoxybenzoyl-OCH3-3′,4′), 2.65 (2H, q, J = 7.6 Hz, CH2CH3-2), 1.61, 1.49 (each 3H, s, CH3-2′), 1.30 (3H, t, J = 7.6 Hz, CH2CH3-2); [α]D −116.3 ° (c = 0.0008, CHCl3); HRMS for (M+ + H): calcd m/z 599.1968, found: 599.1986.
3′R,4′R-Di-(3-cyanolbenzoyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (10)
65% yield (starting from 100 mg of 4); white solid; mp = 253-254 °C; 1NMR δ 8.19 (1H, d, J = 9.0 Hz, H-5), 8.11 (4H, m, benzoyl-3′,4′), 7.86, 7.58 (each 2H, m, benzoyl-3′,4′), 7.02 (1H, d, J = 9.0 Hz, H-6), 7.00 (1H, d, J = 5.4 Hz, H-4′), 6.16 (1H, s, H-3), 5.66 (1H, d, J = 5.4 Hz, H-3′), 2.36 (2H, q, J = 7.8 Hz, CH2CH3-2), 1.69, 1.57 (each 3H, s, CH3-2′), 1.03 (3H, t, J = 7.8 Hz, CH2CH3-2); [α]D −125.0 ° (c = 0.0004, CHCl3); HRMS for (M+ + H): calcd m/z 549.1662, found: 549.1666.
3′R,4′R-Di-(4-methylbenzoyloxy)-2′,2′-dimethyl-2-ethyldihydropyrano[2,3-f]chromone (11)
80% yield (starting from 25 mg of 4); white solid; mp = 144-145 °C; 1NMR δ 8.13 (1H, d, J = 9.0 Hz, H-5), 7.78, 7.78 (each 2H, d, J = 8.1 Hz, COOC(CHCH)2CCH3-3′,4′), 7.17, 7.17 (each 2H, d, J = 8.1 Hz, COOC(CHCH)2CCH3-3′,4′), 6.97 (1H, d, J = 9.0 Hz, H-6), 6.96 (1H, d, J = 4.8 Hz, H-4′), 6.07 (1H, s, H-3), 5.62 (1H, d, J = 4.8 Hz, H-3′), 2.39, 2.38 (each 3H, s, methylbenzoyl-CH3-3′,4′), 2.34 (2H, q, J = 7.5 Hz, CH2CH3-2), 1.67, 1.53 (each 3H, s, CH3-2′), 1.02 (3H, t, J = 7.5 Hz, CH2CH3-2); [α]D −81.3 ° (c = 0.0015, CHCl3); HRMS for (M+ + H): calcd m/z 527.2070, found: 527.2063.
3′R,4′R-Di-(4-ethylbenzoyloxy)-2′,2′-dimethyl-2-ethyldihydropyrano[2,3-f]chromone (12)
70% yield (starting from 65 mg of 4); white powder; mp = 109-102 °C; 1NMR δ 8.13 (1H, d, J = 9.0 Hz, H-5), 7.80, 7.80 (each 2H, d, J = 7.2 Hz, COOC(CHCH)2CCH2CH3-3′,4′), 7.20, 7.17 (2H, d, J = 7.2 Hz, COOC(CHCH)2CCH2CH3-3′,4′), 6.98 (1H, d, J = 9.0 Hz, H-6), 6.97 (1H, d, J = 4.8 Hz, H-4′), 6.07 (1H, s, H-3), 5.63 (1H, d, J = 4.8 Hz, H-3′), 2.69, 2.68 (each 2H, q, J = 7.5 Hz, ethylbenzoyl-CH2CH3-3′,4′), 2.32 (2H, q, J = 7.5 Hz, CH2CH3-2), 1.68, 1.53 (each 3H, s, H-2′), 1.24(3H, t, J = 7.5 Hz, CH2CH3-2), 1.23, 1.03 (each 3H, t, J = 7.2 Hz ethylbenzoyl-CH2CH3-3′,4′); [α]D −93.6 ° (c = 0.01, CHCl3); HRMS for (M+ + H): calcd m/z 555.2383, found: 555.2366.
3′R,4′R-Di-(4-trifluoromethylbenzoyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f] chromone (13)
60% yield (starting from 25 mg of 4); white powder; mp = 114-115 °C; 1NMR δ 8.22 (2H, d, J = 8.1 Hz, trifluoromethylbenzoyl-H), 8.16 (1H, d, J = 8.7 Hz, H-5), 8.99 (2H, d, J = 8.4 Hz, trifluoromethylbenzoyl-H), 7.74, (2H, d, J = 8.1 Hz, trifluoromethylbenzoyl-C(CHCH)2CCF3-3′,4′), 7.65 (2H, d, J = 8.4 Hz, trifuoromethylbenzoyl- C(CHCH)2CCF3-3′,4′), 7.00 (1H, d, J = 8.7 Hz, H-6), 7.00 (1H, d, J = 4.8 Hz, H-4′), 6.12 (1H, s, H-3), 5.67 (1H, d, J = 4.8 Hz, H-3′), 2.33 (2H, q, J = 7.8 Hz, CH2CH3-2), 1.68, 1.56 (each 3H, s, CH3-2′), 1.02 (3H, t, J = 7.8 Hz, CH2CH3-2); [α]D −74.0 ° (c = 0.0025, CHCl3); HRMS for (M+ + H): calcd m/z 635.1504, found: 635.1488.
3′R,4′R-Di-(4-methoxybenzoyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (14)
80% yield (starting from 100 mg of 4); yellow oil; 1NMR δ 8.13 (1H, d, J = 8.7 Hz, H-5), 7.85, 7.85 (each 2H, d, J = 8.7 Hz, COOC(CHCH)2COCH3-3′,4′), 6.97 (1H, d, J = 8.7 Hz, H-6), 6.95 (1H, d, J = 4.8 Hz, H-4′), 6.85, 6.82 (each 2H, d, J = 8.7 Hz, COOC(CHCH)2COCH3-3′,4′), 6.06 (1H, s, H-3), 5.61 (1H, d, J = 4.8 Hz, H-3′), 3.85, 3.84 (each 3H, s, methoxybenzoyl-OCH3-3′,4′), 2.33 (2H, q, J = 7.2 Hz, CH2CH3-2), 1.66, 1.53 (each 3H, s, CH3-2′), 1.03 (3H, t, J = 7.2 Hz, CH2CH3-2); [α]D −105.0 ° (c = 0.0012, CHCl3); HRMS for (M+ + H): calcd m/z 559.1968, found: 559.1954.
3′R,4′R-Di-(4-cyanobenzoyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (15)
80% yield (starting from 100 mg of 4); white powder; mp = 253-254 °C; 1NMR δ 8.17 (1H, d, J = 8.7 Hz, H-5), 7.98, 7.97 (each 2H, d, J = 8.7 Hz, benzoyl-COC(CHCH)2CCN-3′,4′), 7.71, 7.68 (each 2H, d, J = 8.7 Hz, benzoyl-COC(CHCH)2CCN-3′,4′), 6.99 (1H, d, J = 8.7 Hz, H-6), 6.98 (1H, d, J = 5.1 Hz, H-4′), 6.08 (1H, s, H-3), 5.66 (1H, d, J = 5.1 Hz, H-3′), 2.34 (2H, t, J = 7.8 Hz, CH2CH3-2), 1.67, 1.56 (each 3H, s, CH3-2′), 1.01 (3H, t, J = 7.8 Hz, CH2CH3-2); [α]D −104.8 ° (c = 0.0014, CHCl3); HRMS for (M+ + H): calcd m/z 549.1662, found: 549.1648.
3′R,4′R-Di-(4-nitrolbenzoyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (16)
50% yield (starting from 50 mg of 4); white solid; mp = 109-110 °C; 1NMR δ 8.23, 8.22 (each 2H, d, J = 9.0 Hz, COOC(CHCH)2CNO2-3′,4′), 8.14 (1H, d, J = 8.4 Hz, H-5), 8.06, 8.04 (each 2H, d, J = 9.0 Hz, COOC(CHCH)2CNO2-3′,4′), 6.99 (1H, d, J = 8.4 Hz, H-6), 6.98 (1H, d, J = 4.8 Hz, H-4′), , 6.06 (1H, s, H-3), 5.67 (1H, d, J = 4.8 Hz, H-3′), 2.33 (2H, q, J = 7.2 Hz, CH2CH3-2), 1.68, 1.57 (each 3H, s, CH3-2′), 1.00 (3H, t, J = 7.2 Hz, CH2CH3-2); [α]D −74.7 ° (c = 0.0015, CHCl3); HRMS for (M+ + H): calcd m/z 589.1458, found: 589.1437.
3′R,4′R-Di-(2-phenylacetoxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (17)
53% yield (starting from 30 mg of 4); solid powder; mp = 75-77 °C; 1NMR δ 8.07 (1H, d, J = 8.8 Hz, H-5), 7.30 (10H, m, phenyl-3′,4′), 6.85 (1H, d, J = 8.8 Hz, H-6), 6.58 (1H, d, J = 4.8 Hz, H-4′), 6.05 (1H, s, H-3), 5.20 (1H, d, J = 4.8Hz, H-3′), 3.53, 3.46 (each 2H, d, J = 15.9 Hz, acetoxy-COOCH2-3′,4′), 2.25 (2H, q, J = 7.6 Hz, CH2CH3-2), 1.32, 1.25 (each 3H, s, CH3-2′), 1.12 (3H, t, J = 7.6 Hz, CH2CH3-2); [α]D −60.0 ° (c = 0.0015, CHCl3); HRMS for (M+ + H): calcd m/z 527.2070, found: 527.2060.
3′R,4′R-Di-[2-(4-methoxyphenylacetoxy)]-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f] chromone (18)
50% yield (starting from 25 mg of 4); yellow oil; 1NMR δ 8.07 (1H, d, J = 9.0 Hz, H-5), 7.20, 7.17 (each 2H, d, J = 8.7 Hz, phenyl-C(CHCH)2COCH3-3′,4′), 6.87, 6.85 (each, 2H, d, J = 8.7 Hz, phenyl-C(CHCH)2COCH3-3′,4′), 6.78 (1H, d J = 9.0 Hz, H-6), 6.44 (1H, d, J = 4.8 Hz, H-4′), 6.10 (1H, s, H-3), 4.03 (1H, d, J = 4.8 Hz, H-3′), 2.25 (2H, q, J = 7.5 Hz, CH2CH3-2), 1.49, 1.34 (each 3H, s, CH3-2′), 1.13 (3H, t, J = 7.5 Hz, CH2CH3-2); [α]D −47.7 ° (c = 0.0013, CHCl3); HRMS for (M+ + H): calcd m/z 587.2281, found: 587.2266.
3′R,4′R-Di-(3-phenylpropanoyloxy)-2′,2′-dimethyl-2-ethyl -dihydropyrano[2,3-f]chromone (19)
70% yield (starting from 25 mg of 4); clear oil; 1NMR δ 8.09 (1H, d, J = 9.0 Hz, H-5), 7.22 (10H, m, phenyl-C(CHCH)2CH-3′,4′), 6.88 (1H, d, J = 9.0 Hz, H-6), 6.59 (1H, d, J = 4.8 Hz, H-4′), 6.09 (1H, s, H-3), 5.26 (1H, d, J = 4.8 Hz, H-3′), 2.95, 2.95 (4H, t, J = 7.6 Hz, propanoyl-CH2CH2-3′.4′), 2.67, 2.58(each 2H, t, J = 7.6 Hz, propanoyl-CH2CH2-3′,4′), 2.46 (2H, q, J = 7.5 Hz, CH2CH3-2), 1.43, 1.33 (each 3H, s, CH3-2′), 1.67 (3H, t, J = 7.5 Hz, CH2CH3-2); [α]D −74.8 ° (c = 0.0023, CHCl3); HRMS for (M+ + H): calcd m/z 555.2393, found: 555.2378.
3′R,4′R-Di-(E)- phenylacryloyloxy-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3f]chromone (20)
65% yield (starting from 50 mg of 4); white solid; mp = 104-106 °C; 1NMR δ 8.05 (1H, d, J = 9.0 Hz, H-5), 7.63, 7.63 (each 1H, d, J = 15.9 Hz COOCHCH-3′,4′), 7.36-1.40 (5H, m, phenyl-3′,4′), 1.23-1.29 (5H, m, phenyl-3′,4′), 6.88 (1H, d, J = 9.0 Hz, H-6), 6.75 (1H, d, J = 4.8 Hz, H-4′), 6.38 (2H, d, J = 15.9 Hz, COOCHCH-3′.4′), 6.02 (1H, s, H-3), 5.42 (1H, d, J = 4.8 Hz, H-3′), 2.42 (2H, q, J = 6.9 Hz, CH2CH3-2), 1.52, 1.43 (each 3H, s, CH3-2′), 1.17 (3H, t, J = 6.9 Hz, CH2CH3-2); [α]D −75.3 ° (c = 0.0017, CHCl3); HRMS for (M+ + H): calcd m/z 551.2070, found: 551.2074.
3′R,4′R-Di-(E)-4-methoxyphenylacryloyloxy-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3f] chromone (21)
50% yield (starting from 25 mg of 4); white powder; mp = 94-96 °C; 1NMR δ 8.10 (1H, d, J = 9.0 Hz, H-5), 7.66, 7.65 (each 1H, d, J = 15.3 Hz, (E)-acryloxy-OCOCHCH-3′,4′), 7.40 (4H, dd, J = 8.7, 3.0 Hz, 4-methoxyphenyl-C(CHCH)2COCH3-3′,4′), 6.93 (1H, d, J = 9.0 Hz, H-6), 6.85 (4H, dd, J = 8.7, 3.0 Hz, 4-methoxyphenyl-C(CHCH)2COCH3-3′,4′), 6.80 (1H, d, J = 4.8 Hz, H-4′), 6.33, 6.32 (each 1H, d, J = 15.3 Hz, (E)-acryloxy-OCOCHCH-3′,4′), 6.10 (1H, s, H-3), 5.47 (1H, d, J = 4.8 Hz, H-3′), 6.65 (6H, s, OCH3-3′,4′), 2.47 (2H, q, J = 7.5 Hz, CH2CH3-2), 1.58, 1.49 (each 3H, s, CH3-2′), 1.59 (3H, t, J = 7.5 Hz, CH2CH3-2); [α]D −70.0 ° (c = 0.0043, CHCl3); HRMS for (M+ + H): calcd m/z 611.2281, found: 611.2260.
3′R,4′R-Di-(cyclohexanecarbonyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (23)
20 % yield (starting from 25 mg of 4); white solid; mp = 72-74 °C; 1NMR δ 8.08 (1H, d, J = 9.0 Hz, H-5), 6.88 (1H, d, J = 9.0 Hz, H-6), 6.61 (1H, d, J = 4.5 Hz, H-4′), 6.12 (1H, s, H-3), 5.27 (1H, d, J = 4.5 Hz, H-3′), 2.56 (2H, q, J = 7.5 Hz, CH2CH3-2), 2.35 (2H, m, cyclohexanecarbonyl-3′,4′), 2.80 (12H, m, cyclohexanecarbonyl-3′,4′), 1.49, 1.42 (each 3H, s, CH3-2′), 1.35 (8H, m, cyclohexanecarbonyl-3′,4′), 1.23 (3H, t, J = 7.5 Hz, CH2CH3-2); [α]D −15.4 ° (c = 0.005, CHCl3); HRMS for (M+ + H): calcd m/z 511.2696, found: 511.2713.
Synthesis of 3′R,4′R-Dibenzyloxy-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (22)
NaH (20.4 mg, 0.51 mmoL) was suspended in dry DMF. To this suspension were added at 0 °C a solution of compound 4 (50 mg, 0.17 mmoL) in dry DMF (0.5 mL), and benzyl bromide (0.05 mL, 0.43 mmoL), under the protection of N2. After 1 h, the mixture was poured into 5% aqueous NaHCO3 solution and extracted with Et2O for three times. The organic layers were combined and dried over Na2SO4. The crude product was purified by preparative TLC with hexane:EtOAc = 1:1 to afford 21 (16.2 mg, white solid). 57.3 % yield; mp = 77-79 °C; 1NMR δ 8.00 (1H, d, J = 8.7 Hz, H-5), 7.41, 7.28 (each 5H, m, benzyl-3′,4′), 6.81(1H, d, J = 8.7 Hz, H-6), 6.13 (1H, s, H-3), 5.10, 4.95, 4.85, 4.75 (each 1H, d, J = 11.4 Hz, benzyl-CH2-3′,4′)5.09 (1H, d, J = 4.2 Hz, H-4′), 3.75 (1H, d, J = 4.2 Hz, H-3′), 2.60 (2H, q, J = 7.5 Hz, CH2CH3-2), 1.57, 1.50 (each 3H, s, CH3-2′), 1.28 (3H, t, J = 7.5 Hz, CH2CH3-2); [α]D −40.9 ° (c = 0.01, CHCl3); HRMS for (M+ + H): calcd m/z 471.2171, found: 471.2155.
Synthesis of compounds 24-28
Sodium hydride (5 equiv) was added to a solution of compound 4 in dry THF at ice-bath condition and diverse carbamyl chlorides (3 equiv) were added. The reaction mixture was stirred at rt and monitored by TLC. At completion, the reaction mixture was poured carefully onto EtOAc and saturated aqueous NaHCO3. The organic layer was washed with water for three times, dried over anhydrous MgSO4, and evaporated under reduced pressure. The crude product was purifed by prepare TLC.
3′R,4′R-Di-(dimethylcarbamoyloxy)-2′,2′-dimethyl-2-ethyl -dihydropyrano[2,3-f]chromone (24)
30% yield (starting from 100 mg of 4); clear oil; 1NMR δ 8.07 (1H, d, J = 8.8 Hz, H-5), 6.88 (1H, d, J = 8.8 Hz, H-6), 6.52 (1H, d, J = 4.8 Hz, H-4′), 6.11 (1H, s, H-3), 5.22 (1H, d, J = 4.8 Hz, H-3′), 2.97, 2.85 (each 6H, m, dimethylcarbamoyl-CH2-3′,4′), 2.55 (2H, q, J = 7.2 Hz, CH2CH3-2), 1.49, 1.46 (each 3H, s, CH3-2′), 1.24 (3H, t, J = 7.2 Hz, CH2CH3-2); [α]D −28.6 ° (c = 0.01, CHCl3); HRMS for (M+ + H): calcd m/z 433.1957, found: 433.1951.
3′R,4′R-Di-(diethylcarbamoyloxy)-2′,2′-dimethyl-2-ethyl -dihydropyrano[2,3-f]chromone (25)
25% yield (starting from 25 mg of 4); white solid; mp = 125-126 °C; 1NMR δ 8.07 (1H, d, J = 8.8 Hz, H-5), 6.88 (1H, d, J = 8.8 Hz, H-6), 6.59 (1H, d, J = 4.8 Hz, H-4′), 6.11 (1H, s, H-3), 5.25 (1H, d, J = 4.8 Hz, H-3′), 3.35, 3.35, 3.18, 3.18 (each 2H, q, J = 6.8 Hz, diethylcarbamoyl-CH2-3′,4′), 2.55 (2H, q, J = 7.6 Hz, CH2CH3-2), 1.49, 1.47 (each 3H, s, CH3-2′), 1.23 (3H, t, J = 7.6 Hz, CH2CH3-2), 1.16 (6H, t, J = 6.8 Hz, diethylcarbamoyl-CH3-4′), 1.09, 0.98 (each 3H, t, J = 6.8 Hz, diethylcarbamoyl-CH3-3′); [α]D −30.0 ° (c = 0.001, CHCl3); HRMS for (M+ + H): calcd m/z 489.2601, found: 489.2594.
3′R,4′R-Di-(dibutylcarbamoyloxy)-2′,2′-dimethyl-2-ethyl -dihydropyrano[2,3-f]chromone (26)
30% yield (starting from 50 mg of 4); clear oil; 1NMR δ 8.06 (1H, d, J = 8.8 Hz, H-5), 6.86 (1H, d, J = 8.8 Hz, H-6), 6.58 (1H, d, J = 4.8 Hz, H-4′), 6.11 (1H, s, H-3), 5.23 (1H, d, J = 4.8 Hz, H-3′), 3.25 (8H, m, dibutylcarbamoyl-3′,4′), 2.56 (2H, q, J = 7.6 Hz, CH2CH3-2), 1.57 (6H, m, dibutylcarbamoyl-CH2), 1.47, 1.46 (each 3H, s, CH3-2′), 1.33 (8H, m, dibutylcarbamoyl-CH2), 1.25 (3H, t, J = 7.6 Hz, CH2CH3-2), 1.08 (2H, q, J = 7.2 Hz, dibutylcarbamoyl-CH2-3′), 0.95, 0.94 (each 3H, t, J = 7.6 Hz, dibutylcarbamoyl-CH3-4′), 0.86, 0.72 (each 3H, t, J = 7.2 Hz, dibutylcarbamoyl-CH3-3′); [α]D −13.5 ° (c = 0.012, CHCl3); HRMS for (M+ + H): calcd m/z 601.3853, found: 601.3833.
3′R,4′R-Di-(pyrrolidine-1-carbonyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f] chromone (27)
35% yield (starting from 25 mg of 4); white powder; mp = 104-106 °C; 1NMR δ 8.05 (1H, d, J = 8.8 Hz, H-5), 6.87 (1H, d, J = 8.8 Hz, H-6), 6.54 (1H, d, J = 4.8 Hz, H-4′), 6.11 (1H, s, H-3), 5.23 (1H, d, J = 4.8 Hz, H-3′), 3.48, 3.26 (each 4H, m, pyrrolidine-N(CH2)2(CH2)2-3′,4′), 2.55 (2H, q, J = 7.6 Hz, CH2CH3-2), 1.86 (8H, m, pyrrolidine-N(CH2)2(CH2)2-3′,4′), 1.50, 1.47 (each 3H, s, CH3-2′), 1.21 (3H, t, J = 7.6 Hz, CH2CH3-2); [α]D −23.8 ° (c = 0.011, CHCl3); HRMS for (M+ + H): calcd m/z 485.2288, found: 485.2277.
3′R,4′R-Di-(piperidine-1-carbonyloxy)-2′,2′-dimethyl-2-ethyl-dihydropyrano[2,3-f]chromone (28)
32% yield (starting from 25 mg of 4); white solid; mp = 107-109 °C; 1NMR δ 8.06 (1H, d, J = 8.8 Hz, H-5), 6.87 (1H, d, J = 8.8 Hz, H-6), 6.56 (1H, d, J = 4.8 Hz, H-4′), 6.11 (1H, s, H-3), 5.24 (1H, d, J = 4.8 Hz, H-3′), 3.77 (8H, m, piperidine-N(CH2)2(CH2)2CH2-3′,4′), 2.57 (2H, q, J = 7.6 Hz, CH2CH3-2), 1.58 (12H, m, piperidine-N(CH2)2(CH2)2CH2-3′,4′), 1.47, 1.46 (each 3H, s, CH3-2′), 1.26 (3H, t, J = 7.6 Hz, CH2CH3-2); [α]D −28.2 ° (c = 0.0017, CHCl3); HRMS for (M+ + H): calcd m/z 513.2601, found: 513.2620.
Chemosensitization (MDR Modulation) Assay
Three thousand drug-resistant cells, KB-Vin cells (MDR-1) selected using VCR 16 were treated for two or three days with test agent in the presence or absence of VCR. The end-point is cellular protein measured using the sulforhodamine B (SRB) method developed at NIH-NCI for in vitro anticancer drug screening.17 The degree of chemoreversal was determined by fixing the concentration of test agent and measuring the alteration in dose-response of cells to VCR toxicity. Verapamil was used as chemosensitizer control. Data from replicate experiments were plotted, and the apparent GI50 value was determined using statistical software (GraphPad Prizm, CA).
Calcein-AM Loading Assay
KB-Vin (MDR) cells (10,000 per well) were seeded into 96-well plates with medium containing 5% FBS and incubated for one day for adhesion and equilibration. Treatment involved supplementing cultures with distinct concentrations of test compounds for 30 min at 37 °C. After removing medium, fresh medium with both 1 μM calcein-AM (Alexis Biochemicals) and test compounds as needed was then replaced. Following incubation for 30 min at 37 °C, medium was removed and wells were washed gently and quickly with cold PBS buffer in low ambient light conditions. Cells were lysed using 20 mM Tris-HCl buffer and fluorescence was measured using a BioTek TLx800 ELISA reader set to Ex. 494 nm and Em. 517 nm. Composite data obtained from two or more independent experiments were analyzed using Prizm™ (Graphpad Software, San Diego, CA).15
Supplementary Material
Acknowledgement
This investigation was supported by NIH grant CA17625 awarded to K. H. Lee.
Footnotes
Abbreviations: DSP, 3′R,4′R-di-substituted-2′,2′-dimethyldihydropyrano[2,3-f]chrmone; MDR, multi-drug resistance; VCR, vincristine; P-gp, P-glycoprotein; PA, (±)-Praeruptorin A; DMDCK, (±)-3′-O-4′-O-bis(3,4-dimethoxycinnamoyl)-cis-khellactone; HB, hydrogen bond.
Supporting Information Available: HPLC condition and summary of HPLC purity data for final compounds; statistic analysis for cell assay. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dalton WS. Is p-glycoprotein a potential target for reversing clinical drug resistance? Curr Opin Oncol. 1994;6(6):595–600. doi: 10.1097/00001622-199411000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Hindenburg AA, Baker MA, Gleyzer E, Stewart VJ, Case N, Taub RN. Effect of verapamil and other agents on the distribution of anthracyclines and on reversal of drug resistance. Cancer Res. 1987;47(5):1421–1425. [PubMed] [Google Scholar]
- 3.Thomas H, Coley H. Overcoming multidrug resistance in cancer: an updated on the clinical strategy of inhibitinf P-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 4.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 5.Hollt V, Kouba M, Dietel M, Vogt G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem Pharmacol. 1992;43(12):2601–2608. doi: 10.1016/0006-2952(92)90149-d. [DOI] [PubMed] [Google Scholar]
- 6.Wu JY, Fong WF, Zhang JX, Leung CH, Kwong HL, Yang MS, Li D, Cheung HY. Reversal of multidrug resistance in cancer cells by pyranocoumarins isolated from Radix peucedani. Eur J Pharmacol. 2003;473(1):9–17. doi: 10.1016/s0014-2999(03)01946-0. [DOI] [PubMed] [Google Scholar]
- 7.Fong WF, Shen XL, Globisch C, Wiese M, Chen GY, Zhu GY, Yu ZL, Tse AK, Hu YJ. Methoxylation of 3′,4′-aromatic side chains improves P-glycoprotein inhibitory and multidrug resistance reversal activities of 7,8-pyranocoumarin against cancer cells. Bioorg Med Chem. 2008;16(7):3694–3703. doi: 10.1016/j.bmc.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 8.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323(5922):1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pajeva IK, Wiese M. Pharmacophore model of drugs involved in P-glycoprotein multidrug resistance: explanation of structural variety (hypothesis) J. Med. Chem. 2002;45:5671–5686. doi: 10.1021/jm020941h. [DOI] [PubMed] [Google Scholar]
- 10.Zhou T, Shi Q, Chen CH, Zhu H, Huang L, Ho P, Lee KH. Anti-AIDS agents 79. Design, synthesis, molecular modeling and structure-activity relationships of novel dicamphanoyl-2′,2′-dimethyldihydropyranochromone (DCP) analogs as potent anti-HIV agents. Bioorg Med Chem. 2010;18(18):6678–6689. doi: 10.1016/j.bmc.2010.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou T, Shi Q, Lee KH. Anti-AIDS Agent 83. Efficient microwave-assisted one-pot preparation of augular 2,2-dimethyl-2H-chromone containing compounds. Tetrahedron Lett. 2010;51(33):4382–4386. doi: 10.1016/j.tetlet.2010.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baba T, Huang G, MInoru I. Synthesis of the JKLM-ring Fragment of Ciguatoxin. Tetrahedron. 2003;59:6851–6872. [Google Scholar]
- 13.Nguyen TM, Sittisombut C, Boutefnouchet S, Lallemand M-C, Michel S, Koch M, Tillequin F, Mizinghien R, Lansiaux A, David-Cordonnier M-H, Pfeiffer B, KrausBerthier L, Leonce S, Pierre A. Synthesis, antitumor activity, and mechanism of action of benzo[a]pyrano[3,2-h]acridin-7-one analogues of acronycine. J. Med. Chem. 2006;49:3383–3394. doi: 10.1021/jm0602007. [DOI] [PubMed] [Google Scholar]
- 14.Gouaze V, Yu JY, Bleicher RJ, Han TY, Liu YY, Wang H, Gottesman MM, Bitterman A, Giuliano AE, Cabot MC. Overexpression of glucosylceramide synthase and P-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol Cancer Ther. 2004;3(5):633–639. [PubMed] [Google Scholar]
- 15.Armstrong L, Sullivan R, Mechetner E. New functional assays for multidrug resistance. 2004 April 28; http://www.biosciencetechnology.com/ShowPR.aspx?PUBCODE=090&ACCT=9000009885&ISSUE=0404&RELTYPE=PR&Cat=SubCat=1&PRODCODE=00005949&PRODLETT=A&CALLFROM=PR&CommonCount=0.
- 16.Lunney EA, Hagen SE, Domagala JM, Humblet C, Kosinski J, Tait BD, Warmus JS, Wilson M, Ferguson D, Hupe D, et al. A novel nonpeptide HIV-1 protease inhibitor: elucidation of the binding mode and its application in the design of related analogs. J Med Chem. 1994;37(17):2664–2677. doi: 10.1021/jm00043a006. [DOI] [PubMed] [Google Scholar]
- 17.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82(13):1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.