

Abstract

Leinamycin (LNM, 1) biosynthesis is proposed to involve β-alkylation of the polyketide intermediate, catalyzed by LnmKLM. Inactivation of lnmK, lnmL, or lnmM afforded mutant strains that accumulated LNM K-1 (2), K-2 (3), K-3 (4), and isomers LNM K-1′ (5), K-2′ (6), and K-3′ (7) whose polyketide origin were established by feeding experiments with sodium [1-13C]acetate. These findings confirm the indispensability of LnmKLM in 1 biosynthesis and suggest that β-alkylation proceeds on the growing polyketide intermediate while bound to the LNM polyketide synthase.
β-Alkylations contribute to the vast structural diversity displayed by polyketide natural products, with the β-alkyl branches often resulting from the activity of hydroxymethylglutaryl-CoA (HMG-CoA) synthase homologs (HCSs).1 HCS catalyzes condensation of acyl-S-acyl carrier protein (ACP) with the β-carbonyl group of the polyketide synthase (PKS)-ACP-tethered growing polyketide intermediate to afford an β-hydroxymethylacyl-S-ACP intermediate, which can undergo further dehydration, decarboxylation, or both by two enoyl-CoA hydratase homologs (ECH1 and ECH2) to afford a β-alkylated intermediate.1 This pathway for β-methyl branch installation has been extensively studied, and a dedicated set of three proteins has been identified that derives acetyl-S-ACP from malonyl-CoA.1–4
Leinamycin (LNM, 1), a potent antitumor antibiotic, possesses a β-branched C3 unit that is part of the unique five-membered 1,3-dioxo-1,2-dithiolane moiety.5 We previously cloned, sequenced, and characterized the lnm biosynthetic gene cluster from Streptomyces atroolivaceus S-140.6 These studies established that the 18-membered macrolactam backbone of 1 is assembled by a hybrid nonribosomal peptide synthetase (NRPS)-PKS and suggested that the β-branched C3 unit is installed by a set of four proteins: LnmL, an ACP; LnmK, a bifunctional acyltransferase/decarboxylase (AT/DC); LnmM, an HCS homolog; and LnmF, an ECH1 homolog. It has been suggested that LnmM catalyzes condensation of propionyl-S-LnmL with the LnmJ PKS module-8 ACP-tethered polyketide intermediate, followed by LnmF-catalyzed dehydration, to afford the β-branched C3 unit by the same mechanism as that used for β-methyl branch incorporation with the exception of a missing decarboxylation step catalyzed by ECH2.7 However, the origin of propionyl-S-ACP is distinct from that of acetyl-S-ACP.7 The bifunctional AT/DC activity of LnmK has been confirmed to derive propionyl-S-LnmL from methylmalonyl-CoA by acylation of LnmL and subsequent decarboxylation (Figure 1).
Figure 1.

Proposed LNM (1) biosynthetic pathway featuring LnmKLM-catalyzed β-alkylation of the LnmJ PKS module-8 ACP-tethered intermediate in the S. atroolivaceus S-140 wild-type strain (path a) and processive elongation of polyketide intermediates tethered to the LnmJ PKS module-5 and module-8 ACPs as evidenced by accumulation of LNM K-1 (2), K-2 (3), K-3 (4), K-1′ (5), K-2′ (6), and K-3′ (7) in the S. atroolivaceus S-140 mutant strains SB3029 (i.e., ΔlnmK), SB3030 (i.e., ΔlnmL), and SB3031 (i.e., ΔlnmM) (paths b and c).
We now report in vivo characterization of the lnmKLM genes to support their assigned roles in the installation of the β-branched C3 unit for 1 biosynthesis. Inactivation of lnmKLM in S. atroolivaceus S-140 afforded mutant strains SB3029, SB3030, and SB3031. Each of the mutant strains failed to produce 1 but accumulated the same set of six new metabolites (Figure 2). Isolation and structural elucidation of the six new metabolites LNM K-1 (2), K-2 (3), K-3 (4), K-1′ (5), K-2′ (6), and K-3′ (7) support the previously proposed pathway6,7 and unveil new intermediates for the biosynthesis of 1 (Figure 1).
Figure 2.
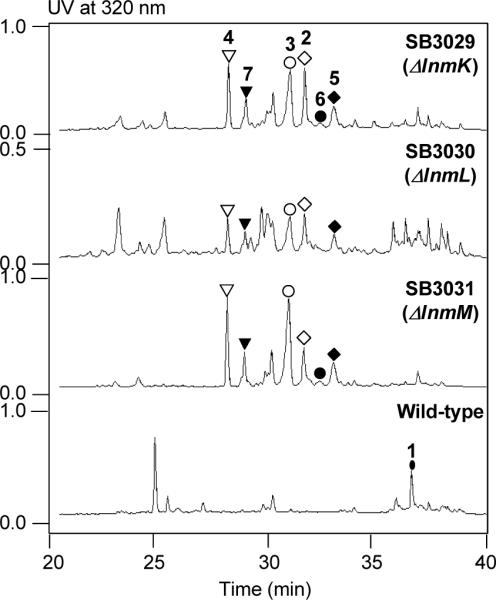
Metabolite profiles of S. atroolivaceus S-140 mutant strains SB3029 (i.e., ΔlnmK), SB3030 (i.e., ΔlnmL), and SB3031 (i.e., ΔlnmM), in comparison with the wild-type, upon HPLC analysis. See Figure 1 for structures of 1–7.
To explore the roles of LnmK, LnmL and LnmM in vivo, inactivation of lnmK, lnmL, and lnmM was accomplished by inserting an apramycin resistance gene cassette into the individual gene within the lnm gene cluster in the S. atroolivaceus S-140 wild-type.8 The resultant mutant strains were named SB3029 (i.e., ΔlnmK), SB3030 (i.e., ΔlnmL), and SB3031 (i.e., ΔlnmM), whose genotypes were confirmed by Southern analysis (Figure S1, Supporting Information).
S. atroolivaceus S-140 mutant strains SB3029, SB3030, and SB3031 were fermented, with the wild-type as a control, and their culture supernatants were extracted with ethyl acetate and extracts analyzed by HPLC.8 Relative to wild-type, all three mutants were unable to produce 1, confirming the indispensability of LnmK, LnmL and LnmM in 1 biosynthesis. Instead, a series of new peaks (2–7) with retention times shorter than that for 1 were observed upon HPLC analysis; all compounds showed a maximum UV absorption at 320 nm, characteristic of the conjugated (Z,E)-thiazol-5-yl-penta-2,4-dienone moiety of 1 (Figure 2). LC-ESI-MS analysis revealed that 2–7 in the three mutant strains SB3029, SB3030, and SB3031 afforded a same set of [M + H]+ ions at m/z of 391, 393, 265, 391, 393, and 265, respectively, suggesting that the three mutant strains accumulated the same series of metabolites. The ΔlnmK mutant strain SB3029 was then cultured under standard conditions and 2–7 were purified by silica gel column chromatography and semipreparative C-18 reverse phase HPLC; their structures were solved by MS and 1H and 13C NMR spectroscopy.8
The molecular formula of 4 was determined to be C20H28N2O4S by high resolution MALDI MS measurement, affording an [M + Na]+ ion at m/z 415.1653 (calculated [M + Na]+ ion at m/z 415.1667). A comparison of the 1H and 13C NMR spectra of 4 with those of 1 revealed that signals in the range of C-9 to C-17 of 1 were similarly present in 4, except for the lack of one H (H-11 at δH 8.03) in 1 and the presence of one additional H (H-11 at δH 7.32) in 4. The existence of the conjugated (E,E)-double bond in the region of C-10 to C-13 was supported by the presence of their unique coupling constants (J > 14.0 Hz), as well as gCOSY and HMBC correlations (Figure 3). The remaining proton and carbon signals suggested the existence of a cyclohexane moiety, which was assigned on the basis of one H-H coupling system (H-7, -2, -3, -4, and -5) and HMBC correlations between H3-18 (δH 1.01) and C-5 (δc 40.1), C-6 (δc 70.1), and C-7 (δc 41.8) (Table S2, Figures S9, S10, Supporting Information). The relative stereochemistry of C-3, C-6, and C-7 was established on the basis of ROESY correlations of H-3 with H-7, and of H3-18 with H-8, while the absolute stereochemistry of C-16 was deduced on the basis that 1 and 4 share the same biosynthetic origins (Figure 1). The structure of 4 was fully consistent with other correlations as observed in gCOSY, gHMBC and HMQC.
Figure 3.
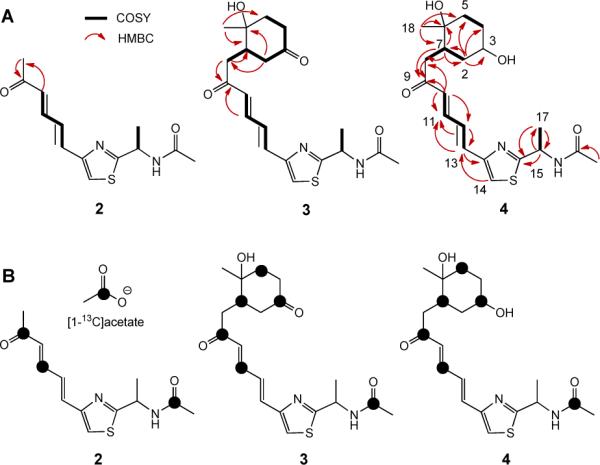
(A) Key HMBC and COSY correlations for LNM K-1 (2), K-2 (3), and K-3 (4). The similar HMBC correlations of 2, 3 with 4 were omitted for clarity. (B) Specific 13C-enrichment of 2, 3 and 4 resulting from feeding experiments with sodium [1-13C]acetate.
The structure of 3 was established in a fashion analogous to 4 by 1H and 13C NMR experiments. The molecular formula of 3 was determined to be C20H26N2O4S on the basis of high resolution MALDI MS measurement, affording an [M + Na]+ ion at m/z 413.1480 (calculated [M + Na]+ ion at m/z 413.1505). In comparison to 4, 3 lacks the oxygenated methine signal (C-3 at δC 68.6 for 4) and has a characteristic ketone carbonyl carbon (C-3 at δC 210.3 for 3) (Table S2, Figures S5, S6, Supporting Information), in agreement with its molecular weight of 2 Da lower than that of 4.
Compounds 2 and 5 cannot be completely separated from each other due to their rapid inter-conversion.8 They both yielded [M + H]+ ions at m/z of 265 upon LCESI-MS analysis, suggesting the existence of a pair of regio-isomers. The molecular formula of 2 and 5 was determined to be C13H16N2O2S by high resolution MALDI MS measurement, yielding an [M + H]+ ion at m/z 265.1001 (calculated [M + H]+ ion at m/z 265.1005), and their structures were assigned on the base of 1H and 13C NMR spectra of the mixture of 2 and 5 (Table S2, Figures S3, S4, Supporting Information). To confirm the inter-conversion of 2 and 5, a mixture of both substances was exposed to UV light (254 nm) for a period of 30 min in CDCl3,8 and 1H NMR spectra before and after irradiation were compared (Figure S2, Supporting Information). Diminished proton signals of 5 corresponded to the compound's conversion to 2, verifying that 5 is the photo-isomer of 2; 2 is the more photo-stable conformation, consistent with the proposed structures (Figure 1).
Similar to 2 and 5, compounds 6 and 7 cannot be fully separated from 3 and 4, respectively, due to rapid conversion of 6 to 3, and 7 to 4. On the basis of LC-ESIMS analyses, which afforded the same [M + H]+ ions at m/z 391 for 3 and 6 and at m/z 393 for 4 and 7, 3/6 and 4/7 were each assigned to be a pair of photo-isomers (Figure 1) with the structure of 6 further confirmed by 1H and 13C NMR spectra of the 3/6 mixture (Table S2, Figures S7, S8, Supporting Information).
The structures of 2–7 differ from 1 most notably by virtue of the absence of the 18-membered macrolactam, acetylation of the D-alanine N-terminus, and the cyclohexane moieties in 3, 4, 6, and 7. To determine their biosynthetic origin and to correlate their intermediacy in 1 biosynthesis, sodium [1-13C]acetate was fed to the ΔlnmK mutant strain SB3029 and 13C-enriched 3 and 4, as well as the mixture of 2 and 5, were isolated and characterized (Table S3, Figures S11–S14, Supporting Information). Thus, as depicted in Figure 3B, specific 13C-enrichments at C-3, C-5, C-7, C-9, and C-11 in 3 and 4 and at C-9 and C-11 in 2 were consistent with the hypothesis that all were biosynthesized by the LNM PKS en route to 1 (Figure 1). The C-2 carbon in 3 and 4 was not labeled, suggesting that it was originated from C-2 of acetate with the loss of the 13C-labeled C-1 carbon. Similarly, the C-8 carbon in 2 was not labeled, indicating that it was also derived from the C-2 of acetate (Figures 1 and 3).
In this study, we have confirmed that lnmK, lnmL and lnmM are essential for 1 biosynthesis, experimentally supporting their predicted roles in β-alkylation steps for 1. These findings shed new light into the initial steps leading to the formation of the dithiolane ring (Figure 1). In brief, following the completion of the final chain extension, the HCS homologue LnmM catalyzes Claisen condensation between the LnmJ PKS module-8 ACP-tethered polyketide intermediate and the propionyl-SLnmL. This results in the attachment of the propionyl group, as a β-alkyl branch, to the ACP-tethered intermediate, which is subsequently dehydrated by the ECH1 homologue LnmF and further modified en route to 1 (Figure 1, path a). Without the β-alkylation steps by LnmKLM, the LnmJ PKS module-8 ACP-bound polyketide intermediate apparently cannot be cyclized by the LnmJ-thioesterase (TE) domain, thereby undergoing hydrolytic release to afford the linear intermediate 8 (Figure 1, path b). This reaction could be catalyzed by either the LnmJ-TE domain, a type I TE, or LnmN, a type II TE within the lnm gene cluster.6 Hydrolytic removal of stalled polyketide or peptide intermediates from PKS or NRPS machinery by both type I and type II TEs are well known in polyketide, peptide, and hybrid polyketide-peptide biosynthesis.9 Finally, the β-ketoacid 8 could undergo a series of transformations to afford 3, 4, 6, and 7, and these reactions are most likely catalyzed by promiscuous enzymes residing outside of the lnm cluster (Figure 1, path b).
Interestingly, the mutant strains SB3029, SB3030, and SB3031 also accumulated 2 and 5. Similar phenotypes have also been observed in mupirocin biosynthesis, where inactivation of the genes encoding for β-methyl incorporation produced shortened shunt metabolites.10 Thus, 2 and 5 are most likely shunt metabolites of the proposed β-ketoacid 9, which undergoes release from LnmJ PKS module-5 in a mechanism similar to the aforementioned (Figure 1, path c). This result suggests the importance of specific protein-protein interactions among LnmKLM and LnmJ PKS, disruption of which leads to premature release of LnmJ PKS ACP-tethered polyketide intermediates, derailing 1 biosynthesis (Figure 1). The above hypothesis was further supported by the observation that the start and stop codons between lnmJ, lnmK, lnmL and lnmM overlap, suggesting the cotranslation of these genes, thereby ensuring the formation of a proper protein complex.
The accumulation of 2–7 in mutant strains SB3029, SB3030, and SB3031 supports a processive mechanism for the LNM NRPS-PKS megasynthase-catalyzed assembly of the LNM hybrid peptide-polyketide backbone from amino acid and short carboxylic acid precursors. Although PKS or hybrid PKS-NRPS systems are widely accepted to assemble polyketides or hybrid peptide-polyketides processively, this has been demonstrated experimentally in only a few cases. For example, inactivation of the amide transferase gene rifF in Amycolatopsis mediterranei abolished rifamycin production, giving rise instead to a series of linear polyketides with chain lengths varying from tetra- to decaketide.11 In bleomycin12 and epothilone13 biosynthesis, various peptides or polyketides representative of varying elongation steps have also been identified. Predicated on these findings and those reported here, the processivity of PKS, NRPS or hybrid PKS-NRPS represents a promising tool by which to generate natural product structural diversity by rational manipulation of these processive biosynthetic machineries.
Supplementary Material
Acknowledgment
We thank Kyowa Hakko Kogyo Co. Ltd. (Tokyo, Japan) for the wild-type S. atroolivaceus S-140 strain and the Analytical Instrumentation Center of the School of Pharmacy, UW-Madison, for support in obtaining MS and NMR data. This work was supported in part by NIH Grants CA106150 and CA113297.
Footnotes
Supporting Information Available Detailed experimental procedures, 1H and 13C NMR spectra for compounds 2–7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Calderone CT. Nat. Prod. Rep. 2008;25:845–853. doi: 10.1039/b807243d. [DOI] [PubMed] [Google Scholar]
- (2).Calderone CT, Kowtoniuk WE, Kelleher NL, Walsh CT, Dorrestein PC. Proc. Natl. Acad. Sci. U.S.A. 2006;103:8977–8982. doi: 10.1073/pnas.0603148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Gu L, Jia J, Liu H, Hakansson K, Gerwick WH, Sherman DH. J. Am. Chem. Soc. 2006;128:9014–9015. doi: 10.1021/ja0626382. [DOI] [PubMed] [Google Scholar]; (b) Buchholz TJ, Rath CM, Lopanik NB, Gardner NP, Hakansson K, Sherman DH. Chem. Biol. 2010;17:1092–1100. doi: 10.1016/j.chembiol.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Simunovic V, Müller R. ChemBioChem. 2007;8:497–500. doi: 10.1002/cbic.200700017. [DOI] [PubMed] [Google Scholar]; (b) Simunovic V, Müller R. ChemBioChem. 2007;8:1273–1280. doi: 10.1002/cbic.200700153. [DOI] [PubMed] [Google Scholar]; (c) Calderone CT, Iwig DF, Dorrestein PC, Kelleher NL, Walsh CT. Chem. Biol. 2007;14:835–846. doi: 10.1016/j.chembiol.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Hara M, Asano K, Kawamoto I, Takiguchi T, Katsumata S, Takahashi KI, Nakano H. J. Antibiot. 1989;42:1768–1774. doi: 10.7164/antibiotics.42.1768. [DOI] [PubMed] [Google Scholar]; (b) Hara M, Takahashi I, Yoshida M, Asano K, Kawamoto I, Morimoto M, Nakano H. J. Antibiot. 1989;42:333–335. doi: 10.7164/antibiotics.42.333. [DOI] [PubMed] [Google Scholar]
- (6).(a) Cheng Y, Tang G, Shen B. J. Bacteriol. 2002;184:7013–7024. doi: 10.1128/JB.184.24.7013-7024.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cheng Y, Tang G, Shen B. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3149–3154. doi: 10.1073/pnas.0537286100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tang G, Cheng Y, Shen B. Chem. Biol. 2004;11:33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]; (d) Cheng Y, Tang G, Shen B. J. Nat. Prod. 2006;69:387–393. doi: 10.1021/np050467t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tang G, Cheng Y, Shen B. J. Bio. Chem. 2007;282:20273–20282. doi: 10.1074/jbc.M702814200. [DOI] [PubMed] [Google Scholar]
- (7).Liu T, Huang Y, Shen B. J. Am. Chem. Soc. 2009;131:6900–6901. doi: 10.1021/ja9012134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).See Supporting Information for experimental details.
- (9).(a) Sieber SA, Marahiel MA. Chem. Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]; (b) Du L, Lou L. Nat. Prod. Rep. 2010;27:255–278. doi: 10.1039/b912037h. [DOI] [PubMed] [Google Scholar]
- (10).Wu J, Hothersall J, Mazzetti C, O'Connell Y, Shields JA, Rahman AS, Cox RJ, Crosby J, Simpson TJ, Thomas CM, Willis CL. ChemBioChem. 2008;9:1500–1508. doi: 10.1002/cbic.200800085. [DOI] [PubMed] [Google Scholar]
- (11).Yu TW, Shen YM, Doi-Katayama Y, Tang L, Park C, Moore BS, Hutchinson CR, Floss HG. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9051–9056. doi: 10.1073/pnas.96.16.9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Takita T, Muroka Y. Biosynthesis and chemical synthesis of bleomycin. Walter de Gruyter; New York: 1990. pp. 289–309. [Google Scholar]; (b) Du L, Sanchez C, Chen M, Edwards DJ, Shen B. Chem. Biol. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- (13).Hardt IH, Steinmetz H, Gerth K, Sasse F, Reichenbach H, Hofle G. J. Nat. Prod. 2001;64:847–856. doi: 10.1021/np000629f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.