

Abstract
Mitochondrial dysfunction has been proposed to play a role in the neuropathology of multiple sclerosis (MS). Previously, we reported significant alterations in the transcription of nuclear-encoded electron transport chain genes in MS and confirmed translational alterations for components of Complexes I and III that resulted in reductions in their activity. To more thoroughly and efficiently elucidate potential alterations in the expression of mitochondrial and related proteins, we have characterized the mitochondrial proteome in postmortem MS and control cortex using Surface-Enhanced Laser Desorption Ionization Time of Flight Mass Spectrometry (SELDI-TOF-MS). Using principal component analysis (PCA) and hierarchical clustering techniques we were able to analyze the differential patterns of SELDI-TOF spectra to reveal clusters of peaks which distinguished MS from control samples. Four proteins in particular were responsible for distinguishing disease from control. Peptide fingerprint mapping unambiguously identified these differentially expressed proteins. Three proteins identified are involved in respiration including cytochrome c oxidase subunit 5b (COX5b), the brain specific isozyme of creatine kinase, and hemoglobin β-chain. The fourth protein identified was myelin basic protein (MBP). We then investigated whether these alterations were consistent in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS. We found that MBP was similarly altered in EAE but the respiratory proteins were not. These data indicate that while the EAE mouse model may mimic aspects of MS neuropathology which result from inflammatory demyelinating events, there is another distinct mechanism involved in mitochondrial dysfunction in gray matter in MS which is not modeled in EAE.
Keywords: Mitochondrial proteomics, multiple sclerosis, experimental autoimmune encephalomyelitis
1. Introduction
Multiple sclerosis (MS) is an inflammatory neurodegenerative disease of the central nervous system resulting in progressive physical and cognitive disability. In the MS brain, white matter lesions are a hallmark of the disease; however, cortical gray matter lesion load has been found to be extensive [1] and damage to neurons and axons correlates with progression and cognitive impairment [2, 3, 4]. Several studies have implicated mitochondrial dysfunction as a possible mechanism in the development of MS related neuropathology. Nuclear magnetic resonance spectroscopy has identified decreases in the neuronal mitochondrial metabolite N-acetyl aspartate (NAA) in MS brain and decreases in NAA appears to precede neuronal atrophy, indicating that dysfunctional mitochondria may precede neurodegeneration [5, 6]. Mitochondrial dysfunction has also been implicated in disease pathology in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS [7]. It has been reported that increased nitration of electron transport chain components leads to decreased mitochondrial activity in EAE [8] and that cyclophilin D, a modulator of the mitochondrial permeability transition pore, is protective [9]. Similarly, analyses of postmortem MS white matter have identified alterations to mitochondrial enzyme activity and damage to mitochondrial DNA in lesions [10, 11, 12]. Other studies have demonstrated defects in mitochondrial electron transport gene expression and function in normal appearing gray matter (NAGM) in postmortem MS cortex [13, 14]. These studies have identified transcriptional changes in important mitochondrial genes; however, translational or post-translational alterations in a multitude of other proteins may also impact mitochondrial function and energy production. Therefore, in the current report, we have applied Surface Enhanced Laser Desorption and Ionization time-of-flight mass spectrometry (SELDI-TOF-MS) to mitochondrial fractions derived from MS cortex to obtain a more complete understanding of protein expression changes that could contribute to the mechanisms of mitochondrial dysfunction in MS. Studies of proteins originating from MS brain tissue may provide new insights into the pathogenesis and etiology of the disease. Previous proteomic analyses of MS tissue have focused on white matter, lesioned tissue, and cerebrospinal fluid (CSF) [15, 16, 17, 18, 19, 20, 21, 22, 23]. However, there is evidence from magnetic resonance imaging studies that the mechanisms involved in tissue damage are different in gray matter and white matter in MS [24, 25]. Therefore studies detailing protein differential expression in normal appearing gray matter are necessary to provide additional insight into disease manifestation and progression as it relates to the neurodegenerative component of MS pathology.
2. Materials and Methods
2.1 Tissue Preparation and Protein Extraction
Reagents used in the preparation of all buffers were obtained from Sigma-Aldrich (St. Louis, MO). Brains were obtained from The Rocky Mountain MS Center Tissue Bank (Englewood, CO), the Brain and Spinal Fluid Resource Center (UCLA) and from The Kathleen Price Bryan Brain Bank (Durham, NC) under IRB protocol. Tissue was obtained by rapid autopsy and frozen at −70°C. This study includes two cohorts, each consisting of four control and four secondary progressive multiple sclerosis (SPMS) donors. Tissue from each cohort was homogenized on separate dates using identical protocols. MS and control brains have been matched primarily for brain region (parietal, Brodmann Area 1–3, and frontal cortex, Brodmann Area 9), and also for age, sex, and post mortem interval (PMI) as closely as possible. Frozen sections, 30 µm thick, from adjacent sides of the tissue blocks were immunostained for myelin proteolipid protein (PLP) to ensure the absence of demyelinated lesions as previously described [26]. Donor demographics are described in Table 1. Eight control and eight MS donors were used for the differential expression proteomic analyses while one additional control (C9) and MS donor (MS9) were used in western blotting for confirmation.
Table 1.
Donor Descriptions
| Sample | Age (yrs.) | Sex | PMI (hrs.) | Brain Region |
|---|---|---|---|---|
| C1 | 57 | M | 4.5 | Fr |
| C2 | 87 | M | 6.5 | Pa |
| C3 | 72 | F | 30 | Pa |
| C4 | 75 | M | 19 | Pa |
| C5 | 80 | M | 14 | Fr |
| C6 | 65 | M | 3.5 | Pa |
| C7 | 86 | F | NA | Pa |
| C8 | 73 | F | 12 | Fr |
| C9 | 74 | F | 5 | Fr |
| MS1 | 48 | M | 4 | Fr |
| MS2 | 30 | M | 5 | Pa |
| MS3 | 62 | F | 6 | Pa |
| MS4 | 36 | F | 3 | Pa |
| MS5 | 30 | M | 5 | Fr |
| MS6 | 78 | M | 15.5 | Pa |
| MS7 | 81 | F | 11.8 | Pa |
| MS8 | 52 | F | 20.6 | Fr |
| MS9 | 51 | F | 5.25 | Fr |
C- control donors; MS- MS donors; M- male; F- female; Fr- frontal cortex; Pa- parietal cortex; NA- not available
EAE was induced in C57Bl/6 mice by subcutaneous injection of MOG35–55 as previously described [27]. EAE brains and brains from control littermates were prepared at The University of Calgary and sent frozen to Kent State University for analysis by Western blotting and SELDI-TOF-MS. Protein was isolated from whole brains and from cortex for Western analyses. EAE-afflicted mice were at peak clinical severity (Grade 4 of the 5 point scale) when sacrificed.
For postmortem human tissue, approximately 250mg tissue from frozen blocks was carefully excised excluding white matter. Tissue was homogenized using a Wheaton homogenizer with a Teflon® pestle in whole cell homogenization buffer (20mM KCl, 3mM MgCl2, 10mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.9, 0.5% NP-40, 5% glycerol with protease inhibitors (P2714, Sigma-Aldrich, St. Louis, MO) in forty strokes. The homogenate was centrifuged for 10 minutes at 500xg at 4°C. The supernatant was removed and centrifuged at 10,000g for 30 minutes at 4°C. The pellet containing the mitochondrially enriched fractions was further purified by washing twice in 20mM phosphate buffered saline (PBS), pH 7.4. The mitochondrial pellet was lysed in mitochondrial lysis buffer (50mM Tris, 7M urea, 3% CHAPS with protease inhibitors) by vortexing for one minute and then incubating for 20 minutes at room temperature. The mitochondrial lysate was centrifuged for 10 minutes at 10,000g at 4°C. A modified Lowry assay was used to quantify the supernatant protein concentration. All samples were stored at −80°C until further analysis.
2.2 Immunofluorescent Staining
Frozen tissue blocks from postmortem MS and control brains were fixed in 4% paraformaldehyde for 24 hours and washed in PBS (2 × 24 hours). Fixed tissue blocks were sliced 75 µm thick perpendicular to the outer edge of the cortex (to include all cell layers) on a media cooled vibratome. Sections were stored in PBS in multiwell plates at 4°C in a humidified chamber until subjected to the staining procedure. Thereafter, free-floating sections were incubated in goat anti-hemoglobin (1:100, Thermo Scientific, Rockford, IL) and mouse anti-neurofilament (1:500, SMI-32, Covance, Princeton, NJ) antibodies in PBS, 5% triton-X 100, 3% normal donkey serum for 24 hours. After 3 washes in PBS (10 minutes each) the tissue was incubated in donkey anti-mouse Alexa 488 (1:250, A-11055 Invitrogen, Carlsbad, CA) and donkey anti-goat Alexa 546 (1:250, A-11056 Invitrogen, Carlsbad, CA) in PBS for 12 hours. Following three ten minute washes in PBS, the sections were incubated in 50 mM ammonium acetate, 10mM cupric sulfate for 30 minutes to quench lipofuscin auto-fluorescence. Sections were next washed three times in PBS (10 minutes each), placed below coverslips under Vectashield® mounting media (Invitrogen, Carlsbad, CA), sealed with clear nail polish, and refrigerated until microscopically imaged. Images were acquired using an Olympus FV500 confocal microscope equipped with four lasers (HeCd 442 nm, Ar 488 nm and 514 nm, HeNe 543 nm and HeNe 633 nm) and a 60X oil objective. Microscopic fields were acquired that clearly contained pyramidal cells as indicated by neurofilament staining. Green and red fluorescence image stacks were captured sequentially to prevent any channel bleed through, and spanned the physical section. All images were acquired with isometric voxels at a resolution of 0.42 microns per pixel. The image stacks were z-projected using ImageJ (NIH, Bethesda, MD) and channels merged to show co-localized signal within pyramidal cells.
2.3 Western Blotting
Mitochondrially enriched or cytoplasmic samples were separated on NuPage® 4–12% Tris gel (Invitrogen, Carlsbad, CA) for 35 minutes at 200V, transferred to nitrocellulose paper for 1 hour at 45V and incubated with the appropriate antibodies, either mouse MBP (AbCam, Cambridge, MA), human MBP (Chemicon, Temecula, CA), creatine kinase (AbCam, Cambridge, MA), COX5b (Invitrogen, Carlsbad, CA), hemoglobin (Thermo Scientific, Rockford, IL), or neurofilament (Chemicon, Temecula, CA). The blots were exposed to the corresponding secondary horseradish peroxidase conjugated secondary IgG (sc2005-mouse/sc2001-rabbit Santa Cruz Biotechnology, Inc. Santa Cruz, CA) and visualized using enhanced chemiluminesence reagents (sc2048, Santa Cruz Biotechnology, Inc. Santa Cruz, CA). Densitometry was performed using ImageJ (NIH, Bethesda, MD) from three MS and control comparisons or EAE and control brain comparisons from at least two experiments. Quantitation was standardized with antibodies against either Complex IV subunit 2 (COX2, 1:2000, MS405, Mitosciences, Eugene, OR), a mitochondrially encoded protein, or the Complex II SDHA subunit (Molecular Probes, Eugene, OR) for mitochondrial fractions, and GAPDH (Chemicon, Temecula, CA) for cytoplasmic fractions. A Student’s T-test was performed to determine statistical significance.
2.4 Ion Exchange Fractionation
The mitochondrial enriched protein extracts were further fractionated using ion exchange chromatography in a spin column format (column- UFC30HV00; centrifuge tube-UFC3000TB Millipore, Billerica, MA). Samples (100µg each) were equilibrated using buffer A (9M Urea, 2% CHAPS in 50mM Tris HCl pH 9.0) and incubated for twenty minutes at room temperature. Buffer B (Buffer A diluted 1 part to 8 parts 50mM Tris HCl, pH 9.0) was used to dilute all samples to 200µL and then stored on ice while the remaining samples were prepared (30 minutes maximum). The quaternary ammonium anion exchange beads (Q ceramic HyperD F, Pall Biosepra, NewYork, NY) were equilibrated three times in buffer B. The samples were loaded onto the column/tube and mixed in an end to end fashion for 30 minutes at room temperature. After this initial incubation, the samples were centrifuged for 1 minute at 1000xg. This eluate was the flow through fraction. The column was removed from the centrifuge tube and placed in the next centrifuge tube labeled according to the wash buffer. The pH 9 elution buffer (50mM Tris HCl, pH 9.0, 0.1% octyl glucopyranoside (OGP), 200µL) was added to the column and mixed in an end to end fashion for 10 minutes, then centrifuged as before. This process was repeated for each buffer at pH 7.0, 5.0, 4.0, and 3.0, and a final organic solvent elution, yielding six fractions with pI ranges of greater than 9.0 in fraction 1, 7.0–9.0 in fraction 2, 5.0–7.0 in fraction 3, 4.0–5.0 in fraction 4, 3.0–4.0 in fraction 5, and less than 3.0 in fraction 6, respectively. Specific buffer compositions were as follows: pH 7.0 elution buffer; 50mM HEPES pH 7.0, 0.1% OGP, pH 5.0 elution buffer; 100mM sodium acetate pH 5.0, 0.1% OGP, pH 4.0 elution buffer; 100mM sodium acetate pH 4.0, 0.1% OGP, pH 3.0 elution buffer; 50mM sodium acetate pH 3.0, 0.1% OGP; organic elution buffer; 33.3% isopropyl alcohol (IPA), 16.7% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA). All protein solutions were stored on dry ice until return to the −80°C freezer.
2.5 SELDI-TOF-MS Acquisition
All samples were mixed with a saturated α-cyano-4-hydroxycinnamic acid matrix solution consisting of 50% ACN and 0.5% TFA by volume with 0.6% OGP by weight in a 1:5 dilution. Each matrix diluted sample (1µL) was applied to a spot on an NP20 Proteinchip® (normal phase chromatographic surface, Ciphergen Biosystems, Fremont, CA) chip in a randomized manner. Mass spectra were acquired with a model PBSIIc SELDI-TOF mass spectrometer manufactured by Ciphergen Biosystems (Fremont, CA). The optimal sensitivity and laser intensity were established on a location not used in transient averaging. These values were then used to develop a spot protocol. This same protocol was applied to all chips. Data were acquired at a digitizer rate of 250.0MHz in positive ion mode with a chamber vacuum of less than 5×10−07 torr. The source voltage was 20kV and the detector voltage was 2,700V. A total of 65 transients were averaged for each spectrum. All spectral processing (smoothing and baseline subtraction) was performed with Proteinchip 3.1 Software (Ciphergen Biosystems, Fremont, CA). Spectra were calibrated externally over the appropriate mass range using at least three of the following standards: recombinant Hirudin BHVK (6,964Da), bovine cytochrome c (12,230 Da) equine cardiac myoglobin (16,952 Da), bovine RBC carbonic anhydrase (29,024 Da) and S. cerevisae enolase (46,670 Da). In order to make appropriate comparisons, all spectra were normalized against the total ion current [15].
2.6 Data Analysis
Manual peak picking, using a signal to noise ratio of greater than 4.0, allowed for unbiased peak selection. Automatic peak detection in Ciphergen Express often neglects those features which appear as shoulders on larger peaks [28] and consequently manual peak selection across all spectra of the same fraction provided a more accurate assessment of the spectral features. Criterion for the selection of peaks across the entire data set required peak occurrence in 25% of the spectra with a signal to noise ratio greater than 4.0. Peak occurrence percentages were selected based upon the expected absence of some peaks from either the control or the MS group. Peaks selected for further evaluation and validation had a signal to noise ratio of greater than 3.0 and were present in the overexpressed sample group (control or MS). All statistical calculations were done with Ciphergen® Express software (Ciphergen Biosystems, Fremont, CA). Univariate analysis included the non-parametric Mann-Whitney test. Applicable multivariate techniques included principal component analysis (PCA) and hierarchical clustering [29]. Peaks considered differentially expressed were those altered by at least 1.8 fold, p< 0.05.
2.7 Immunoprecipitation
Anti-nitrotyrosine antibody (05–233 Millipore, Billerica, MA) was crosslinked to Protein G agarose beads using the Pierce Seize X kit according to manufacturers’ instructions. Briefly, Protein A agarose beads were used to preclear mitochondrially enriched proteins. Protein G beads were covalently crosslinked to the anti-nitrotyrosine antibody using disuccinimidyl suberate and washed to remove any unreacted molecules. The beads were then incubated with mitochondrially enriched NAGM protein and any unbound proteins were removed by washing. The nitrated proteins were then eluted from the beads using a low pH wash. The eluant was then separated using pre-cast Bis-Tris gels (Invitrogen, Carlsbad, CA), stained with blue silver [30] and destained in reverse osmosis water. Bands of interest (those increased in MS) were excised, dehydrated and sent to the University of Pittsburgh Genomics and Proteomics Core Facility for trypsin digestion and peptide fingerprint mapping.
2.8 Antibody Pull Down Confirmation
An antibody against COX5b (Proteintech Group, Chicago, IL, PN 11418-2-AP) were derivatized to 3µm diameter carboxylated polystyrene microspheres (Polysciences PN 09850) using a modified carbodiimide method [31]. Briefly, the beads were washed in 50mM MES (2-(N-morpholino)ethanesulfonic acid, Sigma PN M8250) twice. A 200mg/mL solution of EDC (N-Ethyl-N’-(3-dimethylaminopropyl) carbodiimide hydrochloride Fluka PN-03449) in 50mM MES was prepared. The washed beads (100µL) were incubated with the EDC solution such that the final concentration was 2mg/mL for approximately 2 minutes in a vortexer set on low speed. 200 µg of mitochondrially enriched NAGM protein was added to 500µl of the bead slurry. Protein and beads were incubated with constant mixing at room temperature for 3 hours. Beads were washed in 10mM Tris pH 8.0 0.5%BSA twice. Beads were stored in 150µL 10mM Tris pH 8.0 0.5%BSA at 4°C. Bright field microscopy was used to confirm low self-aggregation and antigen-antibody activity of conjugated beads.
Mitochondrial protein extracts from specific ion exchange fractions (2.5µL) were incubated with bead solution (2.0µL) in 10mM Tris pH 8.0 0.5%BSA to a final volume of 10µL for 1 hour with constant mixing at room temperature. Solutions from both before and after bead incubation were diluted 1/10 in a saturated α-cyano-4-hydroxycinnamic acid (CHCA; Sigma PN 145505-5g) aqueous solution consisting of 50% ACN (Fisher PN A996-4), 0.6% OGP (Sigma PN O8001-5g) and 0.5% TFA (JT Baker PN9470-00). These solutions were randomly spotted onto an NP-20 Proteinchip® at 0.5µL, air-dried and repeated.
2.9 Protein Purification and Peptide Fingerprint Mapping
Individual ion exchange fractions were purified by gel electrophoresis on 16% acrylamide Tris glycine gels at 200V for 45 minutes [32]. The gels were stained with coomassie blue [33] and gel plugs of interest were removed. The gel plugs were destained [34], dehydrated and stored in 1% acetic acid prior to trypsin digestion and peptide fingerprint mapping. Trypsin digestion followed by HPLC-ESI-MS/MS on an LCQ Deca XP mass spectrometer (ThermoFinnigan, San Jose, CA) was performed at the Genomics and Proteomics Core Laboratories at the University of Pittsburgh.
3. Results
3.1 Mitochondrial Protein Differential Expression in Postmortem MS Cortex
Donor demographics are described in Table 1. Mitochondrial samples from either parietal or frontal cortex were obtained from NAGM as confirmed using PLP staining. Analysis of the mitochondrial fractions was divided into two separate groups or cohorts due to tissue sample availability at study onset. The first cohort of samples consisted of mitochondrial fractions obtained from four control and four MS brain slices (C1–4 and MS1–4). As additional brain tissue became available the analysis was extended to incorporate the second group, cohort 2, which also consisted of mitochondrial fractions derived from four control and four MS brain slices (C5–8 and MS5–8). The mitochondrial fractions were separated by ion-exchange fractionation and analyzed by SELDI-TOF-MS. Mass spectra were acquired from fractions with pI values between 9 and 3 and revealed multiple peaks. The Mann-Whitney test was used to identify peaks with significant differences in peak intensities. Filtering the data for proteins altered by at least 1.8 fold at the 0.05 significance level we identified nineteen peaks which were differentially expressed (see Supplemental Data, Table1). Cohort 1 contained twelve differentially expressed peaks, with five of these peaks from fraction 3 (pI 5.0–7.0) and five from fraction 6 (pI<3), and cohort 2 contained seven differentially expressed peaks, three of which originated in fraction 6 (pI <3). These fractions were selected for peptide fingerprint mapping and further investigation. Fractions 4 (pI 4.0 −5.0) and 6 from cohort 2 were also selected for further purification and peptide fingerprint mapping. Table 2 describes the identified differentially expressed peaks from both data sets listing fold change, Mann-Whitney p values, theoretical isoelectric point, theoretical molecular weight, cohort, mass to charge ratio (m/z) and peptide fingerprint mapping results.
Table 2.
SELDI Differential Protein Expression
| Cohort | Fraction pI range |
Mann - Whitney p value |
m/z | Theoretical pI |
Theoretical MW |
fold | UniProt Accession |
Entry Name |
Probability | MASCOT score |
Peptides detected |
Sequence Coverage |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5–7 | 0.05 | 10600 | 6.3 | 10613 | −1.9 | P10606 | COX5b | 3.70E-07 | 20.2 | 2 | 12.24 |
| 1a | 5–7 | 0.009 | 15940 | 11.3 | 21493 | +2.0 | P02686-3 | MBP1 | 4.34E-07 | 80.2 | 10 | 39.59 |
| 1a | 5–7 | 0.009 | 15940 | 11.1 | 20246 | +2.0 | P02686-4 | MBP2 | 4.34E-07 | 80.2 | 10 | 41.94 |
| 1a | 5–7 | 0.009 | 15940 | 11.4 | 18591 | +2.0 | P02686-5 | MBP3 | 4.34E-07 | 80.2 | 10 | 45.61 |
| 1a | 5–7 | 0.009 | 15940 | 11.1 | 17343 | +2.0 | P02686-6 | MBP4 | 4.34E-07 | 80.2 | 10 | 48.75 |
| 2 | 4–5 | 0.021 | 16012 | 6.8 | 15867 | +2.6 | P68871 | Hbb | 9.28E-05 | 60.2 | 6 | 51.37 |
| 2 | < 3 | 0.021 | 42700 | 5.3 | 42513 | +2.0 | P12277 |
Creatine Kinase B |
1.89E-05 | 70.3 | 7 | 27.11 |
Human MBP has 7 isoforms. All ten peptides detected by MS/MS are present in the isoforms listed.
3.2 Analysis of Proteomic Data with Multivariate Statistics
The application of hierarchical clustering to differential protein expression data identifies elements which may be linked in their occurrence. This unsupervised classification method can be used to identify and validate those biomarkers responsible for specific patterns and plays an essential role in current disease investigations. Since it is unlikely that one specific peak is responsible for the differential expression between control and MS NAGM mitochondrially-enriched proteins, a technique which detects correlated sets of variables is needed to analyze this data. In this research, a modified Pearson product-moment correlation coefficient was employed as a distance measure and the average linkage method was used to perform a hierarchical cluster analysis of protein expression and compute a dendrogram. Nodes of the dendrogram join the most similar objects and the relative length of the branches is indirectly proportional to the similarity of those objects. The most significant feature in the hierarchical clustering analysis is the segregation of control and MS donors by disease state (fraction 3 and 6 from cohort 1 and fraction 6 from cohort 2 shown in Figure 1 panels A, B, and C). Other fractions in this data set have only one or two differentially expressed peaks and therefore are not sufficiently complex to warrant the use of multivariate methods such as hierarchical clustering. Fraction 3 of cohort 1 contains two distinct nodes (Figure 1, Panel A). The short length of the branches indicate that the peak intensities of the proteins in each node are highly correlated, suggesting a potential relationship between the peaks in each node. The highly correlated peaks of the upper node, containing peaks at 10.7, 10.6, 10.2, 10.1 and 9.9 kDa, have a correlation coefficient greater than 0.85. The lower node, containing peaks at 15.9, 15.8, 8.0, and 7.9 kDa, has a correlation coefficient greater than 0.94. Of the four peaks, three were differentially expressed at the 95% confidence level, with p values of 0.009, 0.076, 0.028 and 0.028, respectively. Fraction 6 of cohort 1 (Figure 1, Panel B) also shows several highly correlated nodes. The upper middle node containing peaks at 22.9, 22.7, 11.5, 23.7, and 11.4 kDa have a correlation coefficient greater than 0.85, while the correlation coefficient of the lower middle node consisting of peaks at 9.8, 9.7, 12.6, 12.5 and 17.2 kDa is greater than 0.92. The linear correlation of the data combined with significant p values of 0.076, 0.028, 0.028, 0.016, and 0.076, respectively, indicate a group of possible biomarkers. Fraction 6 of Cohort 2 shows a distinct clustering by disease as well as several nodes (Figure 1, Panel C).
Figure 1. Multivariate analysis of cohort 1, fractions 3 and 6 and cohort 2 fraction 6 using principal component analysis and hierarchical clustering.


Multivariate analyses including hierarchical clustering and principal component score scatter plots show the segregation of MS and control samples. Panel A: Cohort 1 fraction 3 analysis. Panel B: Cohort 1 Fraction 6 analysis. Panel C: Cohort 2 fraction 6 analysis. Peaks identified by peptide fingerprint mapping are indicated with asterisks.
Principal component analysis (PCA) is a common technique used in the analysis of large data sets and uses computational methods for pattern recognition [35]. Mathematically, in this particular analysis, a PC is a linear combination of peaks from all samples included in the analysis. The contribution of a specific PC to a sample’s variability is indicated by the sample score. Each sample has a single unique score from each PC. The first PC accounts for the greatest variability in the data set with each subsequent PC accounting for decreasing amounts of variability. Therefore, the first several PCs will reveal which variable contributes the most to the differences between the populations and the sample specific scores will relate that variability to each sample.
Applying PCA to this research enables the identification of key proteins which may be responsible for the differences between control and disease states. We used this particular analysis to generate two dimensional scatter plots of the principal component (PC) scores to aid in the visualization of the data reduction process. Variables with similar scores for a specific PC will segregate together in a scatter plot, allowing the identification of similar variables. Because PCs are uncorrelated by definition [36], this process can be used to verify those factors which independently influence the data segregation. Figure 1, Panel A, B, and C display PC scatter plots of PC1 and PC2. Notice the segregation of the data into MS and control sample groups in each fraction. This technique also confirmed that other parameters such as brain region, gender, age or PMI were not responsible for data segregation. For all controlled variables, PCA verified that disease state was the only donor characteristic responsible for sample segregation.
3.3 Identification and Confirmation of Differentially Expressed Proteins in MS Cortex
The unambiguous identification of specific differentially expressed peaks in cohort 1 by peptide fingerprint mapping was complicated by the low resolution of one dimensional gel electrophoresis and the complex mixture of analytes in the 10 kDa and 16 kDa regions. Each band contained at least two protein species as evidenced by the SELDI-TOF mass spectra. Regardless, the entire band of each sample was excised and submitted for peptide fingerprint mapping on a ThermoFinnigan LCQ Deca XP. Table 2 lists the most probable identifications with Mascot scores, number of peptides, sequence coverage, probabilities and protein characteristics. The list of matches in the database for the 10 kDa protein fragment included cytochrome c oxidase subunit 5b (COX5b) (UniProt accession number P10606), an electron transport chain subunit of cytochrome c oxidase (Complex IV) with a Mascot score of 20.2 from two peptides detected and 12.24 % sequence coverage. The 15.9 kDa peak in fraction 3 was identified as myelin basic protein (MBP) (UniProt accession number P02686) with a Mascot score of 80.21 from 10 peptides with a sequence coverage range of 25.7% for isoform 1 to over 49% for isoform 6 (see Table 2) and almost certainly represents a proteolytic fragment of one of the MBP isoforms. The process and protease leading to this particular differentially expressed MBP fragment is currently under investigation.
In cohort 2, proteins from fraction 4 and 6 were purified by electrophoresis and bands were removed at 16 kDa and 42 kDa, respectively. The protein at 42 kDa was identified as creatine kinase type B (UniProt accession number P12277) with a Mascot score of 70.26 from seven peptides with a sequence coverage of 27.11%. The peak at 16 kDa was identified as hemoglobin β-chain (UniProt accession number P68871) with a Mascot score of 60.25 from six peptides and a sequence coverage of 51.37%. The SELDI-TOF mass spectra for these four proteins are shown in Figure 2, Panels A–D, and demonstrates the overexpression of hemoglobin β, MBP, and creatine kinase B, and decreased expression of COX5b in MS relative to controls.
Figure 2. Proteomic differential expression of identified proteins.
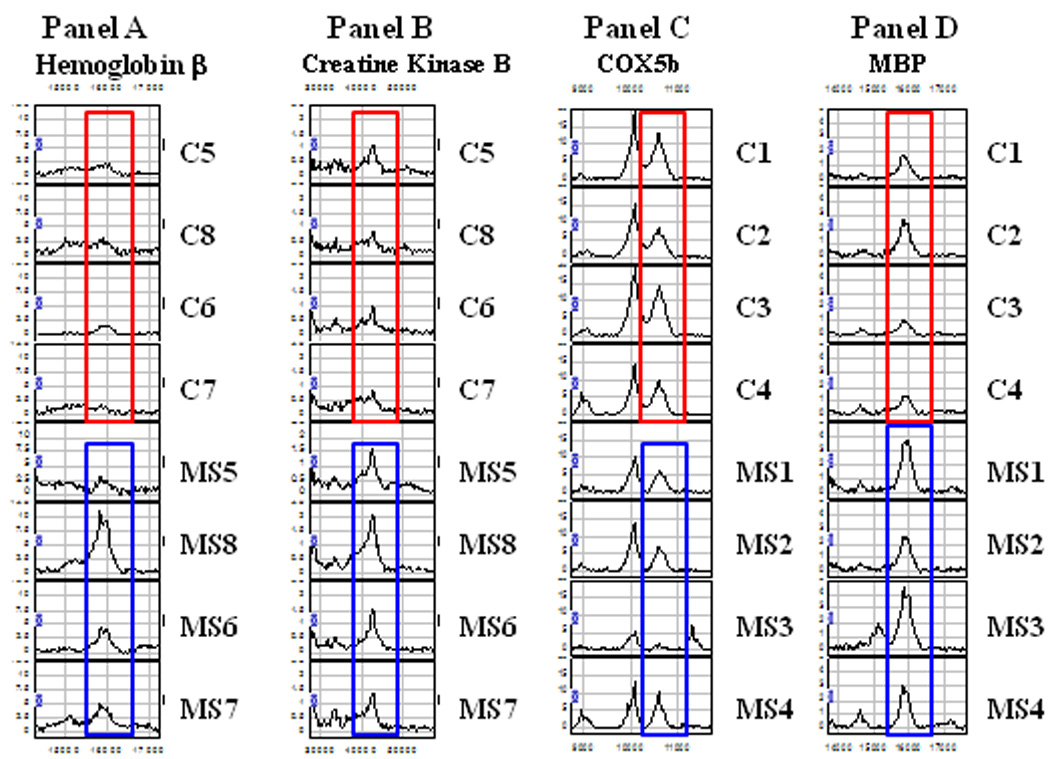
SELDI-TOF mass spectra showing the differential expression of a set of peaks. Panel A: The 16 kDa peak was identified as hemoglobin β chain, Panel B: The 42kDa peak was identified as Creatine Kinase type B, Panel C: The 10.6kDa peak was identified as COX5b, Panel D: The 16kDa peak was identified as MBP. Mass spectra of mitochondrial samples outlined in red are control samples and those outlined in blue are MS samples.
Confirmation of the identification of the peak at 10.6 kDa as COX5b was obtained by employing COX5b antibody labeled beads. SELDI-TOF-MS spectra acquired both before and after protein solutions were exposed to this COX5b antibody confirmed that the differentially expressed peak at 10.6 kDa was COX5b as shown in Figure 3A. Relative peak intensity ratios were used to determine which peak resulted from COX5b. The 10.1 kDa peak was verified as unchanged before and after spectra by one way ANOVA and was therefore used as the normalization peak to determine which peak intensity changed as a result of exposure to the COX5b IgG beads. Representative spectra from one of three trials are shown in Figure 3A. Only the peak at 10.6kDa demonstrated a change in relative peak height. This molecular weight corresponds to the mass of COX5b after signal peptide cleavage.
Figure 3. Confirmation of the identity of differentially expressed proteins.

A. Mitochondrially enriched protein solutions were incubated with an antibody to COX5b covalently bound to beads. Aliquots analyzed by SELDI-TOF-MS before and after incubation confirm the identity of the mass spectral peak at 10.6 kDa as COX5b. The red box highlights the region of interest for these spectra. B. Representative western blot demonstrating the relative purity of the cellular fractionation. Western blots were performed on cytoplasmic (cyto) and mitochondrial (mito) fractions isolated from MS and control cortex, run side by side and blotted with an antibody to the neuron specific protein, neurofilament (NF), and an antibody to the mitochondrial encoded COX2 protein. Multiple NF immunoreactive proteins are denoted by arrows. C. Representative western blots show increased MBP, hemoglobin β (Hbb), and creatine kinase (CKB) and decreased COX5b in mitochondrial fractions isolated from MS cortex. Increased expression of MBP was also observed in the EAE mouse brain while CKB, Hbb, and COX5b were not similarly altered in EAE and MS. To control for protein loading, western blots were reprobed with antibodies to either Complex II (CII) or COX2. D. Representative western blots of hemoglobin β (Hbb) and creatine kinase B (CKB) in cytoplasmic fractions isolated from MS and control cortex and also from EAE and control brains or cortex with GAPDH as a loading control. E. Quantitation was done for MBP, COX5b, Hbb, and CKB expression for MS and control samples and for EAE cortex. Data is expressed as percent of control for MS and EAE samples and densitometry was standardized to either CII or COX2 for mitochondrial fractions or to GAPDH for cytoplasmic fractions. Error bars represent SEM. * p≤ 0.05
We then performed western blotting experiments to confirm the protein expression differences identified by SELDI-MS as shown in Figure 3. In order to demonstrate the purity of our mitochondrial fractionation procedure, we performed western blots for expression of a neuronal marker, neurofilament, and the mitochondrial encoded protein cytochrome c oxidase subunit II (COX2), in MS and control cytoplasmic and mitochondrial fractions (Figure 3B). The altered expression of proteins identified in cohort 1 and in cohort 2, including MBP, COX5b, creatine kinase B (CKB) and the hemoglobin-β chain (Hbb), were confirmed by western blotting in mitochondrial fractions (Figure 3 C and D). While these proteins were not found to be significantly altered in both cohorts by SELDI-TOF-MS, quantitation of western blotting experiments confirmed a significant decrease in COX5b and significant increases in MBP, hemoglobin β, and creatine kinase B expression in mitochondrial fractions from samples across both cohort 1 and 2 and in additional samples (C9 and MS9) (Figure 3E). One control sample not included in the SELDI analysis but which was analyzed by western blot, C9, contained consistently increased mitochondrial content as indicated by the increase in COX2 expression (Figure 3C). MS samples still expressed increased levels of creatine kinase B relative to this control sample in spite of this increase in mitochondria. Further, COX5b expression was significantly decreased in MS samples even when C9 was not included in the analysis. COX5b and MBP were only expressed in mitochondrial fractions. Hemoglobin and creatine kinase B were detected in both mitochondrial and cytoplasmic fractions, but were not significantly increased in cytoplasmic fractions. Western blots were also performed for these proteins in cytoplasmic and mitochondrial fractions isolated from either whole brain or cortex from EAE and control mice. Representative western blots are shown in Figure 3C and D and quantitation in Figure 3E shows that only increased MBP expression is modeled in the EAE mouse. We did observe more than one isoform of MBP increased (two bands ∼ 18–20 kDa) in the EAE mitochondrial fractions rather than the one isoform increased in human MS tissue at ∼ 18 kDa (Figure 3C).
Further, we localized hemoglobin expression to neuronal cell bodies in MS postmortem tissue by immunofluorescent staining with antibodies to hemoglobin (red fluorescence) and neurofilament (green fluorescence) as shown in Figure 4. We observed intense hemoglobin staining in pyramidal neurons in MS cortex supporting our SELDI-TOF data indicating increased expression of hemoglobin in MS samples.
Figure 4. Increased immunofluorescent hemoglobin staining in MS parietal cortex pyramidal cell layer.


A. Confocal images taken at 20X of hemoglobin staining in control and MS postmortem tissue. Micrographs are from control (left column) and MS (right column) postmortem tissue sections stained for hemoglobin (Hbb) (top row) and neurofilament (NF) (middle row). Hemoglobin can be seen to be localized in a number of neurons, denoted by white arrows, including pyramidal cells, colocalized with the neurofilament stain. The last row displays the 2 channels overlayed with the organization of the pyramidal cell layers clearly visible.
B. Images are z-projected multichannel confocal micrographs from gray matter from control and MS parietal cortex. Control (row 1) and MS (row 2) tissue sections were stained for neurofilament (column 1) and hemoglobin (column 2), and image stacks acquired from the pyramidal cell layer using a 60X oil objective and identical acquisition parameters. Note increased hemoglobin staining intensity in the MS sample in agreement with the reported SELDI-TOF-MS results. The third column depicts the hemoglobin (red fluorescence) and neurofilament (green fluorescence) data overlayed, and clearly displays hemoglobin fluorescence within pyramidal cell somas. The scale bar represents 100 µm.
3.4 Increased Nitrotyrosine Modified Proteins in MS
The detection of increased nitrated proteins in MS mitochondrial fractions was performed by immunoprecipitation with an antibody to nitrotyrosine. Eluates from the nitrotyrosine immunoprecipitation of control and MS samples were purified using one dimensional gel electrophoresis and differentially stained bands which were increased in intensity in the MS mitochondrial fraction between 20–25kDa and 40–45kDa were excised. Nitrated proteins identified using peptide fingerprint mapping are shown in Table 3. Identified in the bands exhibiting increased nitrotyrosine immunoreactivity in MS were the mitochondrial encoded catalytic subunit of Complex IV of the electron transport chain, cytochrome c oxidase subunit 2 (COX2), two subunits of Complex I (NDUA8 and NDUV2), a subunit of Complex III (UQCR2), 2’, 3’-cyclic-nucleotide 3’-phosphodiesterase (CNPase), beta actin (ACTB), Creatine kinase B (CKB), and MBP. Several splice variants of MBP have isoforms in the 20kDa range. Ten peptides from MBP isoforms 3 and 5 ( MBP1 and MBP3), were detected and the protein sequence coverage was calculated at 39.1% and 45.0%, respectively (see Table 3).
Table 3.
Nitrotyrosine Modified Proteins
| UniProt Accession |
UniProt Entry Name |
% coverage | probability | Mascot Score |
Peptides detected |
Theoretical MW a |
pIb | Total tyr residues |
tyr location in detected peptides |
|---|---|---|---|---|---|---|---|---|---|
| P02686-3 | MBP1c | 39.1 | 2.82E-07 | 100.2 | 10 | 21493 | 11.3 | 5 | 15, 154 |
| P02686-5 | MBP3c | 45.0 | 2.82E-07 | 100.2 | 10 | 18590 | 11.4 | 4 | 15, 128 |
| P51970 | NDUA8 | 15.2 | 6.44E-05 | 20.2 | 3 | 18857 | 6.7 | 3 | 141 |
| P19404 | NDUV2 | 6.45 | 3.29E-03 | 20.15 | 2 | 23760 | 5.7 | 8 | NONE |
| P00403 | COX2 | 15.9 | 2.37E-04 | 30.19 | 3 | 25550 | 4.7 | 9 | 85 |
| P60709 | ACTB | 19.5 | 1.67E-05 | 50.21 | 5 | 41775 | 5.3 | 15 | 90, 197, 293, 305, 361 |
| P12277 | CKB | 12.4 | 1.21E-06 | 30.3 | 3 | 42513 | 5.3 | 9 | NONE |
| P22695 | UQCR2 | 16.9 | 2.67E-07 | 40.23 | 4 | 46784 | 7.7 | 13 | 168 |
| P09543 | CNPase | 15.7 | 2.28E-07 | 50.26 | 5 | 47578 | 9.2 | 14 | 268 |
MW of processed peptide due to signal peptide and/or initiating methionine cleavage.
pI of processed peptide due to signal peptide or initiating methionine cleavage.
Human MBP has 7 isoforms. All ten peptides detected by MS/MS are present in the isoforms listed.
4. Discussion
In this study, we examined peaks in 96 SELDI-TOF mass spectra (six fractions from sixteen brain samples). Each mass spectrum was represented by over 65,000 data points. To decrease the complexity associated with such a large data set, we reduced the number of variables using principal component analysis and generated PC score scatter plots to more easily visualize the segregation of samples derived from MS cortex compared to those obtained from controls. In our first group (cohort 1), using PCA, we analyzed mitochondrial proteins eluted in fraction 3 and determined that the first two PCs accounted for 80% of the variability in the data set and allowed for segregation of samples obtained from MS or control cortex into two distinct groups with respect to the presence or absence of disease. The ability to separate disease from control suggests that the presence and/or absence of specific groups of peaks correlates with disease expression and that these peaks may be important in elucidating biological processes that are responsible for disease etiology or progression. In addition, we found that of the proteins separated out from fraction 3 of our first cohort, the peaks in the 10 kDa and 16 kDa range contributed the greatest amount to the separation of MS from control donors. The lack of 10 kDa peaks and excess of the 16kDa peak is most prominent in MS donors. Therefore the proteins within these peaks were selected for identification by peptide fingerprint mapping.
The peak at 16kDa was identified by peptide fingerprint mapping as MBP, a component of myelin. This was an unexpected result and one that was consistent in mitochondrial fractions from both controls and MS donors. MBP is a very basic protein which is normally associated with membranes and it is possible that MBP released from myelin during tissue homogenization was bound adventitiously to mitochondrial membranes and remained in the mitochondrial fraction even after the washing steps. However, we consistently found increased levels of MBP associated with MS fractions and its presence is an important distinguishing factor separating controls from MS as determined in our principal component analysis. Other proteomic studies of mouse brain have also shown mitochondrial proteins, including several involved in the electron transport chain, as well as hemoglobin, and creatine kinase B in myelin preparations [37, 38, 39, 40, 41]. However, this is the first time that myelin basic protein has been observed associated with mitochondrial fractions. In addition, we see an increase in the amount of MBP associated with mitochondrial fractions in MS donors relative to controls by Western blot and have also shown that the expression of MBP isoforms is altered in the EAE mouse model of MS. These data suggest that MBP can be localized and associated with mitochondria both in normal and diseased brain, but the significance of this for MS disease pathology is not clear.
Peptide fingerprint mapping identified the protein isolated from fraction 3 of cohort 1 in the 10 kDa region as COX5b, a component of Complex IV of the electron transport chain. The expression of this subunit has also been shown to be decreased by mRNA expression microarray analysis [14]. Nuclear encoded mitochondrial proteins, including subunit 5b, are transcribed with a positively charged N-terminal signal sequence which is cleaved upon import into the appropriate mitochondrial location. The full length COX5b has a molecular weight of 13,696 Da and a pI of 9.07. However, upon removal of the 31 residue transit sequence, the molecular weight is reduced to 10,613 Da and the pI decreases to 6.33. Both physical properties are in strong agreement with the differentially expressed species in fraction 3 (5.0–7.0). The loss of this subunit may play a role in mitochondrial dysfunction since the nuclear encoded subunits function in the assembly and maintenance of Complex IV [42]. Indeed, in null yeast mutants of subunit 4, 5a, 5b, 6c and 7a, Complex IV failed to assemble [43]. Additionally, Complex IV dysfunction may impact the production of free radicals, since it has been shown that various cytochrome c oxidase subunits provide protection from highly reactive peroxynitrite [44].
Our differential protein expression analysis of mitochondrially enriched fractions from MS and control gray matter also demonstrated that the differentially expressed protein in fraction 4 from cohort 2, with a mass of 16 kDa, was consistent with hemoglobin-β. Notably, both hemoglobin-α2 and hemoglobin-β genes are upregulated in mononuclear blood cells from MS patients as reported recently in a study comparing monozygous twins discordant for multiple sclerosis [45]. This phenomenon appears to be extended to the brain, since our data show increased expression of hemoglobin-β in MS cortex. Further, we localized hemoglobin expression to pyramidal neuronal cell bodies by immunofluorescent staining with antibodies to both hemoglobin and neurofilament. The discovery of hemoglobin expression in neurons, until recently considered specific to red blood cells, is novel. Other recent studies support our finding, including studies by Biagoli et al., (2009) [46] and Richter et al., (2009) [47] in which the expression of both α- and β-globin transcripts was shown in dopaminergic neurons from mouse, rat, and human brains. Functionally, alterations in hemoglobin expression may affect cellular energetics, since dopaminergic cell lines over expressing α- and β-globin exhibited changes in oxygen homeostasis and alterations in the expression of oxidative phosphorylation genes. These data support our finding of hemoglobin expression in the brain and support a potential role for hemoglobin in respiration. Hemoglobin may also aid in protecting neurons from oxidative damage, since the ability of hemoglobin α2β2 to scavenge peroxynitrite (ONOO−) in a variety of O2 bound states has been suggested [48].
Lastly, in cohort 2 we identified a protein from fraction 6 as creatine kinase B (42 kDa). It has been observed previously that IgG from MS patients reacts with hemoblobin β, as well as with creatine kinase B and the expression of creatine kinase B has also been found in CSF of MS patients [49, 50]. In myocardial infarction, serum levels of creatine kinase are used as an indicator of heart muscle damage. In MS, it may represent a similar marker for nervous tissue damage. However, we detected an almost 2 fold increase in expression in mitochondrial fractions obtained from nonlesion MS cortex. At this time, the significance and mechanistic importance of these findings are not clear. However, in neurons it has been suggested that creatine kinase B may serve to buffer fluctuations in ATP and levels of inorganic phosphate, as well as serve as a shuttle for high energy phosphate bonds between mitochondria and cytoplasm [51].
Mitochondrial dysfunction and neuroinflammation contribute to neuropathology not only in MS [52], but are also common to other complex neurodegenerative diseases such as Alzheimer’s Disease (AD) and Parkinson’s Disease (PD) [53, 54, 55, 56, 57]. Additionally, several studies suggest that mitochondrial dysfunction is an early occurrence in neurodegeneration and molecular studies have found evidence of mutant proteins interacting with and preventing transport of nuclear encoded proteins to mitochondria [58]. Consistent with our findings in this study and in previous studies [13, 14], the expression of mitochondrial electron transport chain proteins has also been shown to be decreased in PD in the brain with Complex I most consistently reported [59, 60, 61]. In contrast, in AD, fewer mitochondria but increased cytochrome c oxidase (Complex IV) and mitochondrial DNA have been observed in neurons [62]. In AD, reactive oxygen species (ROS) may be a primary event mediating mitochondrial dysfunction [63] whereas in PD, a defect in mitochondria may mediate increased ROS as mitochondrial toxins can elicit neuronal pathology similar to PD [64, 65]. In MS, mitochondrial damage similar to what has been reported in both AD and PD has been observed. Damage to mitochondrial DNA and altered cytochrome c oxidase immunoreactivity similar to what has been observed in AD have been reported in axons in chronic active and inactive lesions [66, 67] while gray matter pathology in MS is more consistent with PD, including reductions in electron transport chain subunits and mitochondrial activity [13, 14]. While the underlying mechanisms and events involved in mitochondrial dysfunction differ between these neurodegenerative diseases, the end result of these mitochondrial perturbations is similar resulting in increased ROS, loss of Ca2+ homeostasis, and necrotic or apoptotic cell death [68]. In MS these processes are exacerbated by demyelination which increases energy demand to maintain ion homeostasis through the energy dependent Na+/K+ ATPase in neurons [69].
We detected increased nitration of electron transport chain subunits, creatine kinase B, and MBP in MS cortex. This is indicative of increased ROS and peroxynitrite mediated oxidative damage in MS and is consistent with previous studies [70, 13] which reported increased levels of nitrated proteins in MS tissue when compared with controls. Nitration of mitochondrial electron transport chain subunits has also been identified in AD postmortem brain and in the spinal cord in EAE [71, 8]. Increased protein nitration suggests an inflammatory mechanism is involved as inflammatory microglial cells express inducible nitric oxide synthase (iNOS) and release nitric oxide (NO) in MS lesions [72, 73]. This NO can diffuse and react with superoxide formed as a byproduct of mitochondrial respiration and create peroxynitrite which can also diffuse through tissue [74] and may account for our finding increased protein nitration even in NAGM in MS [13]. Alternatively, electron transport chain proteins may be oxidatively modified as a result of axonal transport through lesioned axonal segments. Either direct nitration of electron transport chain proteins by peroxynitrite or competitive inhibition of electron transport chain Complex IV by NO can lead to mitochondrial dysfunction [75, 76].
Conclusions
Our data suggest that alterations in MBP expression and protein nitration in MS cortex involve an inflammatory mechanism shared with the EAE mouse model. The EAE mouse model is an autoimmune disease model, in which lesions similar to those observed in MS are observed upon immunization with myelin protein antigens. This model has been helpful in understanding inflammatory disease mechanisms in MS but does not reflect all aspects of MS neuropathology [77]. MS is both an autoimmune and a neurodegenerative disease, and while protein nitration and altered MBP expression are consistent in the EAE mouse model and in MS, we did not observe similar alterations in expression levels of COX5b, hemoglobin β chain, or creatine kinase B in EAE, indicating that a separate mechanism is also involved in MS pathology. This mechanism may involve cellular stress separate from that mediated by neuroinflammation such as metabolic stress which can lead to differential expression of mitochondrial enzymes and subunits of the electron transport chain [78]. The existence of separate mechanisms involved in mitochondrial dysfunction is not surprising given the heterogeneity and complexity of MS pathology [79] and is reflected in the disparate types of mitochondrial abnormalities reported in acute and chronic white matter lesions, in axons, and in gray matter in MS. Increased mitochondrial density and either increased Complex IV activity or protein has been reported in chronic active white matter lesions in axons and astrocytes [66, 12] and also in a subset of demyelinated axons in chronic inactive lesions [67]. In contrast, decreased Complex IV activity and decreased immunoreactivity to Complex IV subunits have been reported in acute white matter lesions [10] and also in injured axons in chronic active lesions [67]. These studies suggest an adaptive response by both neurons and glia to increase mitochondrial activity in response to demyelination, thereby increasing ROS, and also a defect in mitochondrial transport in damaged tissue. Our data however, examined NAGM rather than lesioned tissue and has confirmed our previous studies which found decreased expression of mitochondrial electron transport chain subunit genes in MS [13, 14] and has extended these findings and implicated an imbalance in the regulation of additional respiratory proteins in MS disease pathology. Our findings have important implications for the development of new neuroprotective therapies for MS.
Supplementary Material
Acknowledgements
We would like to thank the Rocky Mountain MS Center, the Kathleen Price Bryan Brain Bank, and the Brain and Spinal Fluid Resource Center at UCLA for tissue. This research was funded by NIH Grant R21NS058921 (JM), a Grant from the Ohio Board of Regents, and by Acorn Ventures, Bellevue, WA (RBG).
Abbreviations
- SELDI-TOF-MS
Surface-Enhanced Laser Desorption Ionization Time of Flight Mass Spectrometry
- PCA
principal component analysis
- NAGM
normal appearing gray matter
- MS
Multiple sclerosis
- EAE
experimental autoimmune encephalomyelitis
- MBP
myelin basic protein
- COX5b
cytochrome c oxidase subunit 5b
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bö L, Geurts JJG, Mörk SJ, van der Valk P. Grey Matter Pathology in Multiple Sclerosis. Acta Neurol. Scand. 2006;113:48–50. doi: 10.1111/j.1600-0404.2006.00615.x. [DOI] [PubMed] [Google Scholar]
- 2.Bjartmar C, Kidd G, Mörk S, Rudick R, Trapp BD. Neurological Disability Correlates with Spinal Cord Axonal Loss and Reduced N-Acetyl Aspartate in Chronic Multiple Sclerosis Patients. Ann. Neurol. 2000;48:893–901. [PubMed] [Google Scholar]
- 3.Mathiesen HK, Tscherning T, Sorensen PS, Larsson HBW, Rostrup E, Paulson OB, Hanson LG. Multi-slice Echo-planar Spectroscopic MR Imaging Provides Both Global and Local Metabolite Measures in Multiple Sclerosis. Mag. Reson. Med. 2005;53:750–759. doi: 10.1002/mrm.20407. [DOI] [PubMed] [Google Scholar]
- 4.Rudick RA, Lee J, Nakamura K, Fisher E. Gray Matter Atrophy Correlates with MS Disability Progression Measured with MSFC but not EDSS. J. Neurol. Sci. 2009;282:106–111. doi: 10.1016/j.jns.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inglese M, Ge Y, Filippi M, Falini A, Grossman RI, Gonen O. Indirect Evidence For Early Widespread Gray Matter Involvement in Relapsing-Remitting Multiple Sclerosis. Neuroimage. 2004;21:1825–1829. doi: 10.1016/j.neuroimage.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 6.Cader S, Johansen-Berg H, Wylezinska M, Palace J, Behrens TE, Smith S, Matthews PM. Discordant White Matter N-Acetylaspartate and Diffusion MRI Measures Suggest that Chronic Dysfunction Contributes to Axonal Pathology in Multiple Sclerosis. Neuroimage. 2007;36:19–27. doi: 10.1016/j.neuroimage.2007.02.036. [7] [DOI] [PubMed] [Google Scholar]
- 7.Mao P, Reddy PH. Is Multiple Sclerosis a Mitochondrial Disease? BBA-Molecular Basis of Disease. 2010;1802:66–79. doi: 10.1016/j.bbadis.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. Mitochondrial Protein Nitration Primes Neurodegeneration in Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2006;281:31950–31962. doi: 10.1074/jbc.M603717200. [DOI] [PubMed] [Google Scholar]
- 9.Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. U S A. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahad D, Ziabreva I, Lassman H, Turnbull D. Mitochondrial Defects in Acute Multiple Sclerosis Lesions. Brain. 2008;131:1722–1735. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalman B, Laitinen K, Komoly S. The Involvement of Mitochondria in the Pathogenesis of Multiple Sclerosis. J. Neuroimmunol. 2007;188:1–12. doi: 10.1016/j.jneuroim.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 12.Witte ME, Bǿ L, Rodenburg RJ, Belien JA, Musters R, Hazes T, Wintjes LT, Smeitink JA, Geurts JJG, De Vries HE, van der Valk P, von Horssen J. Enhanced Number and Activity of Mitochondria in Multiple Sclerosis Lesions. J. Pathol. 2009;219:193–204. doi: 10.1002/path.2582. [DOI] [PubMed] [Google Scholar]
- 13.Pandit A, Vadnal J, Houston S, Freeman E, McDonough J. Impaired Regulation of Electron Transport Chain Subunit Genes by Nuclear Respiratory Factor 2 in Multiple Sclerosis. J. Neurol. Sci. 2009;279:14–20. doi: 10.1016/j.jns.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T, Gudz T, Macklin WB, Lewis DA, Fox RJ, Rudick R, Mirnics K, Trapp BD. Mitochondrial Dysfunction as a Cause of Axonal Degeneration in Multiple Sclerosis Patients. Ann. Neurol. 2006;59:478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 15.Avasarala JR, Wall MR, Wolfe GM. A Distinctive Molecular Signature of Multiple Sclerosis Derived from MALDI-TOF/MS and Serum Proteomic Pattern Analysis. J. Mol. Neurosci. 2005;25:119–126. doi: 10.1385/JMN:25:1:119. [DOI] [PubMed] [Google Scholar]
- 16.Westman-Brinkmalm A, Ruetschi U, Portelius E, Andreasson U, Brinkmalm G, Karlsson G, Hansson S, Zetterberg H, Blennow K. Proteomics/peptodomics Tools to Find CSF Biomarkers for Neurodegenerative Diseases. Front. Biosci. 2009;14:1793–1806. doi: 10.2741/3341. [DOI] [PubMed] [Google Scholar]
- 17.Quintana FJ, Farez MF, Weiner HL. Systems Biology Approaches for the Study of Multiple Sclerosis. J. Cell. Mol. Med. 2008;12:1087–1093. doi: 10.1111/j.1582-4934.2008.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zetterberg H, Rüetschi U, Portelius E, Brinkmalm G, Andressson U, Blennow K, Brinkmalm A. Clinical Proteomics in Neurdegenerative Disorders. Acta Neurol. Scand. 2008;118:1–11. doi: 10.1111/j.1600-0404.2007.00985.x. [DOI] [PubMed] [Google Scholar]
- 19.Han MH, Hwang S, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Proteomic Analysis of Active Multiple Sclerosis Lesions Reveal Therapeutic Targets. Nature. 2008;451:1076–1083. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 20.Selmaj K, Pawlowska Z, Walczak A, Koziolkiewicz W, Raine CS, Cierniewski CS. Corpora Amylacea from Multiple Sclerosis Brain Tissue Consists of Aggregated Neuronal Cells. Acta Biochim. Pol. 2008;55:43–49. [PubMed] [Google Scholar]
- 21.Liu SL, Bai SM, Qin ZY, Yang YR, Cui YZ, Qin YJ. Quantative Proteomic Analysis of Cerebrospinal Fluid of Patients with Multiple Sclerosis. J. Cell. Mol. Med. 2009;13:1586–1603. doi: 10.1111/j.1582-4934.2009.00850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiasserini D, Di Filippo M, Candeliere A, Susta F, Orvietani PL, Calabresi P, Binaglia L, Sarchielli P. CSF Proteome Analysis in Multiple Sclerosis Patients by Two-Dimensional Electrophoresis. Eur. J. Neurol. 2008;15:998–1001. doi: 10.1111/j.1468-1331.2008.02239.x. [DOI] [PubMed] [Google Scholar]
- 23.D’Aguanno S, Barassi A, Lupisella S, Melzi d’eril G, Del Boccio P, Pieragostino D, Pallotti F, Carelli V, Valentino ML, Liguori R, Avoni P, Bernadini S, Gambi D, Urbani A, Federici G. Differential Cerebro Spinal Fluid Proteome Investigtion of Leber Hereditry Optic Neuropathy (LHON) and Multiple Sclerosis. J. Neuroimmunol. 2008;193:156–160. doi: 10.1016/j.jneuroim.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Filippi M, Agosta F. Magnetic resonance techniques to quantify tissue damage, tissue repair, and functional cortical reorganization in multiple sclerosis. Prog. Brain Res. 2009;175:465–482. doi: 10.1016/S0079-6123(09)17531-3. [DOI] [PubMed] [Google Scholar]
- 25.Fisher E, Lee JC, Nakamura K, Rudick RA. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann. Neurol. 2008;64:255–265. doi: 10.1002/ana.21436. [DOI] [PubMed] [Google Scholar]
- 26.Trapp. BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. New Eng. J. Med. 1998;38:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 27.Dasilva AG, Yong VW. Expression and regulation of matrix metalloproteinase-12 in experimental autoimmune encephalomyelitis and by bone marrow derived macrophages in vitro. J. Neuroimmunol. 2008;199:24–34. doi: 10.1016/j.jneuroim.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 28.Carlson SM, Najmi A, Whitin JC, Cohen HJ. Improving feature detection and analysis of surface-enhanced laser desorption/ionization-time of flight mass spectra. Proteomics. 2005;5:2778–2788. doi: 10.1002/pmic.200401184. [DOI] [PubMed] [Google Scholar]
- 29.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster Analysis and Display of Genome-wide Expression Patterns. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Carnemolla B, Orecchia P, Zardi L, Righetti PG. Blue silver: A very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis. 2004;25:1327–1333. doi: 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]
- 31.Hermanson GT. Bioconjugate Techniques. second ed. San Diego, CA: Academic Press; 2008. [Google Scholar]
- 32.Laemmli UK. Cleavage of Structural Proteins During Assembly of Head of Bacteriophage-T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 33.Echan LA, Speicher DW. Protein Detection in Gels Using Fixation. Curr. Protoc. Protein Sci. 2002:10.5.3–11. doi: 10.1002/0471140864.ps1005s29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiménez CR, Huang L, Qui Y, Burlingame AL. In-Gel Digestion of Proteins for MALDI-MS Fingerprint Mapping. Curr. Protoc. Protein Sci. 1998:16.4.1–16.4.3. doi: 10.1002/0471140864.ps1604s14. [DOI] [PubMed] [Google Scholar]
- 35.Lavine B, Workman J. Chemometrics. Anal. Chem. 2010;82:4699–4711. doi: 10.1021/ac101202z. [DOI] [PubMed] [Google Scholar]
- 36.Jolliffe IT. Springer Series Statistics. 2nd ed. Springer, NY; 2002. Principal Component Analysis, Series. [Google Scholar]
- 37.Taylor CM, Marta CB, Claycomb RJ, Han DK, Rasband MN, Coetzee T, Pfeiffer SE. Proteomic mapping provides powerful insights into functional myelin biology. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4643–4648. doi: 10.1073/pnas.0400922101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanrobaeys F, Van Coster R, Dhondt G, Devreese B, Van Beeumen J. Profiling of Myelin Proteins by 2D Gel Electrophoresis and Multidimensional Liquid Chromatography Coupled to MALDI TOF-TOF Mass Spectrometry. J. Prot. Res. 2005;4:2283–2293. doi: 10.1021/pr050205c. [DOI] [PubMed] [Google Scholar]
- 39.Ishii A, Dutta R, Wark GM, Hwang S-I, Han DK, Trapp BD, Pfeiffer SE, Bansai R. Human myelin proteome and comparative analysis with mouse myelin. Proc. Natl. Acad. Sci. U.S.A. 2009;106:14605–14610. doi: 10.1073/pnas.0905936106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mugnaini E, Osen KK, Schnapp B, Friedrich VL. Distribution of Schwann cell cytoplasm and plasmalemmal vesicles (caveolae) in peripheral myelin sheaths. An electron microscopic study with thin sections and freeze-fracturing. J. Neurocytol. 1977;6:647–668. doi: 10.1007/BF01176378. [DOI] [PubMed] [Google Scholar]
- 41.Ravera S, Panfoli I, Calzia D, Aluigi MG, Bianchini P, Diaspro A, MAncardi G, Morelli A. Evidence of aerobic ATP synthesis in isolated myelin vesicles. Ont. J. Biochem. Cell B. 2009;41:1581–1591. doi: 10.1016/j.biocel.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 42.Galati D, Srinivasan S, Raza H, Prabu SK, Hardy M, Chandran K, Lopez M, Kalyanaraman B, Avadhani NG. Role of Nuclear-encoded Subunit Vb in the Assembly and Stability of Cytochrome c Oxidase Complex: Implications in Mitochondrial Dysfunction and ROS Production. Biochem. J. 2009;420:439–449. doi: 10.1042/BJ20090214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capaldi RA. Structure and Assembly of Cytochrome c Oxidase. Arch. Biochem. Biophys. 1990;280:252–262. doi: 10.1016/0003-9861(90)90327-u. [DOI] [PubMed] [Google Scholar]
- 44.Fontanesi F, Soto IC, Horn D, Barrientos A. Barrientos, Assembly of Mitochondrial Cytochrome c-Oxidase, a Complicated and Highly Regulated Cellular Process. Am. J. Physiol. Cell Physiol. 2006;291:C1129–C1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- 45.Särkijärvi S, Kuusisto H, Paalavuo R, Levula M, Airla N, Lehtimäki T, Kaprio J, Koskenvuo M, Elovaara I. Gene Expression Profiles in Finnish Twins with Multiple Sclerosis. BMC Med. Genet. 2006;7:11–20. doi: 10.1186/1471-2350-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biagioli M, Pinto M, Cesselli D, Zaninello M, Lazarevic D, Roncaglia P, Simone R, Vlachouli C, Plessy C, Bertin N, Beltrami A, Kobayashi K, Gallo V, Santoro C, Ferrer I, Rivella S, Beltrami CA, Carninci P, Raviola E, Gustincich S. Unexpected Expression of α- and β-globin in Mesencephalic Dopaminergic Neurons and Glial Cells. Proc. Natl. Acad. Sci. U.S.A. 2009;106:15454–15459. doi: 10.1073/pnas.0813216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richter F, Meurers BH, Zhu C, Medvedeva VP, Chesselet M-F. Neurons Express Hemoglobin α- and β-chains in Rat and Human Brains. J. Comp. Neurol. 2009;515:538–547. doi: 10.1002/cne.22062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herold S, Fago A. Reactions of Peroxynitrite with Globin Proteins and Their Possible Physiological Role. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 2005;142:124–129. doi: 10.1016/j.cbpb.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 49.Nobem J-P, Dumont D, Kwasnikowska N, Verhaert P, Somers V, Hupperts R, Stinissen P, Robben R. Lumbar Cerebrospinal Fluid Proteome in Multiple Sclerosis: Characterization by Ultrafiltration, Liquid Chromatography, and Mass Spectrometry. J. Proteome Res. 2007;5:1647–1657. doi: 10.1021/pr0504788. [DOI] [PubMed] [Google Scholar]
- 50.Almeras L, Lefranc D, Drobecq H, de Seze J, Dubucquoi S, Vermersch P, Prin L. New antigenic candidates in Multiple Sclerosis: identification by serological proteome analysis. Proteomics. 2004;4:2184–2194. doi: 10.1002/pmic.200300732. [DOI] [PubMed] [Google Scholar]
- 51.Andres RH, Ducray AD, Schlattner U, Wallimann T, Widmer HR. Functions and effects of creatine in the central nervous system. Brain Res. Bull. 2008;76:329–343. doi: 10.1016/j.brainresbull.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 52.van Horssen J, Witte ME, Schreibelt G, de Vries HE. Radical changes in Multiple Sclerosis pathogenesis. BBA- Mol. Basis Dis. doi: 10.1016/j.bbadis.2010.06.011. Doi:10.1016/j.bbadis.2010.06.011 in press. [DOI] [PubMed] [Google Scholar]
- 53.Smith MA, Richey-Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 55.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 56.Zhu J, Chu CT. Mitochondrial Dysfunction in Parkinson’s Disease. J. Alzheimer’s Dis. 2010;20:S325–S334. doi: 10.3233/JAD-2010-100363. [DOI] [PubMed] [Google Scholar]
- 57.Di Filippo M, Chiasserini M, Picconi B, Calbresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. 2010;20:S369–S379. doi: 10.3233/JAD-2010-100543. [DOI] [PubMed] [Google Scholar]
- 58.Reddy PM. Mitochondrial Medicine in Aging and Neurodegenerative Diseases. Neuromol. Med. 2008;10:291–315. doi: 10.1007/s12017-008-8044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Kagawa Y. Deficiencies in Complex I subunits of the Respiratory Chain in Parkinson’s Disease. Biochem. Biophys. Res. Co. 1989;163:1450–1455. doi: 10.1016/0006-291x(89)91141-8. [DOI] [PubMed] [Google Scholar]
- 60.Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J. Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 61.Hattori N, Tanaka M, Ozawa T. Immunohistochemical Studies on Complex-I, Complex-II, Complex-III and Complex-IV of Mitochondria in Parkinson’s Disease. Ann. Neurol. 1991;30:563–571. doi: 10.1002/ana.410300409. [DOI] [PubMed] [Google Scholar]
- 62.Hirai K, Aliev G, Nunomura A, Fujioka H, Russel RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PLP, Jones PK, Peterson RB, Perry G, Smith MA. Mitochondrial Abnormalities in Alzheimer's Disease. J. Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moreira PI, Santos MS, Oliveira CR, Shenk JC, Nunomura A, Smith MA, Zhu X, Perry G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol. Disord. Drug Targets. 2008;7:3–10. doi: 10.2174/187152708783885156. [DOI] [PubMed] [Google Scholar]
- 64.Langston JW, Ballard P, Tetrud JW, Irwin I I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 65.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 66.Lu F, Selak M, O’Connor J, Croul S, Lorenzana C, Butunoi C, Kalman B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J. Neurol. Sci. 2000;177:95–103. doi: 10.1016/s0022-510x(00)00343-9. [DOI] [PubMed] [Google Scholar]
- 67.Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, Lassmann H, Turnbull DM. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132:1161–1174. doi: 10.1093/brain/awp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, Rizzuto R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. BBA- Bioenergetics. 2009;1787:335–344. doi: 10.1016/j.bbabio.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat. Rev. Neurosci. 2006;7:932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- 70.Bizzozero OA, DeJesus G, Bixler HA, Pastuszyn A. Evidence of nitrosative damage in the brain white matter of patients with multiple sclerosis. Neurochem. Res. 2005;30:139–149. doi: 10.1007/s11064-004-9695-2. [DOI] [PubMed] [Google Scholar]
- 71.Sultana R, Perluigi M, Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid. Redox Signal. 2006;8:2021–2037. doi: 10.1089/ars.2006.8.2021. [DOI] [PubMed] [Google Scholar]
- 72.Colton CA, Gilbert DL. Microglia, an in vivo source of reactive oxygen species in the brain. In: Seil FJ, editor. Advances in Neurology. Vol. 59. New York: Lippincott Williams and Wilkins or Raven Press; 1993. pp. 321–326. [PubMed] [Google Scholar]
- 73.Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am. J. Pathol. 2001;158:2057–2066. doi: 10.1016/S0002-9440(10)64677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beckman JS. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J Dev Physiol. 1991;15:53–59. [PubMed] [Google Scholar]
- 75.Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- 76.Sarti P, Giuffre A, Barone MC, Forte E, Mastronicola D, Brunori M. Nitric oxide and cytochrome oxidase: reaction mechanisms from the enzyme to the cell. Free Radical Bio. Med. 2003;34:509–520. doi: 10.1016/s0891-5849(02)01326-6. [DOI] [PubMed] [Google Scholar]
- 77.Croxford A, Kurschus FC, Waisman A. Mouse models for Multiple Sclerosis: Historical facts and future implications. BBS-Mol. Basis Dis. 2010 doi: 10.1016/j.bbadis.2010.06.010. doi:10.1016/j.bbadis.2010.06.010. in press. [DOI] [PubMed] [Google Scholar]
- 78.Palmfeldt J, Vang S, Stenbroen V, Pedersen CB, Christensen JH, Bross P, Gregersen N. Mitochondrial proteomics on human fibroblasts for identification of metabolic imbalance and cellular distress. Prot. Sci. 2009;7 doi: 10.1186/1477-5956-7-20. article num. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hu W, Lucchinetti CF. The pathological spectrum of CNS inflammatory demyelinating diseases. Semin. Immunopathol. 2009;31:439–453. doi: 10.1007/s00281-009-0178-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.