

Abstract
Excitation-contraction coupling in the cardiac myocyte is mediated by a number of highly integrated mechanisms of intracellular Ca2+ transport. The complexity and integrative nature of heart cell electrophysiology and Ca2+-cycling has led to an evolution of computational models that have played a crucial role in shaping our understanding of heart function. An important emerging theme in systems biology is that the detailed nature of local signaling events, such as those that occur in the cardiac dyad, have important consequences at higher biological scales. Multi-scale modeling techniques have revealed many mechanistic links between micro-scale events, such as Ca2+ binding to a channel protein, and macro-scale phenomena, such as excitation-contraction coupling gain. Here we review experimentally based multi-scale computational models of excitation-contraction coupling and the insights that have been gained through their application.
Keywords: cardiac myocyte, excitation-contraction coupling, Ca2+-induced Ca2+ release, multi-scale modeling, computational modeling
Overview of cardiac excitation-contraction coupling
Intracellular calcium (Ca2+) concentration plays a number of important regulatory roles in the cardiac myocyte. These include excitation-contraction coupling (ECC)1, intracellular signaling2, regulation of mitochondrial function3, gene expression4, and cell death2, 5. This review article is focused on the role of Ca2+ in ECC in cardiac ventricular myocytes. In particular, we will review experimentally based computational models of ECC, with a specific focus on mechanisms of Ca2+-induced Ca2+ release (CICR), and the insights that have been gained through application of these models.
The nature of ECC is linked closely to both the micro-anatomical structure of the cell, and to the arrangement of contractile proteins within the cell. The basic unit of contraction in the cardiac myocyte is the sarcomere. Individual sarcomeres are bounded on both ends by the t-tubular system1, 6, 7. The t-tubules are cylindrical invaginations of the sarcolemma that extend deep into the cell and approach an organelle known as the sarcoplasmic reticulum (SR). The SR is a luminal organelle located throughout the interior of the cell, and it is involved in the uptake, sequestration and release of Ca2+. SR can be divided into three main components known as junctional SR (JSR), corbular SR, and network SR (NSR). The JSR is that portion of the SR most closely approximating (within 12–15 nm)8 the t-tubules. The close proximity of these two structures forms a micro-domain known as the dyad.
Ca2+-sensitive Ca2+-release channels known as ryanodine receptors (RyRs) are preferentially located in the dyadic region of the JSR membrane. In addition, sarcolemmal voltage-gated L-type Ca2+ channels (LCCs) are preferentially located within the dyadic region of the t-tubules, where they are in close opposition to the RyRs. Early data from freeze fracture electron micrographs suggested that RyRs are arranged in a somewhat regular lattice9. The assumption that RyRs are tightly packed in dyadic junctions led to estimates of 100–300 RyRs per dyad8. However, more recent imaging studies have demonstrated that dyadic clefts are small, where junctions contain an average of 14 RyRs, with some clusters incompletely filled with RyRs, and with a large fraction of clusters closely spaced within 20–50 nm, suggesting that smaller clusters may act together to function as a single site of CICR10, 11.
During the initial depolarization stages of the action potential (AP), LCCs open allowing for the entry of Ca2+. This inward flux of Ca2+ contributes to maintenance of the plateau phase of the AP. As Ca2+ concentration in the dyad increases, Ca2+ binds to the RyRs, increasing their open probability. Opening of RyRs leads to Ca2+ release from the JSR. The amount of Ca2+ released from the JSR is significantly more than the amount of trigger Ca2+ entering via LCCs12. The ratio of release to trigger flux of Ca2+ is known as ECC gain. The Ca2+ release flux is a smooth, continuous function of trigger influx, a behavior observed originally by Fabiato et al13 and known as graded Ca2+ release. The resulting rapid, large increase in dyad Ca2+ concentration ([Ca2+]d) leads to Ca2+ diffusion from the dyad into the cytosol. As cytosolic Ca2+ concentration ([Ca2+]c) increases, Ca2+ binding to troponin C in the myofilaments initiates contraction of the myocyte.
Several mechanisms exist to remove trigger Ca2+ from the myocyte, and for re-uptake of released Ca2+ back into the NSR. During diastole, the Na+-Ca2+ exchanger (NCX) is believed to import three Na+ ions for every Ca2+ ion extruded, yielding a net inward charge movement14 (see15 for recently proposed alternative transport modes). It is the principal means by which Ca2+ is extruded from the myocyte. It can also function in reverse mode during the AP, in which case it extrudes Na+ and imports Ca2+.16 The duration of reverse mode NCX, and hence its effect on AP shape, has been shown to vary with species, heart failure, and other factors17, 18. The sarcolemmal Ca2+-ATPase also transports Ca2+ out of the cell, but flux through this pathway is small (2–3%) relative to other pathways19. The major mechanism by which released Ca2+ is transported back into the SR is the SR Ca2+-ATPase (SERCA pump). Phospholamban (PLB) is normally bound to the SERCA pump, inhibiting its activity. Phosphorylation of PLB functionally up-regulates SERCA by increasing its Ca2+-affinity1, 20. Density of the SERCA pump is highest in NSR. JSR refilling occurs via Ca2+ diffusion from the NSR. As each of these mechanisms restores [Ca2+]c to diastolic values, Ca2+ dissociates from troponin C, terminating the contraction phase of the cardiac cycle.
The elucidation of CICR mechanisms has become possible with the development of experimental techniques for simultaneous measurement of L-type Ca2+ current (ICaL) and Ca2+ transients, and detection of local Ca2+ release events known as Ca2+ sparks21, 22. As indicated by Soeller and Cannell23, the tight regulation of SR Ca2+ release by triggering LCC current became evident with the following observations: (1) ECC gain is ~10 in response to a voltage clamp to 0 mV24, 25; (2) The magnitude of SR release is a smoothly graded function of ICaL amplitude25, 26; and (3) the bell-shaped voltage-dependence of SR release is similar to that for ICaL, but with its peak shifted in the hyperpolarizing direction by ~ 10 mV25, 26. These experimental observations gave rise to and later verified the local control theory of EC coupling. The mechanism of local control predicts that within discrete regions of the dyadic cleft, the opening of an individual LCC will trigger Ca2+ release from a small cluster of RyRs that reside in close proximity directly across the dyad. Tight regulation of CICR is achieved because LCCs and RyRs are regulated by [Ca2+]d rather than by [Ca2+]c. The amplitude and profile of the whole-cell intracellular Ca2+ transient results from the recruitment and temporal summation of the elementary Ca2+ spark release events. The recruitment of Ca2+ sparks via this mechanism allows for graded control of SR Ca2+ release27.
Many advances in integrative modeling of the ventricular myocyte have resulted from quantitative modeling of these components which underlie CICR and ECC in cardiac myocytes. In this review, we will present a range of models that have been developed to understand properties of CICR and ECC in the cardiac ventricular myocyte. We have chosen to focus on development of major new functional components of CICR and ECC, their integration into whole-cell models, and the insights that have been obtained from these advances in modeling capabilities that span multiple biological scales.
Common pool models of CICR
The landmark DiFrancesco-Noble model of the cardiac Purkinje fiber28 was developed from the preceding Beeler-Reuter model29 (first computational model of the cardiac ventricular myocyte) and that of McAllister et al30. This model was ground breaking for many reasons and was the first model to introduce descriptions of the transport of Ca2+ into the NSR by SERCA, extrusion of Ca2+ from the cytosol by NCX, diffusion of Ca2+ from NSR to JSR, and CICR from the JSR. This model established the conceptual framework on which all subsequent models of the myocyte have been built. While this model did reproduce graded SR Ca2+ release, it did so with ECC gain that was non-physiologically low.
The construction of mechanistic mathematical models of CICR which attempt to capture both high gain and graded SR Ca2+ release first lead to a series of “common pool” models, which, in fact, fail to capture these fundamental properties. As defined by Stern27, common pool models are those in which LCC trigger Ca2+ enters the same Ca2+ pool into which JSR Ca2+ is released, and which controls JSR Ca2+ release. The rapid increase of Ca2+ in this pool leads to non-physiological regenerative, all-or-none rather than graded Ca2+ release, a situation where whole-cell SR Ca2+ release escapes control by ICaL once it is initiated26. An additional assumption inherent in this type of model is that the gating dynamics of the entire population of RyRs are controlled by properties of whole-cell ICaL rather than by properties of local LCC channel gating. Stern elegantly demonstrated via the application of linear stability theory that common pool models cannot achieve both high gain and graded Ca2+ release27.
Regardless of this limitation of common pool models, many advances in cardiac myocyte modeling over the past decade have stemmed from improved descriptions of CICR in such models. The Jafri et al31 model of the guinea pig ventricular myocyte was the first to incorporate a Markov model of the LCC (Fig. 1A) into an integrated myocyte model with mechanistic ECC. The LCC model was based on the concept of modal gating introduced by Imredy and Yue32 based on single-LCC recordings in which Ca2+ induced a shift in LCC gating dynamics into a low open probability mode. The model contains subsets of states which reproduce channel behavior prior to (mode-normal), and following (mode-Ca) the occurrence of Ca2+ dependent inactivation (CDI). Openings are rare in mode-Ca, effectively inactivating the channel. Successive LCC models have incorporated the concept of modal gating in Markov models for describing LCC inactivation33–35 as well as other modes such as phosphorylation36. The Jafri et al31 model was also first to implement a “restricted subspace”, a single compartment representing the total volume of all dyads into which all Ca2+ flux through RyRs and LCCs is directed. This model made a number of predictions regarding mechanisms of CICR and Ca2+ cycling (reviewed in37). A major prediction of this model was that experimentally measured interval-force (transient) and frequency-force (steady-state) relationships could be explained by the interplay between RyR inactivation and SR Ca2+ load dynamics. This Ca2+-subsystem was modified for canine in the model of Winslow et al38 from which a model of heart failure was developed based on data obtained from myocytes isolated from dogs with tachycardia pacing-induced heart failure39. The model predicted that in heart failure, down-regulation of the SERCA pump and up-regulation of NCX decreased JSR Ca2+ load, Ca2+ release via the RyRs, and the strength of ICaL CDI. The resulting increase in ICaL acted to prolong AP duration (APD), a characteristic phenotype of heart failure. The model predictions were subsequently validated by experimental studies40, 41, thereby demonstrating that negative feedback on LCCs by Ca2+ release from RyRs can exert significant control of AP properties. Furthermore, the heart failure model could produce early after depolarizations (EADs), a cellular arrhythmia believed to be a possible trigger for arrhythmia at the whole heart level. Upon introduction into a model of the ventricles, the EADs in the heart failure model quickly led to degeneration of electrical propagation into a cardiac arrhythmia, whereas the control myocyte model yielded normal coordinated ventricular activation during pacing.42
Figure 1.

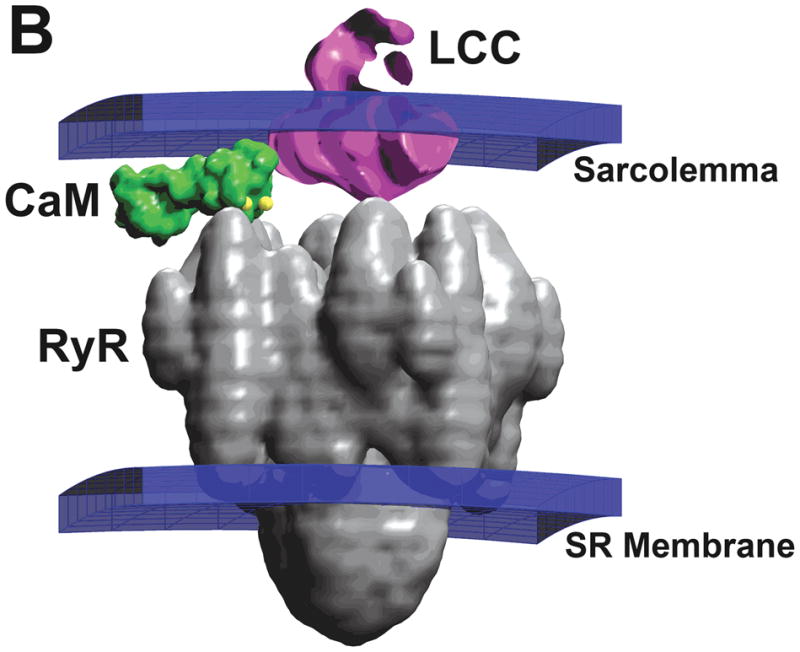
(A) Markov state model for gating of the LCC as proposed by Jafri et al.31 CDI occurs as a result of downward transitions from model-normal to mode-Ca. VDI is described by an independent Hodgkin-Huxley type gate (not shown). (B) Structure of a region of the dyad containing one LCC opposed to a single RyR, and one CaM molecule tethered to the LCC73.
Despite the fact that these common pool models were not able to reproduce graded SR Ca2+ release, they were able to reproduce and predict many important behaviors of the cardiac ventricular myocyte, raising the question of whether capturing the mechanisms of graded release is of primary importance. One way to circumvent this issue is to model SR Ca2+ release flux as a phenomenological function of LCC influx and/or membrane potential. Doing so removes the positive feedback effect inherent to common pool models. Models incorporating a phenomenological mechanism of CICR have been successful in describing many myocyte behaviors (including the widely utilized models of Rudy and colleagues43, 44). However, as with any phenomenological model, care must be taken when interpreting results of these models that pertain to Ca2+ release. The critical relationship between SR Ca2+ release and APD38, 40, 41, and the recognition that the feedback of SR Ca2+ release on LCCs occurs locally in discrete Ca2+ spark events21 suggested that capturing the mechanisms of local interaction between LCCs and RyRs would be essential. Furthermore, experimental data were emerging that supported the idea that LCC inactivation was dominated by CDI rather than voltage-dependent inactivation (VDI)45–47. Strong CDI in combination with weak/incomplete VDI causes instability in common pool models34 (which were therefore typically formulated with strong VDI and weak CDI) because all-or-none SR Ca2+ release induces fast whole-cell CDI which eliminates the late phase of ICaL and destabilizes the AP plateau (see Fig. 10C of ref34). This made it clear that in order to fully capture the core mechanisms of ECC, a model in which LCCs locally control RyR function was needed.
Local control of SR Ca2+ release
In 1992, Stern27 formulated idealized models of “local control” of CICR, and in doing so advanced the local control theory of ECC which underlies how the microscopic properties of CICR translate into macroscopic physiologic phenomena observed in experiments. The approach was to recognize that the non-physiologic positive feedback loop inherent in common pool models of CICR must be broken by separating Ca2+ released from the SR from the trigger Ca2+ influx. This was accomplished by noting that LCCs and RyRs reside in close apposition on opposite sides of the dyadic cleft, which would effectively grant Ca2+ ions permeating an LCC privileged access to the Ca2+ binding sites on nearby RyRs. Importantly, the assumed structural separation of individual clefts8 means that released Ca2+ would not generally act as a trigger to RyRs in distant dyads (though recent studies suggest neighboring RyR clusters may have a functional role in ECC11, 48). Stern’s “calcium-synapse” model, in which each dyad was assumed to contain a single LCC and a single SR release channel, captured these ideas in an elegantly simple way, demonstrating that high gain and graded SR release arise from the local correlation in stochastic LCC and RyR gating due to their communication via a common local Ca2+ signal. In this model, graded release is achieved by statistical recruitment of elementary SR Ca2+ release events. This fundamental idea of local control of SR release spurred a number of modeling studies, some of which focused on detailed dynamics of Ca2+ distribution within the dyadic junction, some of which focused on mechanisms of Ca2+ release termination, and a small handful of which developed fully integrative models that relate macroscopic cellular properties of ECC to the function of independent, locally controlled Ca2+ release units.
Due to the small size of the dyad, it became clear that understanding the spatial and temporal profile of [Ca2+]d would be an important step in elucidating the mechanistic details of local CICR. Langer and Peskoff49 developed one of the first detailed models to numerically simulate the time course of dyadic Ca2+. The model predicted that a single typical LCC opening led to ~ 1 mmol/L [Ca2+]d near the mouth of the LCC. The presence of sarcolemmal buffering sites slowed the decay of the local Ca2+ transient (such that it remained highly elevated long after LCC closure) as compared to the dynamics in the absence of buffers, a finding which was inconsistent with the idea that LCC gating imposes tight control on RyR activity. Soeller and Cannell50 formulated a dyad model which introduced the electrostatic effects of ion movement which predicted that [Ca2+]d actually changes more rapidly in response to LCC gating. In this model, [Ca2+]d was approximately proportional to unitary LCC currents over a wide range of current amplitudes. The model yielded predictions of [Ca2+]d profiles during JSR release, which peaked as high as 300 μmol/L near the center of the dyad. The decay in dyadic Ca2+ occurred in ~ 2 ms following termination of SR release (but this was sensitive to the dyad boundary condition). The high speed of the response of dyadic Ca2+ to LCC and RyR gating in this model helped to explain how tight local control of SR release by LCCs can be achieved.
With the growing acceptance of local control theory of CICR and that Ca2+ sparks are the elementary events underlying SR Ca2+ release (for a review see51), mathematical models of Ca2+ sparks were developed. The summation of Ca2+ sparks could explain the characteristic properties of CICR, ECC gain, and whole-cell Ca2+ transients, therefore the analysis of models of single Ca2+ sparks and ensembles of Ca2+ sparks provided a wealth of information. Local control of CICR requires that SR release be stable, which suggests that Ca2+ sparks, which are locally regenerative, must be terminated rapidly by a robust mechanism. While the mechanisms of Ca2+ spark termination still remain unclear, a number of models have helped elucidate how possible mechanisms may contribute to termination of SR Ca2+ release.
Stern27 originally described three mechanisms that can terminate SR release in a cluster of RyRs: (1) RyR inactivation is a time-dependent process which may be initiated by Ca2+ binding to an inactivation site on the RyR or may occur “fatefully” following opening of the channel. The observed inactivation process has been described as classically inactivating (leading to absolute refractoriness of CICR)52 or taking on the complex properties of adaptation51. (2) Depletion of locally releasable JSR Ca2+ will occur as the result of RyR activity. The emptying of the cisternal Ca2+ store reduces the Ca2+ gradient across the RyRs, and hence the Ca2+ flux. This will diminish the local feedback that underlies regenerative release in a cluster of RyRs, allowing them to close. (3) “Stochastic attrition” occurs as a result of the non-zero probability that all RyRs in a cluster are simultaneously closed. Since Ca2+ diffuses rapidly out of the dyad, the simultaneous closure of all RyRs would lead to a rapid reduction in [Ca2+]d below the level which sustains RyR activity. Active RyR clusters would therefore gradually cease gating spontaneously. This mechanism can only be effective on a reasonable timescale if very few RyRs are activated during a Ca2+ spark, and it does not provide for a mechanism of refractoriness. The stereotypic nature of Ca2+ spark properties suggested that spark generation and termination are likely controlled by regulatory feedback mechanisms and cannot be described by a simple stochastic mechanism alone23. An additional mechanism proposed recently is RyR regulation by luminal SR Ca2+. RyR sensitivity to [Ca2+]d can be altered by the level of JSR luminal [Ca2+] via the interaction of the Ca2+ buffer calsequestrin with RyR accessory proteins triadin and junction within the JSR53. The decrease in RyR open probability with JSR depletion likely contributes to termination of Ca2+ release.
In 1999, Stern et al33 developed a stochastic model of cardiac ECC. The model consisted of a cardiac dyad in which the Ca2+ concentration profile was simulated by solving equations governing diffusion and Ca2+ binding reactions in the cleft. LCCs and RyRs were represented by Markov state models, which were simulated as part of a stochastic system in which the state of each individual channel was tracked. The behavior of six different RyR gating schemes was analyzed in the context of local control of ECC. This study revealed that local control, in a realistic setting of dyad geometry, number, and location of channels, could explain graded release, when the RyR was represented by a simple phenomenological four-state gating scheme which included inactivation (Ca2+-dependent or fateful). This success has led to continued development of this RyR model in more recent work54, 55. However, RyR channel gating schemes that were based on single-channel RyR measurements in lipid bilayers56, 57 failed to give stable ECC (but stability could be restored by introducing allosteric RyR interactions33). The work of Stern et al33 suggested that there must be a sufficiently strong process that substantially reduces RyR open probability shortly following activation such that stochastic attrition can occur on the timescale of physiological ECC.
Whether or not RyR inactivation or adaptation plays an important role in termination of SR Ca2+ release remains controversial (see below). A number of RyR models57, 58 were developed to better understand the observation of RyR adaptation59, whereby an RyR can reopen in response successive increases in Ca2+ concentration, indicating they have not entered a conventional absorbing CDI state. Rice et al60 developed a stochastic model of Ca2+ release in the dyadic junction which demonstrated that RyR adaptation modulated Ca2+ release duration and amplitude but was not required for termination of Ca2+ release (i.e. JSR depletion played an important role). This model achieved high gain and graded release without tracking the spatial gradients of [Ca2+]d. In contrast, the RyR model of Shannon et al54 is a modification of the phenomenological four-state model of Stern et al33, which introduced an important modulatory role for SR luminal Ca2+ in RyR gating but which still depends upon an inactivation/adaptation mechanism for release termination. Variations of this model have been incorporated into a variety of recent models which have successfully described SR load and frequency dependent properties of ECC and APs in rabbit54 and human61, the role of RyR phosphorylation on Ca2+ dynamics and the AP55, and the role of spatial coupling between dyadic release sites in Ca2+ spark-induced sparks and Ca2+ alternans48.
In 2002, Sobie et al62 developed a Ca2+ spark model to further investigate mechanisms underlying termination of SR Ca2+ release. In this model each release site is composed of a large number of two-state RyRs which incorporate two additional experimental observations: (1) elevated SR luminal Ca2+ increases the Ca2+-sensitivity of RyR activation63, and (2) RyR coupling cooperatively influences their gating properties within a cluster64. These mechanisms of RyR gating yield a “sticky cluster” model which reproduces fundamental features of Ca2+ sparks and robust termination of release that arises as a result of the combined effects of these two mechanisms. In this model, the assumption of coupled RyR gating is essential for reliable SR release termination. However, it remains unclear how RyRs would influence the gating properties of all other RyRs in a cluster (as opposed to only adjacent RyRs33) as implemented in this model (see 65, 66 for alternative approaches to modeling the functional effects of RyR coupling). In addition, the >90% SR depletion required for release termination in the Sobie et al62 model may not be realistic as experiments have demonstrated that 40–75% of releasable Ca2+ remains in the JSR following whole-cell triggered SR release67, 68. Despite these issues, the majority of recent experiments68–70 have strengthened the idea that termination of SR release is primarily controlled by luminal SR Ca2+ rather than an RyR inactivation mechanism.
While many controversies as to the details of CICR remain to be resolved, the models of dyadic Ca2+ dynamics, Ca2+ sparks and CICR have helped affirm or disprove a variety of mechanistic hypotheses regarding cardiac ECC. Most of these ECC models, however, have been implemented at the sub-cellular scale, isolated from the physiological environment and influence of other cellular processes. The first mathematical model to incorporate the mechanisms of local control of SR Ca2+ release at the level of the cardiac dyad into an integrative model of the cardiac action potential was published in 2002 by Greenstein and Winslow34 (Greenstein-Winslow model). This model is described in detail below in the section “Multi-scale modeling of CICR”.
Multi-scale modeling of CICR
Quantitative understanding of CICR requires that Ca2+ signaling be understood across a wide range of spatio-temporal scales. The length-scale over which CICR occurs within the dyad is on the order of nanometers, and relevant time-scales are of the order of microseconds. Integration to the level of the cell requires formulation of models that describe behavior at spatio-temporal scales of several hundred microns and tens to hundreds of seconds. Recently, we have developed several new approaches for multi-scale modeling of CICR spanning these spatio-temporal scales34, 71–73. In this section, we present these models and the mathematical techniques for moving across these spatio-temporal scales.
The Nano-Scale Model of CICR
We have recently developed a computational model of Ca2+ signaling in the dyad that simulates motion of Ca2+ ions, binding of ions to RyR receptor sites, and Ca2+ release from RyR. This model was developed in order to understand factors shaping CICR at the nano-scale level. We addressed the following three questions: 1) what is the number of Ca2+ ions that are present in the dyad during CICR?; 2) how does the physical arrangement of large proteins within the dyad influence CICR?; and 3) how does “signaling noise” due to the potentially small number of Ca2+ ions within the dyad affect the nature of CICR? We addressed these questions by developing a molecularly- and structurally-detailed computational model of the cardiac dyad describing motions of Ca2+ ions, as influenced by thermal energy and electrostatic potentials.
The small size of the dyadic cleft (~ 4 × 105 nm3 with ~ 10–40 RyRs)10, 11, in combination with the relatively large proteins that reside in the cleft, results in a space in which Ca2+ diffusion is highly restricted. Figure 1B shows a representation of an LCC, RyR, and a single calmodulin (CaM) molecule in the dyad based directly on structural data74–76. Models predict that during an AP, peak Ca2+ concentration in the dyad ranges from 100 to 1,000 μmol/L49, 50, which corresponds to as few as 20–200 free Ca2+ ions73. Therefore, application of laws of mass-action to analysis of signaling events mediated by this relatively small number of ion may not be justified77. Rather, the stochastic motion of Ca2+ ions in the dyad may impart a degree of signaling noise between LCCs and RyRs, thereby influencing properties of CICR73. In addition, the physical location and dimensions of dyad proteins may have considerable influence on motion of Ca2+ ions and hence, CICR.
Development of the nano-scale model is presented in detail elsewhere73. Briefly, a simplified dyad was modeled using a 200 nm × 200 nm × 15 nm lattice containing 20 RyRs arranged in a 4 × 5 matrix. Four LCCs were positioned randomly across this lattice to achieve a 5:1 RyR:LCC ratio. The geometry of each LCC and RyR protein was modeled as shown in Fig. 1B, with each LCC tethering with one CaM. LCC gating was simulated stochastically using the model of Fig. 1A. The gating kinetics of the RyR were described using the four-state Markov model of Stern33,78. Entry of Ca2+ ions into the dyad via open LCCs and RyRs was modeled as a Poisson process with rate governed by channel permeabilities measured experimentally79. CDI of an LCC required that the two carboxyl-terminal Ca2+ binding sites of the associated CaM were occupied. Opening of an RyR required that Ca2+ was bound to all four activation sites. RyRs were assumed to inactivate when Ca2+ was bound to at least one out of four distinct inactivation sites. Sarcolemmal anionic sites acted as Ca2+ buffers49, 50. The joint positions of Ca2+ ions present in the dyad were modeled as a Brownian motion in a potential field, describing both interactions of Ca2+ ions with other Ca2+ ions as well as electrostatic potentials based on the Fokker-Planck equation (FPE)80. We generated sample paths of Ca2+ ion movement in the dyad using a finite difference representation of the FPE that can be interpreted as a spatially discrete Markov process as described by Wang et al81.
A 10 ms simulation of a model dyad containing only one LCC located at its center at a membrane potential of 0 mV generated an average of ~ 0.5 Ca2+ ions within a 50 nm radius of the LCC (see Fig. 8 of73), with a minimum of zero and a maximum of 7 Ca2+ ions, corresponding to 98.7 μmol/L [Ca2+]d. A similar protocol using a single RyR in the SR membrane (in which the RyR is initially open) generated an average of ~ 22.8 Ca2+ ions. The number of Ca2+ ions peaked at 49 during Ca2+ release, which corresponds to a concentration of ~ 0.7 mmol/L Ca2+, similar to the concentration of Ca2+ found in the JSR82. With a coefficient of variation of 64%, RyR gating produces a high degree of variability in the number of dyadic Ca2+ ions, showing that the LCC-RyR signaling involved in CICR at the dyad level is a noisy process mediated by tens of Ca2+ ions.
Panels A and B of Figure 2 demonstrate fundamental features of a representative Ca2+ release event in a dyad in response to a voltage clamp to 0 mV at time zero. Panel A shows the number of Ca2+ ions in the dyad, and panel B shows the number of LCCs (gray line) and RyRs (black line) that are open as a function of time during the release event. Two LCCs open at ~ 3 ms following the voltage clamp, and Ca2+ influx into the dyad triggers the opening of 3 RyRs 1–2 ms later, which inactivate after ~ 7 ms, ~ 9 ms, and ~ 13 ms, respectively. The temporal shape of the Ca2+ signal in the dyad (Fig. 2A) is closely correlated to the number of open RyRs (Fig. 2B). Figure 2C shows the average dyadic Ca2+ signal, calculated by averaging the number of Ca2+ ions at each point in time across 400 independent dyads. The duration of the average local Ca2+ transient shown in Panel C is ~ 20 ms (at half-maximal amplitude), similar to that measured for Ca2+ spikes in myocytes21, 24, 52.
Figure 2.

A CICR event in a single dyad evoked by a 0 mV voltage clamp step at time zero (from73). (A) The number of free Ca2+ ions in the dyad volume as a function of time. (B) The number of open RyRs (black solid line) and the number of open LCCs (gray solid line) in the dyad during the release event depicted in panel A. (C) The average number of free Ca2+ ions in the dyad as calculated from 400 independent dyads. (D) ECC gain as a function of membrane potential for the baseline model which includes space-filling geometric models of protein structure in the dyad (solid line), for the model with protein structures excluded (dashed line), and for a modified model with dyad height reduced from 15 nm to 13 nm and protein structures excluded (dotted line).
We demonstrated that ECC gain is a robust and reproducible measurement of CICR because the variability in gain that arises from the stochastic gating of LCCs and RyRs is small for a whole-cell population of dyads at potentials near and above 0 mV (see Fig. 11 of73). The functional influence of protein structures on ECC is shown in Fig. 2D. Peak ECC gain obtained for the baseline model (solid line) is compared to that for the model in which LCC, RyR, and CaM molecule structures were omitted (dashed line). Ca2+ binding site locations for the no-structure simulations remain at the same positions in space as when the protein structures are in place. Over a wide range of potentials, the peak ECC gain is decreased upon removal of the protein structures from the dyad. The protein structures occupy ~ 15% of the dyad volume. In order to show that the difference in ECC gain in the presence vs. absence of protein structures is not simply attributable to the difference in Ca2+-accessible dyad volume, peak ECC gain was obtained with protein structures absent and dyad volume reduced by ~ 15% (dotted line). ECC gain values for this model were clearly are not increased to the level obtained in the presence of protein structures. These data indicate that the physical shape and configuration of dyad proteins influences Ca2+ diffusion during CICR in such a way as to enhance ECC gain. In addition, the probability of triggering a release event was 0.152 with protein structures excluded and 0.105 with protein structures intact, a 44% increase. These data suggest that when LCCs are directly apposed to RyRs, the diffusion obstacle imposed by the structure of RyRs leads to an increase in the probability that trigger Ca2+ ions bind to activation sites on the RyRs, and hence increases ECC gain.
These data demonstrate that at the level of single dyads, CICR is a highly noisy process and that models based on laws of mass action may not accurately capture dyadic Ca2+ dynamics. Further, in ensembles of dyads, the fundamental properties of local control of CICR arise de novo as a consequence of the underlying physical structure and channel gating properties, including voltage-dependence of ECC gain. Finally, the physical shape and location of RyRs relative to LCCs is likely to have a major influence on properties of CICR.
The Integrated Local-Control Model of the Cardiac Myocyte
The Greenstein-Winslow34 model of the cardiac ventricular myocyte was the first to fully integrate mechanisms of local control of CICR into a model of the cardiac AP. This was accomplished by updating and extending the common-pool model of Winslow et al.38 to include a population of dyadic Ca2+ release units (CaRUs). In essence, this integrated local control model is generated from the nano-scale model by simplifying the dyad description to that of well-mixed Ca2+ compartment, and incorporating a large population of such dyads into a whole-cell model. Detailed properties of the nano-scale model (e.g. effect of protein structure on ECC gain) are retained in this integrated model by adjusting parameters that influence the effectively equivalent property at this scale (e.g. effective Ca2+ sensitivity of RyRs). In this model, local interactions of individual LCCs with nearby RyRs are simulated stochastically, with these local simulations embedded within the numerical integration of the ordinary differential equations (ODEs) describing ionic and membrane pump/exchanger currents, SR Ca2+ cycling, and time-varying cytosolic ion concentrations.
The Greenstein-Winslow model was formulated to capture fundamental properties such as graded release, while at the same time needed to be computationally tractable. A full mathematical description of the stochastic state models, dynamical equations, parameters, and initial conditions defining the myocyte model are given in Appendix I of 34. Figure 3A shows a schematic of the CaRU model which is intended to mimic the properties of Ca2+ sparks in the t-tubule/SR (T-SR) junction. Figure 3B shows a cross-section of the model T-SR cleft, which is divided into four individual dyadic subspace compartments arranged on a 2 × 2 grid. Each subspace compartment contains a single LCC and 5 RyRs in its JSR and sarcolemmal membranes, respectively83. The division of the CaRU into four subunits allows for the possibility that an LCC may trigger Ca2+ release in adjacent subspaces (i.e., RyR recruitment). Upon activation of RyRs, subspace [Ca2+] will become elevated. This Ca2+ freely diffuses to either adjacent subspace compartments (Jiss) or into the cytosol (Jxfer). The local JSR compartment is refilled via passive diffusion of Ca2+ from the NSR compartment (Jtr). The number of active LCCs was chosen such that the amplitude of the ensemble current summed over all LCCs corresponds to whole-cell measurements in canine myocytes84 which corresponded to 50,000 LCCs (12,500 CaRUs) and agrees with experimental estimates of LCC density79. LCC and RyR gating were based on previous models31, 85, and the channels (ClCh) that carry the Ca2+-dependent transient outward chloride (Cl−) current (Ito2) are included within the CaRU.86 A detailed description of the local control simulation algorithm is given in Appendix II of 34. Stochastic fluctuations in model outputs are the natural result of the ensemble behavior of ion channel and CaRUs, and introduce a degree of variability to simulation output and can be physiologically important to understanding some physiological phenomena such as EADs87.
Figure 3.

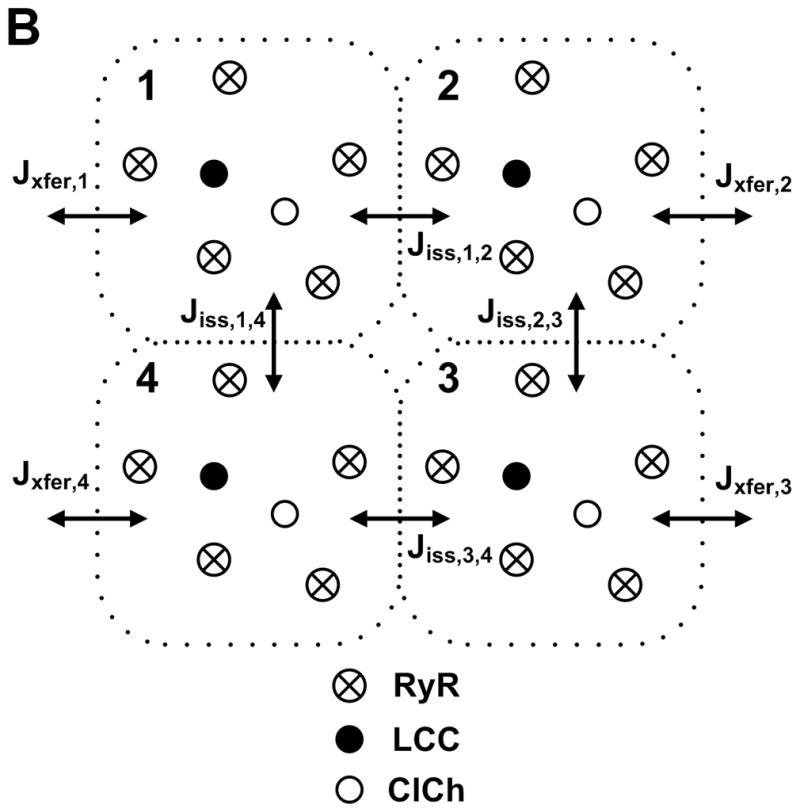
Schematic representation of the CaRU34. (A) Trigger Ca2+ influx through the LCCs enters into the T-SR cleft (dyadic space), RyRs and ClChs open, local Ca2+ passively diffuses into the cytosol, and JSR Ca2+ is refilled via passive diffusion from the NSR. (B) The T-SR cleft (shown in cross-section) is composed of four dyadic subspace volumes, arranged on a 2 × 2 grid, each containing 1 LCC, 1 ClCh, and 5 RyRs. Ca2+ in any subspace may diffuse to a neighboring subspace (Jiss) or to the cytosol (Jxfer).
Panels A and B of Fig. 4 demonstrate the most elementary model release event during CICR, as triggered by a single LCC at 0 mV. Ca2+ flux through an LCC (gray line) and the net SR Ca2+ release flux through the five adjacent RyRs (black line) are shown in Fig. 4A. Local JSR release flux is triggered by the first LCC opening (at ~ 5 ms) and lasts ~20 ms, far longer than the LCC open duration (<1 ms). The amplitude of the release flux varies with the number of open RyRs and the local Ca2+ gradient across the JSR membrane. Figure 4B shows the corresponding subspace [Ca2+], which reaches a peak value of ~ 40 μmol/L. Total Ca2+ influx through the set of four LCCs (gray line) and the net SR Ca2+ release flux through the set of 20 RyRs (black line) that make up a single T-SR junction are shown in Fig. 4C. LCC Ca2+ influx rises to a level consistent with two open channels within a short time following the initiation of the pulse. The spatial average of Ca2+ concentration in the four subspace compartments of the CaRU is intended to represent a Ca2+ spark-like event (Fig. 4D), and has duration of 25 ms (at half-maximal amplitude) similar to that measured in myocytes (20–50 ms) 21, 24, 52.
Figure 4.

Sample results for a single CaRU in response to a 200-ms voltage clamp to 0 mV34. (A) Ca2+ flux through a single LCC (gray line) and through the set of five RyRs (black line) within a single dyadic subspace compartment. Arrows 1 and 2 highlight RyR number and Ca2+ gradient driven changes in SR Ca2+ release flux, respectively. (B) Subspace [Ca2+] associated with the events of panel A. (C) Ca2+ flux through the set of four LCCs (gray line) and the set of 20 RyRs (black line) within a single CaRU. (D). Mean subspace [Ca2+] in the four subspace compartments associated with the events in the CaRU described in panel C.
Previous experimental studies25, 88 and mathematical models27, 33 have shown significant differences between the voltage dependence of ICaL and RyR Ca2+ release flux even though SR Ca2+ release is exclusively controlled by LCC Ca2+ entry. These differences underlie the phenomenon of “variable” EC coupling gain. Figure 5A shows the voltage dependence of LCC (filled circles) and RyR (open circles) Ca2+ flux. In Fig. 5B, the peak fluxes of Fig. 5A have been normalized based on their respective maxima. Maximal LCC Ca2+ influx occurs at 10 mV, whereas maximal RyR Ca2+ release flux occurs at 0 mV. EC coupling gain is plotted as a function of voltage in Fig. 5C (triangles), and is monotonically decreasing with increasing membrane potential in agreement with experimental measurements24, 25, 88. In the negative voltage range LCC open probability is submaximal, leading to sparse LCC openings (i.e. submaximal recruitment of CaRUs), but large amplitude unitary currents efficiently trigger adjacent RyRs, resulting in high values of ECC gain. More CaRUs are recruited with increasing membrane potential as a result of increasing LCC open probability, however ECC gain decreases with increasing membrane potential because the higher LCC open probability leads to redundancy in LCC openings as a result of which a fraction of LCCs are attempting to trigger refractory RyRs and at even higher potentials the decrease in LCC unitary current amplitude reduces the fidelity of local coupling between LCCs and RyRs.
Figure 5.

Ca2+ fluxes and ECC gain for the Greenstein-Winslow model (panels A-C)34 and coupled LCC-RyR model (panels D-F)71. (A) Mean peak LCC (filled circles) and RyR (open circles) Ca2+ flux amplitudes as a function of membrane voltage. (B) Normalized peak Ca2+ fluxes (data of panel A). (C) ECC gain under control conditions. (D) Peak LCC (filled triangles) and RyR (open triangles) Ca2+ flux amplitudes. (E) Normalized peak Ca2+ fluxes (data of panel D). (F) ECC gain for the baseline model (solid line), and models in which dyad size and number of channels per dyad is increased twofold (gray line), and threefold (dashed line).
Figure 6 demonstrates the ability of the Greenstein-Winslow model to reconstruct APs and cytosolic Ca2+ transients of normal canine midmyocardial ventricular myocytes. In Fig. 6A, a normal 1-Hz steady-state AP is shown (solid black line). AP properties are similar to those measured in experiments39, with APD of ~300 ms. Also shown in Fig. 6A are the kinetics of CDI (dashed line) and VDI (dotted line), demonstrating that this model conforms with experimental findings that indicate LCC CDI is strong while VDI is relatively slow and incomplete46, 47. Figure 6B shows cytosolic (black line) and mean subspace (gray line) Ca2+ transients. The cytosolic Ca2+ transient peaks at ~ 0.75 μmol/L and lasts longer than the duration of the AP whereas Ca2+ in the subspace reaches ~11 μmol/L on average, and equilibrates to near-cytosolic levels rapidly, within ~ 100 – 150 ms. The simulations of Figs. 4–6 demonstrate that this multi-scale model faithfully reproduces a variety of experimental observations that span from single-channel to whole myocyte.
Figure 6.

AP and Ca2+ transients at 1-Hz steady state pacing34. (A) Membrane potential (solid line, left axis), and probability that CDI and VDI (dashed and dotted lines, respectively, right axis) has not occurred as a function of time under normal conditions. (B) Cytosolic (black line, left axis) and mean subspace (gray line, right axis) Ca2+ concentrations corresponding to the AP simulated in panel A.
The Coupled LCC-RyR Local Control Model
A limitation of the Greenstein-Winslow34 local control model is that it is far more computationally demanding than deterministic models. To address this, we recently developed an analytical “first-principles” approach for deriving simplified mechanistic models of CICR by applying a carefully chosen set of approximations that allow for the ensemble behavior of CaRUs to be represented by a low dimensional system of ODEs, eliminating the need for computationally expensive stochastic simulations. This model, termed the coupled LCC-RyR model72, retains the biophysically based description of local control of CICR, and captures its key properties including graded SR Ca2+ release and voltage dependent ECC gain and is the basis of a deterministic local control model of the cardiac myocyte71.
Following the methods described by Hinch et al72, LCC and RyR models composed of the minimum number of states necessary to capture fundamental dynamics of channel gating were designed and constrained based on experimental data sets. A simplified CaRU model consisting of only a single LCC and a single RyR was formed. The relatively fast diffusion (i.e. short diffusion distance) of Ca2+ from dyad to cytosol (as compared to the time scale of LCC and RyR gating)62 allows for the application of a steady state approximation for dyadic Ca2+ diffusion. This allows the resulting CaRU to be analytically simplified into a single Markov state model in which each state represents a unique configuration of channel states for the combined set of LCCs and RyRs in the CaRU. In the resulting formulation, the dynamics of a population of CaRUs can be described by a deterministic set of coupled ODEs. Other elegant methods have been derived to improve computational efficiency of ECC simulations as well37, 89, 90, but have not yet been implemented in an integrated myocyte model using biophysically detailed channel models.
Panels D–F of Figure 5 demonstrate that the deterministic coupled LCC-RyR model reproduces the fundamental quantitative properties of CICR in a similar manner to its stochastic predecessor34 in panels A–C. The effect of dyad size on ECC was analyzed using models with 2-fold and 3-fold larger dyads (Fig. 5F, gray and dashed lines, respectively). In each case the number of dyads per cell was reduced such that the total number of LCCs and RyRs per cell was conserved, and the RyR/LCC stoichiometry was unchanged. At potentials more negative than −10 mV, the increase in CaRU size yields a substantial increase in gain such that the ECC gain curve acquires the steep slope similar to those observed experimentally24, 25. This arises as a result of a model-dependent increase in the functional RyR/LCC ratio because it is likely that a single LCC opening will trigger regenerative release from most RyRs in a CaRU. The coupled LCC-RyR local control model reconstructs APs and Ca2+ transients of canine midmyocardial ventricular cells (see Fig. 6 of 71 for details) at a variety of pacing frequencies, and similar to the Greenstein-Winslow model, has an ICaL which exhibits strong CDI and weak VDI. The ability of this model to reproduce all of the core features of the stochastic local control model in an efficient manner has made it a candidate for use in large scale tissue simulations91.
Additional domains of localized Ca2+ signaling
There have been a number of recent models in which Ca2+ signaling in localized compartments other than the dyad have been investigated. Experimental measurements of NCX current18 have provided evidence that this transport mechanism, and possibly the sarcolemmal Ca2+-ATPase, may be driven by subsarcolemmal Ca2+, which is elevated relative to Ca2+ concentration in the bulk cytosol as a result of Ca2+ diffusion limitations. The rabbit ventricular myocyte model of Shannon et al54 was the first to introduce a subsarcolemmal Ca2+ compartment, and a number of recent models now include a subsarcolemmal Ca2+ compartment35, 61, 92, 93. Elevated subsarcolemmal Ca2+ may have a particularly important influence on NCX. It has been suggested that elevated subsarcolemmal Ca2+ during the early phase of the AP may modulate NCX reversal potential, reducing entry of Ca2+.94 More needs to be done to quantify the functional consequences of sensing subsarcolemmal Ca2+ and NCX function. This is particularly important given the role of NCX in shaping AP properties and its possible role in the generation of delayed after-depolarizations. Very interestingly, the Pasek et al95 model of the guinea pig ventricular myocyte includes a diffusive t-tubule system with a heterogeneous distribution of ion channels between tubular and surface membranes, thus introducing a new extracellular compartment. This model predicted that Ca2+ depletion and K+ accumulation in the t-tubule during APs leads to a reduction in SR Ca2+ load, which would impact ECC.
There is now substantial electrophysiological evidence that extra-dyadic processes also influence CICR. Evidence suggests that Ca2+ entry via reverse-mode NCX activity may either trigger or modulate CICR96, 97 and that CICR modulates NCX function98. NCX has also been identified in the t-tubules at a high level. However, the precise location of NCX within99 and/or near100 the dyad remains uncertain. Lines et al101 used a model of the cardiac dyad which accounted for details of both Ca2+ and Na+ diffusion to demonstrate that if fast Na+ channels are in the dyadic cleft then NCX (driven by the local elevation in [Na+]) can influence the timing of local CICR. Recent evidence also suggests NCX may form a functional complex with NKA and its accessory protein phospholemman102. Furthermore, one study suggests that NCX and NKA constitute a physically interacting molecular complex with ankyrin-B and an inositol 1,4,5-phosphate operated Ca2+-releasing channel (InsP3R) in the t-tubules that is distinct from the LCC-RyR complex100. This hypothetical “ankyrin-B complex” may play an important role in the regulation of local Na+ and Ca2+ concentrations and possibly modulate CICR by regulating the boundary conditions on dyadic Ca2+. Again, it is likely that these regulatory interactions are mediated by small numbers of Ca2+ ions. This may be a common theme in cases where proteins interact over nanometer length scales.
Recent work also demonstrates that mitochondria are positioned near dyads103, 104. Electron-dense structures that link the mitochondrial outer membrane with JSR, NSR, and t-tubules have been observed. These structures appear to be preferentially located, with one population forming attachments to NSR, and another set of bridges being near the dyadic cleft. Attachments to t-tubules were observed, but were rare. Ca2+ is taken up by mitochondria via the mitochondrial Ca2+-uniporter105. Therefore, spatial co-localization of mitochondria near the dyadic cleft may give rise to an additional Ca2+-signaling micro-domain involved in the regulation of mitochondrial ATP production, and which modulates dyadic Ca2+ concentrations and Ca2+ diffusion between adjacent RyR couplons106.
Regulation of CICR by cell signaling pathways
Cardiac proteins involved in CICR and ECC are primary targets for regulation via cell signaling pathways and models have played a key role in understanding this regulation (see accompanying article in this series by Yang and Saucerman). The Greenstein-Winslow34 model was extended to study effects of the β-adrenergic signaling pathway by the incorporation of protein kinase A (PKA)-mediated functional changes in target proteins107 and could explain PKA-mediated changes in AP shape, dissected the specific effects of PKA-mediated phosphorylation of LCCs and RyRs on the voltage-dependent gain of CICR, and predicted a mechanism by which increasing levels of LCC phosphorylation could lead to increased frequency of EADs87. Saucerman et al108 developed a differential-algebraic model of the β-adrenergic signaling pathway and incorporated it into the rabbit myocyte model of Puglisi et al109 and used this model to quantitatively understand and predict effects of specific molecular perturbations on cAMP dynamics and contractility via changes in function of target proteins resulting from altered phosphorylation dynamics. Recently, the model of Saucerman et al108 was incorporated into the guinea pig ventricular model of Faber and Rudy110 in a study of the role of β-adrenergic agonists and antagonists in long-QT syndrome111.
An important Ca2+ cycling regulatory mechanism of recent interest is the signaling pathway involving Ca2+/CaM-dependent protein kinase II (CaMKII), whose targets proteins include LCCs, RyRs, PLB, and the SERCA pump, as well as Na+ and K+ channels112. Evidence suggests that CaMKII is directly associated with its target proteins113, 114 (implying that CaMKII signaling occurs locally in the dyadic junction), and that CaMKII activity is elevated in heart failure115. The first model of the cardiac myocyte to integrate the role of CaMKII was presented by Hund and Rudy43, which predicted that CaMKII plays an important role in the rate dependent properties of the cytosolic Ca2+ transient, but not APD. Saucerman and Bers93 incorporated models of CaM, CaMKII, and calcineurin (CaN) into the Shannon et al54 model in order to better understand the functional consequences of the different affinities of CaM for CaMKII and CaN during APs. The model predicted that in the cardiac dyad, Ca2+ levels lead to a high degree of CaM activity which results in frequency-dependent CaMKII activation and constitutive CaN activation, whereas the lower Ca2+ levels in the cytosol only minimally activate CaM, which allows for gradual CaN activation, but no significant activation of CaMKII. Grandi et al116 developed a model of CaMKII overexpression in the rabbit ventricular myocyte, including the role of CaMKII phosphorylation of fast Na+ channels, LCCs, and the transient outward K+ current (Ito1), which revealed a net effect of APD reduction with CaMKII. Recently, Hashambhoy et al36 described dynamic CaMKII phosphorylation of LCCs in an extension of the Greenstein-Winslow34 model. In this model it is assumed that a single CaMKII holoenzyme is tethered to each LCC, each CaMKII monomer can transition among a variety of activity states (see Fig. 2 of Hashambhoy et al36), and CaMKII monomers can catalyze phosphorylation of individual LCCs. This model demonstrated that CaMKII-dependent shifts of LCC gating patterns into high activity gating modes may be the underlying mechanism of a variety of experimentally observed phenomena associated with ICaL facilitation. Hashambhoy et al55 further expanded this model to include CaMKII-dependent regulation of RyRs and demonstrated that under physiological conditions, CaMKII phosphorylation of LCCs ultimately has a greater effect on RyR leak flux and APD as compared with phosphorylation of RyRs (Fig. 7A). APD was shown to correlate well with a CaMKII-mediated shift in modal gating of LCCs (Fig. 7B).
Figure 7.

(A) Simulated Results from a 1-Hz AP pacing protocol under different LCC and/or RyR phosphorylation conditions. (B) APD as a function of average LCC phosphorylation levels. Fully phosphorylated LCCs gate in Mode 2 which exhibit long duration openings.55
Future Directions in ECC Modeling
A number of mechanistic questions remain unanswered in cardiac ECC. As noted above, controversy remains as to mechanisms involved in termination of SR Ca2+ release. The high degree of technical difficulty involved in experimentally assessing RyR and CaRU properties under physiological conditions has resulted in limited and difficult-to-reproduce measures of certain key features of CICR such as RyR gating properties (e.g. coupled gating of RyRs), the degree of local JSR Ca2+ depletion associated with Ca2+ sparks (Ca2+ blinks), and the rate of local refilling of JSR. Models have been able to reveal and demonstrate multiple possible mechanisms of SR Ca2+ release termination, but the lack of consensus on a single experimentally verified mechanism has allowed a variety of mathematical model formulations of SR Ca2+ release to coexist. Improved resolution of both structural and functional imaging of ECC domain ultrastructure and localized Ca2+ dynamics will help to resolve these questions and to further validate and refine nano- and microscale models of ECC10, 11. Additional areas of recent interest, where mechanistic models have yet to be fully developed, include regulation of ECC by mitochondrial Ca2+ dynamics (see above) and functional alteration of key ECC proteins arising from oxidative-stress (e.g. effects of mitochondria-derived reactive oxygen species) associated with heart failure. Recently, Tadross et al117 used a combination of elegant models and experiments to elucidate a mechanism underlying Ca2+ channel CDI, by which CaM in complex with an LCC, can sense and decode local and global Ca2+ signals. The implications of this mechanism on our understanding of dyadic Ca2+ dynamics and cardiac ECC have yet to be studied with models. Finally, the mathematical techniques used in developing models that span multiple biological scales are not adequate for all applications and themselves are an ongoing area of theoretical study (see ref 37 for a recent review of model reduction methods). Mechanistic model reproduction of phenomena such as Ca2+ alternans and delayed after depolarizations depend on the ability of a Ca2+ spark to trigger neighboring release sites and form intracellular Ca2+ waves48. While model reduction techniques have captured important features of local control of ECC (as described above), the lack of low-dimensional models that can capture Ca2+ wave mechanisms has made it difficult to extend such models to the tissue and whole heart level in order to study how these events trigger large scale arrhythmias. The development of models that can accomplish this task in a tractable formulation will greatly improve our ability to understand the mechanistic links between disease-related changes in ECC and arrhythmia.
Conclusion
Integrative modeling of cardiac ECC and myocyte physiology has played a critical role in revealing mechanistic insights at and across a range of biological scales. At the smallest scale, models have shed light on how Ca2+ ions and proteins in the cardiac junction interact during LCC and RyR gating. Expanding our view, simplified models of individual release sites have allowed for detailed simulations that reveal determinants of Ca2+ spark activation and termination properties. On an intermediate scale, models of independent dyadic junctions have revealed how characteristic whole-myocyte properties of ECC emerge from the ensemble behavior of individual release sites. Incorporation of such Ca2+ release sites into whole-cell models elucidates the bidirectional interaction between ECC and membrane potential that determine properties of the cardiac AP. Incorporation of ECC regulatory signaling pathways into models has further refined our understanding of this integrative system (see accompanying article in this series by Yang and Saucerman). At the highest scale, such models can be used as building blocks for large scale tissue and whole heart models which can reveal how wave propagation in a normal heart tissue can degenerate into an arrhythmia as a result of defective ECC (see accompanying articles in this series by Weiss et al and Trayanova et al). The continuum of biological scales spanned by these models leads to the formation of powerful multi-scale approaches whereby macroscopic phenotypes (e.g. steep ECC gain curve) can be understood as resulting from microscopic mechanisms (e.g. Ca2+ funneling by RyR protein geometry). In the same way that Hodgkin and Huxley formed a mathematical framework that now defines the way in which we conceptualize ion channel function, modern multi-scale models of cardiac ECC have provided a framework for understanding complex ECC mechanisms, interpreting experimental data, and making mechanistic predictions that guide experimental design.
Acknowledgments
Sources of Funding
This work was supported by NIH grants HL081427 and HL087345.
Non-standard Abbreviations and Acronyms
- AP
action potential
- APD
action potential duration
- CaM
calmodulin
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CaN
calcineurin
- CaRU
Ca2+ release unit
- CDI
Ca2+-dependent inactivation
- CICR
Ca2+-induced Ca2+ release
- ClCh
Ca2+-dependent Cl− channel
- EAD
early after depolarization
- ECC
excitation-contraction coupling
- FPE
Fokker-Plank equation
- JSR
junctional sarcoplasmic reticulum
- LCC
L-type Ca2+ channel
- LRII
Luo-Rudy phase II
- NCX
Na+-Ca2+ exchanger
- NSR
network sarcoplasmic reticulum
- ODE
ordinary differential equation
- PKA
protein kinase A
- PLB
phospholamban
- RyR
ryanodine receptor
- SERCA pump
SR Ca2+ ATPase
- SR
sarcoplasmic reticulum
- T-SR
t-tubule/sarcoplasmic reticulum
- VDI
voltage-dependent inactivation
Footnotes
Disclosures
None.
References
- 1.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Boston: Kluwer; 2001. [Google Scholar]
- 2.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 3.Balaban RS, Heineman FW. Control of mitochondrial respiration in the heart in vivo. Mol Cell Biochem. 1989;89:191–197. doi: 10.1007/BF00220775. [DOI] [PubMed] [Google Scholar]
- 4.Williams RS, Rosenberg P. Calcium-dependent gene regulation in myocyte hypertrophy and remodeling. Cold Spring Harb Symp Quant Biol. 2002;67:339–344. doi: 10.1101/sqb.2002.67.339. [DOI] [PubMed] [Google Scholar]
- 5.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 6.Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD. [DOI] [PubMed] [Google Scholar]
- 7.Soeller C, Cannell MB. Examinationof the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 8.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev. 1997;77:699–729. doi: 10.1152/physrev.1997.77.3.699. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi T, Martone ME, Yu Z, Thor A, Doi M, Holst MJ, Ellisman MH, Hoshijima M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci. 2009;122:1005–1013. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci U S A. 2009;106:22275–22280. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bers D, Stiffel V. Ratio of ryanodine to dihidropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am J Physiol. 1993;264:C1587–C1593. doi: 10.1152/ajpcell.1993.264.6.C1587. [DOI] [PubMed] [Google Scholar]
- 13.Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reeves JP, Hale CC. The stoichiometry of the cardiac sodium-calcium exchange system. J Biol Chem. 1984;259:7733–7739. [PubMed] [Google Scholar]
- 15.Bers DM, Ginsburg KS. Na:Ca stoichiometry and cytosolic Ca-dependent activation of NCX in intact cardiomyocytes. Ann N Y Acad Sci. 2007;1099:326–338. doi: 10.1196/annals.1387.060. [DOI] [PubMed] [Google Scholar]
- 16.Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu Rev Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- 17.Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O’Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber CR, Piacentino V, 3rd, Ginsburg KS, Houser SR, Bers DM. Na(+)-Ca(2+) exchange current and submembrane[Ca(2+)] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- 19.Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992;453:591–608. doi: 10.1113/jphysiol.1992.sp019246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hicks MJ, Shigekawa M, Katz AM. Mechanism by which cyclic adenosine 3′:5′-monophosphate-dependent protein kinase stimulates calcium transport in cardiac sarcoplasmic reticulum. Circ Res. 1979;44:384–391. doi: 10.1161/01.res.44.3.384. [DOI] [PubMed] [Google Scholar]
- 21.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local stochastic release of Ca2+ in voltage-clamped rat heart cells: visualization with confocal microscopy. J Physiol (Lond) 1994;480:21–29. doi: 10.1113/jphysiol.1994.sp020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soeller C, Cannell MB. Analysing cardiac excitation-contraction coupling with mathematical models of local control. Prog Biophys Mol Biol. 2004;85:141–162. doi: 10.1016/j.pbiomolbio.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H. beta-Adrenergic stimulation synchronizes intracellular Ca(2+) release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001;88:794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- 25.Wier WG, Egan TM, Lopez-Lopez JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. J Physiol. 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannell M, Berlin J, Lederer W. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;283:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- 27.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiFrancesco D, Noble D. A model of cardiac electrical activity incorporating ionic pumps and concentration changes. Philos Trans R Soc Lond B Biol Sci. 1985;307:353–398. doi: 10.1098/rstb.1985.0001. [DOI] [PubMed] [Google Scholar]
- 29.Beeler GW, Reuter H. Reconstruction of the action potential of ventricular myocardial fibres. J Physiol. 1977;268:177–210. doi: 10.1113/jphysiol.1977.sp011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McAllister RE, Noble D, Tsien RW. Reconstruction of the electrical activity of cardiac Purkinje fibres. J Physiol. 1975;251:1–59. doi: 10.1113/jphysiol.1975.sp011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jafri MS, Rice JJ, Winslow RL. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys J. 1998;74:1149–1168. doi: 10.1016/S0006-3495(98)77832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imredy JP, Yue DT. Mechanism of Ca(2+)-sensitive inactivation of L-type Ca2+ channels. Neuron. 1994;12:1301–1318. doi: 10.1016/0896-6273(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 33.Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR, Rios E. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol. 1999;113:469–489. doi: 10.1085/jgp.113.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenstein JL, Winslow RL. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys J. 2002;83:2918–2945. doi: 10.1016/S0006-3495(02)75301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahajan A, Shiferaw Y, Sato D, Baher A, Olcese R, Xie LH, Yang MJ, Chen PS, Restrepo JG, Karma A, Garfinkel A, Qu Z, Weiss JN. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashambhoy YL, Winslow RL, Greenstein JL. CaMKII-induced shift in modal gating explains L-type Ca(2+) current facilitation: a modeling study. Biophys J. 2009;96:1770–1785. doi: 10.1016/j.bpj.2008.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams GS, Smith GD, Sobie EA, Jafri MS. Models of cardiac excitation-contraction coupling in ventricular myocytes. Math Biosci. 2010;226:1–15. doi: 10.1016/j.mbs.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winslow RL, Rice J, Jafri S, Marban E, O’Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res. 1999;84:571–586. doi: 10.1161/01.res.84.5.571. [DOI] [PubMed] [Google Scholar]
- 39.O’Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- 40.Ahmmed GU, Dong PH, Song G, Ball NA, Xu Y, Walsh RA, Chiamvimonvat N. Changes in Ca(2+) cycling proteins underlie cardiac action potential prolongation in a pressure-overloaded guinea pig model with cardiac hypertrophy and failure. Circ Res. 2000;86:558–570. doi: 10.1161/01.res.86.5.558. [DOI] [PubMed] [Google Scholar]
- 41.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winslow RL, Scollan DF, Holmes A, Yung CK, Zhang J, Jafri MS. Electrophysiological modeling of cardiac ventricular function: from cell to organ. Annu Rev Biomed Eng. 2000;2:119–155. doi: 10.1146/annurev.bioeng.2.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hund TJ, Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 2004;110:3168–3174. doi: 10.1161/01.CIR.0000147231.69595.D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 45.Sipido KR, Callewaert G, Carmeliet E. Inhibition and rapid recovery of Ca2+ current during Ca2+ release from sarcoplasmic reticulum in guinea pig ventricular myocytes. Circ Res. 1995;76:102–109. doi: 10.1161/01.res.76.1.102. [DOI] [PubMed] [Google Scholar]
- 46.Linz KW, Meyer R. Control of L-type calcium current during the action potential of guinea-pig ventricular myocytes. J Physiol. 1998;513 (Pt 2):425–442. doi: 10.1111/j.1469-7793.1998.425bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 48.Rovetti R, Cui X, Garfinkel A, Weiss JN, Qu Z. Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ Res. 2010;106:1582–1591. doi: 10.1161/CIRCRESAHA.109.213975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langer GA, Peskoff A. Calcium concentration and movement in the diadic cleft space of the cardiac ventricular cell. Biophys J. 1996;70:1169–1182. doi: 10.1016/S0006-3495(96)79677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J. 1997;73:97–111. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 52.Sham JS, Song LS, Chen Y, Deng LH, Stern MD, Lakatta EG, Cheng H. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci U S A. 1998;95:15096–15101. doi: 10.1073/pnas.95.25.15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hashambhoy YL, Greenstein JL, Winslow RL. Role of CaMKII in RyR leak, EC coupling and action potential duration: a computational model. J Mol Cell Cardiol. 2010;49:617–624. doi: 10.1016/j.yjmcc.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zahradnikova A, Zahradnik I. A minimal gating model for the cardiac calcium release channel. Biophys J. 1996;71:2996–3012. doi: 10.1016/S0006-3495(96)79492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keizer J, Levine L. Ryanodine receptor adaptation and Ca2+(−)induced Ca2+ release-dependent Ca2+ oscillations. Biophys J. 1996;71:3477–3487. doi: 10.1016/S0006-3495(96)79543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng H, Fill M, Valdivia H, Lederer WJ. Models of Ca2+ release channel adaptation. Science. 1995;267:2009–2010. doi: 10.1126/science.7701326. [DOI] [PubMed] [Google Scholar]
- 59.Gyorke S, Fill M. Ryanodine receptor adaptation: control mechanism of Ca(2+)-induced Ca2+ release in heart. Science. 1993;260:807–809. doi: 10.1126/science.8387229. [DOI] [PubMed] [Google Scholar]
- 60.Rice JJ, Jafri MS, Winslow RL. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys J. 1999;77:1871–1884. doi: 10.1016/s0006-3495(99)77030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ, Jafri MS. Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors) Circ Res. 2001;88:1151–1158. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 65.Sobie EA, Guatimosim S, Gomez-Viquez L, Song LS, Hartmann H, Saleet Jafri M, Lederer WJ. The Ca 2+ leak paradox and rogue ryanodine receptors: SR Ca 2+ efflux theory and practice. Prog Biophys Mol Biol. 2006;90:172–185. doi: 10.1016/j.pbiomolbio.2005.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Groff JR, Smith GD. Ryanodine receptor allosteric coupling and the dynamics of calcium sparks. Biophys J. 2008;95:135–154. doi: 10.1529/biophysj.107.119982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 68.Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stevens SC, Terentyev D, Kalyanasundaram A, Periasamy M, Gyorke S. Intra-sarcoplasmic reticulum Ca2+ oscillations are driven by dynamic regulation of ryanodine receptor function by luminal Ca2+ in cardiomyocytes. J Physiol. 2009;587:4863–4872. doi: 10.1113/jphysiol.2009.175547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zima AV, Picht E, Bers DM, Blatter LA. Partial inhibition of sarcoplasmic reticulum ca release evokes long-lasting ca release events in ventricular myocytes: role of luminal ca in termination of ca release. Biophys J. 2008;94:1867–1879. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greenstein JL, Hinch R, Winslow RL. Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J. 2006;90:77–91. doi: 10.1529/biophysj.105.065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hinch R, Greenstein JL, Tanskanen AJ, Xu L, Winslow RL. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys J. 2004;87:3723–3736. doi: 10.1529/biophysj.104.049973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tanskanen AJ, Greenstein JL, Chen A, Sun SX, Winslow RL. Protein geometry and placement in the cardiac dyad influence macroscopic properties of calcium-induced calcium release. Biophys J. 2007;92:3379–3396. doi: 10.1529/biophysj.106.089425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serysheva, Hamilton SL, Chiu W, Ludtke SJ. Structure of Ca2+ release channel at 14 A resolution. J Mol Biol. 2005;345:427–431. doi: 10.1016/j.jmb.2004.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang MC, Collins RF, Ford RC, Berrow NS, Dolphin AC, Kitmitto A. The three-dimensional structure of the cardiac L-type voltage-gated calcium channel: comparison with the skeletal muscle form reveals a common architectural motif. J Biol Chem. 2004;279:7159–7168. doi: 10.1074/jbc.M308057200. [DOI] [PubMed] [Google Scholar]
- 76.Wilson MA, Brunger AT. the 1.0 A crystal structure of Ca2+-bound calcmodulin: an analysis of disorder and implications for functionally relevant plasticity. J Mol Biol. 2000;301(5):1237–1256. doi: 10.1006/jmbi.2000.4029. [DOI] [PubMed] [Google Scholar]
- 77.Bhalla US. Signaling in small subcellular volumes. I. Stochastic and diffusion effects on individual pathways. Biophys J. 2004;87:733–744. doi: 10.1529/biophysj.104.040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang SQ, Stern MD, Rios E, Cheng H. The quantal nature of Ca2+ sparks and in situ operation of the ryanodine receptor array in cardiac cells. Proc Natl Acad Sci U S A. 2004;101:3979–3984. doi: 10.1073/pnas.0306157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rose WC, Balke CW, Wier WG, Marban E. Macroscopic and unitary properties of physiological ion flux through L-type Ca2+ channels in guinea-pig heart cells. J Physiol. 1992;456:267–284. doi: 10.1113/jphysiol.1992.sp019336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Risken H. The Fokker-Planck equation. Berlin: Springer; 1997. [Google Scholar]
- 81.Wang H, Peskin CS, Elston TC. A robust numerical algorithm for studying biomolecular transport processes. J Theor Biol. 2003:221. doi: 10.1006/jtbi.2003.3200. [DOI] [PubMed] [Google Scholar]
- 82.Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J. 2000;78:322–333. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 84.Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling incanine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 85.Keizer J, Smith GD. Spark-to-wave transition: saltatory transmission of calcium waves in cardiac myocytes. Biophys Chem. 1998;72:87–100. doi: 10.1016/s0301-4622(98)00125-2. [DOI] [PubMed] [Google Scholar]
- 86.Collier ML, Levesque PC, Kenyon JL, Hume JR. Unitary Cl-channels activated by cytoplasmic Ca2+ in canine ventricular myocytes. Circ Res. 1996;78:936–944. doi: 10.1161/01.res.78.5.936. [DOI] [PubMed] [Google Scholar]
- 87.Tanskanen AJ, Greenstein JL, O’Rourke B, Winslow RL. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys J. 2005;88:85–95. doi: 10.1529/biophysj.104.051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996;78:166–171. doi: 10.1161/01.res.78.1.166. [DOI] [PubMed] [Google Scholar]
- 89.Williams GS, Huertas MA, Sobie EA, Jafri MS, Smith GD. A probability density approach to modeling local control of calcium-induced calcium release in cardiac myocytes. Biophys J. 2007;92:2311–2328. doi: 10.1529/biophysj.106.099861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Williams GS, Huertas MA, Sobie EA, Jafri MS, Smith GD. Momentclosure for local control models of calcium-induced calcium release in cardiac myocytes. Biophys J. 2008;95:1689–1703. doi: 10.1529/biophysj.107.125948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Flaim SN, Giles WR, McCulloch AD. Contributions of sustained INa and IKv43 to transmural heterogeneity of early repolarization and arrhythmogenesis in canine left ventricular myocytes. Am J Physiol Heart Circ Physiol. 2006;291:H2617–2629. doi: 10.1152/ajpheart.00350.2006. [DOI] [PubMed] [Google Scholar]
- 92.Livshitz L, Rudy Y. Uniqueness and stability of action potential models during rest, pacing, and conduction using problem-solving environment. Biophys J. 2009;97:1265–1276. doi: 10.1016/j.bpj.2009.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Saucerman JJ, Bers DM. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys J. 2008;95:4597–4612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 95.Pasek M, Simurda J, Orchard CH, Christe G. A model of the guinea-pig ventricular cardiac myocyte incorporating a transverse-axial tubular system. Prog Biophys Mol Biol. 2008;96:258–280. doi: 10.1016/j.pbiomolbio.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 96.Goldhaber JI, Lamp ST, Walter DO, Garfinkel A, Fukumoto GH, Weiss JN. Local regulation of the threshold for calcium sparks in rat ventricular myocytes: role of sodium-calcium exchange. J Physiol. 1999;520(Pt 2):431–438. doi: 10.1111/j.1469-7793.1999.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Litwin S, Kohmoto O, Levi AJ, Spitzer KW, Bridge JH. Evidence that reverse Na-Ca exchange can trigger SR calcium release. Ann N Y Acad Sci. 1996;779:451–463. doi: 10.1111/j.1749-6632.1996.tb44820.x. [DOI] [PubMed] [Google Scholar]
- 98.Lewartowski B, Janiak R, Langer GA. Effect of sarcoplasmic reticulum Ca release into diadic region on Na/Ca exchange in cardiac myocytes. J Physiol Pharmacol. 1996;47:577–590. [PubMed] [Google Scholar]
- 99.Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, Sejersted OM, Ottersen OP. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1325–1337. doi: 10.1016/j.yjmcc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 100.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lines GT, Sande JB, Louch WE, Mork HK, Grottum P, Sejersted OM. Contribution of the Na+/Ca2+ exchanger to rapid Ca2+ release in cardiomyocytes. Biophys J. 2006;91:779–792. doi: 10.1529/biophysj.105.072447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bossuyt J, Ai X, Moorman JR, Pogwizd SM, Bers DM. Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ Res. 2005;97:558–565. doi: 10.1161/01.RES.0000181172.27931.c3. [DOI] [PubMed] [Google Scholar]
- 103.Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004;2004:re1. doi: 10.1126/stke.2152004re1. [DOI] [PubMed] [Google Scholar]
- 104.Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr. 2000;32:97–104. doi: 10.1023/a:1005520714221. [DOI] [PubMed] [Google Scholar]
- 105.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 106.Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102:369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Greenstein JL, Tanskanen AJ, Winslow RL. Modeling the actions of beta-adrenergic signaling on excitation--contraction coupling processes. Ann N Y Acad Sci. 2004;1015:16–27. doi: 10.1196/annals.1302.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem. 2003;278:47997–48003. doi: 10.1074/jbc.M308362200. [DOI] [PubMed] [Google Scholar]
- 109.Puglisi JL, Bers DM. LabHEART: an interactive computer model of rabbit ventricular myocyte ion channels and Ca transport. Am J Physiol Cell Physiol. 2001;281:C2049–2060. doi: 10.1152/ajpcell.2001.281.6.C2049. [DOI] [PubMed] [Google Scholar]
- 110.Faber GM, Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ahrens-Nicklas RC, Clancy CE, Christini DJ. Re-evaluating the efficacy of β-adrenergic agonists and antagonists in long QT-3 syndrome through computational modelling. Cardiovascular Research. 2009;82:439–447. doi: 10.1093/cvr/cvp083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631–640. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 113.Currie S, Loughrey CM, Craig MA, Smith GL. Calcium/calmodulin-dependent protein kinase IIdelta associates with the ryanodine receptor complex and regulates channel function in rabbit heart. Biochem J. 2004;377:357–366. doi: 10.1042/BJ20031043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 116.Grandi E, Puglisi JL, Wagner S, Maier LS, Severi S, Bers DM. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys J. 2007;93:3835–3847. doi: 10.1529/biophysj.107.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tadross MR, Dick IE, Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–1240. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]