

Abstract
Penicillin-binding protein 1b (PBP 1b) of the Gram-positive bacterium Streptococcus pneumoniae catalyzes the cross-linking of adjacent peptidoglycan strands, as a critical event in the biosynthesis of its cell wall. This enzyme is representative of the biosynthetic PBP structures of the β-lactam-recognizing enzyme superfamily, and is the target of the β-lactam antibiotics. In the cross-linking reaction, the amide between the -D-Ala-D-Ala dipeptide at the terminus of a peptide stem acts as an acyl donor toward the ε-amino group of a lysine found on an adjacent stem. The mechanism of this transpeptidation was evaluated using explicit-solvent molecular dynamics simulations and ONIOM quantum mechanics/molecular mechanics calculations. Sequential acyl transfer occurs to, and then from, the active site serine. The resulting cross-link is predicted to have a cis-amide configuration. The ensuing and energetically favorable cis- to trans-amide isomerization, within the active site, may represent the key event driving product release to complete enzymatic turnover.
Introduction
Penicillin-binding proteins (PBPs) catalyze the transglycosylase, transpeptidase, and carboxypeptidase activities that create the peptidoglycan structure of the bacterial cell wall.1–3 Within the PBP family, the high-molecular-mass PBPs catalyze both the transglycosylase—glycan lengthening by sequential addition of the N-acetyl-4-O-[2-(acetylamino)-2-deoxy-β-D-glucopyranosyl]-β-muramyl (NAG-NAM) disaccharide—and transpeptidase (glycan cross-linking through the peptide stems on the NAM saccharide) reactions of cell wall synthesis, each in spatially separated domains.4 The high-molecular-mass PBP 1b of the Gram-positive bacterium Streptococcus pneumoniae is a multi-domain enzyme, active during both cell elongation and cell division, and is a representative structure of the Class A PBP members of the β-lactam-recognizing enzyme superfamily.2,5,6 PBP 1b has a helical transmembrane domain for adherence to the outer leaflet of the cell membrane.7 The 2.2 Å resolution crystal structure of a soluble PBP 1b, construct lacking this transmembrane domain, shows three additional domains (PDB Code 2BG1).8,9 These are the transpeptidase and tranglycosylase domains, and an interconnecting domain of unknown function. The transglycosylase domain of this enzyme is adjacent to the membrane surface, and is separated by a structural domain from the transpeptidase domain. The reactions catalyzed by this PBP are summarized in Scheme 1. Despite the central role of these high Mr PBPs in cell wall biosynthesis, and their prominence as antibiotic targets, neither the structures of their substrate complexes nor their catalytic mechanisms are well understood. Due to the complexity of their bifunctional mechanism, there are few experimental studies on the transglycosylase reaction and no experimental studies on the transpeptidase reaction. With respect to transglycosylation, the binding mode of inhibitors has provided a basis for interpreting the occupancy of the growing glycan strand.10 The binding mode for cross-linking by transpeptidase catalysis is much less certain. This shortcoming is particularly vexing since mutational alteration of the transpeptidase active site is a primary mechanism for the development of resistance to β-lactam antibiotics by S. pneumoniae. In the absence of ligand, the crystal structure of PBP 1b displays a “closed” transpeptidase active site, showing a steric impediment toward peptidoglycan entry. In contrast, the structures of PBP 1b inactivated by cephalosporin β-lactams (PDB codes 2UWX and 2UWY) show a more open active site.8,9 The difference is a loop connecting two β-sheets alongside the cleft.9 PBP inactivation by the β-lactams identifies Ser460 as the nucleophile mediating the acyl-transfer reactions, via a presumptive acyl-enzyme intermediate. Serine activation is accomplished (as it is in all PBP transpeptidase/carboxypeptidase active sites) by a proximal lysine.11,12 In order to investigate the events of peptidoglycan binding and cross-linking catalyzed by this Ser460-Lys463 dyad, we evaluated by computation the PBP 1b transpeptidation mechanism. This study involved 991 ONIOM QM/MM MP2/6-31+G(d)//HF/3-21G calculations and 33.6 ns of explicit solvent molecular dynamics (MD) simulations.
Scheme 1.

Summary of the PBP 1b-dependent catalytic reactions used in the synthesis of the S. pneumoniae peptidoglycan.
Methods
Substrate Structure
The PBP 1b transpeptidation reaction was conceptualized as discrete D-alaninyl acyl transfer reactions. Initially, the –D-Ala–D-Ala of one stem is transferred to Ser460 to form an acyl-enzyme, liberating the terminal D-Ala as a leaving group. In the second half-reaction, the D-alaninyl acyl moiety is transferred from the acyl-enzyme to the ε-amino group of the lysine of an adjacent peptidoglycan strand. Following MD identification of the productive conformation of bound substrate, the enzyme-substrate complex was partitioned between QM/MM regions to evaluate the bond-forming and bond-breaking events of catalysis. The peptidoglycan substrate was represented by a NAG-NAM-NAG-NAM tetrasaccharide.12,13 The peptide stem used as the -D-alaninyl acyl donor extends from one of the NAM saccharides into contact with Ser460. Following acyl-enzyme formation, acyl transfer occurs from the Ser460 acyl-enzyme to the stem of a second NAG-NAM-NAG-NAM tetrasaccharide. Identical tetrasaccharide structures were used for the donor and acceptor strands. The peptide stems of this tetrasaccharide are linked to the D-lactyl group of the N-acetylmuramic saccharide, and have a –L-Ala–(γ-D-Glx)–L-Lys–D-Ala–D-Ala pentapeptide structure. Use of a γ-D-Glu in the Glx position rather than the γ-D-Gln structure used in the S. pneumoniae cell wall14,15 simplified the computational preparation of this structure, and is inconsequential with respect to the transpeptidase mechanism. The total charge on the peptide stem is −1 (the sum of the lysine ammonium, the γ-D-Glu a-carboxylate, and the terminal D-Ala α-carboxylate), assuming normal protonation states. As there are two stems on a NAG-NAM-NAG-NAM tetrasaccharide the total charge for the molecule is −2. Coordinates for the substrate were extracted using SYBYL 7.3.16 Three crystal structures of PBP 1b (PDB codes: 2BG1, 1.9 Å resolution; 2UWX, 2.4 Å; 2FFF, 2.2 Å) representing the substrate inaccessible (“closed” loop between the β3 and β4 sheets) and substrate accessible (“open” loop) states of this PBP were examined.9 The 2UWX “open” structure (468 residues) used in this study includes a short N-terminal peptide, the transglycosylase-transpeptidase interdomain (residues 337–396), the transpeptidase (residues 397–709), and the C-terminal regulatory (residues 710–790) domains. Superimposition of the 2UWX and 2FFF structures using an atom-fitting method showed very small differences in the catalytic domain. We used the 2UWX structure due to its nitrocefin-derived acyl-enzyme, used by us to guide substrate positioning. After deletion of the nitrocefin-derived atoms, Lys463 was assigned as a neutral amine and Lys651 was assigned as the protonated amine, based on our PBP 5 study. Subsequent charge assignments, anchor selection, energy minimization, and molecular dynamics procedures were as described previously in our computational study of the E. coli PBP5 carboxypeptidase enzyme.12
Pre-acylation Complex
The non-covalent pre-acylation complex for the acylation reaction was constructed using the Dock 5.4 program.17 A trans-amide conformation was used for the amide of the -D-Ala-D-Ala substrate.12 The ability of the Dock program to use anchor atoms and solid-bonds during initial docking facilitated positioning of the donor peptidoglycan substrate. Docking of the acceptor peptidoglycan substrate to the acyl-enzyme used the single-atom constraint of Gold 3.118 (applied to the Nε of its lysine). The similarity of the PBP 5 and PBP 1b active sites with respect to –D-Ala–D-Ala recognition allowed superimposition of the active sites. Gold provides docking via a single atom constraint, with mobility for the other atoms. Flexibility was allowed by switching “on” intramolecular hydrogen bonds and the carboxylate rotation, and by using torsion distributions based on the Cambridge Structural Database.19 Amide bonds were not allowed to flip during conformational sampling. Chemscore fitting function was used.
Molecular Dynamics
MD simulations were preformed as described previously for PBP 5.12 The Michaelis complex for the initial acyl transfer was neutralized using the Amber Addions program.20 Subsequently, the Amber solvation program generated a solvent box (approximately 88 Å × 114 Å × 92 Å) encompassing the active site. Atomic charges for the substrates were determined by a two-step Resp procedure21 and assigned with in-house scripts and Perl programs. The electrostatic potentials for the NAG-NAM disaccharide, and separately for its pentapeptide stem, were generated using HF/6-31G(d) full optimization.22 Methyl substituents capped the disaccharide and peptide segments during the charge assignments. The charge calculated for the connecting glycosidic oxygen of the disaccharide was assigned to the connecting glycosidic oxygen of the tetrasaccharide.
QM/MM
The enzyme-substrate systems were partitioned into low (MM) and high (QM) layers.22,23 The low layer consisted of 7,319 atoms of the enzyme and the 1,000 water molecules closest to the bound substrate. Their interactions used the MM force fields of the Amber program. The Parm99 and Gaff force-fields determined the atomic harmonic potentials and other force field parameters for the substrate. For formation of the tetrahedral species leading to the Ser460 acyl-enzyme, 38 heavy atoms were included in the QM layer. In the transpeptidation reaction of the acyl-enzyme, 37 heavy atoms were included in the QM layer.
RESULTS
This study commenced with positioning of the NAG-NAM-NAG-NAM donor substrate, in its conformation as seen in NMR analysis,13 into the cavity containing the Ser460-Lys463 dyad. Three anchor atoms (terminal D-Ala α-carbon, amide nitrogen, and the D-Ala carbonyl carbon) and one rigid bond (between the terminal D-Ala α-carbon and its carboxylate carbon) of the pentapeptide stem were selected as the reference points to guide the modeling of the enzyme-donor substrate complex. The computational protocol sampled fifty poses. Each pose was examined with respect to orientation along the cleft and with respect to the distance between the carbon of the D-Ala carbonyl and the Oγ atom of Ser460. A pose showing a productive C–O distance (1.8 Å) for acylation was selected (Figure 1, Panels A–C). In this pose, the pentapeptide stem extends linearly along a cleft leading to Ser460, and positions the D-Glu residue of the stem under the extrusion created by Asn656 of the protein. The α-carboxylate of this Glu is solvent exposed, and shows one hydrogen bond to water and one to the Ser637 on the surface of the catalytic domain. Transformation of this carboxylate (from the NMR structure) to a carboxamide, to give the γ-isoGln sub-structure of the S. pneumoniae peptidoglycan, would preserve this hydrogen bond interaction. The angle of the backbone of the peptide stem entering the active site is nearly perpendicular (approximately 110 ± 10°) to the NAM saccharide, as is also seen in the solution structure of the peptidoglycan.13 The NAG-NAM-NAG-NAM has two stems (on each of the NAM saccharides). The D-Ala terminus of the second peptide stem neighbors Lys367 of the transglycosylase domain (Figure 1, Panel B and red dot in Panel C). Lys367 is 20 Å distant from the transpeptidase active site. The stem cleft of the transglycosylase domain is closed.
Figure 1.
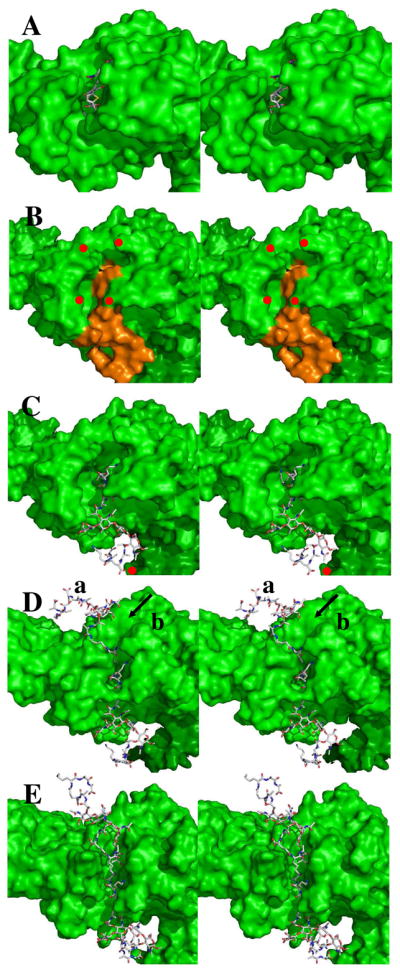
Panel A shows the stereo structure (2UWX) of acylated PBP 1b (depicted as a Connolley surface) with the nitrocefin-derived acyl-enzyme (shown in capped-stick: C, gray; N, blue; O, red; H, cyan; S, yellow). Panel B shows the docked Michaelis complex of the NAG-NAM-NAG-NAM substrate (orange) in the PBP 1b structure (green surface, showing red dots to identify residues Asn500 as the top left red dot; Gln686, top right; Asn494, bottom left; and Asn656, bottom right); Panel C is identical to Panel B with the exception that the docked substrate is in capped-stick representation. The angle of the pentapeptide stems relative to the glycan strand is 110° (± 10°). Panel D shows the probable location (mode a) of the acceptor stem engagement of the acyl-enzyme and an alternative (and less probable) mode b cleft for acceptor stem approach (arrowed). Panel E shows the opened cleft, derived from 10 ns MD simulation of the structure of Panel D. In Panel E the separation between the δNH2 of Asn494 and the δNH2 of Asn656 is 5 Å. The plane of the bacterial membrane to which this enzyme is embedded is located distant to the right edge of these panels.
Conformation of the Michaelis Complex
The energy-minimized Michaelis complex (Fig. 1B) was obtained following an initial 1 ns equilibration. Five structures (extracted at 0.2 ns intervals) served as starting points for five separate 3 ns MD simulations. In the crystal structure of the apo-enzyme9 the neutral ε-amine of Lys463 is hydrogen-bonded (3.2 Å) to the hydroxyl of Ser460. This hydrogen bond was stable (2.6–2.8 Å) throughout the MD simulations. One conformation, chosen on the basis of the orientation of the hydrogen bond (2.7 Å) between the Ser460 hydroxyl and the Lys463 lone pair, was optimized by the two-layer ONIOM method.24 The pentapeptide stem of the resulting Michaelis complex occupies the long cleft between the β3 strand (residues 649 to 654, aligned with the two right-sided red dots of Fig. 1B) and the loop between the a4 (residues 509 to 516) and a5 (residues 518 to 531, aligned with two left-sided red dots). The carbonyl of the scissile amide engages the oxyanion hole defined by the backbone amides of Ser460 (3.2 Å) and Thr654 (2.8 Å) (Figure 2A). The carbon of the carbonyl is 2.2 Å from the Oγ of Ser 460. Additional hydrogen bonds occur between the C-terminal D-Ala carboxylate and Thr652 Nζ (2.6 Å, not shown), the Ser516 Oγ (2.7 Å), and the Lys651 amine (2.7 Å). The optimized length of the Ser460–Lys463 hydrogen bond is 2.6 Å. The NH of the scissile amide is solvent-exposed and engaged in a hydrogen bond (3.0 Å) to a water molecule (Figure 2A).
Figure 2.

Panel A shows a stereo representation of the Michaelis complex. The L-Lys-D-Ala-D-Ala terminus of the stem of the substrate is bound, with a trans-amide conformation for the -D-Ala-D-Ala terminus, into the transpeptidase active site of PBP 1B. Hydrogen bonds are shown as dashed lines (distances are between heteroatoms given in Å, rounded to the nearest tenth). The substrate is nestled between the α3 sheet and the loop connecting the α4 and α5 helices. Panel B shows the QM/MM potential energy surface with respect to the d1 (O–H bond of Ser460) and d2 (distance between the carbonyl carbon and Oγ of Ser460) reaction coordinates. The reaction path from the Michaelis complex (I) to the tetrahedral intermediate (II) is shown with orange arrows. Panel C shows the structure of tetrahedral intermediate II.
Tetrahedral Intermediate
A QM region encompassing 77 atoms (including 26 from the donor substrate) was defined by a 6 Å radius from Oγ of Ser460. The heavy atoms included Cδ, Cε and Nζ of Lys463; Cα, Cβ, Oγ and the N of Ser460; the carbonyl group of Ala459; the Cα and the carbonyl group of Gly653; the backbone NH and Cα atoms of Thr654; the Cβ and Oγ atoms of Ser516; the Cδ, Cε and Nζ of Lys651; the Cβ, Cγ, Oγ and Nδ atoms of Asn518; and the O of the water molecule close to the amide NH group of the substrate. HF/3-21G ONIOM QM/MM geometry optimization initiated the QM/MM calculation of the bond-forming and bond-breaking events leading to the first tetrahedral species. Two reaction coordinates were defined. The first coordinate (d1) is the O–H bond of Ser460, where lengthening of d1 coincides with proton transfer to Lys463. The second reaction coordinate is the distance (d2) between the carbonyl carbon and Oγ of Ser460. Shortening of d2 corresponds to bond formation between the serine oxygen and the carbonyl carbon. A two-dimensional potential energy surface was generated by scanning d1 (at 0.1 Å intervals from 0.9 to 2.0 Å) and d2 (from 1.4 to 2.7 Å), comprising 168 MP2/6-31+G(d) single-point energy calculations. The arrows on the potential energy surface (Figure 2B) indicate progression from the Michaelis complex (Species I at d1 = 1.0 Å, d2 = 2.2 Å) to the tetrahedral (Species II at d1 = 1.8 Å, d2 = 1.5 Å: Figure 2B and 2C). Forward motion along this path is exothermic with species II at −18.1 kcal·mol−1 below the Michaelis complex. The hydrogen bonds of the oxyanion hole shorten (between the substrate O and Ser460 backbone NH by 0.2 Å, between the substrate carbonyl oxygen and the Thr654 backbone NH by 0.1 Å) consistent with increasing negative charge on the O atom.25 Proton transfer to the amine of Lys463 from Oγ of Ser460 results in two bond rotations. The hydrogen of the QM water rotates approximately 110° with loss of the 3.5 Å hydrogen bond to the carbonyl oxygen of the Ser516 backbone, to engage the nitrogen of the D-Ala of the tetrahedral species with a 2.9 Å hydrogen bond. Concomitant with this rotation, a small translational motion (from 3.0 Å to 2.8 Å) of the nitrogen of Lys463 toward the hydroxyl oxygen of Ser516 occurs, as the NH of this nitrogen rotates by approximately 87° to form a 2.7 Å hydrogen bond with the Oβ hydroxyl (not shown) of Thr654.
Formation of the Acyl-enzyme Intermediate
The key event for tetrahedral collapse to the acyl-enzyme is protonation of its nitrogen, enabling this nitrogen to act as a leaving group. The active site residues that are candidates for proton donation are Lys651 (present in the ammonium state in the native protein structure, and also in our Michaelis complex) and Lys463. The possible pathways for proton transfer include direct transfer from Lys651, direct transfer from Lys463, Ser516-mediated proton transfer (from either lysine), or active site water-mediated transfer (also a possibility from either lysine). These possibilities were differentiated using MD trajectories (Figure S1, Supporting Information). A 7.8 ns MD simulation examining the dynamics of Lys651, Ser516 and Lys463 identified two conformations for the Lys463 sidechain. One conformation corresponds to a weak hydrogen bond (2.7–4.2 Å) between the Lys463 ammonium and the nitrogen of the tetrahedral species (Panel A of Figure S2, Supporting Information). The other conformation has two strong hydrogen bonds (Figure S2, Panel B). One is between the nearby water and the scissile bond nitrogen (1.8 Å), and other is between the water and the ammonium group of Lys463 (2.0 Å). These conformations imply two possible paths—with and without participation of a bridging water—for protonation of the tetrahedral species. Starting from the energetically more favorable (the one having the shorter distances) conformation, a QM/MM potential energy surface was constructed for water-mediated protonation of the tetrahedral species II′ (Figure 3A) using the distance between the water hydrogen and the tetrahedral nitrogen (d5), and the distance between the tetrahedral carbon and the nitrogen atom of the scissile bond (d4) as reaction coordinates (Figure 3B). The distance between Lys463 Hη and the water oxygen (d6) was constrained by setting d6 equal to d5, based on previous calculations showing a nearly symmetric energy path for a very similar dual proton transfer in the PBP 5 mechanism.12 Three energy minima appear on the surface (130 points at the MP2/6-31+G(d) level of theory). The first minimum (d4 = 1.5 Å, d5 = d6 = 1.8 Å) is the anionic tetrahedral intermediate (Species II′ of Figure 3A, derived from Species II of Figure 2). The second minimum (d4 = 1.7 Å, d5 = d6 = 1.0 Å) is the zwitterionic tetrahedral Species III (Figure 3C) obtained via synchronous proton transfer to the nitrogen of the tetrahedral species, and acceptance by the water molecule of a proton from Lys463. The barrier for formation of the zwitterionic tetrahedral species from the anionic tetrahedral intermediate is 18.9 kcal·mol−1. The third minimum (d4 = 2.5 Å, d5 = d6 = 1.0 Å) is the acyl-enzyme (Species IV, Figure 3D), obtained by collapse of the zwitterionic tetrahedral species through a small barrier of 5.5 kcal·mol−1. The zwitterionic tetrahedral species is 13.8 kcal·mol−1 above the anionic tetrahedral, and the acyl-enzyme is 5.4 kcal·mol−1 above the zwitterionic tetrahedral species. As d4 lengthens, the terminal D-Ala is expelled as a leaving group, concomitant with formation of the acyl-enzyme. This modeled acyl-enzyme (with the D-Ala leaving group still held in the active site) shows similarity to the crystallographic acyl-enzyme seen for PBP 4a of B. subtilis.26
Figure 3.
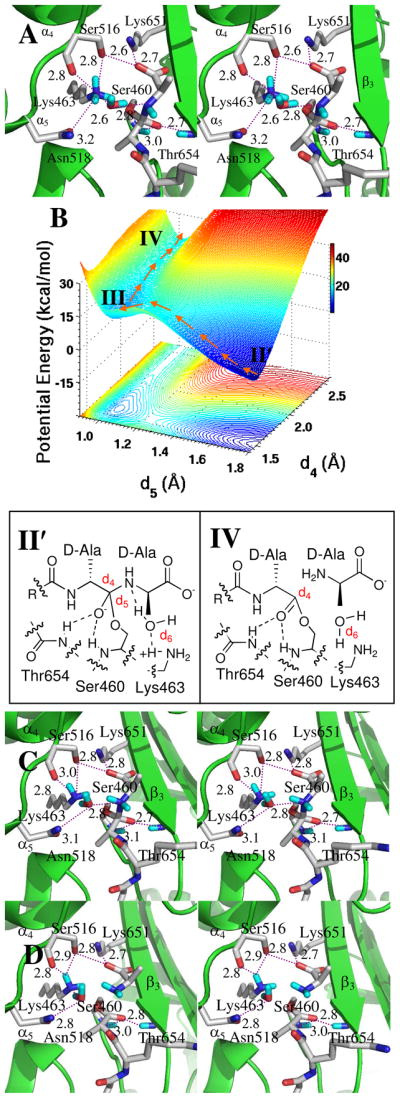
Panel A is a stereoview of tetrahedral intermediate II′, resulting from water-mediated proton transfer from Lys463. Hydrogen bonds are shown as dashed lines (distances between heteroatoms, given as Å). The tetrahedral intermediate is nestled between the β3 sheet and the loop connecting the α4 and α5 helices. Panel B The reaction path from the anionic tetrahedral intermediate II′ through the zwitterionic tetrahedral species III to the acyl-enzyme IV is shown with the orange arrows. Panel C shows the structure of zwitterionic species III. Panel D depicted the structure of acyl-enzyme IV.
In the water-mediated pathway, the PBP 1b acyl-enzyme is formed concurrently with retention of the water molecule hydrogen bonded to the free amine of Lys463. This circumstance, importantly, raises the possibility that the acyl-enzyme might then prove susceptible to hydrolysis, as a competitive event to transpeptidation. For this reason, the location of this water (Figure 3D) was compared carefully to the cognate water of the hydrolytic PBP 5 of E. coli.12 In PBP 5, the water positions between the Lys ammonium nitrogen and the nitrogen of the D-Ala-D-Ala amide. In contrast, the PBP 1b water positions differently between these same two nitrogen atoms. When the D-Ala leaving group is present in the PBP 1b active site, it impedes sterically the approach of this water to the acyl-enzyme. Moreover, water approach to the acyl-enzyme remains improbable even following departure of the D-Ala, as the water is held 3.8 Å from the carbonyl carbon by a strong hydrogen bond (2.8 Å) from the dNH2 of Asn518 (Figure 3D). This hydrogen bond holds the water closer to the protein–solvent interface than to the Lys463–Ser460 catalytic dyad. Further, MD analysis shows periodic escape of this water to solvent. We conclude that the PBP 1b water is not favorably disposed toward hydrolysis of the acyl-enzyme, consistent with the evolution of this enzyme as a transpeptidase (and not as a carboxypeptidase).
Acyl Transfer
Initiation of the second half-reaction requires release of the D-Ala from the active site and the occupancy of its former place—proximal to the acyl-enzyme—by the lysine of the second peptidoglycan strand. This event demands proper positioning of the acceptor peptidoglycan on the PBP 1b surface. A starting pose was identified using the acyl-enzyme of Streptomyces R61 transpeptidase that was inactivated with a cephalosporin with a peptidoglycan stem-like side chain.27 A structure was selected by evaluation of the angle between the glycan and the peptide stem (optimally, approximately 106°) and whether the Nζ nitrogen of the stem L-Lys superimposed with the nitrogen of the ring-opened cephalosporin of the R61 acyl-enzyme. These criteria derive from the solution conformation of peptidoglycan strands and alignment of the lysine side chain from the second stem peptide with the ring-opened cephalosporin, respectively. Three poses met these criteria. The pose having the most hydrogen bonds between the peptide stem and the protein (four hydrogen bonds from the stem and five from the protein, involving residues Lys463, Tyr498, Tyr515, Ser626, Arg627, Thr654 and Gly689 of PBP 1b and with additional hydrogen bonds to solvent) was selected. The distance between the L-Lys Nζ and the carbon of the carbonyl was 2.9 Å. The attack angle for nitrogen to the carbonyl plane was 118°. Moreover, the N–H distance between one of the η-hydrogens of the neutral L-Lys and Nζ of Lys463 is 2.0 Å. The other L-Lys Hη atom orients between the β3-sheet and the α4 C-terminus. This pose was solvated in water, optimized by energy minimization, and evaluated by 2 ns MD simulation. Harmonic restraints on the distances between the L-Lys Nζ and the carbon of the carbonyl, and between the L-Lys Hη and Lys463 Nζ, used a force constant of 20 kcal·mol−1·Å−2. An optimal conformer was selected (from among 10,000) using the distances and angles relating to L-Lys Nζ as the selection criteria. The selected conformation was subjected to a 150,000-step energy minimization. Further QM/MM HF/3-21G optimization (including 1,000 water molecules) yielded an energy-minimized conformation (Figure 4A). In this complex the distance from Nζ of the L-Lys to the carbon of the carbonyl was 3.2 Å and the distance from this same nitrogen to H of the Lys463 Nζ was 3.6 Å. The attack angle of Nζ with respect to the carbonyl plane is 81°. Starting with this energy-minimized structure, amine addition to the carbonyl was evaluated by QM/MM. The resulting potential energy surface (483 MP2/6-31+G(d) points), constructed using the distance between the L-Lys Nζ nitrogen atom and the carbonyl carbon (d7) and the distance between the L-Lys463 Nζ atom and the L-Lys Hη atom (d8) as coordinates, has three minima (Figure 4B). The acyl-enzyme·peptidoglycan complex is minimum V (d7 = 3.2 Å and d8 = 2.6 Å). Minimum VI is the tetrahedral species (d7 = 1.6 Å and d8 = 2.2 Å, Figure 4C). The transition point between acyl-enzyme V and VI (at d7 = 2.3 Å and d8 = 2.3 Å) is 5.4 kcal·mol−1 higher in potential energy relative to the acyl-enzyme. Collapse of VI to product involves transfer of a proton from the nitrogen of the tetrahedral species to the serine oxygen. We observed that as the proton moved toward the Lys463 amine, it was engaged by a bifurcated hydrogen bond involving the tetrahedral nitrogen and the ester oxygen. This transition species VII is located at d7 = 1.5 Å and d8 = 1.5 Å, and is 12.4 kcal·mol−1 higher in potential energy than tetrahedral species VI. Lys463 is necessary for this proton transfer, as it facilitates movement of the proton away from the nitrogen, and thus defines the potential energy surface leading to protonation of the leaving group oxygen. The product complex VIII (Figure 5) is more stable than acyl-enzyme species V by 3.4 kcal·mol−1.
Figure 4.

Panel A shows a stereo representation of the transpeptidation reaction from the acyl-enzyme. The acyl-enzyme is nestled between β3 sheet and the loop connecting α4 and α5 helices. The acyl-enzyme and important active site residues are represented in capped-stick. Hydrogen bonds are shown as dashed lines (distances between heteroatoms in Å, rounded to the nearest tenth). Panel B shows the QM/MM potential energy surface, with the contour over the reaction coordinates represented as a shadow. The reaction path from the acyl-enzyme (V) to the zwitterionic species (VI), and from VI through transition species (VII) to product (VIII), is shown by orange arrows. The location of transition species VII on the energy surface is very close to that of VIII. Panel C shows the conformation of the zwitterionic species (VI).
Figure 5.

Stereo representation of cis-amide product VIII bound in the transpeptidase active site. Hydrogen bonds are shown as dashed lines (distances in Å between heteroatoms, rounded to the nearest tenth). Important active site residues and VIII (Figure 4B) are in capped-stick representation.
Closer examination of the potential energy surface around VIII revealed an alternative pathway for formation of VIII from zwitterionic tetrahedral VI. This second pathway is stepwise transfer of the proton from the nitrogen of VI to the oxygen of Ser460, using the amine nitrogen of Lys463 as a relay. Lys463 accepts the proton from VI to form a transient species IX (Figure S2, Panel A of the Supporting Information), while maintaining its hydrogen bond to the Ser460 oxygen (2.7 Å). The pertinent distances describing the collapse of IX are d10 (distance between Hη of the protonated Lys463 Lys463 and Oγ of Ser460) and d11 (distance between the same Oγ atom and the tetrahedral carbon). The surface (210 MP2/6-31+G(d)//HF/3-21G calculations) shows two minima (Figure S2 Panel B; Supporting Information). The first minimum is tetrahedral species IX (d10 = 1.8 Å, d11 = 1.6 Å). Full transfer of the proton from Lys463 to the ester oxygen, concurrent with tetrahedral collapse (IX to VIII′), ensues through a very small barrier. The designation of this product complex as VIII′ distinguishes it from VIII, and reflects subtle different active site conformations especially with respect to the Lys463 side. The potential energy difference between VIII and VIII′ is inconsequential. A hydrogen bond from Lys463 to the leaving oxygen initiates the formation of VIII′ (Figure S2, Panel B; Supporting Information). In VIII′ (and also in VIII) the four atoms of the amide cross-link (Hη and Nζ from the residue L-Lys in acceptor stem, and the C and O of the carbonyl in the donor stem) reside on the same side of the N–C bond. The dihedral angle (H–N–C=O) defined by these four atoms is 7° (that is, nearly 0°). Therefore, the conformation of the amide formed by acyl transfer from the acyl-enzyme to the lysine is a cis-amide (Figure 5 and Figure S2 Panel C, Supporting Information). Collapse of the tetrahedral species unites the two peptidoglycan peptide stems forming a cis amide bond, while regenerating Ser460 for further turnover.
DISCUSSION
Three recurring structural motifs characterize the active sites of the PBP (and the related serine β-lactamase) families. In PBP 1b, these three motifs have STTK, SWN, and KTG sequences. The SWN and KTG motifs contribute to the width and linearity of the cleft that accommodates the peptide stems. Ser516 of the SWN motif and Lys651–Thr652 of the KTG motif directly engage the carboxylate of the D-Ala at the terminus of the pentapeptide stem of the acyl-donor. Ser460 and Lys463 of the STTK motif are the key catalytic residues in acyl-enzyme creation and transfer. As revealed in this study, the roles for these two residues are essentially identical to the roles of the cognate serine-lysine dyad in our computational study of the E. coli PBP 5 carboxypeptidase.12 In this simple dyad, Lys463 activates the γOH of Ser460 for nucleophilic addition to the amide bond of the D-Ala-D-Ala terminus of the donor stem. The protonated lysine, formed from creation of the tetrahedral species, becomes the catalytic acid enabling tetrahedral collapse to the acyl-enzyme. As with PBP 5, the dominant pathway for proton transfer in tetrahedral collapse involves a water molecule. Although PBP 1b has not been shown to have carboxypeptidase activity, nor E. coli PBP 5 transpeptidase activity (some are PBPs capable of both activities)28–31 the very different positioning of the catalytic water in the PBP 1b active site imparts hydrolytic stability to the PBP 1b acyl-enzyme. A comparison of the structure of the computational PBP 1b acyl-enzyme with four crystallographic PBP acyl-enzyme structures shows strong spatial coincidence both for the acyl-enzyme and surrounding residues (Figure S3, Supporting Information).
The transpeptidase reaction catalyzed by PBP 1b occurs via the two half reactions of acyl-enzyme synthesis and transfer, both evaluated herein. The potential energy progression along the entire reaction path is given in Figure 6, as collected from each potential energy surface. The potential energies are normalized to a value of zero for the Michaelis complex, and the two half-reactions are unified by assigning identical energies to the acyl-enzyme·D-Ala and acyl-enzyme·acceptor peptidoglycan complexes. Key features of the unified reaction coordinate are emphasized. Formation of the first tetrahedral intermediate, by addition of the hydroxyl group of Ser460 to the D-Ala-D-Ala amide, is exothermic by 18 kcal·mol−1. An activation barrier—almost equivalent to this 18 kcal·mol−1 energy—transforms this tetrahedral species to the acyl-enzyme (Figure 6A). The potential energy change for the first acylation half-reaction is +1 kcal·mol−1 with respect to the Michaelis complex (Figure 6A, I to IV). Acyl-transfer follows displacement of the D-Ala leaving group by the stem of the peptidoglycan acyl-acceptor. The overall energetics for the two possible pathways identified by QM/MM for this acyl-transfer are very similar, especially comparing the energy barrier separating VII from VI (Figure 6B and 6C). For both, the barrier for collapse of the second tetrahedral species (V to VII) is 12 kcal·mol−1. This barrier appears to be the rate-limiting chemical step in turnover. The estimated potential energy change in the second half-reaction (V to VIII/VIII′) is exothermic by 2.5 kcal·mol−1. Due to the two energy normalizations and the absence of consideration of entropy, the overall free energy change from the starting Michaelis complex to the product complex is uncertain. Nonetheless, the potential energy barriers for all chemical events are relatively small, and the full profile credibly suggests a thermoneutral potential energy value for transpeptidation. Non-bonding diffusional events, such as substrate binding and product release, likely contribute significantly as rate-limiting events during turnover.
Figure 6.

Potential energy profiles for the acylation (A) and deacylation (B) reactions, where I denotes the complex of the donor peptidoglycan bound into the transpeptidase active site; II and II′, the tetrahedral intermediate (with Lys463 protonated); III, the zwittonionic tetrahedral species (Lys 463 is neutral); IV, the acyl-enzyme·D-Ala complex; V, the complex of the acyl-enzyme with the acceptor peptidoglycan; VI, the anionic tetrahedral species of acyl transfer; VII, the four-atom tetrahedral; VIII and VIII′, the cross-linked product complex; IX, the protonated Lys463 tetrahedral species. The horizontal axis is scaled approximately to the distance changes between the successive energy minima. Entropic factors are not taken into account.
The most interesting mechanistic aspect is the formation of the energetically disfavored cis-amide cross-link (Figures S4 and S5, Supporting Information). While there are several examples of the possible involvement of cis-amide bonds in amido hydrolase catalysis, as discussed previously for PBP 5,12 the definitive implication of a cis-amide in the transpeptidation product was unexpected. There are two compelling reasons, apart from the possible thermodynamic advantage in tetrahedral collapse,32 to regard this cis-amide cross-link as advantageous. The first reason is the possible connection of the thermodynamically favorable cis- to trans-amide isomerization33 as a driving force for the transition from the “closed” state for the stem cleft to the “open” state required for product dissociation.8,9 These states correlate with structural mutations that confer PBP resistance to the β-lactam antibiotics.34–37 The second advantage follows from the reasonable presumption that the biosynthetic PBPs must progressively translocate in order to build the cell wall (a corollary being that the nascent cell wall needs to translocate from the active site). By coupling amide isomerization with product release, a significant kinetic barrier is introduced with respect to product inhibition. The value of such a kinetic barrier is underscored by the relatively low activation barriers accompanying an overall nearly thermoneutral series of bond-transforming events.
The relevance of this cis- to trans-isomerization is also evident at an intuitive level. In order to connect as a cis-amide, the two peptide stems must reach toward each other from opposing sides of the PBP 1b protein (Figure 1D). Whereas each stem diffuses independently into the active site from opposing ends, the product of their union cannot diffuse away from the active site because of its enclosed nature. In essence, two polymers—the PBP protein and the cross-linked peptidoglycan—interlock. The thermodynamic imperative of the cis- to trans-isomerization would force significant conformational change to the cross-link, provoking the release of the cross-linked peptidoglycan product from the active site and conformational change of the loop defining the active site cleft back into a closed conformation to generate the apo state (residues Ile496–Asn500 and Asn656–Asp658: see the bottom pair of red dots in Figure 1B).
Cell wall biosynthesis involves coordination by the bifunctional PBP of transglycosylase-catalyzed glycan strand elongation with the subsequent event of transpeptidase-catalyzed cross-linking of neighboring strands. While the structure of the un-cross-linked peptidoglycan strand13 has proven valuable for the understanding of peptidoglycan strand recognition by other peptidoglycan-binding proteins10,38–41 the basis for the coordination of these two key events, as reflected in the final structure of the peptidoglycan polymer, is unknown. This computational study does not address this question. It does, however, posit specific recognition of the peptide segments of the peptidoglycan by defined clefts crossing an entire face of the transpeptidase domain. Given the recent revelation by Sung et al. of the native structure of the biosynthetic PBP 1b of E. coli, we compared the predicted occupancy of the peptidoglycan-binding clefts as made by this study with the predicted orientation of the peptidoglycan strand emerging from the transglycosylase domain.10 Gratifyingly, the cleft occupancies are compatible. The computational excision of the donor peptidoglycan acyl-enzyme·peptidoglycan acceptor complex from S. pneumomiae PBP 1b, and its computational placement into the active site of E. coli PBP 1b, is a nearly effortless computational exercise (Figure 1E). The comparison of the similarity of cleft selection and occupancy with the one point of difference (Figure 1D, closed cleft; Figure 1E, open cleft; Figure S6, Supporting Information) supports our proposed PBP 1b mechanism.42
The computational mechanism seen here for formation of the initial PBP 1b acyl-enzyme is direct Lys463 activation of Ser460. This mechanism is identical to that seen in our previous computational study of the PBP 5 carboxypeptidase,12 and is the simplest of the proposed mechanisms for PBP catalysis of this event that have been presented over time.43,44 The mechanistic identity between PBP 1b and 5 is significant, as PBP 1b shows a more contracted placement of its active site residues (a typical PBP) compared to the more open active site of PBP 5 (atypical PBP).43 A key remaining question is the mechanistic role of the evolutionarily conserved second serine and second lysine of these PBP active sites. On the basis of the structure of a potent boronate inhibitor of the PBP R39 peptidase Pratt et al. offer a carefully reasoned argument suggesting participation of this second serine in a proton relay, interconnecting the general base lysine and the nucleophilic serine, during hydrolytic deacylation by this enzyme.43,44 We do not find computational evidence for such a proton relay. Rather, the role of this second serine-lysine dyad (Ser516, Lys651) in PBP 1b catalysis is a secondary role—not to be interpreted as unimportant—in substrate positioning, active site residue placement, and possible electrostatic stabilization of the tetrahedral species.
There remains one final point of satisfaction. Coincident with the demonstration that the mechanism of action of the β-lactam antibiotics coincided with release from the bacterium of D-alanine,45 Tipper and Strominger46 proposed penicillin as a mimetic of the conformation of the tetrahedral intermediate of the -D-Ala-D-Ala stem terminus. On the basis of this proposal the active site conformation of the -D-Ala-D-Ala terminus was deduced.48,49 Gratifyingly, a comparison of this predicted conformation with the conformation for the -D-Ala-D-Ala terminus rendered by our computational studies, shows strong similarity (Figure S6, Supporting Information). The prescience of the Tipper-Strominger hypothesis is affirmed.
The molecular events deduced from this mechanistic study are a foundational framework for further study on the relationships among PBP mutation, cleft dynamics and β-lactam resistance; on the processive catalytic events involved in the stepwise synthesis of a polymer;10,50 and lastly on structural models for the three-dimensional architecture of the peptidoglycan.51
Supplementary Material
Footnotes
Supporting Information Available: The six figures of the supporting information include the MD sampling of the tetrahedral species (Figure S1); the potential energy surface leading to Species IX (Figure S2); the computational structure of the acyl-enzyme (Figure S3); a comparison of the open and closed cleft conformations of the active site from MD simulation (Figure S4); the structure of the cis-amide-containing cross-linked peptidoglycan (Figure S5); and the conformation of the N-acyl-D-Ala-D-Ala stem terminus in the Michaelis complex compared to the conformation suggested by the Tipper-Strominger hypothesis (Figure S6). This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Spratt BG, Cromie KD. Rev Infect Dis. 1988;10:699–711. doi: 10.1093/clinids/10.4.699. [DOI] [PubMed] [Google Scholar]
- 2.Macheboeuf P, Contreras-Martel C, Job V, Dideberg O, Dessen A. FEMS Microbiol Rev. 2006;30:673–691. doi: 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- 3.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. FEMS Microbiol Rev. 2008;32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 4.Norris V, den Blaauwen T, Doi RH, Harshey RM, Janniere L, Jimenez-Sanchez A, Jin DJ, Levin PA, Mileykovskaya E, Minsky A, Misevic G, Ripoll C, Saier MJ, Skarstad K, Thellier M. Annu Rev Microbiol. 2007;61:309–329. doi: 10.1146/annurev.micro.61.081606.103348. [DOI] [PubMed] [Google Scholar]
- 5.Morlot C, Zapun A, Dideberg O, Vernet T. Mol Microbiol. 2003;50:845–855. doi: 10.1046/j.1365-2958.2003.03767.x. [DOI] [PubMed] [Google Scholar]
- 6.Contreras-Martel C, Job V, Di Guilmi AM, Vernet T, Dideberg O, Dessen A. J Mol Biol. 2006;355:684–696. doi: 10.1016/j.jmb.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 7.Holtje JV. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macheboeuf P, Di Guilmi AM, Job V, Vernet T, Dideberg O, Dessen A. Proc Natl Acad Sci USA. 2005;102:577–582. doi: 10.1073/pnas.0407186102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovering AL, De Castro L, Lim D, Strynadka NC. Protein Sci. 2006;15:1701–1709. doi: 10.1110/ps.062112106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sung MT, Lai YT, Huang CY, Chou LY, Shih HW, Cheng WC, Wong CH, Ma C. Proc Natl Acad Sci USA. 2009;106:8824–8829. doi: 10.1073/pnas.0904030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Shi Q, Meroueh SO, Vakulenko SB, Mobashery S. Biochemistry. 2007;46:10113–10121. doi: 10.1021/bi700777x. [DOI] [PubMed] [Google Scholar]
- 12.Shi Q, Meroueh SO, Fisher JF, Mobashery S. J Am Chem Soc. 2008;130:9293–9303. doi: 10.1021/ja801727k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meroueh SO, Bencze KZ, Hesek D, Lee M, Fisher JF, Stemmler TL, Mobashery S. Proc Natl Acad Sci USA. 2006;103:4404–4409. doi: 10.1073/pnas.0510182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia-Bustos JF, Chait BT, Tomasz A. J Biol Chem. 1987;262:15400–15405. [PubMed] [Google Scholar]
- 15.Severin A, Tomasz A. J Bacteriol. 1996;178:168–174. doi: 10.1128/jb.178.1.168-174.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sybyl 7.3. Tripos Inc; St. Louis, MO: 2004. [Google Scholar]
- 17.Ewing TJ, Kuntz ID. J Comput Chem. 1997;18:1175–1189. [Google Scholar]
- 18.Gold 3.1, The Cambridge Crystallographic Data Center, 12 Union Rd, Cambridge CB2 1EZ UK, 2007.
- 19.Allen FH. Acta Crystallogr, Sect B: Struct Sci. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 20.Case DA, et al. Amber 8. University of California; San Francisco CA: 2004. [Google Scholar]
- 21.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 22.Frisch MJ, et al. Gaussian 03, revision D.01. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 23.Senn HM, Thiel W. Angew Chem Int Ed. 2009;48:1198–1229. doi: 10.1002/anie.200802019. [DOI] [PubMed] [Google Scholar]
- 24.Vreven T, Morokuma K, Farkas O, Schlegel HB, Frisch MJ. J Comput Chem. 2003;24:760–769. doi: 10.1002/jcc.10156. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Kua J, McCammon JA. J Am Chem Soc. 2002;124:10572–10577. doi: 10.1021/ja020243m. [DOI] [PubMed] [Google Scholar]
- 26.Sauvage E, Duez C, Herman R, Kerff F, Petrella S, Anderson JW, Adediran SA, Pratt RF, Frère JM, Charlier P. J Mol Biol. 2007;371:528–539. doi: 10.1016/j.jmb.2007.05.071. [DOI] [PubMed] [Google Scholar]
- 27.Lee W, McDonough MA, Kotra L, Li ZH, Silvaggi NR, Takeda Y, Kelly JA, Mobashery S. Proc Natl Acad Sci USA. 2001;98:1427–1431. doi: 10.1073/pnas.98.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhazi N, Delmarcelle M, Sauvage E, Jacquemotte F, Devriendt K, Tallon V, Ghosez L, Frère JM. Protein Sci. 2005;14:2922–2928. doi: 10.1110/ps.051641005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar I, Pratt RF. Biochemistry. 2005;44:9971–9979. doi: 10.1021/bi050542z. [DOI] [PubMed] [Google Scholar]
- 30.Adediran SA, Kumar I, Pratt RF. Biochemistry. 2006;45:13074–13082. doi: 10.1021/bi061341d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Josephine HR, Charlier P, Davies C, Nicholas RA, Pratt RF. Biochemistry. 2006;45:15873–15883. doi: 10.1021/bi061804f. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Schofield CJ, Wilmouth RC. J Biol Chem. 2006;281:24024–24035. doi: 10.1074/jbc.M600495200. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen K, Iskandar M, Rabenstein DL. J Phys Chem B. 2010;114:3387–3392. doi: 10.1021/jp1000286. [DOI] [PubMed] [Google Scholar]
- 34.Pernot L, Chesnel L, Le Gouellec A, Croize J, Vernet T, Dideberg O, Dessen A. J Biol Chem. 2004;279:16463–16470. doi: 10.1074/jbc.M313492200. [DOI] [PubMed] [Google Scholar]
- 35.Fuda C, Hesek D, Lee M, Morio K, Nowak T, Mobashery S. J Am Chem Soc. 2005;127:2056–2057. doi: 10.1021/ja0434376. [DOI] [PubMed] [Google Scholar]
- 36.Villegas-Estrada A, Lee M, Hesek D, Vakulenko SB, Mobashery S. J Am Chem Soc. 2008;130:9212–9213. doi: 10.1021/ja8029448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lemaire S, Fuda C, Van Bambeke F, Tulkens PM, Mobashery S. J Biol Chem. 2008;283:12769–12776. doi: 10.1074/jbc.M800079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Dorado I, Campillo NE, Monterroso B, Hesek D, Lee M, Paez JA, Garcia P, Martinez-Ripoll M, Garcia JL, Mobashery S, Menendez M, Hermoso JA. J Biol Chem. 2007;282:24990–24999. doi: 10.1074/jbc.M704317200. [DOI] [PubMed] [Google Scholar]
- 39.Cho S, Wang Q, Swaminathan CP, Hesek D, Lee M, Boons GJ, Mobashery S, Mariuzza RA. Proc Natl Acad Sci USA. 2007;104:8761–8766. doi: 10.1073/pnas.0701453104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Y, Zhang W, Shi Q, Hesek D, Lee M, Mobashery S, Shoichet BK. J Am Chem Soc. 2009;131:14345–14354. doi: 10.1021/ja903773f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Dorado I, Gonzalez A, Morales M, Sanles R, Striker W, Vollmer W, Mobashery S, Garcia JL, Martinez-Ripoll M, Garcia P, Hermoso JA. Nat Struct Mol Biol. 2010;17:576–581. doi: 10.1038/nsmb.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The acceptor amino acid in the biosynthesis of the E. coli peptidoglycan is meso-diaminopimelic acid, as compared to L-lysine for the S, pneumonia peptidoglycan. If the stereochemistry of the acceptor amine of the meso-diaminopimelic acid in the active site was homochiral to the departing D-Ala, its engagement of the acyl-enzyme would give (by microscopic reversibility) a trans-amide cross-link. A comparison of the spatial occupancy by the D-Ala nucleofuge (Fig. 3, Panel D) to the acceptor lysine (Fig. 4, Panel C) for S. pneumoniae PBP 1b suggests that the meso-diaminopimelate does not spatially overlay the D-Ala nucleofuge. We thank a reviewer for bringing this possibility to our attention.
- 43.Dzhekieva L, Rocaboy M, Kerff F, Charlier P, Sauvage E, Pratt RF. Biochemistry. 2010;49:6411–6419. doi: 10.1021/bi100757c. [DOI] [PubMed] [Google Scholar]
- 44.Pratt RF, McLeish MJ. Biochemistry. 2010;49:9688–9697. doi: 10.1021/bi1012222. [DOI] [PubMed] [Google Scholar]
- 45.Wise EM, Jr, Park JT. Proc Natl Acad Sci USA. 1965;54:75–81. doi: 10.1073/pnas.54.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tipper DJ, Strominger JL. Proc Natl Acad Sci USA. 1965;54:1133–1141. doi: 10.1073/pnas.54.4.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silvaggi NR, Josephine HR, Kuzin AP, Nagarajan R, Pratt RF, Kelly JA. J Mol Biol. 2005;345:521–533. doi: 10.1016/j.jmb.2004.10.076. [DOI] [PubMed] [Google Scholar]
- 48.Lee B. J Mol Biol. 1971;61:463–469. doi: 10.1016/0022-2836(71)90393-7. [DOI] [PubMed] [Google Scholar]
- 49.Sweet RM. Chemical and Biological Activity: Inferences from X-ray Crystal Structures. In: Flynn EH, editor. Cephalosporins and Penicillins: Chemistry and Biology. Academic Press; New York: 1972. pp. 281–309. [Google Scholar]
- 50.Perlstein DL, Wang TSA, Doud EH, Kahne D, Walker S. J Am Chem Soc. 2010;132:48–49. doi: 10.1021/ja909325m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vollmer W, Seligman SJ. Trends Microbiol. 2010;18:59–66. doi: 10.1016/j.tim.2009.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.