

Abstract
The process of store-operated Ca2+ entry (SOCE), whereby Ca2+ influx across the plasma membrane is activated in response to depletion of intracellular Ca2+ stores in the endoplasmic reticulum (ER), has been under investigation for greater than 25 years; however, only in the past 5 years have we come to understand this mechanism at the molecular level. A surge of recent experimentation indicates that STIM molecules function as Ca2+ sensors within the ER that, upon Ca2+ store depletion, rearrange to sites very near to the plasma membrane. At these plasma membrane-ER junctions, STIM interacts with and activates SOCE channels of the Orai family. The molecular and biophysical data that have led to these findings are discussed in this review, as are several controversies within this rapidly expanding field.
Keywords: store-operated Ca2+ entry, CRAC, calcium influx, calcium channel, STIM1, STIM2, Orai1, Orai2, Orai3, TRPC
Introduction
The calcium ion is arguably the most ubiquitous, and certainly one of the most diverse signalling entities in the cell [1]. Changes in the intracellular Ca2+ concentration regulate a wide variety of cellular processes that run the gamut from the mitotic birth of a new cell to apoptosis. Elevations in intracellular Ca2+ occur when cells tap into two primary sources: the extracellular medium and intracellular stores, most notably the endoplasmic reticulum (ER). In non-excitable cells the most common route of Ca2+ signal generation results from the activation of cell surface receptors, such as those for various growth factors, hormones and neurotransmitters, which leads to the generation of the second messenger inositol 1,4,5-trisphosphate (IP3). IP3 production results in cytoplasmic Ca2+ elevation that can be separated into two distinct phases. In the first phase, Ca2+ is released from the ER via the IP3 receptor (IP3R). In the second phase, the decrease in ER Ca2+ content that results from IP3R activation signals influx of extracellular Ca2+ via plasma membrane Ca2+ channels in a process known as capacitative or store-operated Ca2+ entry (SOCE) [2]. It should be noted that any reduction in ER Ca2+ content, whether the result of IP3R activation or not, can serve as a stimulus of SOCE; this is, in fact, the defining property of the SOCE mechanism.
The existence of SOCE was first postulated in 1986 [3] and experimental evidence for this concept accrued shortly thereafter [4, 5]. Subsequently, a membrane current that underlies SOCE was described; this current is referred to as Ca2+ release-activated Ca2+ current (ICRAC) [6]. While ICRAC is certainly the best-characterized SOCE current, it remains arguable whether ICRAC is the only current that underlies SOCE. This review will focus primarily on the SOCE pathway that is mediated by ICRAC.
Since the discoveries of SOCE and ICRAC, intense research from numerous laboratories has focused on defining the molecular components of the pathway. The last 5 years have witnessed incredible breakthroughs in this endeavour. It is now evident that SOCE requires members of two families of proteins: the stromal interaction molecule (STIM) molecules (STIM1 and STIM2), which function as Ca2+ sensors in the ER, and the Orais (Orai1, Orai2 and Orai3), which function as pore-forming subunits of SOCE channels. These molecules cooperate in an elegant signalling mechanism whereby a decrease in ER Ca2+ store content causes the ER-resident STIM molecules to reposition themselves very near to the plasma membrane where they activate Orai Ca2+ influx channels.
ICRAC
Our understanding of SOCE has been greatly improved by the biophysical characterization of the underlying ICRAC current [6, 7]. ICRAC is notable for its exceptional Ca2+ selectivity and extremely low single channel conductance [2]. Like voltage-gated Ca2+ channels, CRAC channels achieve their high Ca2+ selectivity through electrostatic repulsion; however, unlike the voltage gated Ca2+ channels, CRAC single channel conductance cannot be measured directly. Approximations of the single CRAC channel conductance have been made using a noise analysis approach, and estimates from Lewis’ lab suggest a Ca2+ conductance of 20–30 fS [8, 9] and a Na+ conductance of 0.7 pS [9]. CRAC channels are also both positively and negatively regulated by Ca2+ itself in a complex manner [7, 10–14].
The biophysical characterization of ICRAC by many laboratories has provided a unique fingerprint of the store-operated channels that aided in the correct molecular identification of the pore-forming subunits of ICRAC, now known as the Orai family of proteins, of which there are three mammalian homologues (Orai1, Orai2 and Orai3). The molecular structures of the Orai proteins are very different from other known Ca2+ channels, which was predictable based on the unique biophysical properties of ICRAC.
Orais
Utilizing gene mapping of a family with a severe combined immunodeficiency (SCID) attributed to a loss of ICRAC, as well as a whole genome RNAi screen in Drosophila S2 cells, Feske et al. discovered a unique family of genes (human: FLJ14466 (Orai1), C7orf19 (Orai2) and MGC13024 (Orai3); drosophila: olf186-F (d-Orai)). The Drosophila gene was absolutely required for ICRAC, and they called the protein encoded by this gene Orai (keepers of the gates of heaven [15]). Shortly thereafter, two other independent laboratories also reported similar results using RNAi in drosophila S2 cells, in which olf186-F (called CRACM1 by Vig et al.) was shown to be essential for ICRAC[16, 17]. In the SCID patients, it was determined that the underlying defect was a single missense mutation in Orai1 (R91W) [15], which increases the hydrophobicity at the transition between the N terminal cytoplasmic portion and the first transmembrane region [18]. This mutation results in a loss of function, but does not affect interactions with STIM1.
The exogenous expression of Orai1 (and Orai2 or Orai3) alone generally causes a suppression of SOCE and ICRAC, presumably due to inappropriate stoichiometry with endogenous STIM1 [19, 20]. However, the co-expression of STIM1 and Orai1 generates very large CRAC currents that are virtually indistinguishable biophysically from native ICRAC[17, 19, 21, 22]. Specifically, these CRAC-like currents are store operated, are highly Ca2+ selective, show inwardly rectifying I-V relationships, have low permeability to Cs+, have undetectable single channel events and are inhibited by low concentrations of lanthanides and 2-APB (except for Orai3, see below) [19, 20, 23–27]. The ability to completely recapitulate ICRAC by co-expressing STIM1 with Orai1 suggests that these two proteins are necessary and sufficient for SOCE, or that any other factor that is required exists in great excess within the cell [28, 29].
The Orai proteins are predicted to have four transmembrane domains, with both the N- and C- termini located on the cytosolic side of the plasma membrane [15, 30]. Orai1 has an N glycosylation site located on the extracellular loop between TM3 and TM4; however, mutations that prevent glycosylation apparently have no deleterious effects on SOCE [31]. That Orai1 is a pore-forming subunit of CRAC channels was demonstrated by several laboratories by mutating specific conserved acidic residues near the beginning of the first extracellular loop. Mutating a glutamate at position 106 (which is conserved in Orai 1, 2 and 3) to alanine results in complete loss of ICRAC, while the conservative mutation to aspartate (E106D) results in a current that is much less Ca2+ selective [30, 32, 33]. Three additional acidic residues located in the extracellular loop between TM1 and TM2 (D110, D112 and D114) in Orai1 have also been shown to influence Ca2+ selectivity of CRAC channels as well as sensitivity to the CRAC channel blocker Gd3+[30, 33, 34]. Increased monovalent permeation seen in the combined mutation of these three residues in Orai1 suggests that they play an important role in the formation of the channel selectivity filter. Unlike E106, these residues differ among Orai1, Orai2 (E84, Q86 and Q88) and Orai3 (E85, D87 and E89).
Recent studies have begun to map the Orai1 domains critical for its activation. The C-termini of all three mammalian Orai homologues contain putative coiled-coil domains. In Orai1 this region has been shown to be critical for the re-arrangement and colocalization of Orai1 with STIM1 in response to ER Ca2+ store depletion [25, 35]. A conserved region of the N terminus just prior to the first transmembrane domain is also essential for SOCE [35], and the N terminus also might play a role in regulating CRAC channel gating properties [36]. For example, Orai2 chimeras containing the N terminal proline/arginine region unique to Orai1 show increased Ca2+ entry compared to wild-type Orai2 [37]. Recent studies identified a minimal domain of STIM1 required to activate CRAC channels (named SOAR for STIM1-Orai activating region). Interestingly, while full length STIM1 could not effectively activate an N terminal truncation mutant of Orai1 (in this case Orai1Δ1-73), the STIM1 SOAR domain could activate the mutated Orai1 [38]. The authors concluded that the Orai1 N terminus interacts with STIM1 in a region that normally inhibits the action of the SOAR domain. Park et al. reported a similar domain structure (which they called CAD [CRAC activation domain]) and concluded that STIM1 functionally interacts not only with the C terminus, but also with the N terminus of Orai1 at the amino acid residues just before TM1 [39]. Interestingly, in a short period of time, four different laboratories all reported minimal Orai-activating domains of STIM1; the amino acid sequences and proposed nomenclature are summarized in Table 1. For the most part, the activating sequences are conserved among STIM1 homologues. While these and other studies have greatly enhanced our understanding of Orai channel activation by STIM1, much more remains to be learned about these unique Ca2+ channels.
Table 1.
Reported Orai-activating domains of STIM1
Orai1 proteins are thought to require multimerization in order to form CRAC channels. Biochemical studies have revealed that Orai1 proteins are capable of forming higher-order multimeric structures [31, 32]. Several recent studies have concluded that the functional CRAC channel pore is made up of Orai1 tetramers [17, 40, 41]; however, it is not clear if these tetramers are constitutively formed, or if STIM1 brings the subunits together. While Penna et al. suggest that functional CRAC channels consist of tetramers formed by STIM1-evoked dimerization of Orai dimers [42], negative stain electron microscopy data do not support this conclusion [39]. Further potential complexity arises from the abilities of different Orai subunits to form hetero-multimeric channels. Interestingly, Orai2 and Orai3 also form store-operated Ca2+ channels with similar, but not identical biophysical and pharmacological properties to Orai1 (see below). Overexpressed Orai2 and Orai3 have also been shown to co-immunoprecipitate with overexpressed Orai1, and the pore dead mutant of Orai1 has been shown to significantly reduce ICRAC in human embryonic kidney (HEK)293 cells expressing STIM1 with either Orai2 or Orai3, suggesting the different Orais might functionally interact [23, 31].
Perhaps the strongest evidence to date suggesting Orai2 or Orai3 form functional CRAC channels comes from studies in Orai1 (in this case called CRACM1) knockout mice that were created using gene trap technology [43]. Ca2+ imaging experiments on mast cells isolated from these Orai1 deficient mice revealed a residual Ca2+ entry pathway that was activated by the addition of thapsigargin (TG) to deplete ER Ca2+ stores. Importantly, the currents remaining in these Orai1 knockout mast cells were ICRAC-like, with strong inward rectification. Quantitative RT-PCR suggested that the residual CRAC-like currents in these cells might be mediated by Orai2 and/or Orai3. Incomplete reduction of SOCE and ICRAC was also observed in T and B cells isolated from Orai1 knockout mice created by the Rao lab (Boston, MA, USA), with a similar conclusion that Orai2 and/or Orai3 may be responsible for the residual SOCE activity [44].
In vitro studies on overexpressed proteins have shown that Orai2 and Orai3 (as well as Orai2 splice variants Orai2L and Orai2S) can also form CRAC channels that require the depletion of internal Ca2+ stores in order to open [19, 20, 23, 24, 45]. Like Orai1, these channels are also highly Ca2+ selective, with a strongly inwardly rectifying current–voltage relationship. Further, the Ca2+ concentrations required to half-maximally block Na+ conductances of Orai2 and Orai3 are similar to that for Orai1, and none of the Orais permeates Cs+ well when expressed as homomeric channels [23–25, 30, 34]. The current densities of the Orai2 and Orai3 Ca2+ currents are several times smaller than the Orai1 CRAC currents in these overexpression assays. This difference in current size is presumably a consequence of expression levels, and also possibly single channel properties. While Orai3-mediated Ca2+ currents are significantly smaller than Orai1 Ca2+ currents, the Na+ currents from Orai3 are much larger in magnitude than Orai1 Na+ currents [23, 24]. It was this difference in Na+ permeation that initially facilitated recording of Orai3-mediated SOC currents, despite being unable to record Ca2+ currents [24]. Orai1, Orai2 and Orai3 apparently show differences in Ca2+ dependent regulatory processes, including fast and slow inactivation [23, 24]. The Orai homologues also differ in their responses to the pharmacological agent 2-APB. While both Orai1- and Orai2-evoked SOCE and CRAC currents are inhibited by 2-APB (albeit Orai2 appears to be somewhat less sensitive to 2-APB), Orai3 is directly activated by the compound [20, 26, 46, 47]. Further, 2-APB-activated Orai3 currents are less Ca2+ selective than ICRAC, showing increased outward Cs+ currents. Whether or not Orai2 and/or Orai3 form endogenous CRAC channels themselves, or in some hetero-multimeric structure with Orai1, is yet to be determined. However, the differences seen with 2-APB on Orais might provide clues to the functions of different Orais in different cells types, and may shed light on the question of whether or not they function as homomeric or heteromeric channel subunits. Our understanding of Orai2 and Orai3 will also greatly be enhanced by the genetic engineering of knockout mice.
STIM1
STIM1 was first cloned in 1996 [48], but it was not until 2005 that its role in SOCE was first realized. A limited RNAi screen of Drosophila S2 cells identified Drosophila STIM as having an essential role in SOCE activation [49], and a similar conclusion was reached almost simultaneously for human STIM1 from a human RNAi screen [50]. Numerous studies since have confirmed the obligate role of STIM1 in SOCE in a variety of cell systems. Substantial molecular and functional analyses have revealed that STIM1 functions as a Ca2+ sensor in the ER that is responsible for communicating depletion of ER Ca2+ stores to Orai channels in the plasma membrane [51].
STIM1 is predicted to be a single-pass transmembrane protein that can localize both to the plasma membrane [52, 53] and the ER membrane [50, 54]. Early evidence suggested that STIM1 is localized within or translocated to the plasma membrane, and that this plays a role in SOCE regulation [53, 55]. However, most subsequent studies have concluded that only ER-localized STIM1 is required [19, 50, 54]. When localized to the ER membrane, STIM1 is oriented such that its N-terminus resides within the ER lumen and its C-terminus in the cytoplasm. The protein comprises several identifiable functional motifs, including an EF-hand Ca2+ binding domain and a sterile-α motif (SAM) in the luminal N-terminus and a pair of coiled-coil domains, a serine/proline rich region and a poly-basic region in the cytoplasmic C-terminus [56]. The SOAR domain, key to activation of Orai channels, is located within the coiled-coil domains [38, 39, 57, 58]. Localization of STIM1 is critical to its SOCE function: when Ca2+ stores are full STIM1 is localized in tubular structures throughout the ER membrane, but when stores are depleted it moves to discrete punctate structures at sites where the ER is closely apposed to the plasma membrane [50, 54, 59] (Fig. 1). It is this relocalization of STIM1 within the ER network towards the plasma membrane that allows it to directly or indirectly interact with and activate Orai channels [60].
Fig 1.
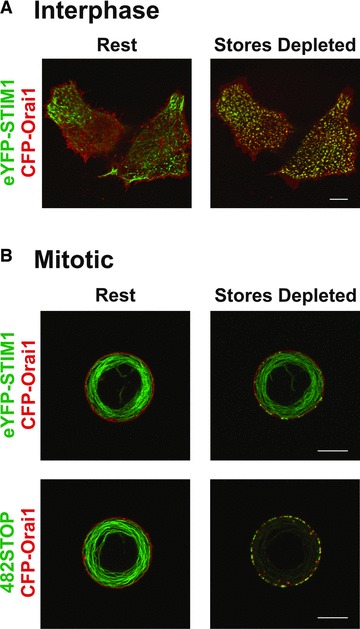
STIM1 and Orai1 colocalize in response to Ca2+ store depletion. (A) eYFP-STIM1 rearranges into near-plasma membrane puncta and colocalizes with CFP-Orai1 in response to Ca2+ store depletion in interphase HEK293 cells. (B) eYFP-STIM1 fails to rearrange into near-plasma membrane puncta in response to Ca2+ store depletion in mitotic HEK293 cells (upper panel). Rearrangement in mitosis is rescued with a STIM1 truncation mutant (482STOP) that lacks mitosis-specific phosphorylation sites (lower panel).
The sequence of molecular reactions and the functional domains of STIM1 that translate changes in ER Ca2+ stores to STIM1 localization and activation of SOCE are becoming clear. Loss of ER Ca2+ results in an initial oligomerization of STIM1 molecules, which is followed by bulk movement of STIM1 oligomers to ER-plasma membrane junctional sites [61]. The most important domain for the Ca2+ sensing function of STIM1 is the EF-hand motif that is localized within the lumen of the ER. The STIM1 EF-hand binds Ca2+ with a Kd of 0.2–0.6 mM [62], well within the range of the physiological ER Ca2+ concentration. The role of Ca2+ binding within the EF-hand was confirmed by experiments demonstrating that mutations of putative Ca2+ binding acidic residues within the domain result in constitutive near plasma membrane STIM1 puncta and constitutive SOCE activity [19, 50, 53, 55]. The EF-hand and nearby SAM domains appear to function in concert in response to changes in ER Ca2+; dissociation of Ca2+ from this region induces a destabilization of the EF-hand/SAM structure, resulting in oligomerization of multiple STIM1 molecules [63]. Elegant experiments from the Lewis Lab further demonstrated that oligomerization via the EF-hand/SAM region is sufficient for STIM1 puncta formation and activation of SOCE [64].
Redistribution of STIM1 oligomers to ER-plasma membrane junctional sites appears to depend largely on functional domains within the cytoplasmic C-terminus of the molecule. Removal of the poly-basic region was shown to prevent STIM1 puncta formation at the plasma membrane [39, 61] without interfering with STIM1 oligomerization in response to Ca2+ store depletion [61]. It was proposed that interaction of these basic residues with plasma membrane phospholipids, particularly PIP2, may account for targeting of STIM1 oligomers to the plasma membrane [61]. However, in a separate report, depletion of plasma membrane PIP2 was found to have no effect on STIM1 redistribution [65, 66], although it is possible that PIP may play a role [66, 67]. More recently it was reported that the poly-basic region is dispensable for redistribution of STIM1 in the context of overexpressed Orai1, suggesting that interaction of STIM1 with Orai1 may contribute to STIM1 puncta formation [39]. However, the question of whether STIM1 directly interacts with Orai channels has been controversial. Several reports have demonstrated FRET between expressed fluorescently tagged STIM1 and Orai1 proteins in response to Ca2+ store depletion [25, 34, 68], and co-immunoprecipitation has also been reported ([16, 33], but see [31]). However, neither of these techniques can unequivocally differentiate between direct or indirect interactions. In light of this, Varnai et al. utilized chemical linkers between the ER and plasma membrane to estimate that Orai1 is part of a molecular complex that protrudes 11–14 nm into the cytoplasm, which is longer than the predicted length of the STIM1 cytoplasmic domain [65]. This result suggests that an intermediary molecule may bridge this gap between STIM1 and Orai1. It has been suggested that iPLA2β may function as such a linker between STIM1 and Orai1 [69, 70] and although a role for iPLA2β in SOCE activation has been substantiated by molecular suppression studies [70], its function as a molecular link between STIM1 and Orai1 has not been proven. It has also been suggested that the primary SOCE-activating function of STIM1 may be to generate a soluble product known as Ca2+ influx factor, which then directly activates Orai1 channels [70]. Full analysis of the physiological role of Ca2+ influx factor in the SOCE activation pathway still awaits full biochemical purification of the compound.
Despite these suggestions that the functional interaction between STIM1 and Orai1 is indirect, the case for direct coupling between these molecules has been significantly bolstered by four nearly simultaneous studies reporting that a minimal sequence of the STIM1 C-terminus that encompasses the putative coiled-coil domain and part of the ERM domain is sufficient to constitutively activate Orai1 (see Table 1 for exact sequences) [38, 39, 57, 58]. This minimal STIM1 domain exhibits a peripheral localization near the plasma membrane only when co-expressed with Orai1 [38, 39, 57, 58], suggesting that Orai1 influences the localization of the minimal STIM1 domain. Store-independent colocalization [38, 57, 58] and FRET [57], as well as co-immunoprecipitation [38, 39, 58] with Orai1 were also demonstrated, suggesting interaction with Orai1. Evidence that this interaction is direct in nature was provided by in vitro biochemical analysis of purified proteins as well as by a split-ubiquitin assay in yeast cells [39]. Interestingly, extension of the minimal STIM1 domain in the C-terminal direction results in a construct that is less effective at activating Orai1-dependent SOCE [57, 58], and in fact direct perfusion of cells with a peptide comprising the 31 amino acids immediately following the minimal sequence (amino acids 339-475) inhibited ICRAC[58]. This suggests that a C-terminal sequence of STIM1 interacts with and inhibits the Orai1-interacting and/or activating domain of STIM1; presumably, Ca2+ dissociation and/or oligomerization of STIM1 relieves this intramolecular interaction, allowing full association with and activation of Orai1 channels. Interestingly, recently three laboratories have reported that a short acidic sequence downstream of the activation domain appears to be involved in Ca2+-dependent inactivation of Orai channels [71–73]. This result was somewhat surprising since it might have been expected that Ca2+ would regulate Orai channels by interacting with the channels themselves.
SOCE is a reversible process that terminates in response to Ca2+ store refilling. Several studies have now revealed that reversal of near-plasma membrane STIM1 puncta in response to store refilling is responsible for termination of SOCE [50, 65, 74]. Reversal of STIM1 puncta by pharmacological agents has also now been demonstrated. In the first such study, the myosin light chain kinase inhibitor ML-9, which was previously shown to inhibit SOCE and ICRAC[75–77] dose-dependently and reversibly inhibited store depletion-induced formation of STIM1 puncta. Furthermore, pre-existing STIM1 puncta, whether formed due to store depletion or due to EF-hand mutations, were also rapidly reversed by the agent. Importantly, it was demonstrated that reversal of STIM1 puncta by ML-9 is responsible for SOCE/ICRAC inhibition by the agent, which represents the first delineation of a molecular mechanism for pharmacological inhibition of SOCE. It should be noted, however, that the direct target of ML-9 responsible for SOCE inhibition appears not to be myosin light chain kinase and remains unknown [74]. A similar reversal of STIM1 puncta was also observed with 2-APB, one of the most commonly employed ICRAC inhibitors [20, 26, 78]. However, in contrast to the case with ML-9, reversal of STIM1 puncta does not appear to be the primary, or at least the only, cause of inhibition of ICRAC[20]. Instead, 2-APB exerts a strong inhibitory effect on the Orai1 channel itself, and this inhibition appears to be independent of STIM1 localization. As discussed previously, the effects of 2-APB on Orai channels are quite complex. Interestingly, in the case of both ML-9 and 2-APB, co-expression of STIM1 with Orai1 significantly attenuates the ability of the drugs to reverse STIM1 puncta [20]. The reason for this is not clear, but could reflect a strong interaction between STIM1 and Orai1 that is accentuated when both molecules are co-expressed in excess.
It was first documented in 1988 [79], and subsequently confirmed [80–82], that SOCE is strongly suppressed during cell division. This represents one of the only known physiological situations in which depletion of Ca2+ stores is dissociated from SOCE activation, and therefore represents an unparalleled system in which to study signalling events that regulate SOCE. Furthermore, the physiological significance of SOCE suppression during mitosis and meiosis is unknown, and delineation of the mechanism by which this suppression occurs will facilitate analysis of broader questions concerning the role of Ca2+ signalling during cell division. A recent study has now demonstrated that during mitosis, STIM1 phosphorylation is up-regulated and rearrangement of STIM1 into near-plasma membrane puncta in response to Ca2+ store depletion is suppressed [83]. In support of the hypothesis that mitosis-specific phosphorylation of STIM1 prevents puncta formation and therefore SOCE activation, expression of STIM1 in which all detectable mitosis-specific phosphorylations were removed by truncation, in combination with Orai1 expression, rescued puncta formation and SOCE in mitotic cells (Fig. 1). These cells in which mitotic SOCE was functional exhibited a slower growth rate and an increased accumulation at the G2/M stage of the cell cycle, consistent with the hypothesis that SOCE negatively impacts mitosis and therefore must be suppressed to ensure the fidelity of cell division [83]. However, it remains possible that phosphorylation may also regulate a novel, mitosis-specific function of STIM1 that is distinct from SOCE. It was also recently demonstrated that suppression of SOCE during meiosis in Xenopus eggs is mediated primarily by Orai1 internalization [84], and decreases in Orai1 protein expression during mitosis have also been noted [83, 85]. Thus, modifications of both STIM1 and Orai1 may contribute to cell cycle-dependent regulation of SOCE. It should also be noted that several mitosis-independent STIM1 phosphorylation sites have been identified [83], suggesting that differential patterns of STIM1 phosphorylation may regulate STIM1 functions in complex ways. Accordingly, it is likely that the C-terminal serine/proline-rich region of STIM1, which contains all STIM1 phosphorylation sites identified to this point, functions as a regulatory domain that modulates STIM1 function according to its state of phosphorylation.
Elucidation of the role of STIM1 in the SOCE signalling pathway has understandably generated a flurry of excitement in the Ca2+ signalling field; however, it is equally exciting to note that STIM1 may be a multi-functional protein with additional roles independent of SOCE. Prior to the realization of its role in SOCE, STIM1 was identified as a surface-expressed protein involved in stromal adhesion of pre-B cells [86]. This function of STIM1 has not been explored further, but interestingly STIM1 was also attributed a tumour-suppressive function, with loss of STIM1 expression associated with several malignancies, most notably embryonic rhabdomyosarcoma [87]. More recently, STIM1 was also shown to be a necessary factor in the activation of the arachidonate-regulated Ca2+ current (IARC), which is similarly activated downstream of PLC-coupled receptors but independently of Ca2+ store depletion. In contrast to ICRAC, IARC appears to depend entirely on STIM1 that is localized in the plasma membrane and does not rely on redistribution of ER-resident STIM1 [88]. Production of cyclic AMP, another second messenger that is generated in response to G protein-coupled receptor stimulation, has also been shown to be regulated by STIM1 in a store-dependent manner, but in this case independently of Ca2+ influx that occurs as a result of activation of SOCE [89]. And finally, several studies that made use of fluorescently tagged STIM1 constructs have noted constitutive, comet-like movements of STIM1, as well as strong colocalization of STIM1 with microtubules [74, 90, 91]. These observations have now been attributed to a novel function whereby STIM1 acts as a link between growing microtubules and the ER and in so doing, regulates microtubule-dependent extension of the ER at the cell periphery [92]. Thus, STIM1 is involved in a diverse array of cellular functions, and it will be interesting to determine the influence and interdependence of each of these functions on one another.
STIM2
STIM2 is the second member of the vertebrate STIM protein family [93]. In human beings, STIM2 is widely expressed in various tissues [93]. However, unlike STIM1, which is localized in both the plasma membrane and the ER membrane, STIM2 has been shown to be exclusively present in the ER membrane [94].
STIM2 has a high amino acid sequence homology and similar domain architecture to STIM1. Like STIM1, STIM2 is a single-pass transmembrane protein with an unpaired N-terminal EF-hand and a SAM domain located in the ER lumen and two C-terminal coiled coil domains, a Pro/Ser-rich region and a Lys-rich region located in the cytoplasm [56, 93]. Nevertheless, the contribution of STIM2 to native SOCE is less obvious than that of STIM1. Liou et al. showed that knockdown of STIM2 partially attenuates SOCE [50], whereas Roos et al. failed to see such an effect [49]. Further, conditional ablation of STIM2 in mice resulted in a small reduction of SOCE in fibroblasts but no effect on CD4+ T cells, whereas STIM1 deficiency led to a complete loss of SOCE in both cell types [95]. In whole-cell patch-clamp recordings, STIM1-deficient T cells also had almost complete loss of ICRAC whereas STIM2 deficient T cells showed no such effect [95]. However, in helper T cells, STIM2 deletion substantially impaired the nuclear transport of NFAT similar to STIM1 deficiency, suggesting an important role of STIM2 in maintaining Ca2+ signalling of T cells.
When STIM1 is overexpressed, SOCE is either modestly increased or unaffected [19, 22, 49]. However, overexpression studies with STIM2 have been less clear. SOCE was dramatically inhibited in HEK293 cells stably overexpressing STIM2 [94] but was slightly increased in cells transiently (9 hrs) overexpressing STIM2 [96]. Our laboratory also observed a modest reduction in SOCE when STIM2 was overexpressed in HEK293 cells (24 to 48 hrs) [97]. Interestingly, unlike STIM1, transient overexpression of STIM2 causes constitutive Ca2+ entry, suggesting a distinct role for STIM2 in resting cells with full Ca2+ stores [22, 96, 98].
Ratiometric Ca2+ imaging and electrophysiological current measurements showed that co-expression of STIM1 and Orai1 recapitulate huge SOCE [19, 21, 22]; however, overexpression of Orai1 in stable STIM2 cells only modestly enhanced SOCE [22]. Furthermore, Parvez et al. suggested that STIM2 plays a dual role in both store-dependent and -independent activation of Orai [99]. These investigators showed that HEK293 cells expressing both STIM2 and Orai1 exhibit a large and transient ICRAC-like current in response to 2-APB even when stores are full. This observation was not seen in cells overexpressing either STIM2 or Orai1 alone, or co-expressing STIM1 and Orai1. Store depletion also generates detectable ICRAC-like currents from cells expressing both STIM2 and Orai1 but only in the absence of the aminoglycoside antibiotic G418, which the authors suggest is a specific inhibitor of STIM2-mediated Orai activation [99].
In an effort to understand the functional roles in SOCE of STIM1 and STIM2, Zheng et al. examined the N terminal Ca2+ binding regions of STIM1 and STIM2 (an EF-hand motif and a SAM domain) by using far UV circular dichroism spectroscopy [100]. These investigators found that both STIM1 and STIM2 EF-SAM exist as monomers in the presence of Ca2+, but that STIM2 EF-SAM forms more stable structures than STIM1 EF-SAM and does not readily aggregate in the absence of Ca2+. Furthermore, unfolding and oligomerization of STIM2 EF-SAM are slower than STIM1 [101]. These differences might play important roles in kinetic differences in redistribution of STIMs into puncta and activation of Ca2+ permeable channels. Indeed, a recent investigation showed that STIM2-mediated Orai1 activation is much slower than that mediated by STIM1 [99]. To address whether STIM2, like STIM1, also redistributes into near-plasma membrane puncta, Soboloff et al. demonstrated that, when overexpressed, STIM2 puncta formation by store depletion is dependent on STIM1 overexpression, suggesting that STIM2 functions as an accessory to STIM1 [94]. However, recent studies utilizing TIRF microscopy demonstrated that overexpressed STIM2 is capable of translocation to ER-PM junctions with endogenous levels of STIM1 following store depletion [98].
Brandman et al. [96] demonstrated that STIM2 is also an ER Ca2+ sensor by showing that the STIM2 EF-hand mutant constitutively forms punctate structures at ER-PM junctions in the absence of store depletion. Interestingly, the same group showed in biochemical assays that STIM2 is partially active even at the resting state and initiates puncta formation rapidly in response to a small degree of Ca2+ depletion [96], consistent with the observation that STIM2 has a lower Ca2+ binding affinity than STIM1 [100]. When the onset of translocation of STIM1 and STIM2 was compared, it was clear that STIM2 began to move almost immediately, while STIM1 began to move after some delay, indicating that because of its higher Ca2+ affinity, a significant threshold of Ca2+ release was needed to reach the concentration range in which STIM1 is activated [96, 98]. However, the lower Ca2+ affinity of STIM2 probably does not explain the slow kinetics of STIM2-mediated Orai1 activation and lower efficiency in activation of Orai1 than STIM1. In an attempt to address this discrepancy, Zhou et al. showed that the short N-terminal domains of STIM proteins localized in the lumen of the ER are responsible for differences in the activation kinetics of Orai1 [102]. These investigators showed by using chimeric replacement that STIM1 with the N-terminal domains of STIM2 slows the kinetics of Icrac activation and suppresses SOCE, whereas STIM2 with the N-terminal domain of STIM1 results in faster Icrac development and increase in SOCE. Taken together, these observations suggest that STIM2 plays a major role in Ca2+ homeostasis by maintaining basal cytosolic and ER Ca2+ levels, rather than being a strong activator of SOCE.
STIM1, STIM2 and Ca2+ oscillations
Much of the work discussed to this point has involved investigation of SOCE by use of experimental conditions that induce maximal or near maximal depletion of Ca2+ stores, for example by use of SERCA inhibitors. However, with physiological stimuli, Ca2+ stores seldom if ever undergo such massive loss of Ca2+. Rather, the [Ca2+]i signals generated with low, physiological levels of receptor agonists most commonly take the form of discrete repetitive discharges of Ca2+, sometimes termed Ca2+ oscillations [103, 104]. These oscillations run down in the absence of extracellular Ca2+, indicating a need for Ca2+ influx to maintain the intracellular stores. Despite some initial debate on the nature of this influx mechanism [105], it now seems clear that in many cell types, it is SOCE that provides this necessary influx [104, 106, 107]. Since the influx associated with Ca2+ oscillations appears to involve very small changes in intracellular Ca2+ store content [106], it would be expected that STIM2 would play the predominant role in signalling SOCE. However, in one study it was clearly shown that the Ca2+ entry supporting muscarinic receptor-driven Ca2+ oscillations depended entirely on STIM1 and not STIM2 [98]. Apparently, STIM2 does not contribute significantly because of its very low efficacy in activating Orai channels. This is consistent with its suggested role in maintaining cellular Ca2+ homeostasis by responding to small changes in store content around the resting level [96]. The authors suggested that STIM1 therefore is specialized for responding to significant changes in ER Ca2+ that are associated with the generation of Ca2+ signals [98]. The threshold for STIM1 is reached in oscillating cells because each oscillation produces a brief drop in ER Ca2+ that is sufficient to activate STIM1. In support of this conclusion, in many cells oscillatory movements of STIM1 towards the plasma membrane were observed slightly following, but in close synchrony with cytoplasmic Ca2+ oscillations [98].
The reason the cell has developed this high threshold mechanism for activation of SOCE becomes clearer when one appreciates the cellular function of the SOCE underlying Ca2+ oscillations. It is clear that SOCE is necessary for maintaining adequate stores that drive the oscillatory Ca2+ release. But more importantly, it is also becoming increasingly clear that Ca2+ entry through plasma membrane channels is capable of providing localized Ca2+ signals that specifically couple to downstream effector pathways [108], and this concept has been shown repeatedly to apply to SOCE [109–115]. Thus, the threshold required for activating STIM1 and SOCE assures that downstream signalling pathways will not be turned on except when bona fide Ca2+ signals are generated, either by high concentrations of agonists that induce substantial Ca2+ store depletion, or by low concentrations that induce spikes of Ca2+ release underlying Ca2+ oscillations. The recently published results of Di Capite et al. [116] demonstrated this concept very well. When mast cells were activated by leukotrienes, Ca2+ oscillations were produced, and this led to activation of the early gene, c-fos. However, by use of the technique of ‘Gd3+ insulation’[106, 117–119] that blocks both Ca2+ influx and efflux, they showed that the global rise in cytoplasmic Ca2+ resulting from each spike or oscillation was not coupled to c-fos activation; rather, influx of Ca2+ through the SOCE (ICRAC) channels was absolutely required.
Previous studies have demonstrated the efficiency of Ca2+ oscillations as signals for downstream effectors [120–123]. However, the experiments have not generally distinguished the relative roles of Ca2+ release and Ca2+ entry through SOCE channels. The more recent findings suggest that in many cases it is the Ca2+ entering through the SOCE channels that is the primary source of signalling Ca2+, and that in such cases the function of the oscillatory discharges of Ca2+ is to produce a sufficiently ample drop in ER Ca2+ levels to activate the signalling Ca2+ sensor, STIM1.
TRPCs and SOCE
Besides the Orai proteins, canonical transient receptor potential (TRPC) proteins have also been suggested to be channels that mediate calcium influx upon store depletion. TRP channels were first identified as cation channels activated in response to photoreceptor stimulation in Drosophila. TRPC channels are the closest mammalian homologues of Drosophila TRP and are activated downstream of phospholipase C activation. Among the seven isoforms of TRPC channels, TRPC1, 3, 4, 5 and 7 have been reported to contribute to SOCE [2]; however, their channel properties are different from those of CRAC channels. While ICRAC is highly selective for Ca2+ ions, endogenously and exogenously expressed TRPC channels are non-selective cation channels. Therefore, it is generally thought that in cases in which TRPCs are activated by store depletion, they form Ca2+-permeable non-selective cation channels distinct from CRAC channels. Following the discovery of STIM and Orai proteins, several groups have begun to address whether TRPCs function in coordination with STIM1 and/or Orai1.
Studies from the Muallem laboratory provided evidence that STIM1 interacts with TRPC1, 2, 4 and 5 but not with TRPC3, 6 and 7 [124]. Subsequently, the same group reported that STIM1 can also regulate TRPC3 and TRPC6 through TRPC1 and TRPC4, respectively [125]. Activation of TRPC1 by STIM1 required the STIM1 C-terminus, which includes the ERM, S/P and polybasic domains [124]. Further studies suggested that gating of TRPC1 is mediated by electrostatic interactions between negatively charged aspartate residues in TRPC1 (639DD640) and positively charged lysine residues in STIM1 (684KK685), at molecular sites distinct from the gating of Orai1 by STIM1 [126]. These results suggested that SOC channels formed by TRPCs, and CRAC channels formed by Orai1, are expressed and function independently on the plasma membrane. However, in our laboratory, we could not find any evidence of TRPC regulation by STIM1 in a variety of cell types [97].
Several studies have also suggested that STIM1, Orai1 and TRPC1 form ternary complexes and that Orai1 knockdown causes suppression of TRPC1-mediated SOCE activity [127, 128]. Further, STIM1 and TRPC1 co-expression resulted in an increase of SOCE compared to TRPC1 expression alone, and this TRPC1-mediated SOCE was sensitive to Gd3+ and 2-APB at the same concentrations as CRAC channels. It should be noted, however, that the current–voltage relationship of the TRPC1-mediated SOCE current was different from that of ICRAC. Therefore, endogenously expressed Orai1 may be involved not only in CRAC channels but also in TRPC1 channel complexes [127]. More recently, Kim et al. showed that simultaneous knockdown of Orai1 and TRPC1 abolished 90% of native SOCE in HEK293 cells, and SOCE was not restored by expression of Orai1 or TRPC1 alone [129]. This result supports the hypothesis that the formation of functional SOCE channels requires both TRPC1 and Orai1. Interestingly, this mutual requirement of Orai1 and TRPC1 is restricted to the TRPC1-mediated SOCE current in HEK293 cells, because endogenous ICRAC was not affected by TRPC1 knockdown in Jurkat T cells. These authors suggest that ICRAC is completely mediated by Orai channels while ISOC is mediated by a TRPC1-Orai1 complex [129]. A problem with these conclusions is that other investigators have investigated the store-operated currents in HEK293 cells, and concluded that the current was identical to ICRAC and essentially completely eliminated by knockdown of Orai1 alone [16, 24].
Birnbaumer’s group has also produced results that support functional and physical coupling between Orais and TRPCs, by demonstrating that expression of exogenous Orai1 increased SOCE and ICRAC in a TRPC-dependent manner [130–132]. Furthermore, Orai1-overexpression in TRPC stable HEK293 cells resulted in an increase of receptor-operated Ca2+ entry which is thought to be predominantly mediated by TRPCs. Interestingly, direct activation of TRPC3 by 1-oleoyl-2-acetyl-sn-glycerol (OAG) was also inhibited by the expression of the SCID mutant form of Orai1 (R180W).
Although several groups have suggested functional interactions between TRPCs, STIM1 and/or Orai1, we have recently reported that TRPC channels function independently of STIM1 and Orai1 [97]. First, co-expression of STIM1 did not induce a significant increase of expressed TRPC activity upon agonist stimulation in HEK293 cells [97], a direct contradiction of the earlier reports from the Muallem lab [124, 125]. Further, in a smooth muscle cell line, endogenous TRPC6 activity was not affected by knockdown of endogenous STIM1. And lastly, although disruption of lipid microdomains by MβCD attenuated OAG-activated TRPC3 activity, it had no effect on TG-induced STIM1 puncta formation or ICRAC development. Based on these results, we concluded that TRPC channels are activated downstream of PLC but not regulated by STIM1.
Numerous studies have reached different and often conflicting conclusions as to the role of TRPCs in SOCE, generally based on somewhat alternative strategies and often different cell backgrounds. Because it is known that TG- or IP3-induced Ca2+ release causes the Ca2+-dependent activation of PLC, careful experimental design is required to discriminate SOCE from Ca2+-activated or PLC-dependent second messenger-activated Ca2+ entry. Furthermore, there are several intriguing observations regarding TRPC1. TRPC1 is known to be expressed on intracellular membrane compartments in addition to the plasma membrane [133, 134]. Knockout of TRPC1 in chicken DT40 cells caused diminished Ca2+ release from internal Ca2+ stores [135]. It has also been reported that TRPC1 knockdown reduced the rate of passive release from stores upon TG treatment [136]. These observations imply the possibility that TRPC1 functions as an intracellular Ca2+ channel [134] and through this function, it might modify SOCE. In fact, the reported STIM1-TRPC1 interaction can be observed even in resting cells and does not change, or increases only modestly, with store depletion or receptor activation [124, 128, 137]. Thus, it will be important to exclude the possibility that TRPC1 and STIM1 form complexes on the ER membrane that regulate luminal ER Ca2+ content, as opposed to a direct role in mediating SOCE.
An additional possibility is that TRPCs might contribute to SOCE in a less direct way, for example if TRPCs were activated in parallel to, or downstream of ICRAC. Despite their clear abilities to be activated by phospholipase C products, TRPCs can also, in some instances, be regulated or activated directly by Ca2+[138–142], or by IP3R [143–146]. An excellent example is the situation in RBL-2H3 cells wherein the standard method for activating ICRAC, whole cell current measurement with IP3 in the pipette, appears to activate both Orai channels as well as TRPC1 [146].
However, it appears that the TRPC1 activation depends on IP3, but not store depletion.
Conclusions
The major goal of this review has been to summarize and highlight recent advances in our knowledge of the molecular mechanisms underlying store-operated or capacitative calcium entry. The phenomenology has been appreciated for well over 20 years, but it was only in the past few years with the development of newer genome-focused strategies that the key molecules and their mechanisms of action and interaction were revealed (Fig. 2). With hindsight this is perhaps not surprising given the rather unique nature of both the signalling mechanism, and channel properties associated with SOCE and ICRAC. Progress is rapidly continuing, even as this review is being written, and we can look forward to soon learning the structural and cell biological characteristics of Orai and STIM molecules – eventually we hope leading to useful information for the treatment of specific diseases.
Fig 2.
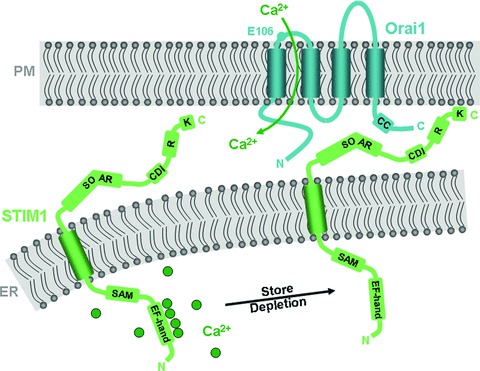
Predicted topologies and functional domains of STIM1 and Orai1 proteins. Shown for STIM1 are the Ca2+-sensing EF-hand domain and the SAM in the ER lumen, and the SOAR, the Ca2+-dependent inactivation (CDI) domain, the regulatory domain that is modified by phosphorylation (R) and the poly-lysine (K) domain in the cytoplasm. As illustrated, STIM1 rearranges within the ER membrane in response to Ca2+ store depletion, allowing it to interact with and activate Orai1 channels in the plasma membrane. Orai1 is predicted to have four transmembrane regions, such that its N- and C-termini are cytoplasmic. Shown are E106, an amino acid within the channel pore that confers Ca2+ selectivity, and a putative coiled-coil in the C-terminus that is required for interaction with and activation by STIM1.
Acknowledgments
Some of the research described in this review was supported by the Intramural Program of the NIH, National Institute of Environmental Health Sciences.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Parekh AB, Putney JW. Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 3.Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 4.Takemura H, Putney JW. Capacitative calcium entry in parotid acinar cells. Biochem J. 1989;258:409–12. doi: 10.1042/bj2580409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takemura H, Hughes AR, Thastrup O, et al. Activation of calcium entry by the tumor promoter, thapsigargin, in parotid acinar cells. Evidence that an intracellular calcium pool, and not an inositol phosphate, regulates calcium fluxes at the plasma membrane. J Biol Chem. 1989;264:12266–71. [PubMed] [Google Scholar]
- 6.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–5. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 7.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–86. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Nat Acad Sci USA. 1993;90:6295–9. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prakriya M, Lewis RS. Regulation of CRAC channel activity by recruitment of silent channels to a high open-probability gating mode. J Gen Physiol. 2006;128:373–86. doi: 10.1085/jgp.200609588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis RS, Dolmetsch RE, Zweifach A. Organellar ion channels and transporters. New York: Rockeffeller University Press; 1996. Positive and negative regulation of depletion-activated calcium channels by calcium; pp. 241–54. [PubMed] [Google Scholar]
- 11.Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–26. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. J Biol Chem. 1995;270:14445–51. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
- 13.Christian EP, Spence KT, Togo JA, et al. Calcium-dependent enhancement of depletion-activated calcium current in Jurkat T lymphocytes. J Membrane Biol. 1996;150:63–71. doi: 10.1007/s002329900030. [DOI] [PubMed] [Google Scholar]
- 14.Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–85. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 16.Vig M, Peinelt C, Beck A, et al. CRACM1 Is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–3. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang SL, Yeromin AV, Zhang XH, et al. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA. 2006;103:9357–62. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Derler I, Fahrner M, Carugo O, et al. Increased hydrophobicity at the N terminus/membrane interface impairs gating of the severe combined immunodeficiency-related ORAI1 mutant. J Biol Chem. 2009;284:15903–15. doi: 10.1074/jbc.M808312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mercer JC, DeHaven WI, Smyth JT, et al. Large store-operated calcium-selected currents due to co-expression of orai1 or orai2 with the intracellular calcium sensor, stim1. J Biol Chem. 2006;281:24979–90. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeHaven WI, Smyth JT, Boyles RR, et al. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem. 2008;283:19265–73. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peinelt C, Vig M, Koomoa DL, et al. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–3. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soboloff J, Spassova MA, Tang XD, et al. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–5. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 23.Lis A, Peinelt C, Beck A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17:794–800. doi: 10.1016/j.cub.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeHaven WI, Smyth JT, Boyles RR, et al. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–56. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
- 25.Muik M, Frischauf I, Derler I, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–22. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- 26.Peinelt C, Lis A, Beck A, et al. 2-APB directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol. 2008;586:3061–73. doi: 10.1113/jphysiol.2008.151365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Inoue R, Shi J, et al. Synergistic actions of diacylglycerol and inositol 1,4,5 trisphosphate for Ca2+-dependent inactivation of TRPC7 channel. Acta Pharmacol Sin. 2008;29:90–7. doi: 10.1111/j.1745-7254.2008.00721.x. [DOI] [PubMed] [Google Scholar]
- 28.Putney JW., Jr New molecular players in capacitative Ca2+ entry. J Cell Sci. 2007;120:1959–65. doi: 10.1242/jcs.03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hewavitharana T, Deng X, Soboloff J, et al. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium. 2007;42:173–82. doi: 10.1016/j.ceca.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Prakriya M, Feske S, Gwack Y, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 31.Gwack Y, Srikanth S, Feske S, et al. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–43. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- 32.Vig M, Beck A, Billingsley JM, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–9. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeromin AV, Zhang SL, Jiang W, et al. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–9. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navarro-Borelly L, Somasundaram A, Yamashita M, et al. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, Lu J, Xu P, et al. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282:29448–56. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- 36.Frischauf I, Muik M, Derler I, et al. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1-3 channels by a STIM1 coiled-coil mutant. J Biol Chem. 2009;284:21696–706. doi: 10.1074/jbc.M109.018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi Y, Murakami M, Watanabe H, et al. Essential role of the N-terminus of murine Orai1 in store-operated Ca2+ entry. Biochem Biophys Res Commun. 2007;356:45–52. doi: 10.1016/j.bbrc.2007.02.107. [DOI] [PubMed] [Google Scholar]
- 38.Yuan JP, Zeng W, Dorwart MR, et al. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–43. doi: 10.1038/ncb1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park CY, Hoover PJ, Mullins FM, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–90. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji W, Xu P, Li Z, et al. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA. 2008;105:13668–73. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586:419–25. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Penna A, Demuro A, Yeromin AV, et al. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–20. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vig M, DeHaven WI, Bird GS, et al. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gwack Y, Srikanth S, Oh-hora M, et al. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–22. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gross SA, Wissenbach U, Philipp SE, et al. Murine ORAI2 splice variants form functional Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem. 2007;282:19375–84. doi: 10.1074/jbc.M701962200. [DOI] [PubMed] [Google Scholar]
- 46.Schindl R, Bergsmann J, Frischauf I, et al. 2-Aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J Biol Chem. 2008;283:20261–7. doi: 10.1074/jbc.M803101200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang SL, Kozak JA, Jiang W, et al. Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem. 2008;283:17662–71. doi: 10.1074/jbc.M801536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parker NJ, Begley CG, Smith PJ, et al. Molecular cloning of a novel human gene (D11S4896E) at chromosomal region 11p15.5. Genomics. 1996;37:253–6. doi: 10.1006/geno.1996.0553. [DOI] [PubMed] [Google Scholar]
- 49.Roos J, DiGregorio PJ, Yeromin AV, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liou J, Kim ML, Heo WD, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol. 2009;11:669–77. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manji SS, Parker NJ, Williams RT, et al. STIM1: a novel phosphoprotein located at the cell surface. Biochim Biophys Acta. 2000;1481:147–55. doi: 10.1016/s0167-4838(00)00105-9. [DOI] [PubMed] [Google Scholar]
- 53.Zhang SL, Yu Y, Roos J, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu MM, Buchanan J, Luik RM, et al. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–3. doi: 10.1083/jcb.200604014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spassova M, Soboloff J, He L-P, et al. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc Nat Acad Sci USA. 2006;103:4040–5. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dziadek MA, Johnstone LS. Biochemical properties and cellular localisation of STIM proteins. Cell Calcium. 2007;42:123–32. doi: 10.1016/j.ceca.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 57.Muik M, Fahrner M, Derler I, et al. A cytosolic homomerization and a modulatory domain within STIM1 C-terminus determine coupling to ORAI1 channels. J Biol Chem. 2009;284:8421–6. doi: 10.1074/jbc.C800229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. doi: 10.1016/j.bbrc.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luik RM, Wu MM, Buchanan J, et al. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815–25. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu MM, Luik RM, Lewis RS. Some assembly required: constructing the elementary units of store-operated Ca2+ entry. Cell Calcium. 2007;42:163–72. doi: 10.1016/j.ceca.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liou J, Fivaz M, Inoue T, et al. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci USA. 2007;104:9301–6. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stathopulos PB, Li GY, Plevin MJ, et al. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: an initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281:35855–62. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 63.Stathopulos PB, Zheng L, Li GY, et al. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135:110–22. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 64.Luik RM, Wang B, Prakriya M, et al. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–42. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varnai P, Toth B, Toth DJ, et al. Visualization and manipulation of plasma membrane-endoplasmic reticulum contact sites indicates the presence of additional molecular components within the STIM1-Orai1 complex. J Biol Chem. 2007;282:29678–90. doi: 10.1074/jbc.M704339200. [DOI] [PubMed] [Google Scholar]
- 66.Korzeniowski MK, Popovic MA, Szentpetery Z, et al. Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J Biol Chem. 2009;284:21027–35. doi: 10.1074/jbc.M109.012252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Broad LM, Braun F-J, LiËvremont J-P, et al. Role of the phospholipase C – inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current (Icrac) and capacitative calcium entry. J Biol Chem. 2001;276:15945–52. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- 68.Barr VA, Bernot KM, Srikanth S, et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: puncta and distal caps. Mol Biol Cell. 2008;19:2802–17. doi: 10.1091/mbc.E08-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gwozdz T, Dutko-Gwozdz J, Zarayskiy V, et al. How strict is the correlation between STIM1 and Orai1 expression, puncta formation, and ICRAC activation. Am J Physiol Cell Physiol. 2008;295:C1133–40. doi: 10.1152/ajpcell.00306.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bolotina VM. Orai, STIM1 and iPLA2beta: a view from a different perspective. J Physiol. 2008;586:3035–42. doi: 10.1113/jphysiol.2008.154997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee KP, Yuan JP, Zeng W, et al. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci USA. 2009;106:14687–92. doi: 10.1073/pnas.0904664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mullins FM, Park CY, Dolmetsch RE, et al. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci USA. 2009;106:15495–500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Derler I, Fahrner M, Muik M, et al. A CRAC modulatory domain (CMD) within STIM1 mediates fast Ca2+-dependent inactivation of ORAI1 channels. J Biol Chem. 2009;284:24933–8. doi: 10.1074/jbc.C109.024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smyth JT, DeHaven WI, Bird GS, et al. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci. 2008;121:762–72. doi: 10.1242/jcs.023903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watanabe H, Takahashi R, Zhang XX, et al. Inhibition of agonist-induced Ca2+ entry in endothelial cells by myosin light-chain kinase inhibitor. Biochem Biophys Res Commun. 1996;225:777–84. doi: 10.1006/bbrc.1996.1250. [DOI] [PubMed] [Google Scholar]
- 76.Norwood N, Moore TM, Dean DA, et al. Store-operated calcium entry and increased endothelial cell permeability. Am J Physiol Lung Cell Mol Physiol. 2000;279:L815–24. doi: 10.1152/ajplung.2000.279.5.L815. [DOI] [PubMed] [Google Scholar]
- 77.Tran QK, Watanabe H, Le HY, et al. Myosin light chain kinase regulates capacitative Ca(2+) entry in human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2001;21:509–15. doi: 10.1161/01.atv.21.4.509. [DOI] [PubMed] [Google Scholar]
- 78.Tamarina NA, Kuznetsov A, Philipson LH. Reversible translocation of EYFP-tagged STIM1 is coupled to calcium influx in insulin secreting beta-cells. Cell Calcium. 2008;44:533–44. doi: 10.1016/j.ceca.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 79.Volpi M, Berlin RD. Intracellular elevations of free calcium induced by activation of histamine H1 receptors in interphase and mitotic HeLa cells: hormone signal transduction is altered during mitosis. J Cell Biol. 1988;107:2533–9. doi: 10.1083/jcb.107.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Preston SF, Sha’afi RI, Berlin RD. Regulation of Ca2+ influx during mitosis: Ca2+ influx and depletion of intracellular Ca2+ stores are coupled in interphase but not mitosis. Cell Regul. 1991;2:915–25. doi: 10.1091/mbc.2.11.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tani D, Monteilh-Zoller MK, Fleig A, et al. Cell cycle-dependent regulation of store-operated I(CRAC) and Mg2+-nucleotide-regulated MagNuM (TRPM7) currents. Cell Calcium. 2007;41:249–60. doi: 10.1016/j.ceca.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Machaca K, Haun S. Store-operated calcium entry inactivates at the germinal vesicle state of Xenopus meiosis. J Biol Chem. 2000;275:38710–5. doi: 10.1074/jbc.M007887200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smyth JT, Petranka JG, Boyles RR, et al. Phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Nat Cell Biol. 2009;11:1465–72. doi: 10.1038/ncb1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu F, Sun L, Machaca K. Orai1 internalization and STIM1 clustering inhibition modulate SOCE inactivation during meiosis. Proc Natl Acad Sci USA. 2009;106:17401–6. doi: 10.1073/pnas.0904651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.El Boustany C, Katsogiannou M, Delcourt P, et al. Differential roles of STIM1, STIM2 and Orai1 in the control of cell proliferation and SOCE amplitude in HEK293 cells. Cell Calcium. 2010;47:350–9. doi: 10.1016/j.ceca.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 86.Oritani K, Kincade PW. Identification of stromal cell products that interact with pre-B cells. J Cell Biol. 1996;134:771–82. doi: 10.1083/jcb.134.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sabbioni S, Barbanti-Brodano G, Croce CM, et al. GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development. Cancer Res. 1997;57:4493–7. [PubMed] [Google Scholar]
- 88.Mignen O, Thompson JL, Shuttleworth TJ. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J Physiol. 2007;579:703–15. doi: 10.1113/jphysiol.2006.122432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lefkimmiatis K, Srikanthan M, Maiellaro I, et al. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11:433–42. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 90.Baba Y, Hayashi K, Fujii Y, et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc Nat Acad Sci USA. 2006;103:16704–9. doi: 10.1073/pnas.0608358103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smyth JT, DeHaven WI, Bird GS, et al. Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J Cell Sci. 2007;120:3762–71. doi: 10.1242/jcs.015735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grigoriev I, Gouveia SM, Van Der Vaart B, et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr Biol. 2008;18:177–82. doi: 10.1016/j.cub.2007.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams RT, Manji SS, Parker NJ, et al. Identification and characterization of the STIM (stromal interaction molecule) gene family: coding for a novel class of transmembrane proteins. Biochem J. 2001;357:673–85. doi: 10.1042/0264-6021:3570673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Soboloff J, Spassova MA, Hewavitharana T, et al. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr Biol. 2006;16:1465–70. doi: 10.1016/j.cub.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 95.Oh-Hora M, Yamashita M, Hogan PG, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–43. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brandman O, Liou J, Park WS, et al. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca(2+) levels. Cell. 2007;131:1327–39. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.DeHaven WI, Jones BF, Petranka JG, et al. TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587:2275–98. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bird GS, Hwang SY, Smyth JT, et al. STIM1 is a calcium sensor specialized for digital signaling. Curr Biol. 2009;19:1724–9. doi: 10.1016/j.cub.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Parvez S, Beck A, Peinelt C, et al. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008;22:752–61. doi: 10.1096/fj.07-9449com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zheng L, Stathopulos PB, Li GY, et al. Biophysical characterization of the EF-hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem Biophys Res Commun. 2008;369:240–6. doi: 10.1016/j.bbrc.2007.12.129. [DOI] [PubMed] [Google Scholar]
- 101.Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284:728–32. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- 102.Zhou Y, Mancarella S, Wang Y, et al. The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of orai1 channels. J Biol Chem. 2009;284:19164–8. doi: 10.1074/jbc.C109.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thomas AP, Bird GStJ, Hajnóczky G, et al. Spatial and temporal aspects of cellular calcium signalling. FASEB J. 1996;10:1505–17. [PubMed] [Google Scholar]
- 104.Putney JW, Bird GS. Cytoplasmic calcium oscillations and store-operated calcium influx. J Physiol. 2008;586:3055–9. doi: 10.1113/jphysiol.2008.153221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shuttleworth TJ. What drives calcium entry during [Ca2+]i oscillations? Challenging the capacitative model. Cell Calcium. 1999;25:237–46. doi: 10.1054/ceca.1999.0022. [DOI] [PubMed] [Google Scholar]
- 106.Wedel B, Boyles RR, Putney JW, et al. Role of the store-operated calcium entry proteins, Stim1 and Orai1, in muscarinic-cholinergic receptor stimulated calcium oscillations in human embryonic kidney cells. J Physiol. 2007;579:679–89. doi: 10.1113/jphysiol.2006.125641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bird GS, Putney JW. Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J Physiol. 2005;562:697–706. doi: 10.1113/jphysiol.2004.077289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2009;59:846–60. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 109.Fagan KA, Mons N, Cooper DMF. Dependence of the Ca2+-inhibitable adenylyl cyclase of C6-2B glioma cells on capacitative Ca2+ entry. J Biol Chem. 1998;273:9297–305. doi: 10.1074/jbc.273.15.9297. [DOI] [PubMed] [Google Scholar]
- 110.Cooper DMF, Yoshimura M, Zhang Y, et al. Capacitative Ca2+ entry regulates Ca2+-sensitive adenylyl cyclases. Biochem J. 1994;297:437–40. doi: 10.1042/bj2970437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Di Capite J, Shirley A, Nelson C, et al. Intercellular Ca2+ wave propagation involving positive feedback between CRAC channels and cysteinyl leukotrienes. FASEB J. 2009;23:894–905. doi: 10.1096/fj.08-118935. [DOI] [PubMed] [Google Scholar]
- 112.Chang WC, Di Capite J, Singaravelu K, et al. Local Ca2+ influx through Ca2+ release-activated Ca2+ (CRAC) channels stimulates production of an intracellular messenger and an intercellular pro-inflammatory signal. J Biol Chem. 2008;283:4622–31. doi: 10.1074/jbc.M705002200. [DOI] [PubMed] [Google Scholar]
- 113.Parekh AB. Local Ca influx through CRAC channels activates temporally and spatially distinct cellular responses. Acta Physiol. 2009;195:29–35. doi: 10.1111/j.1748-1716.2008.01919.x. [DOI] [PubMed] [Google Scholar]
- 114.Ng SW, Di Capite J, Singaravelu K, et al. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+ Influx through Ca2+ release-activated Ca2+ channels. J Biol Chem. 2008;283:31348–55. doi: 10.1074/jbc.M804942200. [DOI] [PubMed] [Google Scholar]
- 115.Parekh AB. Ca2+ microdomains near plasma membrane Ca2+ channels: impact on cell function. J Physiol. 2008;586:3043–54. doi: 10.1113/jphysiol.2008.153460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Di Capite J, Ng SW, Parekh AB. Decoding of cytoplasmic Ca(2+) oscillations through the spatial signature drives gene expression. Curr Biol. 2009;19:385–8. doi: 10.1016/j.cub.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 117.Van Breemen C, Farinas B, Gerba P, et al. Excitation-contraction coupling in rabbit aorta studied by the lanthanum method for measuring cellular calcium influx. Circ Res. 1972;30:44–54. doi: 10.1161/01.res.30.1.44. [DOI] [PubMed] [Google Scholar]
- 118.Kwan CY, Takemura H, Obie JF, et al. Effects of methacholine, thapsigargin and La3+ on plasmalemmal and intracellular Ca2+ transport in lacrimal acinar cells. Am J Physiol. 1990;258:C1006–15. doi: 10.1152/ajpcell.1990.258.6.C1006. [DOI] [PubMed] [Google Scholar]
- 119.Sneyd J, Tsaneva-Atanasova K, Yule DI, et al. Control of calcium oscillations by membrane fluxes. Proc Natl Acad Sci USA. 2004;101:1392–6. doi: 10.1073/pnas.0303472101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dolmetsch RE, Xu KL, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–6. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 121.Li W-H, Llopis J, Whitney M, et al. Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature. 1998;392:936–41. doi: 10.1038/31965. [DOI] [PubMed] [Google Scholar]
- 122.Hu Q, Deshpande S, Irani K, et al. [Ca2+]i oscillation frequency regulates agonist-stimulated NFκB transcriptional activity. J Biol Chem. 1999;274:33995–8. doi: 10.1074/jbc.274.48.33995. [DOI] [PubMed] [Google Scholar]
- 123.Hanley PJ, Musset B, Renigunta V, et al. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc Natl Acad Sci USA. 2004;101:9479–84. doi: 10.1073/pnas.0400733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang GN, Zeng W, Kim JY, et al. STIM1 carboxyl-terminus activates native SOC, Icrac and TRPC1 channels. Nat Cell Biol. 2006;8:1003–10. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 125.Yuan JP, Zeng W, Huang GN, et al. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol. 2007;9:636–45. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zeng W, Yuan JP, Kim MS, et al. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32:439–48. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cheng KT, Liu X, Ong HL, et al. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem. 2008;283:12935–40. doi: 10.1074/jbc.C800008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ong HL, Cheng KT, Liu X, et al. Dynamic assembly of TRPC1/STIM1/Orai1 ternary complex is involved in store operated calcium influx: evidence for similarities in SOC and CRAC channel components. J Biol Chem. 2007;282:9105–16. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim MS, Zeng W, Yuan JP, et al. Native store-operated Ca2+ influx requires the channel function of Orai1 and TRPC1. J Biol Chem. 2009;284:9733–41. doi: 10.1074/jbc.M808097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liao Y, Erxleben C, Yildirim E, et al. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci USA. 2007;104:4682–7. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liao Y, Erxleben C, Abramowitz J, et al. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA. 2008;105:2895–900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Liao Y, Plummer NW, George MD, et al. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci USA. 2009;106:3202–6. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hofmann T, Schaefer M, Schultz G, et al. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Nat Acad Sci USA. 2002;99:7461–6. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Alfonso S, Benito O, Alicia S, et al. Regulation of the cellular localization and function of human transient receptor potential channel 1 by other members of the TRPC family. Cell Calcium. 2008;43:375–87. doi: 10.1016/j.ceca.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 135.Mori Y, Wakamori M, Miyakawa T, et al. Transient receptor potential 1 regulates capacitative Ca2+ entry and Ca2+ release from endoplasmic reticulum in B lymphocytes. J Exp Med. 2002;195:673–81. doi: 10.1084/jem.20011758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cai S, Fatherazi S, Presland RB, et al. Evidence that TRPC1 contributes to calcium-induced differentiation of human keratinocytes. Pflugers Arch. 2006;452:43–52. doi: 10.1007/s00424-005-0001-1. [DOI] [PubMed] [Google Scholar]
- 137.Pani B, Ong HL, Liu X, et al. Lipid rafts determine clustering of STIM1 in endoplasmic reticulum-plasma membrane junctions and regulation of store-operated Ca2+ Entry (SOCE) J Biol Chem. 2008;283:17333–40. doi: 10.1074/jbc.M800107200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zitt C, Obukhov AG, Strübing C, et al. Expression of TRPC3 in Chinese hamster ovary cells results in calcium-activated cation currents not related to store depletion. J Cell Biol. 1997;138:1333–41. doi: 10.1083/jcb.138.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ohki G, Miyoshi T, Murata M, et al. A calcium-activated cation current by an alternatively spliced form of Trp3 in the heart. J Biol Chem. 2000;275:39055–60. doi: 10.1074/jbc.M003606200. [DOI] [PubMed] [Google Scholar]
- 140.Zeng F, Xu SZ, Jackson PK, et al. Human TRPC5 channel activated by a multiplicity of signals in a single cell. J Physiol. 2004;559:739–50. doi: 10.1113/jphysiol.2004.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ordaz B, Tang J, Xiao R, et al. Calmodulin and calcium interplay in the modulation of TRPC5 channel activity. Identification of a novel C-terminal domain for calcium/calmodulin-mediated facilitation. J Biol Chem. 2005;280:30788–96. doi: 10.1074/jbc.M504745200. [DOI] [PubMed] [Google Scholar]
- 142.Shimizu S, Yoshida T, Wakamori M, et al. Ca2+-calmodulin-dependent myosin light chain kinase is essential for activation of TRPC5 channels expressed in HEK293 cells. J Physiol. 2006;570:219–35. doi: 10.1113/jphysiol.2005.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kiselyov K, Xu X, Mozhayeva G, et al. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–82. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- 144.Zhang Z, Tang J, Tikunova S, et al. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Nat Acad Sci USA. 2001;98:3168–73. doi: 10.1073/pnas.051632698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vazquez G, Bird GS, Mori Y, et al. Native TRPC7 channel activation by an inositol trisphosphate receptor-dependent mechanism. J Biol Chem. 2006;281:25250–8. doi: 10.1074/jbc.M604994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zarayskiy V, Monje F, Peter K, et al. Store-operated Orai1 and IP3 receptor-operated TRPC1 channel. Channels. 2007;1:246–52. doi: 10.4161/chan.4835. [DOI] [PubMed] [Google Scholar]