

Abstract
Axon degeneration is a hallmark of neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease. Such degeneration is not a passive event but rather an active process mediated by mechanisms that are distinct from the canonical pathways of programmed cell death that mediate destruction of the cell soma. Little is known of the diverse mechanisms involved, particularly those of retrograde axon degeneration. We have previously observed in living animal models of degeneration in the nigrostriatal projection that a constitutively active form of the kinase, myristoylated Akt (Myr-Akt), demonstrates an ability to suppress programmed cell death and preserve the soma of dopamine neurons. Here, we show in both neurotoxin and physical injury (axotomy) models that Myr-Akt is also able to preserve dopaminergic axons due to suppression of acute retrograde axon degeneration. This cellular phenotype is associated with increased mammalian target of rapamycin (mTor) activity and can be recapitulated by a constitutively active form of the small GTPase Rheb, an upstream activator of mTor. Axon degeneration in these models is accompanied by the occurrence of macroautophagy, which is suppressed by Myr-Akt. Conditional deletion of the essential autophagy mediator Atg7 in adult mice also achieves striking axon protection in these acute models of retrograde degeneration. The protection afforded by both Myr-Akt and Atg7 deletion is robust and lasting, because it is still observed as protection of both axons and dopaminergic striatal innervation weeks after injury. We conclude that acute retrograde axon degeneration is regulated by Akt/Rheb/mTor signaling pathways.
Introduction
The brain pathology observed in the adult-onset neurodegenerative disorders, including Alzheimer's disease and Parkinson's disease (PD), is characterized not only by loss of neurons in select vulnerable brain regions but also by loss of axon projections. Indeed, for both of these disorders some have proposed that axons and their synaptic terminals are the initial locus of the disease process and that it is this pathology, not the loss of neurons, that results in the first manifestations and clinical progression of disease (Hornykiewicz, 1998; Selkoe, 2002; Cheng et al., 2010).
Despite the importance of axon degeneration in these disorders, little is known of the underlying mechanisms. There is an emerging consensus that while the molecular mechanisms of axon degeneration may share some of the components of the canonical pathways of programmed cell death in some contexts (El-Khodor and Burke, 2002; Nikolaev et al., 2009), for the most part these processes are separate and distinct (Finn et al., 2000; Raff et al., 2002). Indeed, in the case of models of degeneration induced in the dopaminergic nigrostriatal system, many experimental approaches targeting the pathways of programmed cell death have succeeded in preserving neuron cell bodies, but not their axon projections (Eberhardt et al., 2000; Chen et al., 2008; Ries et al., 2008). The concept that the molecular mechanisms of axon and cell soma degeneration are distinct has been especially supported by observations in the Wlds mouse, which displays a remarkable resistance to Wallerian anterograde axon degeneration (Lunn et al., 1989). Much of what we do know about the mechanisms of axon degeneration derives from studies of the Wlds mutant protein (for review, see Coleman and Freeman, 2010). However, how this mutant chimeric protein interacts with endogenous signaling pathways remains largely unknown. Additionally, in the nigrostriatal dopaminergic pathway, although Wlds abrogates classic anterograde Wallerian degeneration, it does not prevent retrograde degeneration, the form postulated to occur in PD (Sajadi et al., 2004; Cheng and Burke 2010).
In view of these considerations, we noted with interest the ability of a constitutively active form of the survival-signaling kinase myristoylated Akt (Myr-Akt), which has diverse anti-apoptotic effects, to remarkably preserve the dopaminergic nigrostriatal axon projection in a highly destructive neurotoxin model of PD (Ries et al., 2006). Given not only the extent of axon preservation but also the precise preservation of normal patterns of striatal innervation, we considered that Myr-Akt may provide protection not only of neuron cell bodies (Ries et al., 2006) but also their axons. To examine this possibility directly, we have used a novel confocal imaging approach to visualize dopaminergic axons in the medial forebrain bundle (MFB) in mice expressing green fluorescent protein (GFP) under the tyrosine hydroxylase (TH) promoter. We have used this approach to monitor pathologic changes and quantify axons during the acute injury period in both a neurotoxin model and an axotomy model of induced retrograde axon degeneration in the dopaminergic nigrostriatal system.
Materials and Methods
Animal models of nigrostriatal axon injury.
For these studies, four models of nigrostriatal axon injury were used (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Adult (8 week) male C57BL/6 mice were obtained from Charles River Laboratories. The neurotoxin 6-hydroxydopamine (6OHDA) was used to induce retrograde axonal degeneration by injection into the striatum or anterograde degeneration by injection into the MFB just anterior to the substantia nigra (SN). Axotomy of the MFB was performed either distal to the SN near the striatal target to induce retrograde degeneration or proximal to the SN to induce anterograde degeneration. The intrastriatal 6OHDA model was induced as described previously (Silva et al., 2005). Briefly, a solution of 6OHDA was injected by microliter syringe at a rate of 0.5 μl/min by pump for a total dose of 15.0 μg/3 μl. Injection was performed into the left striatum at the following coordinates: anteroposterior (AP), +0.09 cm; mediolateral (ML), +0.22 cm; dorsoventral (DV), −0.25 cm relative to bregma. After a wait of 2 min, the needle was slowly withdrawn. For lesion of the MFB, 6OHDA was infused at a rate of 0.2 μl/min for 5 min (total dose, 5 μg/1 μl). Axotomy proximal to the SN was performed as described previously (El-Khodor and Burke, 2002) by use of a retractable wire knife (Kopf Instruments) at the following coordinates: AP, −0.10 cm; ML, +0.20 cm. Axotomy distal to the SN was performed in a similar fashion at the following coordinates: AP, −0.030 cm; ML, +0.20 cm. All surgical procedures were approved by the Columbia University Animal Care and Use Committee (New York, NY).
Intranigral injection of adeno-associated virus vectors.
Mice were anesthetized with ketamine/xylazine solution and placed in a stereotaxic frame (Kopf Instruments) with a mouse adapter. The tip of a 5.0 μl syringe needle (26S) was inserted to the following stereotaxic coordinates: AP, −0.35 cm; ML, +0.11 cm; DV: −0.37 cm, relative to bregma. These coordinates place the needle tip dorsal to the posterior SN. Viral vector suspension in a volume of 2.0 μl was injected at 0.1 μl/min over 20 min. After a wait of 5 min, the needle was slowly withdrawn. Successful transduction of dopamine neurons of the SN was confirmed histologically by double immunolabeling for FLAG and TH or by fluorescent single-labeling of TH in combination with fluorescent detection of GFP or DsRed.
Production of adeno-associated virus viral vectors.
All vectors used for these studies were adeno-associated virus (AAV) serotype 1. Myr-Akt was produced as described previously (Ries et al., 2006). A plasmid encoding a 5′ src myristoylation signal in frame with mouse Akt1 was kindly provided by Dr. Thomas Franke (Columbia University, New York, NY) (Franke et al., 1995; Ahmed et al., 1997). The myristoylated Akt1 sequence was modified to incorporate a FLAG-encoding sequence at the 3′ end and inserted into an AAV packaging construct that utilizes the chicken β-actin (CBA) promoter and contains a woodchuck post-transcriptional regulatory element (WPRE) (Olson et al., 2006). AAV1 control injections, as specified for each experiment, were subcloned into the same viral backbone. A plasmid containing GFP-LC3 was kindly provided by Drs. N. Mizushima (Tokyo Metropolitan Institute of Medical Science, Tokyo Japan) and T. Yoshimori (Osaka University, Osaka, Japan) (Kabeya et al., 2000) and subcloned into the same viral backbone. AAV1 DsRed-LC3 was created by exchange of DsRed for GFP in the GFP-LC3 plasmid and subcloned into a pFBGR viral backbone provided by the Gene Transfer Vector Core of the University of Iowa (Iowa City, IA). This backbone utilizes the cytomegalovirus promoter and lacks a WPRE. AAV1 Cre was also produced in the pFBGR viral backbone. AAV1 hRheb(S16H) was produced by the University of North Carolina Vector Core (Chapel Hill, NC) by use of an AAV packaging construct that utilizes the CBA promoter and contains a 3′ WPRE (pBL).
Conditional deletion of Atg7 in SN neurons.
Atg7fl/fl mice were crossed with TH-GFP mice to obtain Atg7fl/wt:TH-GFP mice (where fl is flox and wt is wild type). These mice were then crossed with Atg7fl/fl to obtain the Atg7fl/fl:TH-GFP genotype. Local deletion of Atg7 was achieved by intranigral injection of AAV Cre. Preliminary experiments revealed that AAV Cre achieved expression of Cre in SN neurons and recombination in ROSA26-LacZ mice (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Selective deletion of Atg7 mRNA in the SN was confirmed by nonradioactive in situ hybridization with a riboprobe complementary to the exon 14 sequence (Komatsu et al., 2005) (Figure S2D), performed as described (Oo et al., 2009).
Immunohistochemistry.
Mice were perfused intracardially with 0.9% NaCl followed by 4.0% paraformaldehyde in 0.1 m phosphate buffer, pH 7.1. The brain was removed carefully and blocked into midbrain and forebrain regions. The region containing the midbrain was postfixed for 1 week, cryoprotected in 20% sucrose overnight, and then rapidly frozen by immersion in isopentane on dry ice. A complete set of serial sections then was cut through the SN at 30 μm. Sections were processed free-floating. For TH immunofluorescent staining, the primary antibody was rabbit anti-TH (Calbiochem) at 1:750. Sections then were treated with goat-anti-rabbit Texas Red-conjugated secondary antibody.
For dopamine transporter (DAT), FLAG, phosphorylated 4EBP1, and cathepsin D immunostaining, 30 μm sections were used. For DAT immunohistochemistry, sections were blocked with 0.1 m Tris-buffered saline and 3% normal rabbit serum for 48 h at 4°C. Then they were washed and incubated with rat anti-DAT (Millipore Bioscience Research Reagents) at 1:1000. Sections then were incubated with biotinylated anti-mouse IgG (Vector Laboratories), followed by avidin-biotinylated horseradish peroxidase complexes (ABC; Vector Laboratories). For FLAG, sections were initially treated with Mouse-on-Mouse Blocking Reagent (Vector Laboratories) and then processed free floating with a mouse monoclonal anti-FLAG antibody (Sigma) at 1:1000. Sections then were incubated with biotinylated anti-mouse IgG (Vector Laboratories), followed by ABC (Vector Laboratories). For immunofluorescent staining, fluorescein-conjugated avidin was used after the secondary antibody.
For phosphorylated 4EBP1, immunostaining was performed on 30 μm sections with a rabbit anti-phospho-4EBP1 (Thr37/46) antibody (Cell Signaling Technology) at 1:200. Sections then were treated with biotinylated protein A and ABC (Vector Laboratories). After immunoperoxidase staining, sections were thionin counterstained. For immunofluorescent staining, Texas Red-conjugated goat anti-rabbit secondary antibody or Texas Red-conjugated donkey anti-rabbit secondary antibody was used in double immunofluorescent staining. For cathepsin D immunofluorescent staining, goat-anti-cathepsin D (Santa Cruz Biotechnology) was used at 1:100. Sections were then treated with biotinylated horse anti-goat antibody, followed by fluorescein-conjugated avidin.
Immunohistochemistry for Atg7 was performed with three different rabbit anti-Atg7 antibodies (Sigma; Novus Biologicals; US Biological) at 1:1000 (Sigma) and 1:200 (Novus Biologicals and US Biological). Sections were then treated with biotinylated protein A and ABC (Vector Laboratories). For immunofluorescent staining, Alexa 568-conjugated goat-anti rabbit secondary antibody (Invitrogen) was used. Atg7 recombinant protein (Abnova) and blocking peptide (US Biologicals) were used for primary antibody absorption at the molar ration of 5:1. Those sections were then processed with preabsorbed primary antibody reagent, followed by biotinylated protein A and avidin-biotinylated horseradish peroxidase complexes.
Quantification of dopaminergic axons in the MFB.
Quantification of axons was performed on TH-GFP transgenic mice, which express green fluorescent protein driven by tyrosine hydroxylase promoter (Sawamoto et al., 2001). Mice were perfused intracardially with 0.9% NaCl followed by 4.0% paraformaldehyde in 0.1 m phosphate buffer, pH 7.1. Following postfixation and cryoprotection, the brains were sectioned horizontally at 30 μm. A section containing the posterior third ventricular recess and the A13 dopamine cell group was selected for analysis (supplemental Fig. S1B). Confocal microscopy (Leica TCS SP5 AOBS MP System) was used to acquire images through the entire medial-to-lateral extent of the MFB. Proceeding from a point midway between the anterior A13 cells and the posterior third ventricle recess, images were acquired with a 20× objective with a zoom factor of 8 applied. Depending on the site of the MFB under analysis, five to six contiguous fields (97 × 97 μm) were scanned. Each field was scanned in the z-axis with twenty 0.1 μm thickness optical planes from dorsal to ventral for a total vertical distance of 2.0 μm in the center of the section. These 20 optical planes were then merged to obtain a single maximal projection of the sampled volume. To count the number of axons passing in the rostrocaudal dimension through each sample volume, two horizontal sampling lines were drawn on the image at a separation distance of 10 μm in the center of the maximal projection. Every intact axon crossing both lines was counted as positive.
Quantification of autophagic vacuoles.
AAV GFP-LC3 was injected with AAV Myr-Akt or DsRed into the SN of wild-type mice. AAV DsRed-LC3 was injected with AAV Myr-Akt or vehicle into SN of TH-GFP mice. Mice were then lesioned with 6OHDA or axotomy. Lesioned mice were then killed at different time points as indicated. A complete set of serial sections was then cut through the SN at 30 μm. Beginning with a random section between 1 and 4, every fourth section was selected in keeping with the fractionator method of sampling. Every section was then examined under epifluorescence with a 60× objective lens, and each GFP-LC3 or DsRed-LC3-labeled autophagic vacuole present in SN was counted. AAV DsRed-LC3 was injected in TH-GFP mouse, and DsRed labeled autophagic vacuoles were counted in GFP-positive dopaminergic neurons. Control injections of AAV GFP-LC3, AAV DsRed-LC,3 and AAV DsRed into adult, nonlesioned, wild-type C57BL/6 mice revealed that in the absence of injury, transduced neurons did not reveal distinct puncta or intracellular inclusions (supplemental Fig. S3B,C available at www.jneurosci.org as supplemental material).
Electron microscopy.
At 24 h following injection of 6OHDA into either the striatum or the MFB, adult mice were perfused with 0.9% saline followed by 3% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, for 10 min at 4°C. Brains were postfixed in the same fixative for 1–2 weeks. Sections through the SN were cut on a vibratome and collected into 0.1 m Sorensen's buffer. Sections were stained in 1% OsO4/0.1 m Sorensen's buffer for 60 min and then washed. After dehydration in ascending concentrations of alcohols, sections were flat embedded in Durcupan between two pieces of Aclar. The sections were then trimmed to include the medial portion of the substantia nigra pars compacta (SNpc) mounted on a Durcupan peg and then cut at 8 μm for examination under phase contrast. Sections of interest were then re-embedded, thin sectioned, and examined on a JEOL 1200EX electron microscope as described previously (Oo et al., 1996).
Western analysis.
For analysis of proteins associated with autophagy, mice were unilaterally lesioned with 6OHDA and then killed at indicated time points. Blots were probed with anti-LC3 Ab1 (courtesy of Dr. Y. Uchiyama, Osaka University School of Medicine, Osaka, Japan), anti-LC3 Ab2 (courtesy of Drs. W. Haung Yu, Columbia University Medical Center, New York, NY and R. Nixon, Nathan Kline Institute, Orangeburg, NY), or anti-Rab24 (BD Transduction Laboratories) or anti-Beclin-1 antibodies (Novus Biologicals). Phospho-mTor was detected with anti-p-mTor (Ser2448) (Cell Signaling Technology). Proteins with detected with appropriate secondary antibodies and conjugated with horseradish peroxidase and chemiluminescent substrate (Pierce). Densitometric analysis of band intensity was performed by using a FluorChem 8800 Imaging System (Alpha Innotech).
Statistical analysis.
All data in the figures is presented as mean ± SEM. Multiple group comparisons were performed by one-way ANOVA unless otherwise indicated in the figure legend, followed by a Tukey all pairwise multiple-comparison procedure post hoc analysis. For two group comparisons, a Student's t statistic was performed.
Results
Myr-Akt suppresses retrograde axonal degeneration in the dopaminergic nigrostriatal projection
Following injection of the neurotoxin 6OHDA into the striatum, the principal target of the nigrostriatal dopaminergic projection, intense axon terminal degeneration can be demonstrated by the suppressed silver stain technique within 24–48 h (Ries et al., 2008). The degeneration proceeds retrograde such that by 3–4 d postlesion, degenerating axons can be identified within the medial forebrain bundle, the principal tract conveying dopaminergic axons to the striatum. At this postlesion time, many dopaminergic axons within the MFB have lost expression of the tyrosine hydroxylase phenotype such that their number and morphology cannot be examined by immunostaining (supplemental Fig. S1C). However, in mice that express green fluorescent protein under the TH promoter (TH-GFP) (Sawamoto et al., 2001), it remains possible to continue to monitor these axons by the visualization of GFP by confocal microscopy in horizontal sections through the MFB (supplemental Fig. S1C). This analysis reveals that in mice given a control (DsRed) AAV injection into the SN, there is a 42% loss of GFP-positive axons by 3 d postlesion (Fig. 1A,B). In addition, the injured axons are thickened and fragmented and show the formation of spheroids, a hallmark of axon injury (Fig. 1A). In contrast, in mice given AAV Myr-Akt, there is only a 15% loss of axons (Fig. 1A,B), not a significant change compared with the noninjected, contralateral control side. In addition to preservation of axon number, treatment with AAV Myr-Akt also preserves axon morphology, as fragmentation and spheroid formation are rarely observed (Fig. 1A). We considered the possibility that treatment with AAV Myr-Akt may increase the number of axons in the MFB due to sprouting before 6OHDA injection and thereby confound an analysis of axon preservation. However, we found that this had not occurred (Fig. 1B). Myr-Akt induces an increase in the caliber of dopaminergic axons (Ries et al., 2006), but this effect does not influence the number of axons counted by this methodology, which is based on continuity over a distance of 10 μm.
Figure 1.
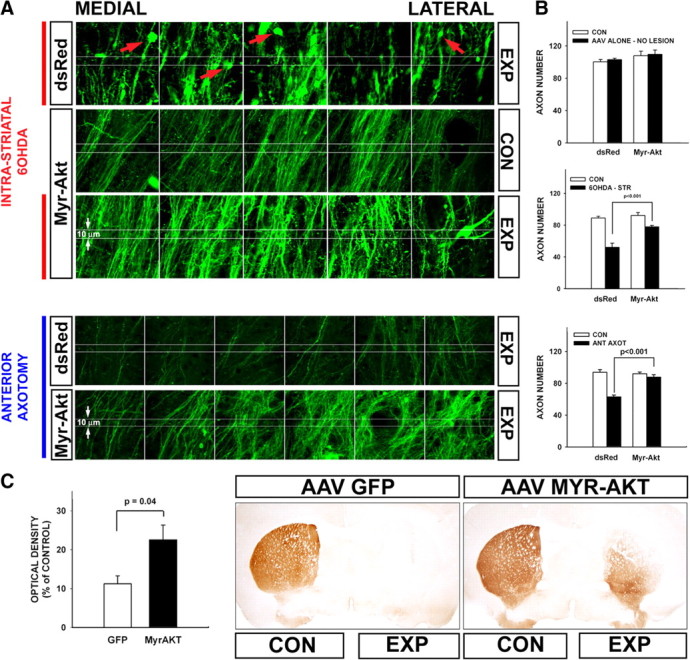
Myr-Akt suppresses retrograde degeneration in dopaminergic axons. A, Confocal images taken through the MFB are shown for representative single mice treated with either a control injection of AAV DsRed or with AAV Myr-Akt, followed 3 weeks later by either ipsilateral injection of 6OHDA or axotomy. Each series of panels represents adjacent optical fields, extending from medial to lateral through the MFB, in a single section. The top group of three sets of panels represents an experiment (EXP) with 6OHDA; the bottom group of two sets of panels represents an experiment with axotomy. For comparison to lesion conditions, the contralateral, noninjected control (CON) side of a Myr-Akt mouse is shown in the middle set of panels in the top group (the noninjected sides of AAV DsRed-injected mice were comparable, as shown by axon counts in B). In the top set of panels in the top group there is a loss of axons and the appearance of axonal spheroids (red arrows) at 3 d following intrastriatal 6OHDA in the mouse treated with AAV DsRed. In the mouse treated with Myr-Akt, shown as the bottom set of panels in the top group, there is a relative preservation of axons and minimal spheroid pathology. In the bottom group of panels there is a loss of axons at 6 d following unilateral anterior MFB axotomy in a mouse treated with AAV DsRed. Relative preservation of axons is observed in the Myr-Akt condition. The central white lines in each image are separated by 10 μm. An axon was counted only if it is traversed both of these lines. B, In the top graph, MFB axons were counted in mice that had received either AAV DsRed (n = 4) or AAV Myr-Akt (n = 4) without 6OHDA lesion. Neither control AAV DsRed injection nor Myr-Akt had an effect on the number of GFP-visualized axons. In the middle graph, the effect of 6OHDA lesion is shown (6OHDA-STR, intrastriatal 6OHDA). Mice treated with AAV DsRed as a control (n = 8) demonstrated a mean loss of 37 (or 42%) of their MFB axons, whereas mice treated with Myr-Akt (n = 5) demonstrate only a mean loss of 14 axons (or 15%), a highly significant difference compared with the AAV DsRed control condition (p < 0.001, ANOVA). The 15% loss of axons in the mice treated with Myr-Akt was not significant compared with the contralateral CON noninjected side (p = 0.13, NS). In the bottom graph, the effect of unilateral anterior axotomy is shown. Mice treated with AAV DsRed (n = 9) demonstrated a mean loss of 31 (or 33%) of their axons by postlesion day 6, whereas mice treated with Myr-Akt (n = 9) showed only a mean loss of 4 (or 4%) of their axons, a highly significant difference (p < 0.001, ANOVA). The 4% loss of axons in the mice treated with Myr-Akt was not significant compared with contralateral CON nonaxotomized MFB (p = 0.7, NS). C, Myr-Akt provides a robust lasting protection of dopaminergic striatal terminals following intrastriatal 6OHDA. At 4 weeks following lesion, mice were processed for immunostaining of DAT to assess striatal dopaminergic innervation. Representative coronal sections reveal that DAT immunostaining is eliminated following unilateral 6OHDA in AAV GFP (control)-injected mice, whereas there remains a moderate degree of preservation, particularly in the ventromedial quadrant in mouse treated with Myr-Akt. This result is shown quantitatively as optical density of staining on the lesioned side as a percentage of the contralateral, nonlesioned side. Among mice treated with AAV GFP, optical density was reduced to 11% of control, whereas among AAV Myr-Akt-treated mice 22% of staining remained (p = 0.04, t test; AAV GFP, n = 5; Myr-Akt, n = 7).
Given the wide spectrum of cellular effects of Akt, we considered the possibility that it may act by preventing the primary neurotoxicity of 6OHDA, rather than by acting specifically on mechanisms of axon degeneration. To address this possibility, we examined effects on retrograde degeneration induced by direct axotomy injury. In this model, made by knife cut in the anterior projection of the MFB, a postlesion interval of 6 d is required to observe a substantial loss of axons caused by retrograde degeneration. At this time, mice injected with control AAV demonstrate a 33% loss of axons (Fig. 1A,B). In contrast, mice injected with AAV Myr-Akt demonstrate only a 5% loss (Fig. 1A,B), not a significant difference compared with the contralateral, nonlesioned side. We conclude that the ability of constitutively active Akt to suppress retrograde axon degeneration is general to diverse forms of axon injury and, in the case of 6OHDA, is unlikely to be due to prevention of neurotoxicity.
To ascertain whether the axon protection provided by Myr-Akt in the acute period of degeneration following 6OHDA affords a robust and lasting preservation of the dopaminergic nigrostriatal projection, we performed immunostaining for the high-affinity dopamine transporter, a specific marker for dopaminergic terminals, at 4 weeks postlesion in the striatum. This analysis demonstrated that acute protection did indeed result in a lasting preservation of striatal dopaminergic innervation (Fig. 1C).
Macroautophagy occurs in the axons and cell bodies of dopamine neurons of the SN following axon injury and is suppressed by Myr-Akt
We have shown previously that activation of caspase-3 occurs in axons in the immature nervous system following either axotomy (El-Khodor and Burke, 2002) or intrastriatal injection of 6OHDA, but there is neither activation of caspase-3 nor calpain following intrastriatal 6OHDA in the mature nervous system (Ries et al., 2008). Given the evidence of a role for macroautophagy (hereafter referred to as “autophagy”) in the degeneration of axons and their terminals in the context of injury in tissue culture (Larsen et al., 2002; Yang et al., 2007) and both axon pruning (Song et al., 2008) and degeneration (Wang et al., 2006) in vivo, we performed ultrastructural analysis of the striatum following intrastriatal injection of 6OHDA. This analysis revealed the presence of numerous autophagic vacuoles (AVs) in striatal neuropil (Fig. 2). AVs were identified as vesicular structures with a delimiting double membrane, often containing subcellular organelles such as mitochondria (Mizushima, 2004). A number of these vesicles also contained multilamellar membranous structures, another characteristic ultrastructural feature of AVs (Hornung et al., 1989; Jia et al., 1997; Hariri et al., 2000; Borsello et al., 2003; Nixon et al., 2005) (Fig. 2B,C). Occasional AVs were observed within nerve terminals, identified by adjacent synaptic vesicles (Fig. 2B). We considered the possibility that the formation of AVs within the striatum may be a response unique to the proximate injection of 6OHDA rather than a general feature of the degeneration of nigrostriatal axons. To assess this possibility, we also performed an analysis following a distant injection of 6OHDA into the MFB, which induces anterograde degeneration. In these mice as well, AVs were identified within striatal neuropil (Fig. 2B,C). Examination of numerous striatal sections from control nonlesioned adult C57BL/6 mice failed to reveal AVs (supplemental Fig. S3A).
Figure 2.
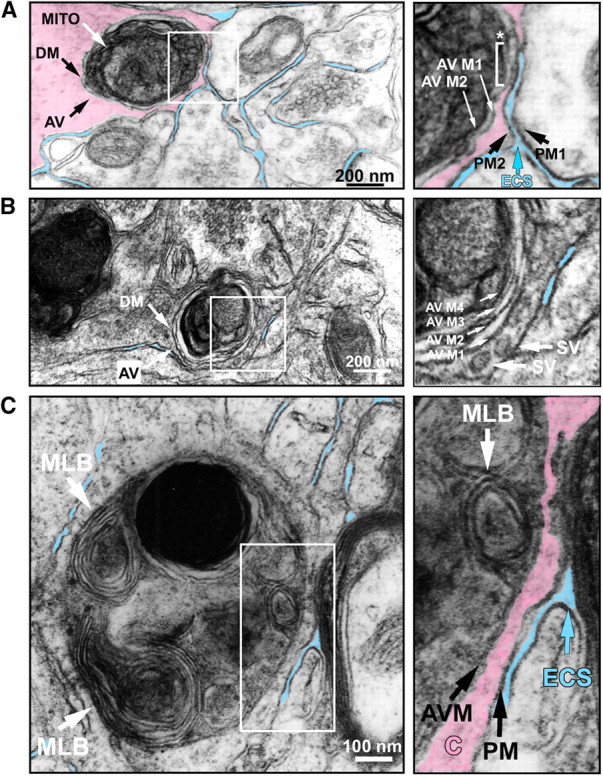
Ultrastructural features of autophagy in striatal neuropil following 6OHDA injection. A, A single autophagic vacuole, AV, is observed in a striatal neurite 1 day following intrastriatal 6OHDA. The AV is defined by a characteristic double membrane (DM), and it contains a degenerating mitochondrion (MITO). To assist the visual presentation, the cytoplasm of the cell has been colorized in semitransparent pink, and adjacent extracellular space (ECS) is in blue. Only the definitively clear ECS adjacent to the profile of interest has been colorized. In the right panel, the cellular features of the boxed area in the left panel are shown in detail. ECS (blue arrow) separates the plasma membrane of an adjacent cell (PM1) from the plasma membrane (PM2) of the AV-containing cell. A thin rim of cytoplasm (pink) separates PM2 from the outer double membrane of the AV (AV M1) for most of its extent, except at one region where AV M1 abuts the inner surface of PM2 (*, white bracket). The inner membrane of the AV double membrane (AV M2) runs parallel to AV M1. B, An AV is observed in a neurite following injection of 6OHDA into the MFB. A characteristic double membrane (DM) is again observed. The right panel, an enlarged view of the boxed area in the left panel, reveals that the outermost membrane of the AV (AV M1) is adjacent to two synaptic vesicles (SV) in the cytoplasm of the neurite. This AV is multilamellar, with four concentric membranes (AV M1–4) indicated by white arrows in the right panel. The formation of multiple concentric lamellae is highly characteristic of AVs (Hornung et al., 1989; Jia et al., 1997; Hariri et al., 2000; Nixon et al., 2005). C, An AV is observed in striatal neuropil following injection of 6OHDA into the MFB. This AV contains several multilamellar bodies (MLB). The right panel, an enlarged view of the boxed area in the left panel, shows that ECS separates the plasma membrane (PM) of the AV-containing cell from the external myelin layer of a myelinated axon. A thin rim of cytoplasm (C, colored pink) separates the membrane of the AV (AVM) from the PM. A single MLB is subjacent to the outermost AV membrane (white arrow).
To assess whether autophagy occurs specifically within defined dopaminergic neurons in these neurotoxin models and assess the general occurrence of autophagy following axon injury in these neurons, we made use of a viral vector approach to transduce them with DsRed-LC3 in TH-GFP mice. Microtubule-associated protein 1 light chain 3 (LC3) is a mammalian homolog of yeast Apg8p, a critical component of a ubiquitin-like conjugation system required for autophagosome formation (Kabeya et al., 2000; Mizushima, 2004), and it associates with isolation membranes in an Atg5-Atg12-dependent manner and through a process catalyzed by Atg7 (Mizushima, 2004). The use of DsRed-LC3 therefore permits demonstration of AVs as intracellular red puncta in dopaminergic neurons. Induction of retrograde axon degeneration by intrastriatal 6OHDA caused the appearance of multiple discrete puncta in the axons and cell bodies of dopaminergic neurons (Fig. 3A; Movie S1, available at www.jneurosci.org as supplemental material). Such puncta were also observed following induction of retrograde axon degeneration by anterior MFB axotomy and anterograde degeneration by posterior axotomy (Fig. 3A). Thus, autophagy occurs in both axons and cell bodies of dopaminergic neurons following either neurotoxin or physical injury. DsRed-LC3 red puncta were not observed in these neurons in the absence of 6OHDA or axotomy lesion (supplemental Fig. S3C)
Figure 3.

Autophagy occurs in dopamine neuron cell bodies and axons in diverse models of axon injury. A, The presence of AVs in dopaminergic axons and cell bodies is identified in the intrastriatal 6OHDA and in both the anterior axotomy (AXOT ANT) and posterior axotomy (AXOT POST) models by DsRed-LC3 labeling in TH-GFP mice. In the top panels, two clusters of AVs (red arrows) are identified in a GFP-labeled dopaminergic axon in the MFB 2 d following intrastriatal 6OHDA (6OHDA STR). Localization of the AVs within the axon was confirmed by virtual rotation of the set of Z-stack images (see Movie S1, available at www.jneurosci.org as supplemental material). AVs are also observed in dopaminergic cell bodes in the SNpc. A single example is identified by a red arrow. In the bottom two sets of panels, red punctuate AVs are identified in dopaminergic neurons of the SNpc following both anterior and posterior axotomy. Single examples are indicated by red arrows. Bar,10 μm. ANT, Anterograde; RET, retrograde. B, To monitor the number of AVs following lesions, mice were preinjected into the SN with AAV GFP-LC3 in a 1:1 mixture with either AAV Myr-Akt or AAV DsRed as control. After 3 weeks, all mice received a unilateral intrastriatal 6OHDA injection and were then killed at the indicated postlesion day (PLD) to quantify AVs in SNpc neurons as green GFP-LC3-positive puncta. The top graph shows that Myr-Akt decreased the number of AVs at all PLDs. On PLD 1, Myr-Akt reduced the number of neurons with AVs by 79% (p < 0.001, n = 6 both groups). The middle graph shows similar results when expressed as the total number of AVs in each SN section. To monitor the number of AVs in dopamine neurons following anterior axotomy, we used DsRed-LC3 in TH-GFP mice as illustrated in A. The reason for this change in methodology was that the axotomy lesion, unlike the 6OHDA lesion, is not selective for dopamine neurons, and our experimental goal was to monitor AV number specifically in this population. Therefore, TH-GFP mice were preinjected into the SN with AAV DsRed-LC3 in a 1:1 mixture with Myr-Akt or DsRed-LC3 alone. After 3 weeks, all mice received a unilateral anterior axotomy and were killed at 2 d postlesion to quantify the number of GFP-positive SNpc neurons with discrete red puncta, as shown in A. Myr-Akt reduced the number of neurons with AVs by 78% (p < 0.001, n = 6 both groups).
To further establish and characterize the occurrence of autophagy in the nigrostriatal system following intrastriatal 6OHDA lesion, we examined the expression of several proteins that have been associated with autophagy. LC3 exists in two forms in cells: LC3-I is identified at an apparent molecular weight of 18 kDa and is localized to the cytosol; LC3-II is identified at 16 kDa, is enriched in the AV fraction, and is present on both sides of AV membranes (Kabeya et al., 2000). Increase in conversion of LC3-I to LC3-II is an indicator of activation of autophagy. Utilizing two different antibodies to LC3, we observed that the LC3-II isoform is identified in mouse SN homogenates following intrastriatal 6OHDA, but not vehicle, injection (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material). Rab24 is a GTP-binding protein that has also been localized to AVs (Munafó and Colombo, 2002), and identified in the CNS following axon injury (Egami et al., 2005). There is an induction of Rab24 protein following intrastriatal 6OHDA (supplemental Fig. S4B). This induction is maximal at postlesion day 2 (Fig. S4B). Beclin-1, a novel Bcl-2 interacting protein, is homologous to the yeast autophagy gene Atg6 and is a mediator of autophagy (Liang et al., 1999). Beclin-1 protein expression is also induced in SN following 6OHDA and, like Rab24, induction is maximal at postlesion day 2 (supplemental Fig. S4C).
To explore the relationship between the ability of Myr-Akt to preserve nigrostriatal dopaminergic axons following distal injury and the occurrence of autophagy during the degenerative process, we quantified both the number of neurons in the SNpc with AVs and the total number of AVs in the SNpc neuron population following Myr-Akt in models of retrograde degeneration induced by intrastriatal 6OHDA and anterior axotomy. For this analysis, wild-type rather than TH-GFP mice were used in the 6OHDA model, and AVs were identified following transduction with AAV GFP-LC3 (supplemental Fig. S3B). Following treatment with Myr-Akt, fewer SNpc neurons contain AVs at postlesion days 1–3 after 6OHDA (Fig. 3B). Similar results were observed in an analysis of the total number of AVs in the SNpc neuron population (Fig. 3B). Treatment with Myr-Akt also diminished the number of dopaminergic neurons with AVs at postlesion day 2 following anterior axotomy (Fig. 3B), determined as the number of GFP-positive neurons with red puncta following transduction with AAV DsRed-LC3.
Inhibition of retrograde axonal degeneration by Myr-Akt is associated with mTor signaling
In diverse contexts, the ability of Akt to inhibit autophagy has been attributed to signaling through the mTor protein complex (Fig. 4A) (Meijer and Codogno, 2004; Lum et al., 2005; Mizushima et al., 2008). We determined that transduction of SN with AAV Myr-Akt results in increased abundance of phosphorylated mTor protein (Fig. 4B). At the cellular level, expression of p-4E-BP1, an mTor substrate, was observed exclusively in the injected SN following transduction with AAV Myr-Akt (Fig. 4C). Following transduction, immunostaining for p-4E-BP1 was observed not only in the cell bodies of neurons in the SNpc, but also in their axons within the MFB (Fig. 4D). Expression of p-4E-BP1 was observed exclusively in neurons that also expressed Myr-Akt (Fig. 4C). In such p-4E-BP1-positive neurons there was a significant increase in neuron size (Fig. 4C), confirming a biologic effect of enhanced Akt/mTor signaling. We observed a punctate appearance of p-4E-BP1 immunostaining in neurons due to a precise colocalization of p-4E-BP1 immunostaining with that for cathepsin-D, a lysosomal marker (Fig. 4C).
Figure 4.

Myr-Akt mediates axon protection through mTor signaling. A, Akt regulates mTor signaling by phosphorylation and inhibition of the tuberous sclerosis complex 2 (TSC2) GTPase-activating protein for Rheb-GTPase. Released from inhibition by TSC, Rheb-GTPase activates mTORC1, which phosphorylates downstream targets such as 4E-BP1. In addition, mTor signaling negatively regulates autophagy. B, Western analysis of phospho-mTor(Ser2448) in ventral mesencephalon whole tissue extracts 3 weeks following unilateral AAV SN injection shows an increase in abundance of phosphorylated mTor protein on the side of AAV injection [E (blots) and EXP (graph), Experimental] compared with the noninjected contralateral control [C (blots) and CON (graphs)] for AAV Myr-Akt, but not AAV DsRed. The increase induced by Myr-Akt is shown quantitatively in the bar graph depicting the optical density of p-mTor bands normalized for the optical density of the corresponding β-actin band (n = 3). C, Immunoperoxidase staining for p-4E-BP1 is observed in neurons in the SNpc (arrows) following transduction with AAV Myr-Akt on the injected side, but not on the contralateral control side. At a cellular level, positive staining for p-4E-BP1 is observed exclusively in neurons expressing Myr-Akt, identified by positive FLAG staining. A total of 50 p-4E-BP1-positive neurons were identified, and all were also positive for FLAG. Among p-4E-BP1-positive neurons, there was a 1.7-fold increase in cross-sectional area (p < 0.001, n = 25 neurons in each group). At a subcellular level, p-4E-BP1 expression colocalizes with that of cathepsin-D, a lysosomal marker. The white rectangles in the lower magnification images are shown at higher magnification in the right-hand panels. D, Both Myr-Akt-FLAG and p-4E-BP1 are expressed in axons in the MFB and the striatum. E, To assess the role of mTor signaling in the ability of Myr-Akt to protect axons, we created an AAV vector expressing a constitutively active form of human Rheb, hRheb(S16H). Transduction of the SNpc is demonstrated by immunoperoxidase staining for FLAG in the top panel. Successful transduction of dopamine neurons (with an efficiency ranging from 80 to 90%) is demonstrated by fluorescent double-labeling for TH and FLAG. As for AAV Myr-Akt, transduction with AAV hRheb(S16H) induces an abundant increase in the number of p-4E-BP1-positive neurons, demonstrated by immunoperoxidase staining. F, Following transduction with AAV hRheb(S16H), axons of dopaminergic neurons are resistant to retrograde degeneration induced by intrastriatal 6OHDA (6OHDA-STR). A mean loss of 26 axons (or 36%) occurred in the mice treated with DsRed (n = 6), whereas a mean loss of only 12 axons (or 16%) occurred in the mice treated with hRheb(S16H) (n = 7; p < 0.001, ANOVA). The 16% loss in the MFB following 6OHDA in the hRheb(S16H)-treated mice was not significant (p = 0.09, NS).
To assess whether the ability of Myr-Akt to protect axons is mediated through mTor signaling, we examined the possibility of also achieving protection by activation of Rheb, a GTPase that is activated by Akt and mediates activation of mTor (Fig. 4A). We created a mutant form of human Rheb, hRheb(S16H), that had been identified in a screen for constitutively active mutants in yeast and confirmed to be active in mammalian cells (Yan et al., 2006). Like Myr-Akt, transduction of dopamine neurons of the SNpc with hRheb(S16H) resulted in an increased abundance of p-4E-BP1 protein (Fig. 4E). Transduction of dopamine neurons with hRheb(S16H) also conferred resistance of their axons to retrograde degeneration induced by intrastriatal 6OHDA (Fig. 4F), suggesting that Myr-Akt acts, at least in part, through Rheb-mTor signaling to achieve axon protection.
Abrogation of autophagy signaling within dopamine neurons by regionally selective deletion of Atg7 confers resistance to retrograde axonal degeneration
Many previous investigations have identified the occurrence of autophagy in the presence of axon degeneration (Larsen et al., 2002; Wang et al., 2006; Komatsu et al., 2007; Yang et al., 2007; Chen et al., 2009), but its role has been unclear. Based on our observation that Myr-Akt may be acting through Rheb-mTor signaling to provide axon protection, we considered that there may be a relationship between the ability of Myr-Akt to suppress autophagy through mTor signaling and to protect axons. To assess this possible relationship, we sought to selectively disrupt autophagy signaling by genetic deletion of an essential autophagy mediator, Atg7, in mice with a floxed allele (Komatsu et al., 2005). Furthermore, to avoid disturbance of the development of the nigrostriatal system, we sought to ablate Atg7 in mature mice because there is extensive evidence that intact autophagy signaling is required during development for normal axon maintenance (Hara et al., 2006; Komatsu et al., 2006, 2007). In normal adult mice we observed Atg7 protein expression in numerous axons in brain (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material), but in dopaminergic axons of the MFB, protein expression was observed only following injury (Fig. S5B). Regionally selective deletion of Atg7 in mature mice was achieved by intranigral injection of AAV Cre (supplemental Fig. S2A–D). In mice homozygous for the Atg7fl allele and injected with AAV Cre but not control mice, there was protection from retrograde axon degeneration induced by either intrastriatal 6OHDA (Fig. 5A) or axotomy (Fig. 5B). This axon protection was observed not only as preservation of axon number but also by the absence of axonal pathology such as fragmentation and spheroid formation (Fig. 5A,B). Deletion of Atg7 provided a lasting protection of axons as shown by an increase in their remaining number at 4 weeks after 6OHDA lesion (Fig. 5C). A comparable degree of protection was observed for striatal dopaminergic innervation, demonstrated by immunostaining for DAT (data not shown).
Figure 5.
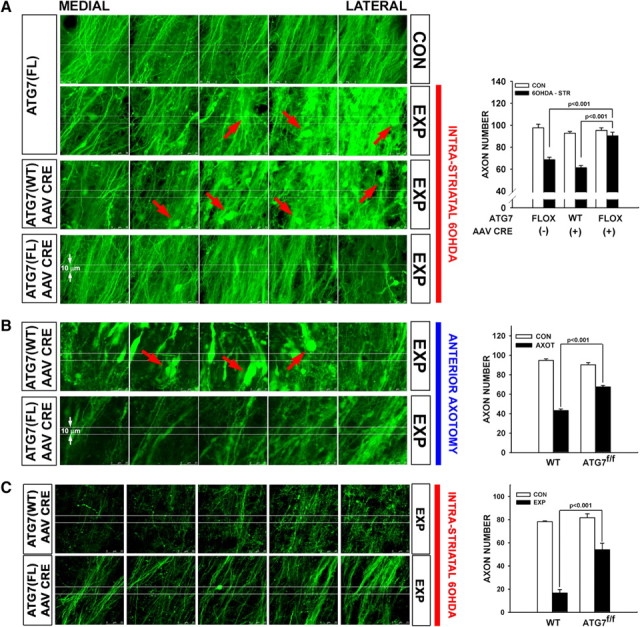
Following deletion of Atg7, axons of SNpc dopamine neurons are resistant to retrograde axon degeneration. A, In the absence of AAV Cre injection, Atg7fl/fl:TH-GFP mice show a loss of MFB dopaminergic axons (visualized by confocal microscopy of endogenous GFP) and the appearance of axonal spheroid pathology (red arrows) following unilateral 6OHDA injection. Atg7w/w:TH-GFP mice injected with AAV Cre show a similar axon loss and pathology. However, following injection of AAV Cre, Atg7fl/fl:TH-GFP mice show minimal axon loss and pathology following 6OHDA injection. Following 6OHDA injection, Atg7fl/fl:TH-GFP mice without AAV Cre (n = 5) and Atg7w/w:TH-GFP mice given AAV Cre (n = 6) show a mean loss of 31 (or 32%) and 32 (or 34%) MFB axons respectively, whereas Atg7fl/fl:TH-GFP mice treated with AAV Cre (n = 6) show a mean loss of only 5 (or 5%) (p < 0.001, ANOVA). The minor loss of axons (5%) on the 6OHDA-treated side is not significant compared with the contralateral, noninjected control (CON) side (p = 0.7). 6OHDA-STR, Intrastriatal 6OHD. B, Following deletion of Atg7, nigrostriatal axons in Atg7fl/fl:TH-GFP mice show less pathology and relatively preserved number following anterior MFB axotomy (AXOT) compared with Atg7wt/wt:TH-GFP mice. In the Atg7wt/wt:TH-GFP mice, numerous axon spheroids are observed (red arrows). These mice (n = 6) demonstrated a mean loss of 53% of their axons by PLD 6, whereas Atg7fl/fl:TH-GFP mice injected with AAV Cre (n = 4) show only a 26% loss, a highly significant difference (p < 0.001, ANOVA). C, Abrogation of autophagy by deletion of Atg7 provides a robust and lasting neuroprotection of axons following intrastriatal 6OHDA lesion. At 4 weeks following lesion, Atg7wt/wt:TH-GFP mice (n = 3) demonstrate a mean loss of 78% of their axons, whereas Atg7fl/fl:TH-GFP mice injected with AAV Cre (n = 3) reveal still only a 34% loss (p < 0.001, ANOVA). FL or fl, Flox; WT or wt, wild type; EXP, experimental.
Discussion
Akt has a diverse array of cellular phenotypes, including effects on cell death, growth, and metabolism (Manning and Cantley, 2007). Within postmitotic neurons it not only has these general cellular effects on survival (Brunet et al., 2001) and size (Kwon et al., 2006; Ries et al., 2006), but also effects that are unique to neurons, including regulation of axon sprouting (Namikawa et al., 2000; Ries et al., 2006; Park et al., 2008), caliber and branching (Markus et al., 2002), synaptic strength (Wang et al., 2003), and dendritic growth (Kwon et al., 2006). We herein describe a novel neuronal phenotype of Akt, the ability to regulate axon degeneration. Demonstration of this phenotype was achieved with an imaging approach that allows quantification of dopaminergic axons despite loss of endogenous TH protein expression due to injury and permits the conclusion that the apparent protection of axons is due to their actual structural preservation. This conclusion is supported by the observation that Akt diminishes the occurrence of axon swelling and fragmentation. In addition, because this method of axon quantification requires identification of individual fibers and is not a population measure, it is not influenced by either amount of GFP protein expression or caliber of the axons, both of which may be affected by Akt. We therefore conclude that Akt acts to preserve the structural integrity of axons following distal injury. Our observations have been made with a mutant, constitutively active form of Akt that incorporates a myristoylation signal at its N terminus (Ahmed et al., 1997). This mutation is believed to mediate constitutive activity by targeting Akt protein to cellular membranes, where it is then activated by phosphorylation (Ahmed et al., 1997). While Myr-Akt shares most cellular phenotypes with both overexpressed wild-type and other nonmyristoylated constitutively active forms of Akt, nevertheless some phenotypes are unique to the myristoylated form (Dufner et al., 1999). Whether the myristoylation signal is necessary for the axon protection phenotype that we have observed will require future study.
Little is yet known about the mechanisms of axon degeneration, but there is now abundant evidence that they are subject to cellular regulation and control (Coleman, 2005; Luo and O'Leary, 2005). There is also growing evidence that these pathways are diverse. For example, while the mutant protein Wlds suppresses axon degeneration caused by injury, it does not affect normal developmental axon pruning (Hoopfer et al., 2006). The mechanisms of axon degeneration also change with maturation. Although we observed activation of caspase-3 in nigrostriatal axons in the intrastriatal 6OHDA model in immature mice, it is not activated in mature animals (Ries et al., 2008). In mature mice, we observe a robust induction of autophagy following 6OHDA. AVs were identified by ultrastructural analysis (Mizushima, 2004) in striatal neuropil and specifically within nerve terminals. Autophagy was further confirmed by the identification of both GFP-LC3 and DsRed-LC3-positive puncta (Kabeya et al., 2000; Mizushima, 2004), the latter specifically within the cell soma and axons of dopaminergic neurons following four different types of axon injury. These observations are in keeping with those of other investigators who have reported the occurrence of autophagy in the course of neurite or axon degeneration (Larsen et al., 2002; Wang et al., 2006; Komatsu et al., 2007; Yang et al., 2007; Plowey et al., 2008; Chen et al., 2009).
However, the precise role that autophagy plays in the setting of axon injury has been debated. Some investigators have proposed that it plays a direct role in axon destruction (Yang et al., 2007; Plowey et al., 2008), whereas other have proposed that AVs passively accumulate in injured axons due to impaired axon transport (Chen et al., 2009) or that the induction of autophagy represents an attempted protective response (Larsen et al., 2002; Komatsu et al., 2007). To explore a possible relationship between the ability of Myr-Akt to protect axons and the robust occurrence of autophagy in these models of axon injury, we examined the effect of Myr-Akt expression on the prevalence of AVs and observed that it resulted in a marked decrease in their number. This observation is in keeping with extensive evidence in other contexts that Akt signaling negatively regulates autophagy (Meijer and Codogno, 2004; Lum et al., 2005; Mizushima et al., 2008). To further evaluate a possible relationship between the ability of Myr-Akt to protect axons and its ability to suppress autophagy, we examined effects of a constitutively active form of Rheb, a GTPase that mediates downstream effects of Akt by direct activation of mTor. For this purpose, we used hRheb(S16H), a mutant that is resistant to negative regulation by TSC1/2 (Yan et al., 2006). Transduction of SN neurons with hRheb(S16H) resulted in a robust increase in the number of p-4E-BP1-positive neurons, and it successfully recapitulated the axon protection phenotype of Myr-Akt, thus supporting the possibility of a relationship between the ability of both Myr-Akt and hRheb(S16H) to protect axons and signaling through mTor.
mTor activation has a wide range of cellular effects (Swiech et al., 2008). To examine whether suppression of autophagy by mTor specifically mediates the axon protection phenotype of Myr-Akt and hRheb(S16H), we used a genetic approach to abrogate autophagy signaling selectively within SN neurons by transduction with AAV Cre in Atg7fl/fl mice (Komatsu et al., 2005). Deletion of Atg7 signaling in these neurons results in a striking resistance of their axons to retrograde degeneration. This resistance is robust and lasting, as it is observed as an increased number of preserved axons and striatal dopaminergic nerve terminals at 4 weeks after toxin injury. Thus, we conclude that the ability of Myr-Akt to protect axons from retrograde degeneration is likely to be due, at least in part, to its ability to suppress autophagy through mTor signaling.
Our observations in these in vivo models are consistent with those of Yang and colleagues, who noted in several in vitro models of injury that both pharmacologic and genetic disruption of autophagy signaling protected from axon degeneration (Yang et al., 2007). They are also in keeping with the findings of Plowey and colleagues, who observed that RNA interference-mediated knockdown of Atg7 expression prevented degeneration of neurites induced by a mutant form of LRRK2 (leucine-rich repeat kinase 2) that causes PD (Plowey et al., 2008). While our findings would appear to be at variance with several observations that genetic ablation of autophagy signaling leads to neurodegeneration (Hara et al., 2006; Komatsu et al., 2006) and specifically to axon degeneration in the Atg7fl/fl mice (Komatsu et al., 2007), all of these observations were made in a context in which autophagy signaling had been disrupted during development. Perhaps the most universal emerging theme for the cellular role of autophagy is that it is highly context dependent (Levine and Yuan, 2005; Cherra and Chu, 2008). We postulate that while autophagy may be essential for normal axon homeostasis during development, in the setting of the mature brain in the context of injury or neurodegenerative disease, it can lead to destruction.
Our conclusion that Myr-Akt provides protection from retrograde axon degeneration in the mature brain has implications for the pathogenesis and treatment of neurodegenerative disease, the most important being that the cellular consequences of activation of autophagy can be deleterious or beneficial, depending on the precise context. Autophagy has been postulated to play an important role in the clearance of pathogenic protein aggregates in neurodegenerative diseases (Mizushima et al., 2008). Of particular relevance in relation to our studies is that it has been postulated to play a critical role in the clearance of α-synuclein (Cuervo et al., 2004), which has been postulated to form toxin aggregates in dopaminergic axons. However, our observations in acute models of retrograde axon degeneration suggest that, to the extent that similar mechanisms may underlie axon degeneration in chronic neurodegenerative diseases, activation of autophagy signaling may be deleterious. Our findings also imply that activation of Akt signaling pathways not only provides important neuroprotection of the cell soma level by blockade of apoptosis, but also a novel ability to forestall degeneration of axons. Thus, if activated at the appropriate time in select cellular compartments, these pathways may provide attractive candidates for the development of therapeutics in the treatment of neurodegenerative diseases.
Footnotes
This work was supported by National Institutes of Health Grants NS26836 and NS38370, the Parkinson's Disease Foundation, and the RJG Foundation (R.E.B.). We thank D. Sulzer, A. M. Cuervo, and A. Yamamoto for critical review of the manuscript and useful discussions. We also thank D. Sulzer, R. A. Nixon, W. H. Yu, and A. Tagliaferro for helpful examination of the electron micrographs. We also wish to express our gratitude in memoriam to Anne M Cataldo, who generously offered her assistance in the interpretation of these micrographs. We thank R. A. Nixon and W. H. Yu for making available to us their antibody to LC3, and Y. Uchiyama and M. Shibata for making available to us their antibody to LC3. We express our gratitude to N. Mizushima and T. Yoshimori for kindly providing us with their GFP-LC3 clone. We gratefully acknowledge the expert technical assistance of Mary Schoenebeck in the performance of electron microscopy.
References
- Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc Natl Acad Sci U S A. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T, Croquelois K, Hornung JP, Clarke PG. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–485. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Chen Q, Peto CA, Shelton GD, Mizisin A, Sawchenko PE, Schubert D. Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J Neurosci. 2009;29:118–130. doi: 10.1523/JNEUROSCI.3985-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Rzhetskaya M, Kareva T, Bland R, During MJ, Tank AW, Kholodilov N, Burke RE. Antiapoptotic and trophic effects of dominant-negative forms of dual leucine zipper kinase in dopamine neurons of the substantia nigra in vivo. J Neurosci. 2008;28:672–680. doi: 10.1523/JNEUROSCI.2132-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Burke RE. The Wid(S) mutation delays anterograde, but not retrograde, axonal degeneration of the dopaminergic nigro-striatal pathway in vivo. J Neurochem. 2010;113:683–691. doi: 10.1111/j.1471-4159.2010.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715–725. doi: 10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008;3:309–323. doi: 10.2217/14796708.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dufner A, Andjelkovic M, Burgering BM, Hemmings BA, Thomas G. Protein kinase B localization and activation differentially affect S6 kinase 1 activity and eukaryotic translation initiation factor 4E-binding protein 1 phosphorylation. Mol Cell Biol. 1999;19:4525–4534. doi: 10.1128/mcb.19.6.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt O, Coelln RV, Kugler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E, Haid S, Isenmann S, Gravel C, Srinivasan A, Bahr M, Weller M, Dichgans J, Schulz JB. Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci. 2000;20:9126–9134. doi: 10.1523/JNEUROSCI.20-24-09126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egami Y, Kiryu-Seo S, Yoshimori T, Kiyama H. Induced expressions of Rab24 GTPase and LC3 in nerve-injured motor neurons. Biochem Biophys Res Commun. 2005;337:1206–1213. doi: 10.1016/j.bbrc.2005.09.171. [DOI] [PubMed] [Google Scholar]
- El-Khodor BF, Burke RE. Medial forebrain bundle axotomy during development induces apoptosis in dopamine neurons of the substantia nigra and activation of caspases in their degenerating axons. J Comp Neurol. 2002;452:65–79. doi: 10.1002/cne.10367. [DOI] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hariri M, Millane G, Guimond MP, Guay G, Dennis JW, Nabi IR. Biogenesis of multilamellar bodies via autophagy. Mol Biol Cell. 2000;11:255–268. doi: 10.1091/mbc.11.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Hornung JP, Koppel H, Clarke PG. Endocytosis and autophagy in dying neurons: An ultrastructural study in chick embryos. J Comp Neurol. 1989;283:425–437. doi: 10.1002/cne.902830310. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Biochemical aspects of Parkinson's disease. Neurology. 1998;51(Suppl 2):S2–S9. doi: 10.1212/wnl.51.2_suppl_2.s2. [DOI] [PubMed] [Google Scholar]
- Jia L, Dourmashkin RR, Allen PD, Gray AB, Newland AC, Kelsey SM. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol. 1997;98:673–685. doi: 10.1046/j.1365-2141.1997.2623081.x. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus A, Zhong J, Snider WD. Raf and akt mediate distinct aspects of sensory axon growth. Neuron. 2002;35:65–76. doi: 10.1016/s0896-6273(02)00752-3. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafó DB, Colombo MI. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic. 2002;3:472–482. doi: 10.1034/j.1600-0854.2002.30704.x. [DOI] [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci. 2000;20:2875–2886. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Olson VG, Heusner CL, Bland RJ, During MJ, Weinshenker D, Palmiter RD. Role of noradrenergic signaling by the nucleus tractus solitarius in mediating opiate reward. Science. 2006;311:1017–1020. doi: 10.1126/science.1119311. [DOI] [PubMed] [Google Scholar]
- Oo TF, Blazeski R, Harrison SM, Henchcliffe C, Mason CA, Roffler-Tarlov SK, Burke RE. Neuron death in the substantia nigra of weaver mouse occurs late in development and is not apoptotic. J Neurosci. 1996;16:6134–6145. doi: 10.1523/JNEUROSCI.16-19-06134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo TF, Marchionini DM, Yarygina O, O'Leary PD, Hughes RA, Kholodilov N, Burke RE. Brain-derived neurotrophic factor regulates early postnatal developmental cell death of dopamine neurons of the substantia nigra in vivo. Mol Cell Neurosci. 2009;41:440–447. doi: 10.1016/j.mcn.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE. Oncoprotein Akt/PKB: trophic effects in murine models of Parkinson's disease. Proc Natl Acad Sci U S A. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries V, Silva RM, Oo TF, Cheng HC, Rzhetskaya M, Kholodilov N, Flavell RA, Kuan CY, Rakic P, Burke RE. JNK2 and JNK3 combined are essential for apoptosis in dopamine neurons of the substantia nigra, but are not required for axon degeneration. J Neurochem. 2008;107:1578–1588. doi: 10.1111/j.1471-4159.2008.05713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi A, Schneider BL, Aebischer P. Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol. 2004;14:326–330. doi: 10.1016/j.cub.2004.01.053. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Nakao N, Kobayashi K, Matsushita N, Takahashi H, Kakishita K, Yamamoto A, Yoshizaki T, Terashima T, Murakami F, Itakura T, Okano H. Visualization, direct isolation, and transplantation of midbrain dopaminergic neurons. Proc Natl Acad Sci U S A. 2001;98:6423–6428. doi: 10.1073/pnas.111152398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JW, Misgeld T, Kang H, Knecht S, Lu J, Cao Y, Cotman SL, Bishop DL, Lichtman JW. Lysosomal activity associated with developmental axon pruning. J Neurosci. 2008;28:8993–9001. doi: 10.1523/JNEUROSCI.0720-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Findlay GM, Jones R, Procter J, Cao Y, Lamb RF. Hyperactivation of mammalian target of rapamycin (mTOR) signaling by a gain-of-function mutant of the Rheb GTPase. J Biol Chem. 2006;281:19793–19797. doi: 10.1074/jbc.C600028200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci. 2007;26:2979–2988. doi: 10.1111/j.1460-9568.2007.05914.x. [DOI] [PubMed] [Google Scholar]