

Abstract
Magnetic resonance imaging (MRI) has long been used clinically and experimentally as a diagnostic tool to obtain three-dimensional, high resolution images of deep tissues. These images are enhanced by the administration of contrast agents such as paramagnetic Gd(III) complexes. Herein we describe the preparation of a series of multimodal imaging agents in which paramagnetic Gd(III) contrast agents are conjugated to a fluorescent tetrapyrrole, namely a porphyrazine (pz). Zinc metaled pzs conjugated to one, four, or eight paramagnetic Gd(III) complexes are reported. Among these conjugates, Zn-Pz-8Gd(III) exhibits an ionic relaxivity four times that of the monomeric Gd(III) agent, presumably because of increased molecular weight, and a molecular relaxivity that is approximately thirty times larger, while retaining the intense electronic absorption and emission of the unmodified pz. Unlike current clinical MR agents, Zn-Pz-1Gd(III) is taken up by cells. This probe demonstrates intracellular fluorescence by confocal microscopy and provides significant contrast enhancement in MR images, as well as marked phototoxicity in assays of cellular viability. These results suggest that pz agents possess a new potential for use in cancer imaging by both MRI and near-infrared (NIR) fluorescence, while acting as a platform for photodynamic therapy.
INTRODUCTION
In the past decade, magnetic resonance imaging (MRI) has become a powerful diagnostic tool in clinical uses such as brain imaging, angiography, and tumor detection. The strength of MRI lies in its ability to produce 3-D anatomical information of human structures non-invasively with exceptional soft tissue contrast.(1–3) Typically, MR contrast agents are applied to enhance the image contrast. Clinical MR contrast agents are primarily Gd(III) complexes that can effectively reduce the relaxation time of water protons, rendering a locally brighter region in an MR image (T1 weighted). Low sensitivity and the lack of cell specificity, however, remain as barriers to realizing the potential of MR probes.
One approach to improve MR agents is through co-registration of cells and tissues by creating a multimodal agent. Exploiting the strengths of two different imaging modalities, such as MRI and near-infrared (NIR) fluorescence imaging, can increase the accuracy of analysis.(4–8) Conjugating probes that are detectable by different techniques creates agents of multimodality. For example, covalent attachment of a fluorescent probe to a MR agent results in a conjugate that reports cellular and subcellular localization via fluorescence while providing deep anatomical information by MRI.(9–10) In this report, we focus on employing a class of NIR molecules known as the porphyrazines (pzs) to serve as the platform for the attachment of Gd(III) MR contrast agents.
Porphyrazines, or tetraazaporphyrins, resemble the porphyrins in structure, but the meso-CH groups linking the pyrrole rings are replaced by nitrogen atoms. The pzs are prepared via a different synthetic route than the porphyrins; this synthetic pathway enables the pzs to be more easily functionalized than the porphyrins.(11) Pzs exhibit intense optical absorption (ε>105) and fluorescence at short wavelengths (~300–400 nm) and NIR wavelengths (~700–900 nm).(12) The NIR emission bands of pzs appear beyond the absorption of hemoglobin (λ~700 nm) and before the onset of absorption by water (λ ~900 nm), minimizing interference in the imaging of tissues.(13) In addition, certain pzs accumulate preferentially in tumor tissues and show marked phototoxicity due to their excellent quantum yield of singlet oxygen generation. These properties make pzs extremely attractive as potential tumor optical imaging and photodynamic therapy (PDT) agents. (14–17)
Herein we exploit the structural flexibility of pzs and apply click chemistry to covalently attach a specific number of Gd(III) complexes to the periphery of zinc metaled pzs. The photophysical properties that make pzs advantageous for biological applications are preserved upon the attachment of Gd(III) complexes, and the resulting Zn-pz-Gd(III) conjugates have improved magnetic relaxivities relative to current clinical MR contrast agents. Furthermore, these modified Zn-pz-Gd(III) complexes exhibit enhanced water solubility compared to previously studied pz agents and can be tailored to exhibit selective cellular uptake. Thus they represent a promising new class of multimodal and multifunctional diagnostic agents for tumor imaging and therapy.
EXPERIMENT PROCEDURES
Materials
All solvents and chemicals were of reagent grade quality, purchased from Sigma-Aldrich Chemical Co., and used without further purification unless otherwise noted. Thin-layer chromatography (TLC) and column chromatography were performed with 25 DC-Plastikfolien Kieselgel 60 F254 (Merck), and Baxter silica gel 60 Å (230–400 mesh), respectively.
Instrumentation
Nuclei magnetic resonance (NMR) spectra were obtained using a Varian Inova 500 MHz spectrometer. Matrix-assisted laser desorption ionization time-of-flight mass spectra (MALDI-TOF-MS) were recorded on a PE Biosystems Voyager System 6050, using 2,5-dihydroxybenzoic acid as the matrix. High performance liquid chromatography (HPLC) analyses and preparation were performed on a Varian Prepstar system (Varian Instruments Inc., USA) using a reverse phase HPLC column (Atlantis C18, Waters, US). Electrospray ionization mass spectra (ESI-MS) were recorded using a Micromass Quattro II Electronic HPLC/MS/MS mass spectrometer. UV-Vis spectra were recorded on an Agilent 8453 UV-Vis spectrometer. Fluorescence measurements were conducted on a Hitachi F-4500 fluorescence spectrophotometer. Relaxivity was measured on a Bruker 60 MHz, at 37 ºC. Doubly distilled deionized water was obtained from a Millipore-Q system to 18 MΩ· cm resistivity.
Synthesis
1-azido-3-aminopropane, Gd(III)-DOTA derivative (MW=595, Gd595), and Pzs 1 and 11 were prepared according to literature. (18–20)
Pz-1acid. H2[Pz(A2); (SC4O2Pr)3(SC4O2H)1], A = 3,6 Bis(isopropyloxy)-1,2-benzo (5)
30 mL 0.4M LiOH aqueous solution was added to a solution of pz 1 (260 mg, 0.20 mmol) in 20 mL THF and stirred at room temperature for 1.5 h. THF was removed by rotary evaporation and 0.1M HCl(aq) was added drop wise until the solution was acidic and followed by extraction with CH2Cl2. The organic phase was dried with Na2SO4 and evaporated under reduced pressure. The resulting residue was purified by column chromatography on silica gel (eluent AcOEt/CH2Cl2 = 5:95, MeOH/CH2Cl2 = 1/99) to give 5 as a green solid (50 mg, 20 %). 145 mg starting material 1 was recovered from the column chromatography. 1H NMR (400 MHz, CDCl3) δ −0.52 (br s, 2H), 0.73 (m, 9H), 1.40 (m, 6H), 1.79 (m, 24H), 1.98 (m, 8H), 2.57 (m, 8H), 3.83 (m, 6H), 4.21 (m, 8H), 5.26 (sept, J = 5.6 Hz, 4H), 7.71 (d, J = 5.6 Hz, 4H); APCI-MS m/z 1245.5 (M + H+) calcd for C61H81N8O12S4 1245.5.
Pz-1azide. H2[Pz(A2); (SC4O2Pr)3(SC4ONHC3N3)1], A = 3,6 Bis(isopropyloxy)-1,2-benzo
Pz-1acid 5 (40 mg, 0.032 mmol), N-hydroxysuccinimide (NHS) (18 mg, 0.16 mmol), and N, N′-dicyclohexylcarbodiimide (DCC) (33 mg, 0.16 mmol) were dissolved in 5 mL of THF and stirred at room temperature overnight. The mixture was concentrated to dryness and re-dissolved in CH2Cl2. The mixture was filtered, and the solid was washed with CH2Cl2. The filtrate and washings were combined, evaporated to dryness under reduced pressure, and dissolved in 4 mL of THF. 1-Azido-3-aminopropane (16 mg, 0.16 mmol), dissolved in 1 mL of THF, was added, and the mixture was stirred at room temperature for 2 hrs. The final mixture was concentrated and purified by TLC on silica gel (eluent CH3OH/CH2Cl2 = 5/95) to give Pz-1azide as a green solid (34 mg, 80%). 1H NMR (500 MHz, CDCl3) δ −0.52 (br s, 2H), 0.75 (m, 9H), 1.45 (m, 8H), 1.81 (m, 24H), 2.00 (sept, J = 7.0 Hz, 8H), 2.57 (t, J = 7.5 Hz, 6H), 2.61 (t, J = 7.5 Hz, 2H), 3.01 (m, 4H), 3.85 (m, 6H), 4.01 (t, J = 6.5 Hz, 2H), 4.22 (t, J = 7.0 Hz, 4H), 4.29 (t, J = 7.0 Hz, 2H), 5.30 (m, 4H), 6.35 (t, J = 6.0 Hz, 1H), 7.59 (m, 4H); 13C NMR (500 MHz, CDCl3) δ 10.5, 22.0, 23.01, 23.03, 23.1, 25.8, 28.9, 30.0, 33.0, 33.2, 34.9, 35.0, 36.9, 49.2, 66.1, 66.3, 72.5, 72.6, 73.6, 118.8, 118.6, 118.4, 120.1, 128.7, 137.6, 139.2, 140.0, 149.4, 149.9, 150.4, 173.0, 173.3, 173.5; MALDI-MS m/z 1327.6 (M + H+) calcd for C64H87N12O11S4 1327.5.
Zn-Pz-1azide. Zn[Pz(A2); (SC4O2Pr)3(SC4ONHC3N3)1], A = 3,6 Bis(isopropyloxy)-1,2-benzo (6)
Pz-1azide (34 mg, 0.026 mmol) and 10 equiv. of zinc chloride were dissolved in 5 mL of DMF. The solution was stirred at room temperature under nitrogen. The metalation procedure was monitored by UV-Vis spectroscopy (24–36 hrs). The mixture was evaporated to dryness under reduced pressure, dissolved in CH2Cl2, and twice washed with H2O. The organic phase was concentrated and purified by TLC on silica gel (eluent CH3OH/CH2Cl2 = 5/95) to give 6 as a green solid (28 mg, 86%). 1H NMR (500 MHz, CDCl3) 0.71 (m, 9H), 1.40 (m, 8H), 1.82 (m, 24H), 1.98 (m, 8H), 2.51 (m, 8H), 3.34 (m, 4H), 3.67 (m, 6H), 4.16 (m, 8H), 5.31 (m, 4H), 6.15 (s, 1H), 7.79 (d, J = 5.0 Hz, 2H), 7.56 (s, 2H); 13C NMR (500 MHz, CDCl3) δ 10.4, 10.5, 22.0, 22.96, 23.04, 23.1, 25.8, 25.9, 30.0, 33.1, 35.0, 36.3, 48.5, 66.1, 66.2, 72.2, 72.4, 73.6, 128.9, 130.0, 140.9, 149.7, 149.9, 150.6, 172.7, 173.55, 173.61; ESI-MS m/z 1389.8 (M + H+) calcd for C64H85N12O11S4Zn 1389.5.
Zn-Pz-1Gd(III). Zn[Pz(A2); (SC4O2Pr)3(SC4ONHC3Gd)1], A = 3,6 Bis(isopropyloxy)-1,2-benzo
Gd595 (17.0 mg, 0.029 mmol), copper sulfate (1.9 mg, 0.012 mmol), sodium ascorbate (3.4 mg, 0.044 mmol) and pz 6 (13.0 mg, 0.009 mmol) were dissolved in 4 mL of water/DMF (v/v=1:1). The reaction mixture was heated in an oil bath at 70 °C overnight. After cooling, the reaction mixture was loaded into a dialysis bag with MWCO 100 and dialyzed for 48 hrs against Millipore water. The final reaction mixture was re-concentrated and passed through a sephadex LH20 column using MeOH as the eluent to give Zn-Pz-1Gd(III) as a green solid (86.1%). MALDI-MS m/z 1985.8 (M + H+) calcd for C83H113GdN17O18S4Zn 1985.6.
Pz-4azide. H2[Pz(A2); (SC4ONHC3N3)4], A = 3,6 Bis(isopropyloxy)-1,2-benzo
Pz 2 (44 mg, 0.039 mmol), N-hydroxysuccinimide (NHS) (90 mg, 0.79 mmol), and N, N′-dicyclohexylcarbodiimide (DCC) (55 mg, 0.79 mmol) were dissolved in 10 mL of THF and stirred at room temperature overnight. The mixture was concentrated to dryness and re-dissolved in CH2Cl2. The resulting suspension was filtered, and the solid was washed with CH2Cl2. The filtrate and washings were combined, evaporated to dryness under reduced pressure, and redissolved in 5 mL of THF. 1-Azido-3-aminopropane (53 mg, 0.79 mmol) in 5 mL of THF was added and the mixture was stirred at room temperature for 2 hrs. The mixture was concentrated and purified by column chromatography on silica gel (eluent CH3OH/CH2Cl2 = 2/98) to give Pz-4azide as a green solid (31 mg, 54%). 1H NMR (500 MHz, CDCl3) δ −0.54 (br s, 2H), 1.49 (m, 8H), 1.79 (d, J = 6.5 Hz, 24H), 1.96 (m, 8H), 2.53 (t, J = 7.5 Hz, 8H), 3.01 (m, 16H), 4.15 (m, 8H), 5.26 (sept, J = 6.0 Hz, 4H), 6.36 (s, 4H), 7.57 (s, 4H); 13C NMR (500 MHz, CDCl3) δ 22.1, 23.1, 26.2, 28.8, 30.0, 35.0, 35.5, 37.0, 49.3, 73.2, 119.5, 129.0, 139.1, 149.9, 172.9; ESI-MS m/z 1447.9 (M + H+) calcd for C64H87N24O8S4 1447.6.
Zn-Pz-4azide. Zn[Pz(A2); (SC4ONHC3N3)4], A = 3,6 Bis(isopropyloxy)-1,2-benzo, (3)
Pz-4azide (29 mg, 0.019 mmol) and 10 equivalents of zinc chloride were dissolved in 5 mL of DMF. The resulting solution was heated to 70 ºC under nitrogen. The metalation procedure was monitored by UV-Vis spectroscopy (24–36 hrs). The mixture was evaporated to dryness under reduced pressure and re-dissolved in CH2Cl2. The solution was washed with equal volume of brine twice. The organic phase was dried over Na2SO4, concentrated under reduced pressure, and purified by TLC on silica gel (eluent CH3OH/CH2Cl2 = 5/95) to give 3 as a green solid (26 mg, 86%). 1H NMR (500 MHz, CDCl3) δ 1.12 (m, 8H), 1.37 (m, 8H), 1.71 (d, J = 6.0 Hz, 24H), 2.00 (m, 8H), 2.51 (m, 8H), 2.83 (t, J = 6.0 Hz, 8H), 3.75 (m, 8H), 5.27 (sept, J = 6.0 Hz, 4H), 6.29 (t, J = 5.5 Hz, 4H), 7.52 (s, 4H); 13C NMR (500 MHz, CDCl3) δ 23.0, 25.7, 28.4, 34.4, 35.2, 36.8, 49.0, 73.3, 119.1, 129.2, 149.7, 154.6, 156.1, 172.8; ESI-MS m/z 1509.3 (M + H+) calcd for C64H85N24O8S4Zn 1509.5.
Zn-Pz-4Gd(III). Zn[Pz(A2); (SC4ONHC3Gd)4], A = 3,6 Bis(isopropyloxy)-1,2-benzo
Gd595 (44 mg, 0.074 mmol), copper sulfate (3.8 mg, 0.024 mmol), sodium ascorbate (8.7 mg, 0.044 mmol) and Zn-Pz-4azide 3 (25.0 mg, 0.017 mmol) were dissolved in 8 ml of water/DMF (v/v=1:1) mixture. The reaction mixture was heated in an oil bath at 70 ºC overnight. After cooling, the reaction mixture was loaded into a dialysis bag with MWCO 100 and dialyzed for 48 hrs against Millipore water. The resulting solution was re-concentrated and run through a Chelax column to ensure the complete removal of copper ions. Finally the product was purified with reverse phase HPLC using water/acetonitrile gradient to give Zn-Pz-4Gd as a green solid (40 mg, 62%). MALDI-TOF-MS m/z 3893.1 (M + H+) calcd for C140H197Gd4N44O36S4Zn 3894.0.
Bis(2-azidoethyl) amine
A solution of bis(2-chloroethyl) amine hydrochloride (3.57 g, 20.0 mmol) and sodium azide (7.80 g, 120 mmol) in 20 mL H2O was heated to 80 ºC for 15 hrs. After removing the water by distillation, the reaction mixture was cooled in an ice bath. Diethyl ether (20 mL) and KOH (pellets 1.6 g) were added in small portions. The solution was stirred at room temperature for 2 hrs. After separation of the organic phase, the aqueous layer was extracted with diethyl ether. The combined organic layers were dried over Na2SO4, concentrated, and purified by column chromatography (eluent AcOEt/CH2Cl2 = 5/95) to give 6 as a pale yellow liquid (2.05 g, 66 %). 1H NMR (500 MHz, CDCl3) δ 1.56 (s, 1H), 2.81 (t, J = 6.0 Hz, 4H), 3.42 (t, J = 6.0 Hz, 4H); 13C NMR (500 MHz, CDCl3) δ 48.4, 51.6, 53.7; ESI-MS m/z 156.2 (M + H+) calcd for C4H9N7 156.1.
Pz-8azide. H2[Pz(A2); (SC4ON(C2N3)2)4], A = 3,6 Bis(isopropyloxy)-1,2-benzo
Pz 2 (64 mg, 0.057 mmol), 1-hydroxybenzotriazol hydrate (HOBT) (62 mg, 0.46 mmol), dicyclohexylcarbodiimide (DCC) (94 mg, 0.46 mmol), and 6 (178 mg, 1.15 mmol) were dissolved in 10 mL of THF and stirred at room temperature for 2 days. The suspension was filtered and washed with cold THF. The combined filtrates were evaporated to dryness and purified by column chromatography on silica gel (eluent CH3OH/CH2Cl2 = 2/98) followed by TLC (eluent CH3OH/CH2Cl2 = 2/98) to give Pz-8azide as a green solid (53 mg, 56%). 1H NMR (500 MHz, CDCl3) δ 0.53 (br s, 2H), 1.80 (d, J = 6.0 Hz, 24H), 1.97 (m, 8H), 2.66 (t, J = 7.0 Hz, 8H), 3.33 (m, 32H), 4.28 (t, J = 6.5 Hz, 8H), 5.27 (sept, J = 6.0 Hz, 4H), 7.58 (s, 4H); 13C NMR (500 MHz, CDCl3) δ 23.1, 25.6, 31.5, 35.2, 46.4, 48.2, 49.5, 50.1, 72.8, 128.8, 129.9, 138.9, 149.9, 173.0; ESI-MS m/z 1667.5 (M + H+) calcd for C68H91N36O8S4 1667.7.
Zn-Pz-8azide. Zn[Pz(A2); (SC4ON(C2N3)2)4], A = 3,6 Bis(isopropyloxy)-1,2-benzo, (4)
Pz-8azide (37 mg, 0.022 mmol) and 10 equivavelents of zinc chloride were dissolved in 5 mL DMF. The solution was heated to 70 ºC under nitrogen protection. The metalation procedure was monitored by UV-Vis spectroscopy (24–36 hrs). The mixture was evaporated to dryness under reduced pressure, and re-dissolved in CH2Cl2. The solution was washed with equal volume of H2O twice. The organic phase was concentrated and purified by thin-layer chromatography on silica gel (eluent CH3OH/CH2Cl2 = 5/95) to give 4 as a green solid (33 mg, 86%). 1H NMR (500 MHz, CDCl3) δ 1.71 (m, 8H), 1.79 (d, J = 6.5 Hz, 24H), 2.44 (m, 8H), 2.95 (m, 16H), 3.10 (m, 16H), 4.10 (m, 8H), 5.33 (sept, J = 6.0 Hz, 4H), 7.58 (s, 4H); 13C NMR (500 MHz, CDCl3) δ 23.0, 25.5, 31.3, 35.2, 46.2, 48.1, 49.2, 49.8, 72.5, 128.9, 149.8, 154.6, 155.8, 173.0; ESI-MS m/z 1729.8 (M + H+) calcd for C68H89N36O8S4Zn 1729.6.
Zn-Pz-8Gd(III). Zn[Pz(A2); (SC4ON(C2Gd)2)4], A = 3,6 Bis(isopropyloxy)-1,2-benzo
Gd595 (97 mg, 0.16 mmol), copper sulfate (5.6 mg, 0.035 mmol), sodium ascorbate (19 mg, 0.098 mmol) and Zn-Pz-8azide 5 (31.0 mg, 0.019 mmol) were dissolved in 10 ml of water/DMF (1:1) mixture. The reaction mixture was heated in an oil bath at 70 ºC overnight. After cooling, the reaction mixture was loaded into a dialysis bag with MWCO 100 and dialyzed for 48 hrs against Millipore water. The resulting solution was re-concentrated and run through a Chelex column to ensure the complete removal of copper ions. The product was purified with reverse phase HPLC using water/acetonitrile gradient to give Zn-Pz-8Gd as a green solid (60 mg, 50%). MALDI-TOF-MS m/z 6496.5 (M + H+) calcd for C220H313Gd8N76O64S4Zn 6498.6.
Relaxivity (r1)
Solutions of Zn-Pz-nGd(III) conjugates were prepared in 500 μL of Millipore water for T1 acquisition. T1 (spin lattice relaxation time) was determined at 60 MHz (1.41 T) and 37 °C using an inversion recovery pulse sequence on a Bruker mq60 Minispec NMR spectrometer with 4 averages, 15 sec repetition time, and 10 data points (Bruker Canada; Milton, Ontario, Canada). The starting and final Gd(III) concentrations of the solutions were determined using inductively coupled plasma-mass spectrometer (ICP-MS). The inverse of the longitudinal relaxation time (1/T1, s−1) was plotted against Gd(III) concentration (mM) and fitted to a straight line with R2 > 0.99. The slope of the fitted line was recorded as the relaxivity, r1.
General Cell Culture
Cell lines were obtained from American Type Culture Collection (Manassas, VA, USA), and all media, Dulbecco’s phosphate buffered saline (DPBS), and 0.25% trypsin/EDTA solutions were purchased from Invitrogen (Carlsbad, CA, USA). Human pulmonary adenocarcinoma cell line (A549) and a SV40 transformed embryonic cell line (WI-38 VA13) were used in this study. The A549 cell line was grown in RPMI 1640 media supplemented with 10% Fetal Calf Serum heat inactivated at 56 °C for 30 minutes, 2 mM L-Glutamine, 100 μg/mL Streptomycin, 100 U/mL Penicillin, and 2.5 mcg/ml Amphotericin B solution. The WI-38 VA13 cell line was grown in Minimum Essential Medium (MEM) with Earle’s salts supplemented with 10% Fetal Calf Serum heat inactivated at 56 °C for 30 minutes, 2 mM L-Glutamine, 100 μg/mL Streptomycin, 100 U/mL Penicillin, and 2.5 mcg/ml Amphotericin B solution, as well as 100 μM MEM nonessential amino acids and 1 mM Sodium Pyruvate. All experiments were performed in the aforementioned cell-specific media in a 5.0% CO2 incubator operating at 37 °C.
Confocal Laser Scanning Microscopy (CLSM)
Cells were seeded on glass cover-slips in round 10 cm2 plates and allowed to grow to 60–80% confluency. Media solutions for staining cells were prepared from a 5 mM solution in water for Zn-Pz-4Gd(III) and Zn-Pz-8Gd(III), in DMSO for Zn-Pz-1Gd(III). After 24 hrs incubation with pz-Gd(III) complexes, cover slips were rinsed twice with PBS and placed onto microscope slides with imaging buffer for live cell imaging. The imaging buffer used for live-cell imaging was prepared using a literature procedure.(21) Images were acquired on a Zeiss LSM 510 inverted microscope (computer controlled using Zeiss Zen software) equipped with a modelocked Mai Tai DeepSee® Ti:sapphire two-photon laser (Spectra Physics, Mountain View, CA, USA). Specifically, pz-Gd(III) conjugates were excited using the 750 nm wavelength of the twophoton laser and imaged with a PMT through a band-pass 390–465 nm emission filter. Images of cells stained with pz-Gd(III)s and associated vehicle controls were taken at the same gain and amplitude settings.
MR Imaging
For cell phantoms, ~1.5×106 cells were incubated with 50 or 100 μM [Gd(III)] concentrations of pz-Gd(III) conjugates for 18 hrs at 37 °C, rinsed three times with DPBS buffer, and harvested with trypsin. After the addition of complete media (1.0 mL total volume), cells were added to flame-sealed 5¾′ Pasteur pipettes and centrifuged at 4.0 °C and 100 × g for 5 min to form pellets with a diameter of approximately 1 mm.
Cell pellets were arranged around a tube of water and imaged on a 4.7 T (200 MHz) Bruker Biospec 4740 MR imager (Bruker Corporation, Billerica, MA). The radiofrequency coil used was a volume coil with 38 mm internal diameter (Rapid MR International, Columbus, OH). Images were acquired by a standard T2-weighted multi-slice multi-echo sequence with TR=750 ms, TE ranging from 15–60 ms, matrix size 128×128, FOV=25×25×1 mm3.
Inductively Coupled Plasma-Mass Spectrometry (ICP-MS)
ICP-MS was performed on either a computer-controlled (Plasmalab software) Thermo PQ ExCell ICP-MS (Thermo Fisher Scientific, Waltham, MA) equipped with a CETAC 500 autosampler or a computer-controlled (Plasmalab software) Thermo X series II ICP-MS equipped with an ESI SC-2 autosampler (Omaha, NE, USA). Each sample was acquired using 1 survey run (10 sweeps) and 3 main (peak jumping) runs (100 sweeps). The isotopes selected were 156,157Gd for acquisition, while 115In, 165Ho, and 209Bi as internal standards for data interpolation and machine stability.
Cell Viability Assays
Zn-Pz-nGd(III) solutions were prepared as 5 mM stock solutions in either DMSO [Zn-Pz-1Gd(III)] or H2O [Zn-Pz-4Gd(III), Zn-Pz-8Gd(III)]. A549 and WI-38 VA13 cells were seeded into 96-well microtiter plates and grown until they were approximately 70% confluent. The plates were treated in the dark with 50, 25, or 12.5 μM pz,-Gd(III) or a volume of DMSO or H2O equivalent to the volume of compound added at these concentrations. The plates were incubated for 72 hours, after which time the media was removed and replaced with 100 μL of a 2 g/L solution of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) in DPBS. The plates were then incubated for 5 hours. During this incubation, purple formazan crystals formed due to the cleaving of the tetrazolium ring of the MTT molecules by the mitochondrial dehydrogenases of viable cells. Following the 5 hour incubation, the supernatant was removed and 100 μL of DMSO was added to each well to dissolve the remaining formazan crystals. A SpectraMax® Plus 384 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA) was used to record the absorbance at 540 nm for each well. Each data point represents the average of at least four microtiter wells for each plate, and a minimum of two independent trials were carried out for each experiment. Data was normalized and averaged such that the final reported values represent at least eight independent values for each reported condition, for each cell line. Data points lying outside of two standard deviations from the mean were removed.
Phototoxicity Assays
Zn-Pz-nGd(III) solutions were prepared as 5 mM stock solutions in either DMSO [Zn-Pz-1Gd(III)] or H2O [Zn-Pz-4Gd(III) and Zn-Pz-8Gd(III)]. Two identical 96-well microtiter plates were seeded with A549 and WI-38 VA13 cells; the cells were established to ~70% confluency. The cells were treated with 50 μM pz-Gd(III), and the plates were incubated for an additional 24 hours. A volume of DMSO or H2O equivalent to 50 μM were used as negative controls, and Photofrin (HpD, 50 μM) was used as a positive control. Following the 24 hours incubation, aluminum foil was wrapped around the sides and top of the first plate, leaving the bottom of the plate uncovered for illumination. The second plate was completely wrapped in aluminum foil to inhibit light from reaching the cells and served as the dark control. The two plates were then placed on top of a standard X-ray illuminator (consisting of four 15 W bulbs, ~3600 total lumens) and exposed to 10 minutes of white light. The plates were placed back into the incubator for an additional 24 hours, after which time MTT cytotoxicity assays were performed as described above. Each data point represents the average of at least four microtiter wells for each plate. Data was normalized and averaged such that the final reported values represent at least four independent values for each reported condition, and for each cell line. Data points lying outside of two standard deviations from the mean were removed.
RESULTS AND DISCUSSION
Synthesis
Since the synthesis of the first optical/MRI contrast agent, a monomeric Gd(III) complex conjugated with tetramethylrhodamine,(22) polymeric MR-optical agents were developed based on polylysine and dextran to increase the sensitivity of the agents.(10, 23) Conjugation of Gd(III) complexes with polymer backbones were carried out under peptide coupling conditions in most cases.(24) These methods, however, normally resulted in a polydispersed mixture with poor batch-to-batch reproducibility, rendering the determination of the Gd(III)-to-fluorophore ratio difficult. In the present work, the coupling procedure adopts click chemistry where multiple Gd(III)-alkyne complexes are simultaneously attached to a pz-azide in a high yield to produce monodispersed MR-optical agents.(19, 25, 26) This click chemistry approach provides a facile means to generate new MRI contrast agents with high relaxivities and discrete molecular weights.
The synthesis starts with A2B2 pz 1, prepared following literature procedures, where ‘A’ has an appended sulfide moiety and ‘B’ is a fused benzo group (Scheme 1).(20) Hydrolysis with excess LiOH(aq) affords 2 with four terminal acids while sub-stoichiometric hydrolysis produces 5 with one terminal acid. Peptide coupling of 2 and 5 with 1-azido-3-aminopropane followed by metalation with zinc chloride yields Zn-Pz-4azide 3 and Zn-Pz-1azide 6, respectively. In a similar fashion, coupling of 2 with bis(2-azido ethyl)amine yields Zn-Pz-8azide 4.
Scheme 1.

The synthetic scheme for the preparation of pz azide substrates. Conditions: (a) LiOH, H2O, THF; (b) DCC, NHS, THF; (c) 1-azido-3-aminopropane, THF; (d) bis(2-azidoethyl) amine, THF; (e) ZnCl2, DMF. (The inserted figure shows the building blocks of A2B2 pz).
Initial attempts to apply click chemistry to metal-free azido-pzs showed that pzs would sequester copper ions, resulting in a mixture of metalated and non-metalated products. Therefore, the pz azides were metalated with zinc(II) prior to conjugation with Gd(III) complexes via click chemistry. Trans-metalation of Zn-Pzs in the presence of catalytic copper salts was not observed. In later attempts, stoichiometric copper salt was used to ensure that pzs were fully metalated and Cu-Pz-nGd(III) (n=4, 8) conjugates were characterized. Cu-Pz and Zn-Pz possess different optical properties, but their Gd(III) conjugates exhibit similarities in relaxivity (Tabel 2 and supporting information, Table SI-1).
Click chemistry between the Gd(III) alkyne analogue, Gd595, and pz substrates 3, 4, and 6 was carried out in a DMF/water (v/v=1:1) mixture (Scheme 2). The reaction was highly efficient in conjugating one, four, and eight Gd595 molecules at the pz periphery in 50–60% yield. The resulting Zn-Pz-4Gd(III) and Zn-Pz-8Gd(III) complexes are freely water soluble suggesting that the conjugates could be administered intravenously without the addition of a co-solvent. Zn-Pz-1Gd(III) exhibited limited water solubility but could be dissolved in a water/DMSO mixture.
Scheme 2.

The synthetic scheme for the preparation of Zn-Pz-nGd(III) (n=1, 4, 8) via click chemistry.
UV absorption and fluorescence
The electronic absorption and emission features of Zn-Pz-nGd(III) (n=1, 4, 8) are summarized in Table 1. Similar to previously reported pzs, Zn-Pz-nGd(III) conjugates display an intense Soret band (340 nm) and a pair of Q–bands (~670 nm, ~780 nm). Both Zn-Pz-4Gd(III) and Zn-Pz-8Gd(III) exhibit dual fluorescence in water (Table 1 and supporting information Figure SI-5). Short wavelength fluorescence was observed at λ~440 nm, and long wavelength near-infrared (NIR) fluorescence occurred at λ~815 nm. With Zn-Pz-1Gd(III), both emission peaks were present in DMSO solution, but the 813 nm emission peak was quenched in aqueous solution (Figure 1 and Figure SI-6).
Table 1.
Summary of UV-Vis/NIR absorption and emission peaks of Zn-Pz-nGd(III) complexes (n=4, 8, in water; n=1, in DMSO).
| Compounds | Absorption/nm | Emission/nm | νa - νf (cm−1) |
|---|---|---|---|
| Zn-Pz-1Gd(III) | 348, 668, 765 | 432, 813 | 772 |
| Zn-Pz-4Gd(III) | 346, 678, 783 | 460, 813 | 471 |
| Zn-Pz-8Gd(III) | 347, 683, 790 | 458, 816 | 403 |
| * Free Pz (1) | 340, 654, 794 | 444, 826 | 489 |
measured in CH2Cl2.
Figure 1.

UV-Vis/NIR absorption and emission spectrum of Zn-Pz-1Gd(III) in DMSO. (dotted line: absorption; continuous line: fluorescence emission; excited at 340 nm).
The NIR emission from a hydrophobic pz such as 1 is typically sensitive to the local polar environment and is quenched by water due to a combination of hydrogen-bonding and aggregation.(27) The observation of NIR fluorescence in water from Zn-Pz-nGd(III) (n=4, 8) compounds suggests that the hydrophilic Gd(III) complexes tethered to the pz periphery protect the pz core from interaction with water and consequent quenching. According to the Lippert-Mataga relationship, a fluorophore’s stokes shift increases with the polarity of its local environment.(28) The stokes shift of the pz-Gd(III) conjugates decrease with added Gd(III)-chelates to the periphery with Zn-Pz-8Gd(III) exhibiting the smallest effective stokes shift (in H2O). This is in agreement with the notion that appended Gd(III) complexes protect the pz core from non-radiative de-excitation in aqueous solutions. Cu-pz-nGd(III) (n=4, 8) conjugates showed weak if at all NIR fluorescence, presumably due to enhanced spin-orbit coupling to the triplet state by the paramagnetic Cu(II). This characteristic of Cu-Pz-Gd(III) limits application in fluorescence imaging (see supporting information Table SI-2, Figure SI-5). However, Cu-Pz-Gd(III) conjugates can serve as model compounds for the preparation of MR-PET (positron emission topography) agents, that use 64Cu as the radionuclide.
Relaxivity properties
The efficiency of a contrast agent in reducing the longitudinal relaxation time of water protons is measured by the property known as relaxivity (mM−1s−1). Several major parameters influence the relaxivity, including number of coordinated water molecules, q; rotational correlation time, τR; and mean water residence time, τm. Solomon-Bloembergen-Morgan theory predicts that an increase in rotational correlation time, τr, by slowing down the molecular tumbling of an agent in solution will result in an increase in relaxivity.(29) A common approach to reduce molecular tumbling is by attaching small Gd(III) complexes to a macromolecule through rigid linkages.(19, 25, 30)
The relaxivities of Zn-Pz-nGd(III) (n=1, 4, 8) at 60 MHz, 37 °C are summarized in Table 2. As expected, the molecular relaxivities of these conjugates increase nonlinearly with the number of appended Gd(III) complexes, presumably because of increasing molecular weights. Zn-Pz-1Gd(III) showed an ionic relaxivity of 4.2 mM−1s−1 per Gd(III), which is slightly higher than the monomeric Gd595. Zn-Pz-4Gd(III) and Zn-Pz-8Gd(III) exhibited the enhanced ionic relaxivities of 10.5 mM−1s−1 and 12.8 mM−1s−1, respectively. These ionic relaxivities are ~3–4 times higher than that of the clinically employed agent DOTA-Gd(III) at the same magnetic field strength (Dotarem, r1 = 2.7–3.1 mM−1 s−1 at 60 MHz, 37 °C; DOTA=1, 4, 7, 10-tetraazacyclododecane-N, N′, N″, N‴-tetraacetic acid), while the molecular relaxivities range up to more than thirty-fold greater.(30) A slight difference in relaxivities was observed between Cu-Pz-nGd(III) and Zn-Pz-nGd(III) (n=4, 8) which is likely due to the paramagnetism of Cu(II) (supporting information Table SI-1). Overall, these conjugates exhibit similar relaxivities to the literature reported polymer-Gd(III) conjugates.(32–34)
Table 2.
Molecular and Gd(III) ionic relaxivity of Zn-Pz-nGd(III) (n=1, 4, 8) complexes in water at 37 °C, 60 MHz.
| Compounds | MW (g/mol) | Gd(III) ionic r1(mM−1 s−1) | No. of Gd(III) | Molecular r1(mM−1 s−1) |
|---|---|---|---|---|
| Gd595 | 595.7 | 3.21 | 1 | 3.21 |
| Zn-Pz-1Gd(III) | 1985.6 | 4.2* | 1 | 4.2* |
| Zn- Pz-4Gd(III) | 3893.9 | 10.5 | 4 | 42 |
| Zn- Pz-8Gd(III) | 6496.9 | 12.8 | 8 | 102.4 |
measured in DMSO/Water, v/v=1:5
Confocal Fluorescence Microscopy
Initial cellular localization and uptake studies of Zn-Pz-nGd(III) conjugates were performed using confocal fluorescence microscopy in WI-38 VA13 cells, a non-tumorigenic, embryonic fibroblast-like human cell line. Cells were incubated with 50 μM Zn-Pz-nGd(III) (n=1, 4, 8) for 24 hours and imaged for both short wavelength (390–465 nm) and NIR (>700 nm) fluorescence. Cells that were stained with Zn-Pz-1Gd(III) displayed the short-wavelength pz fluorescence but very little NIR emission (Figure 2). This result revealed that Zn-Pz-1Gd(III) is incorporated inside the cells. Intracellular pz fluorescence is not observed from Zn-Pz-nGd(III) (n=4, 8) under the same condition, suggesting no cellular uptake of these two conjugates (see supporting information Figure SI-7).
Figure 2.
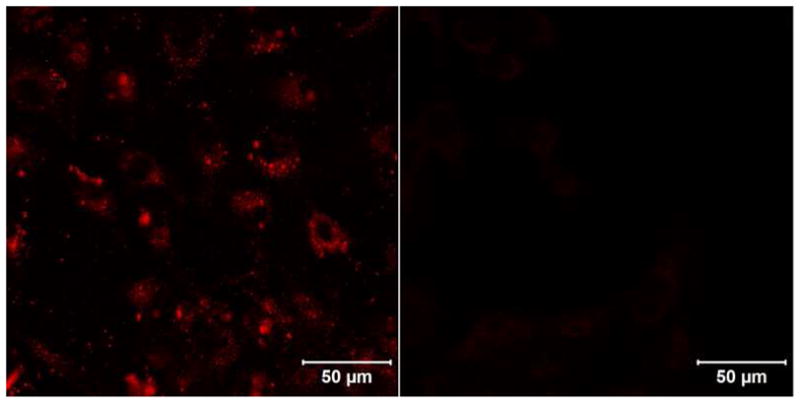
False-red images taken with confocal fluorescence microscopy of WI38-VA13 human embryonic fibroblast-like cells treated with 50 μM Zn-Pz-1Gd(III) for 24 hrs (left) versus vehicle control (right); (750 nm two-photon excitation, 390–465 nm emission).
Although the mechanism for cellular uptake and sub-cellular localization of the pz-Gd(III) conjugates has not been investigated, evidence has shown that low density lipoprotein (LDL) can facilitate cellular uptake of pz analogs.(17) In these previous studies, the hydrophobic portions of pzs were found to interact with LDL, leading to receptor-mediated cellular uptake. It is likely that Zn-Pz-1Gd(III) follows a similar uptake mechanism, involving the hydrophobic pz core of the compound. The fact that Zn-Pz-nGd(III) (n=4, 8) showed no observable cellular uptake further indicates that introducing a higher number of Gd(III) complexes can prohibit cellular uptake, either because of the hindered interaction of the pz with LDL or the increased hydrophilicity. Therefore, MR imaging of cell pellets was carried out with Zn-Pz-1Gd(III).
MR imaging and cell uptake
Cell pellets labeled with the Zn-Pz-1Gd(III) over a range of concentrations were imaged on a Bruker Biospec 4.7 T MR imager. Each cell pellet imaged was ~1 mm in diameter and contained ~1.5×106 cells. Image contrast was highly dependent on the incubation concentration (Figure 3A), with cells incubated at 100 μM of Zn-Pz-1Gd(III) producing a more pronounced contrast enhancement than those incubated at 50 μM in the T1 weighted image.
Figure 3.
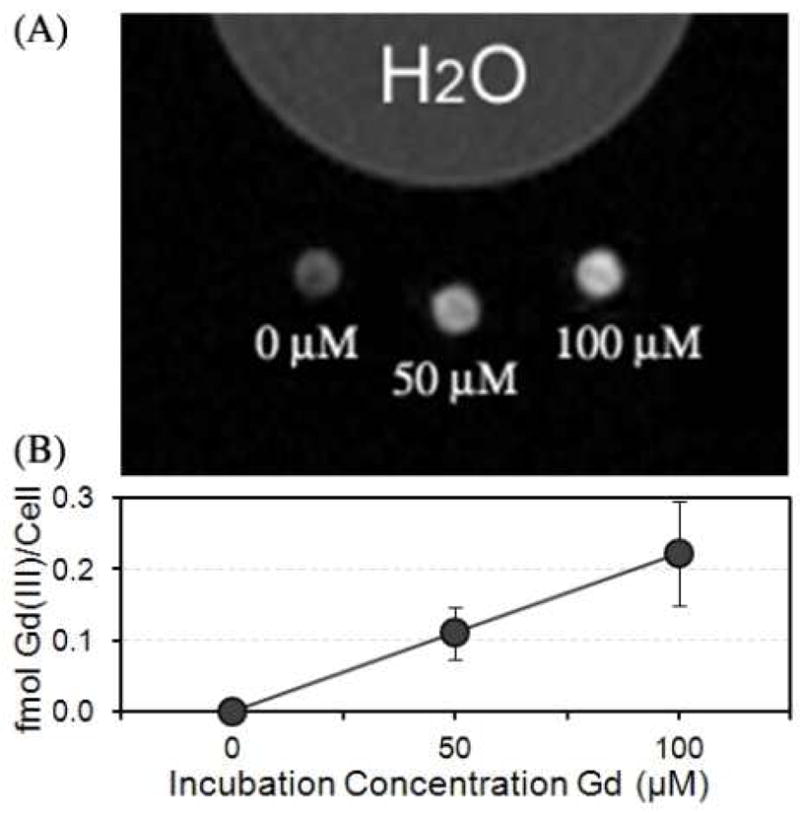
(A) The T1 weighted MR image of WI-38 VA13 cells incubated with 0, 50, and 100 μM of Zn-Pz-1Gd(III) for 24 hours at 4.7 T; (B) cellular uptake of Zn-Pz-1Gd(III) measured by ICP-MS corresponding to the cell pellets in the MR image of (A).
To quantify the Gd(III) content in each cell pellet, ICP-MS was employed to determine the amount of Gd(III) per cell. As shown in Figure 3B, Gd(III) per cell increases linearly with the increase in cell incubation concentration. In addition, we have compared the cellular uptake between Zn-Pz-1Gd(III) and DOTA-Gd(III), which is known to be exogenous (supporting information Figure SI-8). The result shows that WI-38 VA13 cell uptake was ten-fold greater for Zn-Pz-1Gd(III) than for DOTA-Gd(III) when treated under the same conditions. This finding is consistent with cellular incorporation of Zn-Pz-1Gd(III).
Cellular viability
MTT assays were performed with WI-38 VA13 cells exposed to varying concentrations of Zn-Pz-nGd(III) (n=1, 4, 8) over a 72 hour period (Figure 4). Control cells were exposed to water or DMSO according to the solvent of the pz-Gd(III) stock solutions. Zn-Pz-nGd(III) (n=4, 8) showed little effect on cell viability at all three concentrations, namely 12.5, 25, 50 μM, over 72 hours (~90% viable vs. control). Zn-Pz-1Gd(III) exhibited dose dependent toxicity where only 60% of cells remained viable upon 50 μM exposure for 72 hours.
Figure 4.

Concentration-dependent dark toxicity of Zn-Pz-nGd(III) (n =1, 4, 8) in WI-38 VA13 cells. Cells were incubated for 72 hours for all three agents.
Phototoxicity was measured by exposing cells to the conjugates followed by irradiation with white light for 10 minutes (Figure 5). Given that the Zn-Pz-nGd(III) all contain the same core structure, it would be expected that all three compounds would generate similar levels of singlet oxygen and therefore exhibit similar phototoxicity effects when incorporated into cells. However, only Zn-Pz-1Gd(III) was found to show phototoxicity. Approximately 50% of the cells were killed upon treatment of Zn-Pz-1Gd(III) for 24 hours and white light for ten minutes. In contrast, nearly all of the cells were killed upon exposure to Photofrin under the same conditions. The lack of phototoxicity observed for Zn-Pz-4Gd(III) and Zn-Pz-8Gd(III) corresponds to our observation with confocal microscopy and MR contrast that these two conjugates are not taken up by cells, whereas Zn-Pz-1Gd(III) is.
Figure 5.

Phototoxicity of Zn-Pz-nGd(III) (n=1, 4, 8) in WI-38 VA13 cells upon 50 μM pz exposure for 24 hours, followed by 0 or 10 minutes of white light exposure. Photofrin was used as a positive control. It is notable that Zn-Pz-1Gd(III) is the only pz-Gd(III) conjugate showing significant phototoxicity.
CONCLUSION
The measurements of pz-based-MRI multimodal probes described herein highlight numerous properties of this unique class of complexes that make them superior to Gd(III)-based MR contrast agents and optical probes. Unlike previously developed polymeric MR-Optical multimodal agents, the pzs can be characterized and possess strict batch-to-batch consistency. Furthermore, the photophysical properties that make the pz class of molecules attractive for biological applications are preserved upon addition of the Gd(III) complexes, and the water solubility properties of these compounds are enhanced by the addition of the hydrophilic Gd(III) moieties. The pz-Gd(III) conjugates possess improved ionic and molecular relaxitivites relative to well known single molecule MRI agents. Perhaps most importantly, we show that the biological properties of the conjugates can be tailored to achieve cellular uptake of an MR agent, as exhibited by Zn-Pz-1Gd(III).
These studies show that we have the potential to create a single pz [containing Gd(III) complexes] that can not only function as an MRI agent, but also possess a second moiety that can be used to target specific cell types (i.e., tumors). Furthermore, changing the core pz structure and/or metal ion allows us to control the anti-tumor functionality of the compound, suggesting these compounds could be used as PDT agents. Thus, multimodal and multifunctional pzs, such as those described herein, have the potential to serve as dual MRI-NIR/anti-tumor therapeutic agents capable of targeting specific tumor types.
Supplementary Material
Acknowledgments
We thank Ellen K. Kohlmeir and Allison S. Harney for helpful discussions. This work was supported by National Institutes of Health Grant 2R01EB005866-05A1and 5 U54 CA1193 41-02 and by the Baxter Healthcare Corporation through the Baxter-Northwestern Alliance (BMH).
Footnotes
Supporting Information Available: Information on Zn-Pz-Gd(III) conjugates in terms of characterization, cell uptake, toxicity details are free of charge via Internet at http://pubs.acs.org.
LITERATURE CITED
- 1.Mulder WJM, Strijkers GJ, Griffioen AW, van Bloois L, Molema G, Storm G, Koning GA, Nicolay K. A Liposomal system for contrast-enhanced magnetic resonance imaging of molecular targets. Bioconjugate Chem. 2004;15:799–860. doi: 10.1021/bc049949r. [DOI] [PubMed] [Google Scholar]
- 2.Josephson L, Tung CH, Moore A, Weissleder R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjugate Chem. 1999;10:186–191. doi: 10.1021/bc980125h. [DOI] [PubMed] [Google Scholar]
- 3.Mazooz G, Mehlman T, Lai TS, Greenberg Charles S, Dewhirst Mark W, Neeman M. Development of magnetic resonance imaging contrast material for in vivo mapping of tissue transglutaminase activity. Cancer Res. 2005;65:1369–75. doi: 10.1158/0008-5472.CAN-04-2269. [DOI] [PubMed] [Google Scholar]
- 4.Ntziachristos V, Ripoll J, Wang LV, Weissleder R. Looking and listening to light: the evolution of whole-body photonic imaging. Nat Biotechnol. 2005;23:313–320. doi: 10.1038/nbt1074. [DOI] [PubMed] [Google Scholar]
- 5.Bremer C, Ntziachristos V, Weissleder R. Optical-based molecular imaging: contrast agents and potential medical applications. European radiology. 2003;13:231–43. doi: 10.1007/s00330-002-1610-0. [DOI] [PubMed] [Google Scholar]
- 6.Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JWM, Amedio JC, Looby RJ, Supkowski RM, Horrocks WD, McMurry TJ, Lauffer RB. The interaction of MS-325 with human serum albumin and its effect on proton relaxation rates. J Am Chem Soc. 2002;124:3152–62. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- 7.Allen MJ, Meade TJ. Magnetic resonance contrast agents for medical and molecular imaging. Metal Ions in Biological Systems. 2004;42:1–38. [PubMed] [Google Scholar]
- 8.Allen MJ, MacRenaris KW, Venkatasubramanian PN, Meade TJ. Cellular Delivery of MRI Contrast Agents. Chem Biol. 2004;11:301–7. doi: 10.1016/j.chembiol.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Modo MM, Bulte JWW. Molecular and Cellular MR Imaging. CRC Press; FL: 2007. [Google Scholar]
- 10.Frullano L, Meade TJ. Multimodal MRI contrast agents. J Biol Inorg Chem. 2007;12:939–49. doi: 10.1007/s00775-007-0265-3. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Morgade MS, Stuzhin PA. The chemistry of porphyrazines: an overview. J Porphyrins Phthalocyanines. 2004;8:1129–65. [Google Scholar]
- 12.Michel SLJ, Hoffman BM, Baum SM, Barrett AGM. Peripherally functionalized porphyrazines: novel metallomacrocycles with broad, untapped potential. Prog Inorg Chem. 2001;50:473. [Google Scholar]
- 13.Fuchter MJ, Zhong C, Zong H, Hoffman BM, Barrett AGM. Porphyrazines: Designer Macrocycles by Peripheral Substituent Change. Aust J Chem. 2008;61:235–55. [Google Scholar]
- 14.Trivedi ER, Vesper BJ, Weitman H, Ehrenberg B, Radosevich JA, Barrett AGM, Hoffman BM. Chiral bis-Acetal Porphyrazines as Near-Infrared Optical Agents for Detection and Treatment of Cancer. Photochem Photobiol. 2010;86:410–417. doi: 10.1111/j.1751-1097.2009.00681.x. [DOI] [PubMed] [Google Scholar]
- 15.Vesper BJ, Lee S, Hammer ND, Elseth KM, Barrett AGM, Hoffman BM, Radosevich JA. Developing a structure-function relationship for anionic porphyrazines exhibiting selective anti-tumor activity. J Photochem Photobiol, B. 2006;82:180–6. doi: 10.1016/j.jphotobiol.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Hammer ND, Lee S, Vesper BJ, Elseth KM, Hoffman BM, Barrett AGM, Radosevich JA. Charge Dependence of Cellular Uptake and Selective Antitumor Activity of Porphyrazines. J Med Chem. 2005;48:8125–33. doi: 10.1021/jm050466y. [DOI] [PubMed] [Google Scholar]
- 17.Trivedi ER, Harney AS, Olive MB, Podgorski I, Moin K, Sloane BF, Barrett AGM, Meade TJ, Hoffman BM. Chiral porphyrazine near-IR optical imaging agent exhibiting preferential tumor accumulation. Proc Natl Acad Sci. 2010;107:1284–88. doi: 10.1073/pnas.0912811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carboni B, Benalil A, Vaultier M. Aliphatic Amino Azides as Key Building-Blocks for Efficient Polyamine Syntheses. J Org Chem. 1993;58:3736–41. [Google Scholar]
- 19.Song Y, Kohlmeir EK, Meade TJ. Synthesis of Multimeric MR Contrast Agents for Cellular Imaging. J Am Chem Soc. 2008;130:6662–63. doi: 10.1021/ja0777990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S, White AJP, Williams DJ, Barrett AGM, Hoffman BM. Synthesis of Near-IR Absorbing/Emitting Porphyrazine Derivatives with Tunable Solubility. J Org Chem. 2001;66:461–5. doi: 10.1021/jo001220y. [DOI] [PubMed] [Google Scholar]
- 21.Brown PS, Wang E, Aroeti B, Chapin SJ, Mostov KE, Dunn KW. Definition of distinct compartments in polarized Madin-Darby canine kidney (MDCK) cells for membrane-volume sorting, polarized sorting and apical recycling. Traffic (Copenhagen) 2000;1:124–40. doi: 10.1034/j.1600-0854.2000.010205.x. [DOI] [PubMed] [Google Scholar]
- 22.Hueber MM, Staubli AB, Kustedjo K, Gray MHB, Shih J, Fraser SE, Jacobs RE, Meade TJ. Fluorescently Detectable Magnetic Resonance Imaging Agents. Bioconjugate Chem. 1998;9:242–49. doi: 10.1021/bc970153k. [DOI] [PubMed] [Google Scholar]
- 23.Modo M, Cash D, Mellodew K, Williams Steven CR, Fraser Scott E, Meade Thomas J, Price J, Hodges H. Tracking transplanted stem cell migration using bifunctional, contrast agent-enhanced, magnetic resonance imaging. Neuroimage. 2002;17:803–11. [PubMed] [Google Scholar]
- 24.Modo M, Mellodew K, Cash D, Fraser Scott E, Meade Thomas J, Price J, Williams Steven CR. Mapping transplanted stem cell migration after a stroke: a serial, in vivo magnetic resonance imaging study. Neuroimage. 2004;21:311–7. doi: 10.1016/j.neuroimage.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 25.Prasuhn DE, Jr, Yeh RM, Obenaus A, Manchester M, Finn MG. Viral MRI contrast agents: coordination of Gd by native virions and attachment of Gd complexes by azide-alkyne cycloaddition. Chem Commun. 2007;28:1269–1271. doi: 10.1039/b615084e. [DOI] [PubMed] [Google Scholar]
- 26.Bryson JM, Chu WJ, Lee JH, Reineke TM. A β-Cyclodextrin “Click Cluster” Decorated with Seven Paramagnetic Chelates Containing Two Water Exchange Sites. Bioconjugate Chem. 2008;19:1505–1509. doi: 10.1021/bc800200q. [DOI] [PubMed] [Google Scholar]
- 27.Maiti NC, Mazumdar S, Periasamy N. J- and H-Aggregates of Porphyrin-Surfactant Complexes: Time-Resolved Fluorescence and Other Spectroscopic Studies. J Phys Chem B. 1998;102(9):1528–1538. [Google Scholar]
- 28.Mataga N, Kaifu Y, Koizumi M. Solvent effects upon fluorescence spectra and the dipole moments of excited molecules. Bull Chem Soc Japan. 1956;29:465–70. [Google Scholar]
- 29.Toth E, Helm L, Merbach AE. Relaxivity of MRI contrast agents. Top Curr Chem. 2002;221:6–101. [Google Scholar]
- 30.Datta A, Hooker JM, Botta M, Francis MB, Aime S, Raymond KN. High relaxivity gadolinium hydroxypyridonate-viral capsid conjugates: Nanosized MRI contrast agents. J Am Chem Soc. 2008;130:2546–52. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- 31.Rohrer M, Bauer H, Mintorovitch J, Requardt M, Weinmann HJ. Comparison of magnetic properties of MRI contrast media solutions at different magnetic field strengths. Invest Radiol. 2005;40:715–24. doi: 10.1097/01.rli.0000184756.66360.d3. [DOI] [PubMed] [Google Scholar]
- 32.Kiessling F, Heilmann M, Lammers T, Ulbrich K, Subr V, Peschke P, Waengler B, Mier W, Schrenk HH, Bock M, Schad L, Semmler W. Synthesis and Characterization of HE-24.8: A Polymeric Contrast Agent for Magnetic Resonance Angiography. Bioconjugate Chem. 2006;17:42–51. doi: 10.1021/bc0501909. [DOI] [PubMed] [Google Scholar]
- 33.Kaneshiro Todd L, Ke T, Jeong EK, Parker Dennis L, Lu ZR. Gd-DTPA L-cystine bisamide copolymers as novel biodegradable macromolecular contrast agents for MR blood pool imaging. Pharm Res. 2006;23:1285–94. doi: 10.1007/s11095-006-0024-0. [DOI] [PubMed] [Google Scholar]
- 34.Lu ZR, Ye F, Vaidya A. Polymer platforms for drug delivery and biomedical imaging. J Controlled Release. 2007;122:269–277. doi: 10.1016/j.jconrel.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.