

Abstract
Background
Neuroinflammation is involved in several acute-onset neuropathologies such as meningitis, encephalitis, stroke and traumatic brain injury as well as in neurodegenerative diseases. All of these patholologies are associated with cognitive deficits. Using a model of pure neuroinflammation (intracisternal injection of endotoxin in mice), we tested the hypothesis that brain regions involved in cognition are the most vulnerable to inflammatory insults, and this vulnerability is an inherent property of neocortical neurons.
Methods
Mice (N=10/group) injected with endotoxin (LPS) or saline in the cisterna magna underwent neurobehavioral and cognitive testing followed by quantitative autoradiographic assessment of regional neuroinflammation with [3H]PK11195, an established marker of microgliosis. In parallel, co-cultures of cortical and striatal neurons taken from embryonic day 19 rat embryos or postnatal day 1 mice expressing green fluorescent protein were exposed for 24 h to the proinflammatory cytokine TNFalpha, glutamate or a combination of the two agents.
Results
LPS treated mice exhibited significant deficits in memory and significant increases in specific PK11195 binding in cortical and hippocampal regions, but not in striatum. Cultured neurons of cortical origin showed significantly lower survival rate relative to striatal neurons in response to TNFalpha, glutamate or a combination of the two agents. Furthermore, TNFalpha exerted neuroprotective rather than neurotoxic effects in the striatal but not in the cortical neurons.
Conclusions
These results suggest that the cortex is inherently more sensitive than the striatum to the deleterious effects of neuroinflammation, and may offer an explanation for the preponderance of cognitive deficits in neuropathologies with a neuroinflammatory component.
Keywords: Neuroinflammation, Regional sensitivity, Autoradiography, Cognitive deficits, LPS, Peripheral benzodiazepine receptors, translocator protein
Introduction
Neuroinflammation is believed to play a major role in several acute-onset human neuropathologies such as meningitis, encephalitis, stroke and traumatic brain injury as well as chronic neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease. Cognitive deficits, especially memory loss of varying intensity and duration are common to all of these pathologies (Arvin et al., 1996; Floyd et al., 2000; Giulian and Vaca, 1993; Pikis et al., 1996; Raghavendra Rao et al., 2000). In rodents, administration of LPS via different routes was shown to produce changes in behavior (Rosi et al., 2005; Sayyah et al., 2003; Sparkman et al., 2005a; Sparkman et al., 2005b), a loss of cholinergic neurons (Willard et al., 1999) and a loss of NMDA receptors (Biegon et al., 2002). Reports from our (Biegon et al., 2002; Grossman et al., 2003) and other groups (Kim et al., 2000; Raghavendra Rao et al., 2000; Wenk and Barnes, 2000) suggest that the intensity of the neuroinflammatory response to various challenges and the ensuing neuronal damage is highly region dependent, although the mechanisms responsible for this regional sensitivity have not been elucidated to date.
The regional intensity of the neuroinflammatory response can be examined by [3H]PK11195 autoradiography, which was found to be an excellent quantitative marker for neuroinflammation and microgliosis in the rodent and human brain (Chauveau et al., 2008; Chen and Guilarte, 2008; Venneti et al., 2006). The ligand binds to peripheral benzodiazepine receptors (PBR), cholesterol pores located primarily on the mitochondrial membrane of non-neuronal cells in the brain. The density of these receptors is increased when glial cells undergo activation, hypertrophy, and proliferation. Immunohistochemical and histological studies performed in parallel with autoradiography of [3H]PK11195 confirmed a close anatomical and temporal association between the increased expression of these receptors in the central nervous system, the presence of activated microglia and enhanced neuronal death (Maeda et al., 2007; Miyazawa et al., 1995; Raghavendra Rao et al., 2000; Stephenson et al., 1995). Elevation of PBR levels, studied with [11C]PK11195 PET, has been documented in humans with encephalitis, multiple sclerosis, Alzheimer’s disease, Parkinson’s disease and stroke as well as in animal models of brain injury, stroke and intoxication (Banati et al., 1997; Banati et al., 2000; Biegon et al., 2002; Dubois et al., 1988; McGeer et al., 1988; Raghavendra Rao et al., 2000).
The present study was designed to test whether injection of endotoxin (LPS) into the cisterna magna of mice (an animal model of meningitis) is more conducive to neuroninflammation and behavioral deficits related to cognition rather then other neurological function and whether this phenomenon reflects intrinsic differences between cortical and striatal neurons which can be detected in primary neuronal culture.
Materials and Methods
The study was conducted according to the international guidelines and approved by the Institutional Animal Care and Use Committee of the Hebrew University. Adult male Sabra mice (8–9 weeks old) weighing 35–45 g were used in all experiments. Animals were kept under controlled light and dark conditions and given food and water ad libitum. The animals were randomized 1:1 to receive LPS or saline.
Induction of neuroinflammation
Mice (N=10/group) were anesthetized with isofluorane and immobilized manually. Injections of 10μl LPS (16.6 μg Escherichia coli lipopolysaccharide, Serotype 0127:B8, Sigma, USA, in saline; ~0.4mg/Kg body weight) or 10μl saline were given into the cisterna magna by puncture of the alanto-occipital membrane covering the cisterna magna using a 25 μl Hamilton syringe, as previously described (Biegon et al., 2002).
Neurobehavioral evaluation
The functional status of the mice was evaluated according to the Neurological Severity Score (NSS, (Beni-Adani et al., 2001). This score consists of a 10-point scale that assesses the functional neurological status based on the presence of reflexes and the ability to perform various motor and behavioral tasks such as beam walking, beam balance, and spontaneous locomotion. Animals are awarded one point for failure to perform a task, such that scores range from zero to 10, increasing with the severity of dysfunction. The NSS was obtained 24h, 48h 3 and 7 days following intra cisternal injections.
Cognitive function evaluation with the Novel Object Recognition Test
The novel object recognition test (ORT) was performed 48h and 7 days after intra cisternal injection as previously described (Ennaceur and Delacour, 1988). In the first day of the test mice (N=10/group) were placed in the testing cage (a glass container measuring 60 × 25 × 40 cm) for 1 h habituation. On the following day, they were put back into the same enclosure with two identical objects. The objects were all of similar size, surface complexity and material. The cumulative time spent by the mouse at each of the objects was recorded manually and calculated using a specific computer program, during a total of 5-min. Four hours later the mice were reintroduced into the cage, where one of the two objects was replaced by a new one. The time (out of 5 min) spent at each of the objects was recorded. The percent of the total exploration time spent by the mice in exploring the two objects during the testing period was calculated. A normal mouse will spend relatively more time exploring a new object than a familiar, that is, ‘memorized’ object.
In vitro autoradiography
Mice were decapitated 8 days after intra cisternal injection and brains quickly removed and frozen in powdered dry ice. Brains were stored at −80°C, until sectioned in a cryostat (Leica cm1850) in the coronal plane. Six consecutive series of sections (10 μm) were produced at a cutting temperature of −19°C from the prefrontal cortex to cerebellum and thaw- mounted onto coated glass slides. On the day of the assay, sections were removed from the −80°C freezer and allowed to reach room temperature. Peripheral benzodiazepine receptors (PBR) were labeled with [3H] PK11195, (specific activity 85.5 Ci/mmole, Perkin Elmer USA) using a methodology adapted from the literature (Guilarte et al. 1995; Raghavendra Rao et al. 2000) as previously described (Biegon et al 2002). Briefly, sections were first pre-incubated in PBS for 15 min at room temperature, followed by 30 min incubation at room temperature with the radioactive ligand. Total binding was determined with 1 nM [3H] PK11195. Non-specific binding was determined on consecutive sections in the presence of excess (20 μM) unlabeled PK11195. Sections were then washed two times for 6 min in 4°C PBS, then dipped in 4°C deionized water remove buffer salts prior to drying on a slide warmer set to 60 °C. The dried sections were exposed to low-energy radiation sensitive film (Biomax MR film, Kodak) for 3 weeks alongside calibrated tritium scales (Amersham Pharmacia Biotech, Inc., Piscataway, NJ, USA). The films were developed in Kodak D-19, fixed, and dried.
Quantitative image analysis
After film development, all brain sections were stained with Hematoxylin/eosine for histological and anatomical reference. The films were scanned using a flatbed scanner, digitized and saved in 8-bit Tiff format for analysis with ImageJ (NIH) software. Using ImageJ routines, the standard curve was measured and used to calibrate regional brain measurements. Brain regions including amygdala (Bregma −1.70--2.06)., cerebellum (Bregma −5.52--6), cingulate cortex (Bregma 1.34--0.10), dentate gyrus (dorsal), dorsal hippocampus CA1, dorsal hippocampus CA3 (all three Bregma −1.70--2.06), entorhinal cortex (Bregma −2.92--3.40), hypothalamus (Bregma −1.70--2.06), mammillary nucleus (Bregma 1.70–2.06), perirhinal cortex (Bregma −1.70–3.40), preoptic area (Bregma), striatum (Bregma 1.34-0.10), subiculum, substantia innominata/Ventral pallidum, substantia nigra (Bregma-2.92--3.40), temporal cortex (Bregma-1.70--3.40), thalamus (Bregma −1.70--2.06) and ventral hippocampus (Bregma-2.92--3.40) were identified from the histologically stained sections using the Paxinos and Watson (1998) mouse brain atlas and outlined on the autoradiographic image to provide mean radioactive density in nci/mg tissue. Each region was defined by its anatomical border and measured in each section. The mean density for a region was calculated from all the sections in which it was measured, for each animal separately.
Primary Neuronal culture
Mixed striatal-cortical cultures were prepared as previously described (Segal et al., 2003) with some modifications. Briefly, embryonic day 19 (E19) embryos were removed from cervically dislocated pregnant rats. Embryonic striatum or cortex regions were dissected and dissociated, and plated separately on poly-L-lysine-coated glass coverslips, covered with glia, and placed in 24-well plates. The same dissection procedures were carried out in parallel on postnatal day 1 (p1) mice expressing green fluorescent protein (GFP, strain B5/BGFP, where all the cells express GFP). GFP cortical or striatal neurons were then plated on 3-day-old E19 rat striatal or cortical cultures respectively. The number of fluorescent and nonflourescent cells, determined by cell counting before plating, was adjusted to 5×105 nonflourescent cells per well and 5×104 fluorescent cells per well. Cultures were fed with 10% horse serum and were incubated at 37°C with 5% CO2. Two weeks-old cultures were exposed for 24 h to glutamate (10−4M) and/or to the pro-inflammatory cytokine TNFalpha (0.5×10−6 μg/μl) and neuronal survival was assessed by immunohistofluorescence as an endpoint. Using two different species, one of which is expressing GFP enables us to differentiate the origin of the neuron in a co-culture.
Immunofluorescence
Cultures were fixed in 4% paraformaldehyde for 15 min, washed 3 times with phosphate-buffered saline (PBS), and incubated for 1 h in 5% goat serum with 0.03% triton at room temperature and overnight at 4°C with the primary antibody NeuN (1:1000 in 3% goat serum, Chemicon MAB377 500 μg). After washing 3 times with PBS, cultures were incubated for 1h at room temperature with secondary antibody (1:200 in 3% goat serum, Alexia flour 546, goat anti mouse Molecular probes). Cultures were washed with PBS and coverslips were dried and fixed on slides using mounting medium containing Dapi nuclear stain (Santa Cruz biotechnology Inc, USA). Cells were visualized on a IX 81 Olympus inverted microscope, and digitized pictures were taken using velocity software. Eight random fields were taken per 12-mm coverslip for statistical analysis.
Neuronal survival analysis
The in vitro experiments were repeated 3 times with triplicate wells used for each treatment. Each field was represented by 4 different pictures using 3 filters for each color (Dapi-blue for nuclear staining, NeuN- Red for all neurons, green for neurons taken from green fluorescent protein (GFP)- expressing mice and yellow- for merged green and red). The stained neurons were counted in each of the 8 fields from each coverslip and the mean cell number calculated.
Statistical Analysis
The data are expressed as the mean ± SEM. NSS values were analyzed by the Mann-Whitney test. ORT results were examined by one-way analysis of variance (ANOVA). Data from quantitative image analysis and neuronal survival measurements were analyzed by two way ANOVA (treatment and region) followed by post-hoc comparisons with Fisher’s PLSD test. A value of P< 0.05 was considered statistically significant for all comparisons.
Results
Neurological andcognitive impairment in LPS treated mice
At 24h post LPS, NSS of the mice treated with LPS or saline were 4.2±0.5 and 2.9±0.45, respectively (p=0.05). One of the saline-treated mice died shortly after injection, so the results represent 9 control and 10 LPS treated mice. NSS values decreased with time in both groups indicating spontaneous recovery (Fig 1). NSS measured 48h, 3 and 7 days post- LPS was no longer significantly different (p=0.11, 0.13, 0.08 respectively) and there was no further mortality in the LPS or vehicle treated groups
Figure 1. Effect of intracisternal LPS on motor performance.

Motor function was assessed and expressed using the neurological severity score, NSS, as described in Materials and methods. NSS was evaluated from day 1 to 7 after i.c injection of LPS (N=10) or saline (N=9).
*p= 0.05 by Mann-Whitney test vs saline.
Cognitive function was assessed by the ORT 48h and 7 days following intra cisternal injection. When tested 2 days after injection, both groups of mice spent a similar proportion of time (~50%) exploring two identical objects at baseline (Figure 2). However, in the test situation performed 4h later, after a novel object replaced a familiar one, vehicle treated mice spent more of their exploration time at the new object (66.0%±4.2%, p=0.0005 versus baseline). In contrast, the LPS-treated mice failed to prefer the ‘novel’ object over the familiar one (exploration time of the new object 44.8%±4.2%; not different from baseline). Similar results were obtained a week after injection whereby both groups spent equal time at the two objects at baseline, but only the saline group showed significant preference for the novel object in the test situation (saline: 67.1%±2.2%, p=0.0003 versus baseline, LPS: 58.8%%±4.1%, not different from baseline) (Fig 2).
Figure 2. Effect of intracisternal LPS on cognitive performance.
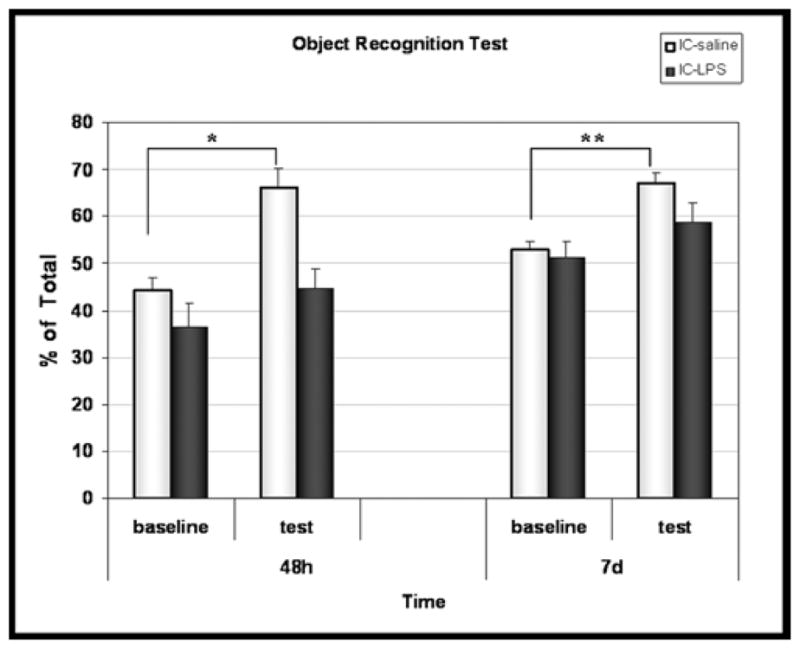
Novel object recognition test (ORT) was performed 2 and 7 days post i.c saline (N=9) or i.c LPS (N=10).
*p=0.0005 **p=0.0003 by Mann-Whitney test, test vs. baseline.
Intracisternal LPS increases PBR density in cortex and hippocampus more than in striatum
The intensity of the neuroinflammatory response to intra-cisternal LPS was assessed by measuring the density of PBR labeled with [3H]PK11195, 8 days after LPS injection. Specific [3H]PK11195 binding was significantly increased in LPS compared to saline treated animals in all brain regions but the increase was highly region dependent (2 way ANOVA by treatment and region, p<0.0001 for both main effects and a significant treatment by region interaction, p<0.0001). Since there were no left-right differences in binding in any of the regions (2 way ANOVA by side and region, p<0.0001 for region, no effect of side) the mean of the right and left side measurements for each animal/region was calculated and used for further analysis of treatment effects within regions (Table 1). The most sensitive regions were the ventral hippocampus (52% increase, p<0.0001), mammillary nucleus (49% increase, p<0.0001) substantia nigra (37% increase, p<0.0001), frontal association cortex (34% increase, p<0.0001), entorhinal cortex (33% increase, p<0.0001), and temporal cortex (30% increase, p=0.0014). Smaller but still significant increases were found in dorsal hippocampus CA1 (23% increase p=0.003), dentate gyrus (21% increase p=0.006), thalamus (22% increase p=0.003), hypothalamus (21% increase p=0.004), amygdale (20% increase p=0.006), and the Perirhinal cortex (19% increase, p=0.001). All other regions investigated showed non-significant increases in PBR, including the striatum, which was one of the least sensitive regions with an increase of 1.3% (non-significant, see Table 1, Fig 3).
Table 1.
Effects of intracisternal LPS on PBR density in various brain regions
| Brain region | Saline | LPS | % increase |
|---|---|---|---|
| Ventral hippocampus | 19.6±1 | 29.7±1.3** | 52 |
| Mammillary nucleus | 32.3±1.8 | 48±3.3** | 49 |
| Substantia nigra | 22.2±2.3 | 30.4±1.8* | 37 |
| Frontal association cortex | 22.8±0.9 | 27.6±1.7* | 34 |
| Entorhinal cortex | 17.4±0.6 | 23.4±1.1** | 33 |
| Temporal cortex | 17.6±0.8 | 22.9±0.8** | 30 |
| Dorsal hippocampus CA1 | 19.5±0.5 | 24.0±1.1* | 23 |
| Thalamus | 18.3±0.4 | 22.4±1* | 22 |
| Dentate gyrus (dorsal) | 31.5±1 | 38.1±1.7* | 21 |
| Hypothalamus | 20.9±0.6 | 25.3±1.7* | 21 |
| Amygdala | 18.9±0.8 | 22.7±0.9* | 20 |
| Perirhinal cortex | 18.1±0.6 | 21.7±0.6** | 19 |
| Cerebellum | 38.7±3.9 | 49.6±4.3 | 28 |
| Cingulate cortex | 20.7±1 | 24±1.2 | 16 |
| Preoptic area | 19.4±1 | 23.8±1.9 | 22 |
| Dorsal hippocampus CA3 | 18.6±1.9 | 21.7±1.6 | 17 |
| Substantia innominata/Ventral pallidum | 17.9±0.6 | 19.2±1.1 | 7 |
| Striatum | 18.1±0.8 | 18.4±0.8 | 1.3 |
| Dorsal Subiculum | 27.3±1.3 | 27.6±2.2 | 1.1 |
Neuroinflammation was induced by injection of LPS into the cisterna magna. Sham controls where injected with saline. Animals were killed 8 days later. Results are mean±SEM of 9 control and 10 LPS-treated animals. PBR specific binding (nCi/mg, derived from tritium standards) was calculated by subtracting non-specific binding from total binding. The regions are arranged in descending order of sensitivity to LPS injection, as expressed by % increase in PBR. Two way ANOVA revealed significant effects of treatment (p<0.0001), region (p<0.0001) and treatment by region interaction (p<0.0001). Within-region comparisons were conducted using Student’s t-test.
p< 0.05
p<0.001
Figure 3. Effect of intracisternal LPS on the regional density of [3H]PK11195 binding.

A. Left column=anatomy(histology), right column=autoradiography. The autoradiograms in the right column compare hemispheres from 2 different animals. The hemispheres on the left are from brains of saline treated animals and the ones on the right form LPS treated mice. Autoradiograms were pseudo-colored using the rainbow spectrum, with the lowest levels in purple and highest levels in red. Images show total binding. Non-specific binding (not shown) was low and homogenous throughout gray matter regions.
CG = cingulate cortex; CPu = caudate-putamen (striatum); TeA = temporal association cortex; Ect = ectorhinal cortex; PRH = perirhinal cortex; LEnt = lateral entorhinal cortex
Differential survival of cortical and striatal neurons after exposure to glutamate
After demonstrating regional sensitivity in-vivo, we proceeded to in-vitro study, using mixed striatal-cortical cultures exposed to the major components of the neuroinflammatory response, namely, the proinflammatory cytokine, TNFalpha, and the excitotoxic neurotransmitter, glutamate.
Two-way ANOVA of percent neuronal survival by region of neuronal origin (cortex or striatum) and pharmacological treatment (TNFalpha, glutamate, TNFalpha+glutmatae or buffer) revealed significant effects of both region (p<0.0001) and treatment (p< 0.0001) as well as a significant region by treatment interaction (p < 0.001). Exposure to glutamate alone reduced mean cortical neuronal survival to 5.6±1.5% of control while striatal neurons, exposed to the same concentration of the toxic mediator demonstrated a nearly four-fold higher survival rate (21±4.9%, p=0.01). Exposure of cortical neurons to the combination of both glutamate and TNFalpha reduced mean survival to (5.2±1%), probably reflecting a floor effect. In contrast, striatal neurons exposed to the combination of glutamate and TNFalpha showed a nearly 8 fold higher survival rate compared to cortical neurons (39.6±9.5%), which was also significantly higher than the survival rate of striatal neurons exposed to glutamate alone (p=0.03). This observation reveals a qualitative, rather than just quantitative, difference between striatal and cortical neurons in response to TNFalpha, whereby TNFalpha exerts a neuroprotective action against glutamate toxicity in striatum but not in cortex. The possible region-specific neuroprotective effect of TNFalpha was also detected in cultures exposed to TNFalpha alone: While treatment with TNFalpha in the cortex reduced survival to about 70% of control, the density of neurons in the striatum increased by about 16% relative to controls (p=0.002, Fig. 4).
Figure 4. Effects of glutamate and TNFalpha on survival of neurons in mixed cortical/striatal neuronal cultures.

A: Co-cultured cortical and striatal neurons were exposed for 24 h to buffer (control, top left panel) glutamate (10-4 M, bottom left panel), TNFalpha (0.5ng/ml, top right panel) or a combination of the two agents (bottom right panel). Cortical cells in this experiment were derived from GFP mice, showing green fluorescence. Mixed Cultures were stained for the neuronal marker NeuN (red). Thus Cortical neurons show up in yellow and striatal neurons show up in red
Note the virtual disappearance of yellow (cortical) neurons and partial survival of red (striatal) neurons in mixed cultures exposed to glutamate alone or with TNFalpha.
B. Neurons were counted in 8 random fields (×10 magnification) from 3 coverslips/treatment. The whole experiment was repeated twice, with mice contributing the cortex in one experiment and the striatum in the second experiment. Results were expressed as % survival relative to the relevant control cultures. Two-way ANOVA showed a significant effect of region (p<0.0001), treatment (p < 0.0001) and region by treatment interaction (p < 0.001).
* p<0.05 Cortex compared to striatum, Student’s t-test, two-tailed
Discussion
Using an animal model of global brain inflammation, we found that injection of LPS into the cisterna magna resulted in a sustained memory deficit measured by the novel object recognition test and relatively minor and transient motor deficits as detected by the NSS.
The NSS was originally developed for head-injured mice by Beni-Adani et al. (Beni-Adani et al., 2001). This 10-point scale was later found to be a reliable measure of injury severity and a predictor of outcome after experimental closed head injury (Tsenter et al., 2008). Intracisternal injection of LPS as well as vehicle resulted in motor deficits equivalent to those seen after mild traumatic brain injury when measured in the first 2 days after injection, probably due to a volume/pressure effect on the spinal cord. Severe deficits were not expected and indeed hemiparesis was not found in any of the mice treated with i.c saline or LPS. The early NSS was higher in the LPS injected mice compared to saline injected controls, but both groups showed rapid spontaneous improvement over time and the difference between the groups was not significant beyond the first day after LPS administration..
Functional damage due to neuroinflammation has been investigated in several studies in mice and rats. Administration of LPS via different routes was shown to produce short-term changes in behavior (“sickness behavior”) including reduced food and water intake and decreased exploration and social interactions (Boje, 1995; Sparkman et al., 2005a; Sparkman et al., 2005b). These behavioral changes can be accompanied by transient motor deficits as measured by decreased swim speed in the Morris water maze (Sparkman et al., 2005b).
In contrast with the transient nature of the motor deficits, the performance of LPS injected mice on the ORT was significantly impaired 2 and 7 days after injection. Unlike saline injected mice, the LPS group showed no significant preference for the novel object on both test days. To our knowledge, this is the first observation of cognitive deficits in this relatively pure and non-invasive model of neuroinflammation, since previous studies of intra cisternal LPS in mice and rats did not employ cognitive or behavioral endpoints (Biegon et al., 2002; Boje, 1995). Cognitive deficits were reported following intraventricular injection of LPS, (Hauss-Wegrzyniak et al., 2000) which involves intraparenchymal damage, and with intraperitoneal injection, which is complicated by peripheral organ involvement and sepsis (Noble et al., 2007; Semmler et al., 2007; Sparkman et al., 2005a; Sparkman et al., 2005b). Our findings are similar to those reported in human brain inflammation, as it is well documented that meningitis and encephalitis in children and adults is accompanied by long lasting deficits in learning ability, memory, language, attention and executive function (Anderson et al., 1997; Carter et al., 2003; Hoogman et al., 2007)
Those higher cognitive functions are intimately associated with areas such as the hippocampus, frontal, entorhinal and temporal cortices in both humans and rodents. Therefore, we have focused next on exploring the regional distribution of neuroinflammation in the same animals. Neuroinflammation was measured eight days post injection since this time point was shown to coincide with peak microglial activation and macrophage infiltration in acute-onset models (Miyazawa et al., 1995). As expected in a global neuroinflammation model, we found evidence of microgliosis (increased binding of [3H]PK11195) throughout the brain of LPS injected mice. However, the size of the increase in PK11195 bindings was region-dependant, with the largest increases observed in hippocampus and cortex and the smallest increase, which did not reach statistical significance in this sample, found in the striatum.
This enhanced sensitivity of hippocampal and cortical regions and relative resistance of the striatum, is similar to our published observations in rats subjected to the same treatment (Biegon et al., 2002) suggesting that this phenomenon is not species specific and may be common in mammalian brain including humans.
Regional sensitivity to neuroinflammation was also reported by Kim et al (2000), who investigated a small number of regions and showed regional differences in the number of resident microglia cells and their activational state. Hauss et al. (2000) reported large increases in microglial number and reactivity in dorsal hippocampus, subiculum, entorhinal, and piriform cortices following chronic intracerebroventricular infusion of LPS. Indeed, in the present work, we, like others (Dubois et al., 1988; Raghavendra Rao et al., 2000) found that the distribution of PBR in normal mouse brain is heterogeneous, although the regional distribution of the response to LPS could not be explained by this difference alone (Biegon et al., 2002)
Several putative explanations are compatible with such regional sensitivity. Since the inflammogen is delivered through the cisterna magna, extraparenchymal factors such as proximity to CSF, the regional density of the vascular network or the effectiveness of the blood brain barrier may contribute to the intensity of neuroinflammation and ensuing neuronal damage. Conversely, the differences may be due to intrinsic properties of the brain cells comprising various brain regions; which may be established in early brain development. To address this possibility, we chose to examine the sensitivity of striatal and cortical neurons to neuroinflammatory mediators in primary culture, where extraparenchymal factors do not play a role. Activated glia are known to secrete glutamate and proinflammatory cytokines, most notably TNFalpha (Ghoshal et al., 2007; Pickering et al., 2005; Qin et al., 2007); so these two agents were chosen for investigation. In this experimental setup, we found that neurons of striatal origin were significantly protected from glutamate and TNFalpha toxicity compared to cortical neurons. Furthermore, TNFalpha concentrations which were toxic to cortical neurons, exerted neuroprotective effects against glutamate excitotoxicity and spontaneous neuronal death in striatal neurons in culture. While enhanced sensitivity of cortical, relative to striatal, neurons to excitotoxicity could be expected based on the higher concentration of glutamate receptors in the cortex relative to striatum (Betarbet et al., 2000; Biegon et al., 2002), the qualitative difference in the response to TNFalpha is novel and unexpected. Since TNFalpha is known to interact with two receptor types, one which mediates cytotoxicity and apoptosis and one that can be cytoprotective (Pickering et al., 2005; Quintana et al., 2005), it is possible that the relative density of these receptor subtypes also varies among brain regions. This possibility as well as alternative or additional explanations needs to be explored in future studies, including investigation of the fate of the other cell types (astrocytes, microglia) present in these regions.
In summary, neuroinflammation is a common pathway in many different acute and chronic human neuropathologies. A better understanding of the regional determinants governing sensitivity or resistance to neuroinflammation may help to identify new targets for treatment or prevention of neuroinflammatory damage in human disease.
Acknowledgments
Supported by a grant from the BSF# 2005-021-01
The authors thank Menahem Segal and Varda Greenberger, of the Weizmann Institute, for their help with the primary neuronal cultures.
Contributor Information
Sigal Liraz-zaltsman, Email: s.lirazaltsman@gmail.com.
Victoria Trembovler, Email: trembovlerv@yahoo.com.
Ianai Fishbein, Email: ianai.fish@gmail.com.
Rami Yaka, Email: Yaka-ramiy@ekmd.huji.ac.il.
Esther Shohami, Email: esty@cc.huji.ac.il.
Anat Biegon, Email: Biegon@bnl.gov.
References
- Anderson V, Bond L, Catroppa C, Grimwood K, Keir E, Nolan T. Childhood bacterial meningitis: impact of age at illness and acute medical complications on long term outcome. J Int Neuropsychol Soc. 1997;3(2):147–158. [PubMed] [Google Scholar]
- Arvin B, Neville LF, Barone FC, Feuerstein GZ. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev. 1996;20(3):445–452. doi: 10.1016/0149-7634(95)00026-7. [DOI] [PubMed] [Google Scholar]
- Banati RB, Myers R, Kreutzberg GW. PK (‘peripheral benzodiazepine’)--binding sites in the CNS indicate early and discrete brain lesions: microautoradiographic detection of [3H]PK11195 binding to activated microglia. J Neurocytol. 1997;26(2):77–82. doi: 10.1023/a:1018567510105. [DOI] [PubMed] [Google Scholar]
- Banati RB, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F, Price G, Wegner F, Giovannoni G, Miller DH, Perkin GD, Smith T, Hewson AK, Bydder G, Kreutzberg GW, Jones T, Cuzner ML, Myers R. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123 (Pt 11):2321–2337. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- Beni-Adani L, Gozes I, Cohen Y, Assaf Y, Steingart RA, Brenneman DE, Eizenberg O, Trembolver V, Shohami E. A peptide derived from activity-dependent neuroprotective protein (ADNP) ameliorates injury response in closed head injury in mice. J Pharmacol Exp Ther. 2001;296(1):57–63. [PubMed] [Google Scholar]
- Betarbet R, Porter RH, Greenamyre JT. GluR1 glutamate receptor subunit is regulated differentially in the primate basal ganglia following nigrostriatal dopamine denervation. J Neurochem. 2000;74(3):1166–1174. doi: 10.1046/j.1471-4159.2000.741166.x. [DOI] [PubMed] [Google Scholar]
- Biegon A, Alvarado M, Budinger TF, Grossman R, Hensley K, West MS, Kotake Y, Ono M, Floyd RA. Region-selective effects of neuroinflammation and antioxidant treatment on peripheral benzodiazepine receptors and NMDA receptors in the rat brain. J Neurochem. 2002;82(4):924–934. doi: 10.1046/j.1471-4159.2002.01050.x. [DOI] [PubMed] [Google Scholar]
- Boje KM. Cerebrovascular permeability changes during experimental meningitis in the rat. J Pharmacol Exp Ther. 1995;274(3):1199–1203. [PubMed] [Google Scholar]
- Carter JA, Neville BG, Newton CR. Neuro-cognitive impairment following acquired central nervous system infections in childhood: a systematic review. Brain Res Brain Res Rev. 2003;43(1):57–69. doi: 10.1016/s0165-0173(03)00192-9. [DOI] [PubMed] [Google Scholar]
- Chauveau F, Boutin H, Van Camp N, Dolle F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35(12):2304–2319. doi: 10.1007/s00259-008-0908-9. [DOI] [PubMed] [Google Scholar]
- Chen MK, Guilarte TR. Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008;118(1):1–17. doi: 10.1016/j.pharmthera.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois A, Benavides J, Peny B, Duverger D, Fage D, Gotti B, MacKenzie ET, Scatton B. Imaging of primary and remote ischaemic and excitotoxic brain lesions. An autoradiographic study of peripheral type benzodiazepine binding sites in the rat and cat. Brain Res. 1988;445(1):77–90. doi: 10.1016/0006-8993(88)91076-1. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31(1):47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Floyd RA, Hensley K, Bing G. Evidence for enhanced neuro-inflammatory processes in neurodegenerative diseases and the action of nitrones as potential therapeutics. J Neural Transm. 2000;(Suppl 60):387–414. doi: 10.1007/978-3-7091-6301-6_28. [DOI] [PubMed] [Google Scholar]
- Ghoshal A, Das S, Ghosh S, Mishra MK, Sharma V, Koli P, Sen E, Basu A. Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia. 2007;55(5):483–496. doi: 10.1002/glia.20474. [DOI] [PubMed] [Google Scholar]
- Giulian D, Vaca K. Inflammatory glia mediate delayed neuronal damage after ischemia in the central nervous system. Stroke. 1993;24(12 Suppl):I84–90. [PubMed] [Google Scholar]
- Grossman R, Shohami E, Alexandrovich A, Yatsiv I, Kloog Y, Biegon A. Increase in peripheral benzodiazepine receptors and loss of glutamate NMDA receptors in a mouse model of closed head injury: a quantitative autoradiographic study. Neuroimage. 2003;20(4):1971–1981. doi: 10.1016/j.neuroimage.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Vannucchi MG, Wenk GL. Behavioral and ultrastructural changes induced by chronic neuroinflammation in young rats. Brain Res. 2000;859(1):157–166. doi: 10.1016/s0006-8993(00)01999-5. [DOI] [PubMed] [Google Scholar]
- Hoogman M, van de Beek D, Weisfelt M, de Gans J, Schmand B. Cognitive outcome in adults after bacterial meningitis. J Neurol Neurosurg Psychiatry. 2007;78(10):1092–1096. doi: 10.1136/jnnp.2006.110023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20(16):6309–6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, Zhang MR, Suzuki K, Suhara T. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007;1157:100–111. doi: 10.1016/j.brainres.2007.04.054. [DOI] [PubMed] [Google Scholar]
- McGeer EG, Singh EA, McGeer PL. Peripheral-type benzodiazepine binding in Alzheimer disease. Alzheimer Dis Assoc Disord. 1988;2(4):331–336. doi: 10.1097/00002093-198802040-00001. [DOI] [PubMed] [Google Scholar]
- Miyazawa N, Diksic M, Yamamoto Y. Chronological study of peripheral benzodiazepine binding sites in the rat brain stab wounds using [3H] PK-11195 as a marker for gliosis. Acta Neurochir (Wien) 1995;137(3–4):207–216. doi: 10.1007/BF02187195. [DOI] [PubMed] [Google Scholar]
- Noble F, Rubira E, Boulanouar M, Palmier B, Plotkine M, Warnet JM, Marchand-Leroux C, Massicot F. Acute systemic inflammation induces central mitochondrial damage and mnesic deficit in adult Swiss mice. Neurosci Lett. 2007;424(2):106–110. doi: 10.1016/j.neulet.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90(5):663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Pikis A, Kavaliotis J, Tsikoulas J, Andrianopoulos P, Venzon D, Manios S. Long-term sequelae of pneumococcal meningitis in children. Clin Pediatr (Phila) 1996;35(2):72–78. doi: 10.1177/000992289603500204. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55(5):453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A, Giralt M, Rojas S, Penkowa M, Campbell IL, Hidalgo J, Molinero A. Differential role of tumor necrosis factor receptors in mouse brain inflammatory responses in cryolesion brain injury. J Neurosci Res. 2005;82(5):701–716. doi: 10.1002/jnr.20680. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp Neurol. 2000;161(1):102–114. doi: 10.1006/exnr.1999.7269. [DOI] [PubMed] [Google Scholar]
- Rosi S, Ramirez-Amaya V, Vazdarjanova A, Worley PF, Barnes CA, Wenk GL. Neuroinflammation alters the hippocampal pattern of behaviorally induced Arc expression. J Neurosci. 2005;25(3):723–731. doi: 10.1523/JNEUROSCI.4469-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyah M, Najafabadi IT, Beheshti S, Majzoob S. Lipopolysaccharide retards development of amygdala kindling but does not affect fully-kindled seizures in rats. Epilepsy Res. 2003;57(2–3):175–180. doi: 10.1016/j.eplepsyres.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Segal M, Greenberger V, Korkotian E. Formation of dendritic spines in cultured striatal neurons depends on excitatory afferent activity. Eur J Neurosci. 2003;17(12):2573–2585. doi: 10.1046/j.1460-9568.2003.02696.x. [DOI] [PubMed] [Google Scholar]
- Semmler A, Frisch C, Debeir T, Ramanathan M, Okulla T, Klockgether T, Heneka MT. Long-term cognitive impairment, neuronal loss and reduced cortical cholinergic innervation after recovery from sepsis in a rodent model. Exp Neurol. 2007;204(2):733–740. doi: 10.1016/j.expneurol.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Sparkman NL, Kohman RA, Garcia AK, Boehm GW. Peripheral lipopolysaccharide administration impairs two-way active avoidance conditioning in C57BL/6J mice. Physiol Behav. 2005a;85(3):278–288. doi: 10.1016/j.physbeh.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Sparkman NL, Kohman RA, Scott VJ, Boehm GW. Bacterial endotoxin-induced behavioral alterations in two variations of the Morris water maze. Physiol Behav. 2005b;86(1–2):244–251. doi: 10.1016/j.physbeh.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15(7 Pt 2):5263–5274. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsenter J, Beni-Adani L, Assaf Y, Alexandrovich AG, Trembovler V, Shohami E. Dynamic changes in the recovery after traumatic brain injury in mice: effect of injury severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J Neurotrauma. 2008;25(4):324–333. doi: 10.1089/neu.2007.0452. [DOI] [PubMed] [Google Scholar]
- Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006;80(6):308–322. doi: 10.1016/j.pneurobio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk GL, Barnes CA. Regional changes in the hippocampal density of AMPA and NMDA receptors across the lifespan of the rat. Brain Res. 2000;885(1):1–5. doi: 10.1016/s0006-8993(00)02792-x. [DOI] [PubMed] [Google Scholar]
- Willard LB, Hauss-Wegrzyniak B, Wenk GL. Pathological and biochemical consequences of acute and chronic neuroinflammation within the basal forebrain cholinergic system of rats. Neuroscience. 1999;88(1):193–200. doi: 10.1016/s0306-4522(98)00216-4. [DOI] [PubMed] [Google Scholar]