

Abstract
The growing evidence that nitroxyl (HNO) has a rich pharmacological potential that differs from that of nitric oxide (NO) has intensified interest in HNO donors. Recently, the diazeniumdiolate (NONOate) based on isopropylamine (IPA/NO; Na[(CH3)2CHNH(N(O)NO)]) was demonstrated to function under physiological conditions as an organic analogue to the commonly used HNO donor Angeli’s salt (Na2N2O3). The decomposition mechanism of Angeli’s salt is dependent on pH, with transition from an HNO to an NO donor occurring abruptly near pH 3. Here, pH is shown to also affect product formation from IPA/NO. Chemical analysis of HNO and NO production led to refinement of an earlier, quantum mechanically based prediction of the pH-dependent decomposition mechanisms of primary amine NONOates such as IPA/NO. Under basic conditions, the amine proton of IPA/NO is able to initiate decomposition to HNO by tautomerization to the nitroso nitrogen (N2). At lower pH, protonation activates a competing pathway to NO production. At pH 8, the donor properties of IPA/NO and Angeli’s salt are demonstrated to be comparable, suggesting that at or above this pH, IPA/NO is primarily an HNO donor. Below pH 5, NO is the major product, while IPA/NO functions as a dual donor of HNO and NO at intermediate pH. This pH-dependent variability in product formation may prove useful in examination of the chemistry of NO and HNO. Furthermore, primary amine NONOates may serve as a tunable class of nitrogen oxide donor.
Keywords: nitroxyl, nitric oxide, NONOate, diazeniumdiolate, IPA/NO, Angeli’s salt
Introduction
Interest in the pharmacological utility of nitroxyl (HNO) has been elevated recently by the observations that HNO induces positive inotropy, vasodilation, and cardioprotection through mechanisms distinct from those of nitric oxide (NO).1, 2 In addition to cardiovascular activity, HNO has been successfully used in pharmacological treatment of alcoholism3 and has been reported to inhibit tumor growth and angiogenesis.4 The growth of the field is evident in the rising numbers of reviews describing both the chemistry and pharmacology of HNO (e.g., references 5–7).
A significant issue in the study of HNO is that dimerization to hyponitrous acid is irreversible due to rapid dehydration (Eq. 1)8, 9 (8 × 106 M−1 s−1)10.
| (1) |
The intrinsic metastability of HNO both complicates direct detection and necessitates that HNO is generated in situ, typically by donor compounds. The most commonly used donors of HNO are Angeli’s salt (Na2N2O3) and derivatives of sulfohydroxamic acid, particularly Piloty’s acid (benzenesulfohydroxamic acid, C6H5SO2NHOH)11. Several clinically used compounds such as cyanamide and hydroxyurea can also be bioactivated to HNO (for reviews on HNO donors, see references 12, 13). While such compounds have provided considerable insight into the chemistry and biology of HNO, their pharmacological utility is limited by formation of by-products with discrete biological activities (e.g., nitrite) or by lack of structural versatility.
Amine-based diazeniumdiolates, or NONOates, were originally synthesized by Drago and colleagues14 and have come into wide use as NO donors15, 16. Secondary amine NONOates are an attractive class of NO donor due to the dependence of the rate of spontaneous decomposition exclusively on amine identity, pH and temperature.17, 18 The ability to produce NO on the second to day timescale has led to extensive use of NONOates in chemical, biochemical, cellular and in vivo experiments.19 The vast majority of NONOates are synthesized from secondary amines,17 and the only primary amine NONOates reported in the literature are based on isopropylamine (IPA/NO) and cyclohexylamine.14 We recently demonstrated that analogously to the oxide-based NONOate Angeli’s salt, decomposition of IPA/NO produces HNO as well as NO.20 Here, we extend the experimental examination of product profile from IPA/NO as a function of pH in order to develop an enhanced understanding of structural attributes that contribute to the decomposition mechanism. The goal of this analysis is to assist in designing more selective donors of HNO or NO for pharmacological applications.
Materials and Methods
Alert: Recommended Preparative Method for the Sodium Salt of IPA/NO
(Na[(CH3)2CHNH(N(O)NO)], sodium 1-(N-isopropylamino)diazen-1-ium-1,2-diolate; modified from reference 15). A solution of 35 g (0.60 mol) of isopropylamine in 60 mL of ether is placed in a 250-mL Parr bottle. The solution is degassed by successive cycles of argon and vacuum exposure, cooled in dry ice, exposed to 40 psi of NO2-free NO and allowed to stir at −80°C for 6 h. The pressure is released, and the product is collected by filtration while still cold. The collected powder is washed with ether and air-dried to give ~10 g (this varies somewhat from run to run) of isopropylammonium 1-(isopropylamino)diazen-1-ium-1,2-diolate. The isopropylammonium salt is placed in a beaker containing 10 mL of methanol, whereupon 12.2 mL (one equivalent) of 25% methanolic sodium methoxide is added, and the resulting mixture is stirred for about 5 min to effect cation exchange. Using exactly one equivalent of base is a crucial step if generation of highly reactive diazoate ion and/or diazoalkane is to be avoided (see below). The resulting solution is treated with 200–500 mL of ether to induce precipitation of 2 g of the sodium salt. The sodium salt of IPA/NO is collected by vacuum filtration, washed with ether and dried under vacuum. Cold storage of solids and stock solutions is recommended for all NONOates but is especially important for primary amine NONOates. Additionally, it is suggested that <250 mg of IPA/NO is stored in a single container due to the potential for decomposition.
Angeli’s salt (Na2N2O3, sodium trioxodinitrate was synthesized and utilized as previously described.21 Concentrations of NONOate stock solutions (>10 mM), prepared in 10 mM NaOH and stored at −20°C, were determined directly prior to use from the extinction coefficients at 250 nm (ε of 8000 M−1 cm−1 for Angeli’s salt15 and 10,000 M−1 cm−1 for IPA/NO20).
Unless otherwise noted, chemicals were purchased from Sigma-Aldrich and used without further purification. Stock solutions other than nitrogen oxide donors were prepared fresh daily at 100× (e.g, 100-fold higher stock concentration than the final reaction concentration) in MilliQ or Barnstead Nanopure Diamond filtered H2O, unless specified. Typically, the assay buffer consisted of the metal chelator diethylenetriaminepentaacetic acid (DTPA, 50 μM) in calcium- and magnesium-free Dulbecco’s phosphate-buffered saline (PBS, pH 7.4). Addition of DTPA has been demonstrated to sufficiently sequester contaminating metals, such that the concentration of NO from Angeli’s salt is less than <0.1%.22 All reactions were performed at 37°C except those measured with the NO-specific electrode, which were run at room temperature. Figures are representative data sets, each from n= 2 individual experiments.
Instrumentation
UV-visible spectroscopy was performed with a Hewlett-Packard 8453 diode-array spectrophotometer. Fluorescence measurements were acquired on a Perkin Elmer LS50B fluorometer or a Thermo Spectronic Aminco Bowman Series2 Luminescence Spectrometer. Electrochemical detection was accomplished with a World Precision Instruments Apollo 4000 system equipped with NO, O2 and H2O2 sensitive electrodes (Sarasota, FL). Chemiluminescent detection of NO was performed with a Sievers 280i NO analyzer (Ionics, Boulder, CO). pH measurements were made with a ThermoElectron Orion 420A+ pH meter.
Rates of NONOate Decomposition
The rate constants of decomposition were measured spectrophotometrically by monitoring the decrease in absorbance near 250 nm, which is characteristic of the NONOate functionality. Assay buffer pH was adjusted prior to use by adding NaOH or HCl as necessary. Although PBS is a relatively weak buffer, this triprotic system was sufficient to maintain the desired pH value from 3–13 at the low concentrations of donor compound used for spectroscopic analysis. The rate profile at 37°C for Angeli’s salt was comparable to that previously published at 25°C using citrate and acetate buffers. Since buffer composition can significantly impact measurable signals from HNO,23 we choose to use phosphate buffer whenever possible. To maintain deaerated conditions, all solutions were transferred using gas-tight Hamilton syringes, and the reaction buffer was sparged with ultra-high purity argon at the rate of ≥1 min for each mL of buffer. Aliquots (2–3 mL) were removed by Hamilton syringe and transferred to an argon-flushed, graded seal quartz cuvette (Spectrocell; Oreland, PA) stoppered with a Suba-Seal septum (Sigma-Aldrich). The buffer within the cuvette was again gently purged with argon for 5 min, and the cuvette was kept under an argon atmosphere for the duration of the experiment.
The spectrophotometer was blanked after warming the cuvette containing assay buffer at the appropriate pH in the instrument heat block (37°C) for 5 min. The reaction was initiated by introducing a small volume of NONOate (≤10 μL of stock in cold 10 mM NaOH), the cuvette was capped and inverted to mix (a pipette was used to mix for measurement of higher rates), and spectra were collected at 0.5–180 s intervals for 1–300 min or until A∞ < 0.05. Kinetic analysis was performed by fitting the data to an exponential decay (A = ΔAe−kt + A∞).
Chemiluminescence and Electrochemical Assays
The relative yields of NO and HNO from Angeli’s salt or IPA/NO were examined via NO-selective chemiluminescence detection24 and with an NO-specific electrode. In the chemiluminescence assay, IPA/NO (10 nmol) was injected by Hamilton syringe into the argon-purged, temperature-controlled reaction vessel, which contained ~6 mL of assay buffer maintained at 37°C. Gases formed during donor decomposition were purged into the detection chamber of the instrument. Triplicate runs were performed in series following signal return to the original baseline. To compensate for varied decay rates, the signal area was estimated by multiplying the maximum peak height (mV) by the peak width at half maximum peak height (s) (integration of the peak is also feasible). Production of HNO was examined by addition of a large excess of glutathione (GSH; 1 mM) to the assay buffer, which was returned to pH 7.4 with 1 M NaOH.
For the electrochemical assay, IPA/NO or Angeli’s salt (5 μM) was injected into the reaction vessel after a baseline for the NO-specific electrode was obtained in room temperature assay buffer (20 mL) of the desired pH. After the signal reached a maximum and began to decline, the assay buffer was replaced, and the process was repeated to obtain several maximum signal values per condition. The production of HNO during decomposition of each donor was estimated by addition of the oxidant ferricyanide (1 mM) to the assay buffer.25
Fluorescence Assay
Two-electron oxidation was evaluated by formation of the fluorescent dye rhodamine 123 from dihydrorhodamine 123 (DHR; Molecular Probes, Eugene, OR) as described previously.26 Briefly, IPA/NO or Angeli’s salt (10 μL of 100× stock) was added to 1 mL of PBS (pH 7.4, 50 μM DTPA) containing DHR (2–100 μM), and the solution was immediately vortexed and incubated for 1 h at 37°C. Subsequently, 1 mL of H2O was added, and fluorescence intensity at 530 nm was measured following excitation at 500 nm. The concentrations of rhodamine produced were determined from a standard curve of fluorescence from authentic rhodamine 123. Double-reciprocal plots of the concentration of total rhodamine produced vs. initial DHR can provide the relative reactivity toward DHR of each oxidative intermediate as described.23, 26 Here, such plots were used for qualitative assessment only to compare the intermediates of IPA/NO and Angeli’s salt.
Results and Discussion
Dissociation Rates of Angeli’s salt and IPA/NO
That NONOates are typically relatively stable in the solid state and in highly alkaline solution is critical for both storage and quantification. Decomposition is generally initiated by dilution of basic stock solutions into less alkaline media. The first-order rate constants for spontaneous decomposition of Angeli’s salt27 or IPA/NO as a function of pH are shown in Figure 1. At 25°C, the rate constants for Angeli’s salt decomposition vary by 20-fold through the pH range studied. Upon measurement at 37°C (data not shown), a similar curve shape was obtained, but the rate constants only varied from 0.0069 s−1 at pH 3 to 0.0015 s−1 at pH 10. At pH 3, Angeli’s salt sharply transitions from an HNO donor to an NO donor,28–30 which is consistent with the sharp rise in decomposition rate below pH 4. In contrast, the rate profile for IPA/NO shows a single broad transition, a slight acceleration at high pH, and a 250-fold variance in rate constant. Additionally, although the rate constants at pH 7.4 and 37°C for Angeli’s salt and IPA/NO are comparable (2.7 × 10−3 s−1, 31 and 1.7 × 10−3 s−1, respectively), IPA/NO functions as both an HNO and NO donor.20 Based on this information and quantum mechanical calculations, Houk and colleagues predicted the pH-dependent mechanisms of decomposition of Angeli’s salt and primary amine NONOates.32, 33 Here, we provide further experimental evidence to both support and expand upon these proposed mechanisms.
Figure 1.

The pH-dependence of the first-order rate constants of decomposition of A) Angeli’s salt (from Hughes and Wimbledon27 (25°C)) or B) IPA/NO (100 μM) at 37°C in PBS (+ 50 μM DTPA) measured at 250 nm (mean ± SEM, n ≥ 3; all R2 values >0.995, log scale inset).
Product Analysis as a Function of pH
Direct detection of HNO requires highly specialized conditions, and quantitation is currently not possible, in part due to metastability (Eq 1). We have published a protocol based on four analytical assays and one biological method for qualitatively analyzing the production of HNO or NO from prospective donor compounds.12 The analytical methods rely on trapping of HNO by a chromophore or fluorophore, on chemiluminescence detection, or on conversion of HNO to NO followed by use of an NO-specific electrode. This protocol was used to demonstrate that unlike Angeli’s salt, IPA/NO is both an HNO and NO donor under physiological conditions.20 Coupled with the dissimilar rate profiles (Figure 1), these data suggest a unique, pH-dependent mechanism for decomposition of primary amine NONOates. Due to sensitivity to pH extremes, the spectroscopic and biological methods are of highest utility near neutral pH. Here, production of HNO and NO as a function of pH was investigated via the electrochemical, chemiluminescence and fluorescence assays.
Electrochemical and chemiluminescence methods are often employed to detect NO due to the relative sensitivity and specificity compared to other techniques.35, 36 Conversion of HNO into NO in the presence of an oxidant such as ferricyanide (Eq. 2) allows use of these techniques for NO detection,25, 34 at least qualitatively.
| (2) |
Outer sphere electron transfer is supported by the relative inertness of ferricyanide and the lack of significant effect on the rate of decomposition of NONOates (data not shown).
In metal-free buffer, significant current intensity from an NO-specific electrode was only observed during decomposition of Angeli’s salt below pH 3 (Figure 2A, blue bars). Addition of ferricyanide elevated the signal intensity at all pH values examined, indicating that at and above pH 3 the major product from Angeli’s salt is HNO (red bars, indicative of both NO and HNO production). These results support prior reports that Angeli’s salt converts from an HNO donor to an NO donor at pH 3.28–30 At pH 2, enhancement of the signal over that in the absence of ferricyanide may suggest minor production of HNO. Reaction of HNO with NO (Eqs. 3–5)10 has a similar initial rate constant to HNO dimerization (Eq. 1) and thus could lower the concentration of detectable product.
Figure 2.

The pH-dependence of maximum current intensity from an NO-specific electrode during decomposition of A) Angeli’s salt (5 μM) or B) IPA/NO (5 μM) in PBS/DTPA (blue bars, NO alone, baseline signal at pH 8, 9) ± 1 mM ferricyanide (red bars, HNO + NO) at room temperature (mean ± SEM, n = 2).
| (3) |
| (4) |
| (5) |
The maximum current intensity in the presence of ferricyanide is presumably dependent on the ratio of the rates of HNO production and consumption, primarily by dimerization. The similar current intensity maxima below pH 8 are indicative of the constant decomposition rate (Figure 1A). At elevated pH, the small deceleration in HNO production may impede dimerization, thus increasing the maximum current. Additionally, it may be that in basic conditions, release of NO as a minor product is inhibited.
The pH-dependence of detectable NO from IPA/NO (Figure 2B) was similarly analyzed. The decrease in maximum NO signal mimics the decrease in decomposition rate with increasing pH. Decomposition of the NO donor diethylamine NONOate (DEA/NO) is sensitive to pH24 similarly to IPA/NO, although with a more than 500-fold variance in rate constant from pH 5 to 10 (reference 37 and data not shown). Observation of a similar trend in signal intensity decline with pH for DEA/NO (data not shown; comparable signals with or without ferricyanide) suggests that the primary influence on signal intensity for IPA/NO, at least to pH 7, is decay rate rather than decreased production of NO. Unfortunately, signal integration, which would indicate total NO production, is not as reliable as maximum signal measurement from the commercial NO electrode.
The current maxima in the presence of ferricyanide are informative. The comparable signals up to pH 5 suggest that IPA/NO is primarily an NO donor at or below pH 5. At pH 7, the elevated signal maximum with ferricyanide suggests significant production of HNO. At pH 8 and 9, signal above baseline was only apparent with ferricyanide, indicating that at alkaline pH production of HNO from IPA/NO increases at the expense of NO release.
Unlike Angeli’s salt, the decomposition rate of IPA/NO is relatively constant at high pH, suggesting that the consumption of HNO by dimerization is relatively constant. Additionally, the rate constant for decomposition is more than six-fold lower for IPA/NO than Angeli’s salt at pH 9 at 37°C. A comparable difference is assumed at 25°C, which explains the lower maximum signal intensity at alkaline pH for IPA/NO. The decrease in maximum current with elevated pH in the presence of ferricyanide is likely a result of an accelerated rate of HNO consumption. Full understanding of the data requires a more detailed study of the interaction of HNO and NO at varied concentrations and ratios. Additionally, at alkaline pH deprotonation of HNO needs to be considered (for full discussion see reference 5). That NO− reacts with NO nearly three orders of magnitude faster (2 × 109 M−1 s−1) than HNO,10, 38 may be significant. Nevertheless, from the data in Figure 2B, the conclusion can be made that unlike Angeli’s salt28, the product crossover for IPA/NO is not sharply delineated; rather, two decomposition pathways are competitive such that IPA/NO is a dual donor of HNO and NO near neutral pH.
NO can also be measured with a commercial chemiluminescence analzyer, which is both substantially more sensitive than the electrode and produces signals that are more amenable to integration. Additionally, the reaction vessel of the analyzer is sparged with argon to sweep product into the detector, which would be expected to increase the lifetimes of reactive, volatile products. To equate product formation from different donors or reaction conditions, the signal area can be approximated by multiplying the maximum peak height by peak width at half maximum height (mV s), which normalizes for varied decomposition rates. The area obtained from IPA/NO was relatively constant from pH 2–6.5 (upon further examination, the decrease at pH 3.5 in the data set was found to be anomalous), but decreased significantly from pH 7 (Figure 3).
Figure 3.

The pH-dependence of chemiluminescence signal area during decomposition at 37°C of IPA/NO (10 nmol; instrument detection is independent of volume) in PBS/DTPA (blue bars, HNO + NO) ± 1 mM GSH (red bars, NO) (mean ± SEM, n = 3).
Interestingly, the commercial instrument is not entirely specific for NO,20 and thus the signal may actually include HNO. The relatively slow reaction rate of HNO with ferricyanide is not amenable to distinguishing HNO from NO in the chemiluminescent analyzer. However, HNO can be scavenged in the analyzer vessel by excess GSH, which reacts directly with HNO (6 × 106 M−1 s−1)39 but not NO. Assuming a trend from pH 2–5 of comparable area in the presence or absence of GSH and decreased area in the presence of GSH compared to buffer alone beginning at pH 5.5 suggests the point where HNO production becomes significant.
A variety of analyses have suggested that HNO reacts with thiolates faster than thiols. Thus, the observed difference in signal area could conceivably be a function of the pKa of GSH (9.0)40. However, the decreased total area beginning at pH 7 even in the absence of GSH suggests the point at which consumption becomes significant compared to sparging from the reaction vessel to the detector. The loss of signal at elevated pH, presumably as the ratio of HNO to NO increases may suggest that rather than HNO itself, the instrument is detecting a product of the reaction of HNO and NO (Eqs. 3–5), although this remains to be determined.
Above pH 3 decomposition of Angeli’s salt is generally accepted to be initiated by protonation of the dianion to produce HNO and nitrite.28, 41, 42 Whereas the pKa for HN2O3− is 9.7,43 electrochemical and chemiluminescence data (Figures 2B and 3) suggest that the most basic pKa for IPA/NO is between 5 and 7. A pKa value of 5.8 was suggested by HCl titration of IPA/NO (10 mM) in 10 mL of water (Figure 4A). Given that NONOate decomposition is initiated by protonation, the titration was performed expeditiously using ice cold solutions to inhibit degradation. Similar results were obtained when beginning with a 10 mM NaOH stock solution of IPA/NO (Figure 4B), which produced a higher starting pH. This titration curve also shows the neutralization of NaOH. These data, which were highly reproducible, indicate that the onset at pH 5.5 of a GSH effect on the chemiluminescence signal (Figure 3) is more indicative of the pKa of IPA/NO rather than of GSH.
Figure 4.

Determination of the pKa for A) solid IPA/NO dissolved in 10 mL of cold nanopure water to produce a 10 mM solution or B) a 10 mM solution of IPA/NO in 10 mM NaOH. Solutions were immediately titrated with cold 10 mM HCl while on ice to limit decomposition. The titration curves showing the first neutralization of IPA/NO are representative of three trials.
Further verification of the pKa for IPA/NO was obtained from the rapid shift in the absorption maximum of the diazeniumdiolate chromophore from 250 nm to 229 nm, which occurs prior to decomposition and is attributed to protonation of the terminal oxygen.37 A simple sigmoidal fit of the data in Figure 5A yielded a pKa of 4.9. Finally, treating the IPA/NO decomposition mechanism as a simple two-term rate law for reaction through protonated and non-protonated states37 and fitting the measured rate constants for decomposition at 37°C (Figure 1B) to equation 6 also yields a kinetically derived value for the IPA/NO pKa of 4.9 (Figure 5B). Given the importance of the rate constant at pH 3 to the fit, the value of 0.103 s−1 was verified in various buffer systems including 100 mM phosphate and 10 or 100 mM citrate. For comparison, similar analysis (not shown) for Angeli’s salt indicated a pKa of 9.2 via initial absorbance maxima and yielded a calculated pKa of 9.1. As for IPA/NO, these values are lower but reasonably similar to the published experimental value of 9.7 for HN2O3−.43
Figure 5.

Variation of A) λmax and B) kobs for IPA/NO as a function of solution pH. Initial absorbance maxima were extracted from the data sets used to produce Figure 1B, and experimental rate constants were fit to equation 6 (kAH = 0.105 M−1 s−1, kA = 0.00137 s−1, pKa = 4.91, R = 0.9985).
| (6) |
Dissociation Mechanisms of Angeli’s Salt and IPA/NO
Based on the rate data in Figure 1 and quantum mechanical calculations, Houk and colleagues predicted that upon NONOate dissolution, equilibria are established between stable and unstable, higher energy isomers of varied protonation states.32, 33 The proposed pH-dependent mechanisms provided derived rate expressions that qualitatively agree with the kinetic data in Figure 1. Here, Schemes 1 and 2 are adapted from references 32 and 33 to accommodate the data shown in Figures 2–4.
Scheme 1.

Dual decomposition mechanisms available for Angeli’s salt leading to release of HNO or NO.
Scheme 2.

Dual decomposition mechanisms available for primary amine NONOates leading to release of HNO or NO.
For Angeli’s dianion (Scheme 1), the most basic position is predicted to be the nitroso oxygen.32 Protonation at this position produces an anion that is thermodynamically stable and kinetically inert to decomposition to HNO and nitrite, and thus a normal acid-base equilibrium is established. In contrast, the higher energy tautomer protonated at the nitroso nitrogen is unstable to cleavage of the N-N bond due to charge distribution, and therefore, although this tautomer is not accumulated significantly in the equilibrium, it is presumed to be the important species leading to HNO formation. The mechanism of interchange between these monobasic species has yet to be established but may involve tautomerization, complex acid-base equilibria or possibly an intermolecular proton transfer.
With increased acidity, a second protonation is again predicted to shift the equilibrium to a stable tautomer, protonated at both the nitroso oxygen and nitrogen. Interchange from this hydroxylamine derivative to a higher energy tautomer doubly protonated at a nitro oxygen leads to formation of NO as the only nitrogen-containing product via spontaneous dehydration.
Product identity in this scheme is dependent on the relative rates of decomposition compared to protonation of monobasic species. The insensitivity of rate to pH between 4–8 (Figure 1A)27 and the sharp transition from HNO to NO donation at pH 3 suggest a mechanism more complex than a simple kinetic competition between two pathways. Based on pKa2 of 9.7,43 the stable dianion is no longer favored in the equilibrium below pH 8. Scheme 1 indicates that a second protonation (pKa1 of 2.5)43 of either of the two important monobasic tautomers will stabilize against HNO formation by impeding both formation and decomposition of the reactive intermediate. It is precisely the complexity of the system that allows for shuttling toward NO production rather than simply producing competing pathways of HNO and NO release (Figure 2A; reference 28). That the decomposition rate increases rapidly with decreased pH indicates the involvement of acid-base equilibria rather than a simple tautomerization of diprotic species.
Substitution of a primary amine for the oxide in Angeli’s salt leads to expected similarities in the decomposition pathways of these NONOates. However in contrast to Angeli’s salt, which can be fully deprotonated to the dianion (Scheme 1), the amine is predicted33 to not deprotonate in aqueous solution (Scheme 2). Thus, tautomerization of the proton from the amine nitrogen to the nitroso nitrogen, which is unstable to cleavage of the NO dimer bond, can occur even in highly alkaline solution as indicated by the pH-independence of the decomposition rate at high pH (Figure 1B). Although decomposition to HNO and a diazoate ion is relatively slow in alkaline solution, primary amine NONOates are inherently less stable than NONOates that do not similarly contain a tautomerizable proton. As illustrated in Scheme 3, the diazoate ion generated in the HNO-producing reaction is expected to protonate and lose water to form the diazonium ion,35,44 which can then lose nitrogen to produce a carbonium ion. This is turn can either deprotonate at the β-carbon to yield an olefin or react with water or other nucleophiles to generate stable adducts such as an alcohol. This type of reactivity underscores the importance of using fully decomposed NONOate solutions as controls.
Scheme 3.

Postulated mechanism of dissociation of IPA/NO to HNO and other products in aqueous media
With increased acidity, the IPA/NO anion is preferentially protonated at the terminal oxygen (Scheme 2).33 Tautomerization to the amine nitrogen induces instability through cleavage of the amine-NO dimer bond, presumably in the reverse of the synthetic pathway. With only a single protonation involved, the acid-base equilibrium is less complicated than for Angeli’s salt. Thus, the pH-dependence of the decomposition rate at intermediate pH is a function of the kinetic competition between tautomerizations either before or after protonation. Primary amine NONOates are thus indicated to function as dual donors of HNO and NO, with product ratios dependent on pH (Figures 2B and 3) and the basicity of the nitroso oxygen (Figure 4). In contrast, secondary amine NONOates will only undergo decomposition through the protonation pathway45 and can be considered to exclusively donate NO.
Donor Chemistry of Angeli’s Salt and IPA/NO
The utility of Angeli’s salt as an HNO donor is thus far unparalleled. That Angeli’s salt functions as solely an HNO or an NO donor is particularly useful. However, new donors are needed to investigate chronic exposure to HNO and to verify that the effects of Angeli’s salt are based on HNO production and not due to nitrite or the dianion itself.
The primary amine substituent not only provides a pH-independent tautomerization pathway to HNO but also lowers the basicity of the nitroso oxygen relative to Angeli’s salt.33 However, production of NO at a higher pH from IPA/NO can be explained by the higher basicity of the IPA/NO nitroso oxygen (pKa of ~5; prior prediction of pKa of 433) compared to the nitroso nitrogen (pKa of 2.5)43 of Angeli’s salt. Based on this analysis, IPA/NO is expected to function as an HNO donor above pH 8, an NO donor below pH 4 and a dual donor at intermediate pH.
The effectiveness of IPA/NO as an HNO donor was assessed by the extent of two-electron oxidation of DHR to rhodamine.26 This fluorescent assay is the final analytical method in our described protocol.12 Oxidation of DHR has been used extensively if somewhat nonspecifically to signal for the presence of reactive oxygen and nitrogen species. The autoxidation products of NO or HNO both react with DHR, but since NO donors induce a nearly negligible signal compared to HNO donors at equimolar concentrations, this assay can be used to distinguish HNO from NO.26
The relative efficiencies of DHR oxidation are readily compared via double-reciprocal plots (Figure 6). Since the rates of decomposition of Angeli’s salt and IPA/NO are comparable,15 the steeper slope23 at pH 7.4 from IPA/NO indicates that less HNO is trapped by O2 compared to Angeli’s salt, presumably due to partial formation of NO. In contrast, the double-reciprocal plots for both NONOates are superimposed at pH 8, suggesting production of similar levels of HNO. Thus, to maximize production of HNO from IPA/NO, a solution of pH 8 is recommended. On the other hand, donation of both HNO and NO at pH 7.4 may prove to be an interesting capability.
Figure 6.

Oxidation of DHR (2–100 μM) by Angeli’s salt (squares; 10 μM) or IPA/NO (circles; 10 μM) at pH 7.4 (open symbols) or 8 (closed symbols, dashed lines). The experiment was performed as described in the Experimental section.
The acid-base relationship of HNO and NO− is rather unique in that proton transfer is spin forbidden. An estimated pKa of >11 for HNO suggests that NO− is the dominant species only in highly alkaline solution.10, 46, but the highly unusual slowness of proton transfer in either direction (kf = 5 × 104 M−1 s−1 and kr = 1 × 102 s−1)10 dictates that any resulting chemistry is actually restricted by these rate constants rather than the pKa (see reference 5 for full discussion). Nonetheless, decomposition of HNO donors including Angeli’s salt at high pH has been indirectly demonstrated to produce NO−, based on spectrophotometric observation of peroxynitrite.10, 42
| (7) |
Decomposition of IPA/NO at pH > 9 results in reasonably clean production of an intermediate with a maximum near 300 nm (Figure 7A), which is attributed to ONOO− (λmax 302 nm),47 via Eq. 7. Gradual diminution of the 300 nm peak in the final two spectra was accompanied by the appearance of an ~220 nm peak, indicative of ONOO− decomposition to nitrite.48 Deaeration, which does not affect the rate of decomposition of IPA/NO, eliminates growth of the ~300 nm peak and the subsequent higher energy absorbance (Figure 7B), supporting identification of the product as ONOO−. Based on ε302 of 1670 M−1 cm−1, 47 the yield of ONOO− in Figure 7A is 70%, presumably due to partial consumption of HNO by dimerization (Eq. 1). The rate of loss of this product is comparable to that of synthetic ONOO− under the same conditions (9.5 and 7.2 × 105 s−1, respectively).
Figure 7.

Decomposition of IPA/NO (100 μM) at 37°C in PBS (+ 50 μM DTPA) of pH 13 either A) in air or B) in deaerated solution. Spectra are shown for A) at 3 min intervals to 21 min then at 27, 33, 39, 48, 57, 69, 105 and 270 min and for B) at 3 min intervals to 21 min then 27, 33, 39, 48, 57, 75, 87 and 165 min.
Synthesis and Storage of Bulk IPA/NO: A Cautionary Note
It has been our experience that IPA/NO preparations can be unstable in the solid state, sometimes decomposing suddenly and without warning or apparent provocation long after isolating the material as a solid. The primary risk is pressure build-up leading to forceful removal of the tops of storage vessels. To minimize explosive hazards, the synthesis is carried out behind a shield, and the solid product is typically stored in amounts of ≤250 mg in vials with a relatively large head space. The accompanying fizzing occasionally heard might be expected for an acid-sensitive diazeniumdiolate that decomposes on protonation according to Scheme 3 to produce a variety of gases including N2O (by HNO dimerization, Eq 1), N2 (by dissociation of the highly energetic diazonium ion), and NO (the solvolysis product favored at low pH in the solution phase). Therefore, we at first sought to prevent such acid-catalyzed decomposition by deliberately adding small amounts of excess base when converting the initial isopropylammonium salt of IPA/NO to the sodium salt. This strategy had previously worked well for stabilizing secondary amine diazeniumdiolates such as DEA/NO in the solid phase, but unfortunately, it generally proved counterproductive for primary amine diazeniumdiolates. For instance, since early attempts at producing straight chain primary amine diazeniumdiolates demonstrated low stability,14 excess base was added in an attempt to improve the yield. For n-butylamine diazeniumdiolate, this procedure accelerated decomposition, resulting in destruction of the storage vessel a few minutes after filtration and transfer to the vial.
Such explosive behavior under basic conditions led us to postulate the formation of a diazoalkane intermediate according to Eq 8. If protonation in the solid state of the diazeniumdiolate ion were to lead to the diazoate, as is assumed in solution, further prototropic shifts as in Scheme 3 could result in diazoalkane production. To verify the presence of diazoate ions in the solid state, we took advantage of their known propensity to form nitrosamines (Eq. 9) on reaction with alkylating agents.49 A sample of IPA/NO whose reactivity had dramatically changed after nine months of storage at −10°C but that was nevertheless visually indistinguishable from the freshly isolated pure material was slurried as the dry powder (51 mg) in 1 mL of iodomethane. After standing at room temperature for 3 d in a dry nitrogen atmosphere, the solid was filtered off, the filtrate was evaporated, and the residue was taken up in dichloromethane for analysis by gas chromatography. Two peaks were observed but neither corresponded to the retention time of O2-methyl IPA/NO (the known methylation product of the IPA/NO anion)50. NMR analysis characterized the major product as N-nitroso-N-isopropylmethylamine (nitrosamine 1, Eq. 9), formed by reaction of isopropanediazoate ion with the iodomethane, and the minor product as N-nitrosodiisopropylamine (nitrosamine 2, Eq. 10). Subsequent coelution with authentic samples confirmed the identities. Nitrosamine 2 is assumed to have formed from the reaction of the diazoate ion with isopropyl cations produced on protolysis of the diazoate as in Scheme 3. We conclude that the initially pure IPA/NO sample in question was completely converted to the corresponding diazoate during the nine months of low-temperature storage, and that some of that diazoate further decomposed during storage to the diazonium ion, which then alkylated adjacent diazoate ions.
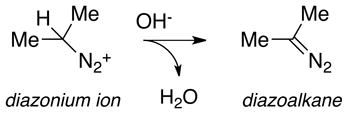 |
(8) |
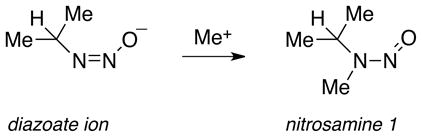 |
(9) |
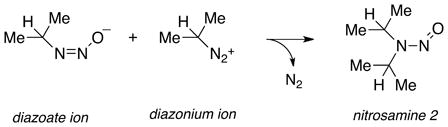 |
(10) |
Given the demonstrated sensitivity of primary amine diazeniumdiolates such as IPA/NO to both acid- and base-induced decomposition, we strongly recommend exactly neutralizing the initially produced alkylammonium salt with sodium methoxide during the ion exchange step of the synthesis procedure. An illustrative protocol for doing so is provided above in the Materials and Methods section.
Conclusion
Here, the primary amine-based NONOate IPA/NO has been shown to release both HNO and NO in a pH-dependent manner. These results, which extend prior physiological data,20 provide experimental evidence for the previously determined theoretical decomposition mechanisms to release HNO or NO.32, 33 It is evident that the nature and basicity of the heteroatom to which the diazeniumdiolate functional group is attached are critical to the dissociation pathways. These analyses may aid in design of primary amine NONOates that release varied ratios of HNO and NO. Such compounds may serve as viable alternatives to Angeli’s salt as HNO donors for pharmacological and biomedical applications. Previous studies on the pharmacological efficacy of IPA/NO in the cardiovascular system20 have demonstrated the ability of this compound to donate HNO in vivo1 to positive effect. Other primary amine NONOates may be useful for investigation of the concomitant effects of HNO and NO. As such, the structural versatility of primary amines is attractive in the pursuit of a series of HNO donors to complement existing NO donors based on secondary amines, which are highly utilized in investigation of fundamental chemistry and design of therapeutic agents.
Acknowledgments
This work was supported by the National Institutes of Health (R01-GM076247 to KMM; 1F31AA018069-01A1 to DJS), by the National Cancer Institute under contract HHSN261200800001E, and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We thank Gail Willette (UA) for technical assistance.
Abbreviations
- Angeli’s salt
sodium trioxodinitrate
- DHR
dihydrorhodamine 123
- DTPA
diethylenetriaminepentaacetic acid
- GSH
glutathione
- HNO
nitroxyl
- IPA/NO
isopropylamine NONOate
- NONOate
diazeniumdiolate
- NO
nitric oxide
- PBS
phosphate-buffered saline
References
- 1.Paolocci N, Saavedra WF, Miranda KM, Martignani C, Isoda T, Hare JM, Espey MG, Fukuto JM, Feelisch M, Wink DA, Kass DA. Proc Natl Acad Sci USA. 2001;98:10463–10468. doi: 10.1073/pnas.181191198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pagliaro P, Mancardi D, Rastaldo R, Penna C, Gattullo D, Miranda KM, Feelisch M, Wink DA, Kass DA, Paolocci N. Free Radical Biol Med. 2003;34:33–43. doi: 10.1016/s0891-5849(02)01179-6. [DOI] [PubMed] [Google Scholar]
- 3.Nagasawa HT, DeMaster EG, Redfern B, Shirota FN, Goon DJ. J Med Chem. 1990;33:3120–3122. doi: 10.1021/jm00174a001. [DOI] [PubMed] [Google Scholar]
- 4.Norris AJ, Sartippour MR, Lu M, Park T, Rao JY, Jackson MI, Fukuto JM, Brooks MN. Int J Cancer. 2008;122:1905–1910. doi: 10.1002/ijc.23305. [DOI] [PubMed] [Google Scholar]
- 5.Miranda KM. Coord Chem Rev. 2005;249:433–455. [Google Scholar]
- 6.Fukuto JM, Switzer CH, Miranda KM, Wink DA. Annu Rev Pharmacol Toxicol. 2005;45:335–355. doi: 10.1146/annurev.pharmtox.45.120403.095959. [DOI] [PubMed] [Google Scholar]
- 7.Wink DA, Miranda KM, Katori T, Mancardi D, Thomas DD, Ridnour LA, Espey MG, Feelisch M, Colton CA, Fukuto JM, Pagliaro P, Kass DA, Paolocci N. Am J Physiol-Heart Circul Physiol. 2003;285:H2264–H2276. doi: 10.1152/ajpheart.00531.2003. [DOI] [PubMed] [Google Scholar]
- 8.Kohout FC, Lampe FW. J Am Chem Soc. 1965;87:5795–5796. [Google Scholar]
- 9.Lin MC, He YS, Melius CF. Int J Chem Kinet. 1992;24:489–516. [Google Scholar]
- 10.Shafirovich V, Lymar SV. Proc Natl Acad Sci USA. 2002;99:7340–7345. doi: 10.1073/pnas.112202099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piloty O. Ber Dtsch Chem Ges. 1896;29:1559–1567. [Google Scholar]
- 12.Miranda KM, Nagasawa HT, Toscano JP. Curr Top Med Chem. 2005;5:649–664. doi: 10.2174/1568026054679290. [DOI] [PubMed] [Google Scholar]
- 13.King SB. Curr Top Med Chem. 2005;5:665–673. doi: 10.2174/1568026054679362. [DOI] [PubMed] [Google Scholar]
- 14.Drago RS, Karstetter BR. J Am Chem Soc. 1961;83:1819–1822. [Google Scholar]
- 15.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie JA, Keefer LK. J Med Chem. 1991;34:3242–3247. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 16.Hrabie JA, Klose JR, Wink DA, Keefer LK. J Org Chem. 1993;58:1472–1476. [Google Scholar]
- 17.Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135–1154. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 18.Thomas DD, Miranda KM, Espey MG, Citrin D, Jourd’heuil D, Paolocci N, Hewett SJ, Colton CA, Grisham MB, Feelisch M, Wink DA. Methods Enzymol. 2002;359:84–105. doi: 10.1016/s0076-6879(02)59174-6. [DOI] [PubMed] [Google Scholar]
- 19.Keefer LK. Annu Rev Pharmacol Toxicol. 2003;43:585–607. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]
- 20.Miranda KM, Katori T, Torres de Holding CL, Thomas L, Ridnour LA, McLendon WJ, Cologna SM, Dutton AS, Champion HC, Mancardi D, Tocchetti CG, Saavedra JE, Keefer LK, Houk KN, Fukuto JM, Kass DA, Paolocci N, Wink DA. J Med Chem. 2005;48:8220–8228. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- 21.Smith PAS, Hein GE. J Am Chem Soc. 1960;82:5731–5740. [Google Scholar]
- 22.Lopez BE, Shinyashiki M, Han TH, Fukuto JM. Free Radical Biol Med. 2007;42:482–491. doi: 10.1016/j.freeradbiomed.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 23.Miranda KM, Yamada K, Espey MG, Thomas DD, DeGraff W, Mitchell JB, Krishna MC, Colton CA, Wink DA. Arch Biochem Biophys. 2002;401:134–144. doi: 10.1016/S0003-9861(02)00031-0. [DOI] [PubMed] [Google Scholar]
- 24.Keefer LK, Nims RW, Davies KM, Wink DA. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 25.Wink DA, Feelisch M, Fukuto J, Christodoulou D, Jourd’heuil D, Grisham MB, Vodovotz Y, Cook JA, Krishna M, DeGraff WG, Kim S, Gamson J, Mitchell JB. Arch Biochem Biophys. 1998;351:66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- 26.Miranda KM, Espey MG, Yamada K, Krishna M, Ludwick N, Kim S, Jourd’heuil D, Grisham MB, Feelisch M, Fukuto JM, Wink DA. J Biol Chem. 2001;276:1720–1727. doi: 10.1074/jbc.M006174200. [DOI] [PubMed] [Google Scholar]
- 27.Hughes MN, Wimbledon PE. J Chem Soc Dalton Trans. 1976;8:703–707. [Google Scholar]
- 28.Bonner FT, Ravid B. Inorg Chem. 1975;14:558–563. [Google Scholar]
- 29.Cambi L. Ber Dtsch Chem Ges. 1936;B69:2027–2033. [Google Scholar]
- 30.Veprek-Siska J, Pliska V, Smirous F, Visely F. Collect Czech Chem Commun. 1959;24:687–693. [Google Scholar]
- 31.Miranda KM, Dutton AS, Ridnour LA, Foreman CA, Ford E, Paolocci N, Katori T, Tocchetti CG, Mancardi D, Thomas DD, Espey MG, Houk KN, Fukuto JM, Wink DA. J Am Chem Soc. 2005;127:722–731. doi: 10.1021/ja045480z. [DOI] [PubMed] [Google Scholar]
- 32.Dutton AS, Fukuto JM, Houk KN. J Am Chem Soc. 2004;126:3795–3800. doi: 10.1021/ja0391614. [DOI] [PubMed] [Google Scholar]
- 33.Dutton AS, Miranda KM, Wink DA, Fukuto JM, Houk KN. Inorg Chem. 2006;45:2448–2456. doi: 10.1021/ic051505z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma XL, Gao F, Liu GL, Lopez BL, Christopher TA, Fukuto JM, Wink DA, Feelisch M. Proc Natl Acad Sci USA. 1999;96:14617–14622. doi: 10.1073/pnas.96.25.14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Archer S. FASEB J. 1993;7:349–360. doi: 10.1096/fasebj.7.2.8440411. [DOI] [PubMed] [Google Scholar]
- 36.Christodoulou D, Kudo S, Cook JA, Krishna MC, Miles A, Grisham MB, Murugesan R, Ford PC, Wink DA. Methods Enzymol. 1996;268:69–83. doi: 10.1016/s0076-6879(96)68010-0. [DOI] [PubMed] [Google Scholar]
- 37.Davies KM, Wink DA, Saavedra JE, Keefer LK. J Am Chem Soc. 2001;123:5473–5481. doi: 10.1021/ja002899q. [DOI] [PubMed] [Google Scholar]
- 38.Gratzel M, Taniguchi S, Henglein A. Ber Bunsenges Phys Chem. 1970;74:1003–1010. [Google Scholar]
- 39.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, Wink DA. Proc Natl Acad Sci USA. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falkner KC, Clark AG. Insect Biochem Molec Biol. 1992;22:917–923. [Google Scholar]
- 41.Angeli A, Angelico F. Gazz Chim Ital. 1903;33:245–252. [Google Scholar]
- 42.Donald CE, Hughes MN, Thompson JM, Bonner FT. Inorg Chem. 1986;25:2676–2677. [Google Scholar]
- 43.Sturrock PE, Ray JD, McDowell J, Hunt HR. Inorg Chem. 1963;2:649–650. [Google Scholar]
- 44.Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS, Keefer LK. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 45.Dutton AS, Fukuto JM, Houk KN. Inorg Chem. 2004;43:1039–1045. doi: 10.1021/ic0349609. [DOI] [PubMed] [Google Scholar]
- 46.Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. Proc Natl Acad Sci USA. 2002;99:10958–10963. doi: 10.1073/pnas.162095599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes MN, Nicklin HG. J Chem Soc A. 1968:450–452. [Google Scholar]
- 48.Pfeiffer S, Gorren AC, Schmidt K, Werner ER, Hansert B, Bohle DS, Mayer B. J Biol Chem. 1997;272:3465–3470. doi: 10.1074/jbc.272.6.3465. [DOI] [PubMed] [Google Scholar]
- 49.Keefer LK, Wang S-M, Anjo T, Fanning JC, Day CS. J Am Chem Soc. 1998;110:2800–2806. [Google Scholar]
- 50.Saavedra JE, Bohle DS, Smith KN, George C, Deschamps JR, Parrish D, Ivanic J, Wang YN, Citro ML, Keefer LK. J Am Chem Soc. 2004;126:2880–12887. doi: 10.1021/ja031538i. [DOI] [PubMed] [Google Scholar]