

Abstract
The contributions of genetics research to the science of normal and defective color vision over the previous few decades are reviewed emphasizing the developments in the 25 years since the last anniversary issue of Vision Research. Understanding of the biology underlying color vision has been vaulted forward through the application of the tools of molecular genetics. For all their complexity, the biological processes responsible for color vision are more accessible than for many other neural systems. This is partly because of the wealth of genetic variations that affect color perception, both within and across species, and because components of the color vision system lend themselves to genetic manipulation. Mutations and rearrangements in the genes encoding the long, middle, and short wavelength sensitive cone pigments are responsible for color vision deficiencies and mutations have been identified that affect the number of cone types, the absorption spectrum of the pigments, the functionality and viability of the cones, and the topography of the cone mosaic. The addition of an opsin gene, as occurred in the evolution of primate color vision, and has been done in experimental animals can produce expanded color vision capacities and this has provided insight into the underlying neural circuitry.
Keywords: color vision, cone photoreceptor, colorblindness, cone mosaic, opsin genes, evolution, comparative color vision, cone photopigments, circuitry
Introduction
Elucidating the neural machinery underlying normal and defective color perception is a difficult problem. It requires an understanding of mechanisms and events at the level of genes, proteins, neurons and circuits; all of which are relatively inaccessible. Most of the available information 25 years ago was measurements of the perceptual responses to visual stimuli and electrical responses of neurons in the visual pathway. The questions in color vision concern the mechanisms responsible for the appearance, detection and discriminability of stimuli of varying wavelength composition. The amount of information about the biology underlying color vision has been greatly increased through the application of the tools of molecular genetics; however, our understanding is still incomplete. In this review, we have made an effort to highlight new results that have come to light. Where possible, we have included new hypotheses that seek to incorporate new discoveries of the last few decades and we have tried to indicate future research directions.
This review focuses on the impact of genetic research on understanding mechanisms of normal and defective color vision. For other aspects of color vision, please refer to the other review articles in this issue. In addition, for a recent review emphasizing human psychophysics, we refer the reader to Stockman and Brainard (2010), for one highlighting primate physiology, Solomon and Lennie (2007), and for comparative primate color vision, Jacobs (2008).
Genes and Photopigments
In 1986, Nathans and colleagues isolated and sequenced the genes encoding the human long wavelength (L), middle wavelength (M) and short wavelength (S) cone opsins and took the first steps toward testing the long-held, two-part hypothesis that (1) variation in the amino acid sequences of the cone opsins are responsible for the spectral differences among the photopigments that all share the same 11-cis retinal chromophore, and (2) alterations in the cone opsin genes underlie inherited color vision deficiencies (Nathans, Piantanida, Eddy, Shows & Hogness, 1986a, Nathans, Thomas & Hogness, 1986b). Findings from these studies both confirmed what previous genetic studies had suggested, and they produced several surprises.
As predicted by inheritance patterns of red-green (Waaler, 1968) and blue-yellow (Kalmus, 1955) color vision deficiencies, the genes for human long-wavelength (L) and middle-wavelength (M) cone opsins were localized to the X-chromosome at Xq28, and the gene for the short-wavelength (S) cone opsin to an autosome, chromosome 7 at 7q32 (Nathans et al., 1986a). The official genetic designations for the L, M and S opsin genes are OPN1LW, OPN1MW, and OPN1SW, respectively. OPN1LW and OPN1MW are arranged in a tandem array (Nathans et al., 1986b, Vollrath, Nathans & Davis, 1988). Among individuals with normal color vision there is variability in the number of OPN1LW and OPN1MW genes per X-chromosome array, with more variability in the number of OPN1MW than in OPN1LW genes; thus, contrary to expectation, most people with normal color vision do not have just one L and one M gene (Drummond-Borg, Deeb & Motulsky, 1989, Macke & Nathans, 1997, Nathans et al., 1986a, Nathans et al., 1986b, Neitz & Neitz, 1995, Neitz, Neitz & Grishok, 1995a, Ueyama, Hayashi, Tanabe, Tanaka, Hayashi, Deeb, Yamade & Ohkubo, 2001). OPN1LW and OPN1MW are nearly identical to one another, sharing more than 98% nucleotide sequence identity, whereas they share only about 40% nucleotide sequence identity with OPN1SW, indicating that OPN1LW and OPN1MW arose via a relatively recent gene duplication (Nathans et al., 1986b). Because of their similarity, the L and M opsin genes are prone to unequal homologous recombination, which has profound implications for visual function.
When Nathans and colleagues began their pioneering work, it was expected that all people with normal color vision would share the same L and the same M pigment. However, as a consequence of recombination that has intermixed the L and M opsin genes, there is variation in the amino acid sequences of both L and M opsins among individuals with normal color vision (Neitz et al., 1995a, Winderickx, Battisti, Hibibya, Motulsky & Deeb, 1993). Several studies have identified amino acid differences that shift the spectral peaks of the L and M cone photopigments and have correlated color vision behavior to variation in the L and M opsin genes (Neitz, Neitz & Kainz, 1996, Neitz, Carroll, Renner, Knau, Werner & Neitz, 2004, Neitz, Neitz & Jacobs, 1995b, Sanocki, Shevell & Winderickx, 1993, Sharpe, Stockman, Jägle, Knau, Klausen, Reitner & Nathans, 1998, Shevell, He, Kainz, Neitz & Neitz, 1998, Winderickx, Lindsey, Sanocki, Teller, Motulsky & Deeb, 1992b).
All eutherian mammalian pigments share the same 11-cis retinal chromophore (Wald, 1967, Wald, 1968). It had been pretty well agreed that binding of the chromophore to opsin red-shifted the chromophore's absorption spectrum, and that amino acid sequence differences among the opsins were responsible for the spectral characteristics of each of the cone pigments (Chen, Nakamura, Ebrey, Ok, Konno, Derguini Nakanishi & Honig, 1989, Kosower, 1988, Wald, 1967). More recent technical innovations made it possible to measure spectral sensitivities of individual cone classes (Baylor, Nunn & Schnapf, 1987, Dartnall, Bowmaker & Mollon, 1983b, Kraft, Neitz & Neitz, 1998, Schnapf, Kraft & Baylor, 1987) and to evaluate the effects of amino acid sequence differences on spectral sensitivity (Asenjo, Rim & Oprian, 1994, Carroll, Neitz & Neitz, 2002, Merbs & Nathans, 1992, Merbs & Nathans, 1993, Merbs, 1992, Neitz et al., 1995b, Sharpe et al., 1998, Stockman, Sharpe, Merbs & Nathans, 2000).
Figure 1 summarizes what is known about spectral tuning of the human L and M cone pigments. The L and M opsin genes each have six exons. The first and sixth exons do not typically vary among or between L and M opsin genes. Exon 5 encodes amino acid dimorphisms at positions 277 and 285 that together are responsible for the majority of the spectral difference between human L and M pigments. Exons 2, 3 and 4 also encode amino acid dimorphisms that produce additional smaller spectral shifts. There is considerable normal variability in the amino acid sequences of the L and M pigments, which in turn produces variability in the absorption spectra (Figure 1A and 1B). The shortest normal M pigment variant is the most common. There is more normal variability in the L pigment and versions with one of two different spectral sensitivities occur with high frequency (Neitz & Jacobs, 1986). The longest L pigment, with a peak of approximately 559 nm, is somewhat more common than the version with the slightly shorter spectral peak (555.5 nm). The identities of the seven spectral tuning codons for the gene encoding the longest normal L and shortest normal M pigment are shown in Figure 1C along with the consequences for spectral sensitivity of substituting each exon from the L pigment into the M pigment and vice versa. Substituting L exons individually into the M pigment tends to produce smaller spectral shifts compared to substituting M exons into L pigments. Exon 2 encoded differences shift the spectra of human L pigments, but do not measurably shift the peak of M pigments. Exons 3 and 4 encode differences that shift the spectra of both human L and M pigments, with relatively slightly larger shifts when substitutions are made in L compared to M pigments (Asenjo et al., 1994, Merbs & Nathans, 1992).
Fig.1.
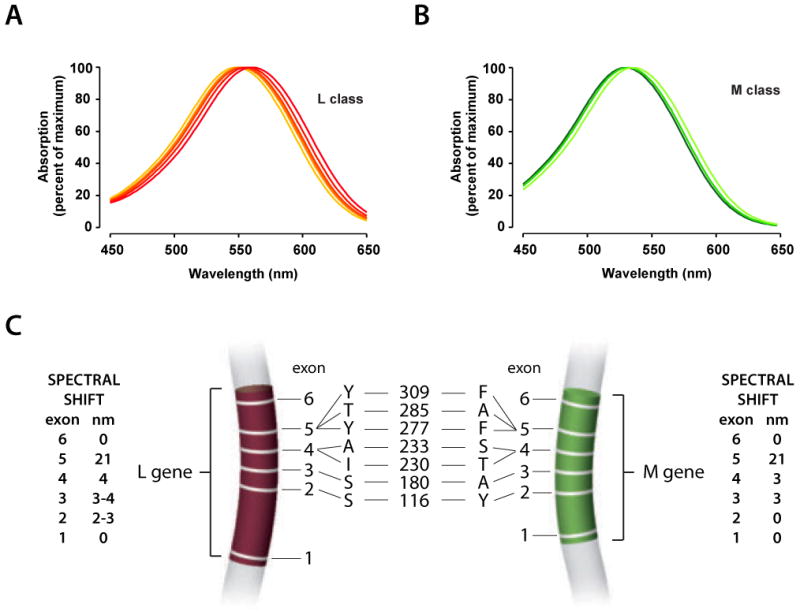
A) Spectral tuning of human L and M cone photopigments. A)L-class pigments absorb maximally near 560 nm. B) M-class pigments absorb maximally near 530 nm. C) Genes encoding the L and M opsins each have 6 exons represented by narrow white bars and numbered 1 through 6. The colored regions indicate the exons in the L and M opsin genes. The genes are drawn to scale. Codons that specify amino acids involved in spectral tuning are indicated using the single letter amino acid code and the codon number/amino acid position is indicated by the numbers in the middle of the panel. The single letter amino acid code is as follows: Y = tyrosine, T=threonine, A=alanine, I=isoleucine, S=serine, F=phenylalanine. The magnitude of the spectral shift in nanometers (nm) produced by the indicated amino acid differences specified by each exon are indicated on the far right and far left.
Seven amino acids, all encoded by exon 5, are typically different between human L versus M opsins, of which 3, indicated in Figure 1C, are involved in spectral tuning (Asenjo et al., 1994, Merbs & Nathans, 1992, Neitz, Neitz & Jacobs, 1991). L pigments have the amino acid tyrosine (Y) at positions 277 and 309, and threonine (T) at position 285; M pigments have phenylalanine (F) at positions 277 and 309, and alanine (A) at 285. Thus, the identity of exon 5 separates the pigments into two clusters, the L class and the M class (Figure 1A & B). Depending on the amino acids at other tuning sites, pigments with Y277 and T285 have peak sensitivities as long as 559 nm and pigments with F277 and A285 have peak sensitivities as short as 530 nm. Together, the substitutions at positions 277 and 285 account for about 20 nm of the difference in peak sensitivity of human L versus M pigments. F versus Y at position 309 shifts the peak sensitivity by only about 1 nm (Asenjo et al., 1994).
Exons 2, 3 and 4 all encode amino acid differences responsible for variability within L-class and M-class pigments. Exon 2 encodes 3 amino acid polymorphisms at positions 65, 111, and 116. The dimorphism at position 116 is the spectrally active one of the three (Asenjo et al., 1994). L pigments with serine at position at 116 (S116) are red-shifted by 2 to 3 nm compared to those with tyrosine. As introduced above, there are context effects such that size of the spectral shift produced by a particular substitution depends on the identity of the other amino acids in the pigment. Thus, even though a T116S substitution in an L class pigment produces a shift, an S116T substitution in the M pigment does not produce a measureable shift.
Of the 5 dimorphic amino acid positions specified by exon 3, the most variable exon, only serine (S) versus alanine (A) at position 180 produces a measurable shift in the spectrum. Pigments with S180 have peak sensitivities at longer wavelengths relative to pigments with A180. As with all the amino acids that produce variability within L and M pigment classes, the absolute magnitude of the spectral shift is a little larger when an S180A substitution occurs in an L pigment versus when an A180S substitution occurs in an M pigment (Asenjo et al., 1994, Merbs, 1992, Neitz et al., 1991). The variability in normal L pigments, introduced above, is evident in Rayleigh color matches (Neitz & Jacobs, 1986) and it is principally the result of the S/A difference at position 180 of the L pigment (Neitz, Neitz & Jacobs, 1993, Piantanida & Gille, 1992, Winderickx et al., 1992b).
Exon 4 specifies dimorphisms at three amino acid positions, 230, 233 and 236, two of which have an identified role in spectral tuning. Pigments with isoleucine at position 230 and alanine at position 233 are red shifted compared to pigments with threonine 230 and isoleucine 233, and the magnitude of the spectral shifts are larger in L than in M pigments (Asenjo et al., 1994, Carroll et al., 2002, Merbs, 1992).
Because OPN1LW and OPN1MW genes are on the X-chromosome, females who are heterozygous, for example, having OPN1LW genes that encode two spectrally distinct L pigments, would have four different cone types -- two different L, plus M and S -- and thus the potential for having tetrachromatic color vision (Bosten, Robinson, Jordan & Mollon, 2005, Jordan & Mollon, 1993, Jordan & Mollon, 1997). Prior to the discovery of variation in peak sensitivity of pigments underlying normal color vision, it had been appreciated that female carriers of a red-green color vision deficiency have the potential for tetrachromatic color vision (Nagy, MacLeod, Heeyndermann & Eisner, 1981). However, because of the normal variability in the L and M pigments more than half of all women express more than three spectrally different photopigments (Neitz, Kraft & Neitz, 1998). There has been a persistent fascination with the possible existence of tetrachromatic females over the last quarter of a century, but experimental evidence has been mostly negative. Jordan and colleagues (Jordan, Deeb, Bosten & Mollon, 2010) have conducted a study in which they were able to analyze DNA from nine subjects to confirm the genetic potential for tetrachromacy. Intriguingly, 1 of 24 obligate carriers of deuteranomaly exhibited tetrachromatic behavior on all their tests. However, they conclude that the overwhelming majority of carriers of color anomaly who have the potential to have four cone subtypes, do not exhibit four-dimensional color vision and that it is unlikely that anomalous trichromacy is maintained by an advantage to the carriers in discriminating colors. Human females who have an extra pigment most commonly have two L pigments, one with S180 and another with A180, in addition to S and M pigments. In female platyrrhine monkeys, having two L pigments with this difference compared to having only a single X-encoded pigment is associated with very clearly demonstrable trichromacy vs. dichromacy (Rowe & Jacobs, 2007). It appears that the addition of an extra subpopulation of cones to a dichromat has a more dramatic effect on color vision than making the same addition to an already trichromatic retina. As discussed in later sections, the evolution of trichromacy from dichromacy may have occurred through opportunities afforded by specific features of the preexisting circuitry in dichromats. This same circuitry in trichromats may be nearer its limits for supporting additional color vision capacity, and thus, in turn, may not lend itself so readily to tetrachromacy.
The genetics of color vision defects
Protan, deutan and tritan defects are characterized by the absence of a contribution to vision from L, M and S cones, respectively. Twenty five years ago, the accepted model for the genetics of red-green color vision deficiencies (Piantanida, 1974, Pokorny & Smith, 1977) held that the dichromatic forms, protanopia and deuternaopia, were caused by the replacement of the normal M gene with one that encoded an L pigment for deuteranopia and the replacement of the normal L gene with one that encoded an M pigment for protanopia. These gene replacements were thought to cause the inappropriate expression of M opsin in L cones for protanopia, and of L opsin in M cones for deuteranopia. Anomalous trichromatic forms were thought to be caused by genes for anomalous photopigments replacing either the normal L or M pigment gene. Replacing one normal pigment with an anomalous one resulted in having peak sensitivities that were closer together than the peak sensitivities of the normal L and M cones. For each of the anomalous trichromacies, protanomaly and deuteranomaly, two forms, simple and extreme, were proposed to differ in the magnitude of the spectral difference between the anomalous pigment and the normal one with anomalous pigments being more shifted from normal in extreme forms.
Prior to the molecular genetics work, the genes for the L and M cone pigments were thought to exist at independent loci with another gene, glucose-6-phoshphate dehydrogenase, located between them. It was believed that allelic diversity for the two independent genes was the cause for unrelated protan and deutan color vision deficiencies. To everyone's great surprise, results from molecular genetics revealed that the L and M opsin genes were adjacent to one another with no intervening genes and the high frequency of recombination between these two genes is responsible for many peculiarities of red green color vision including the extraordinarily high frequency of color vision defects.
Inherited color vision deficiencies can be explained by gene rearrangements that arose through unequal homologous recombination in females during meiosis (Nathans et al., 1986a), as illustrated in Figure 2. The DNA between the L and M genes is nearly identical to the DNA that follows the last gene in the array so an intergenic crossover (in-between the genes) is possible. Such a crossover (Figure 2A) produces daughter chromosomes in which one daughter has one additional opsin gene compared to the parents, and the other chromosome has one fewer than the parents. As shown in Figure 2A, the products are an X-chromosome with one opsin gene and a second with a tandem array of three opsin genes. A majority of human deuteranopes are “single gene dichromats” having been reduced to an L gene as the only opsin gene on the X-chromosome. The most frequent arrangement of opsin genes in humans with normal color vision is to have one L and two M genes arranged as shown in Figure 2A. There are about 50 times more people with 2 M and one L genes than there are deuteranopes with one L gene, even though they would be produced in equal numbers from ancestors with one L and one M gene by the crossover mechanism. Selective pressure against color blindness, particularly in ancestors to modern humans, could explain the much lower frequency of deuteranopes.
Fig.2.
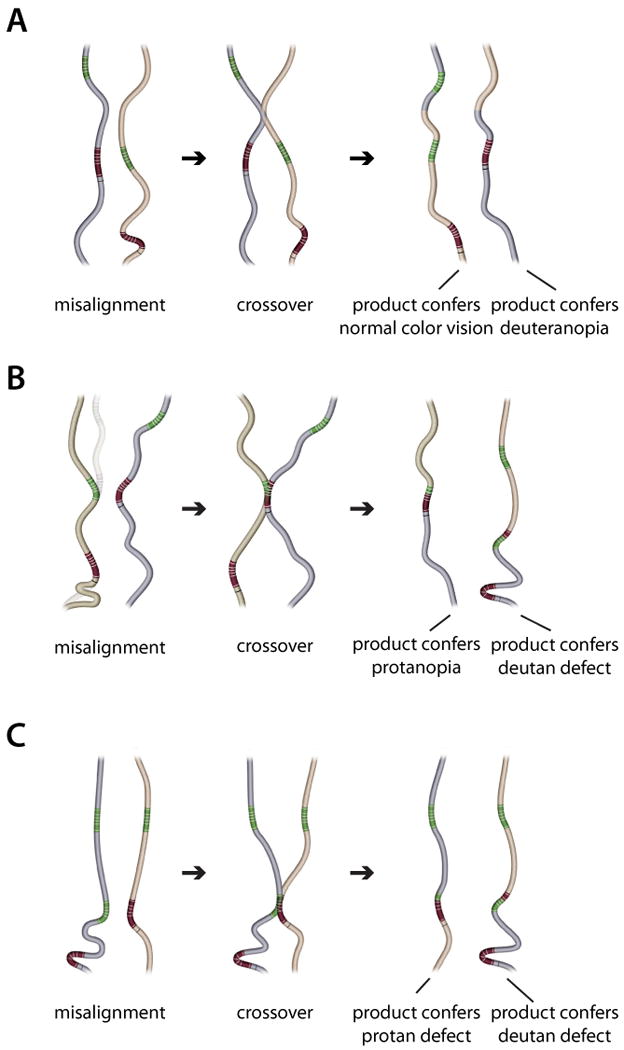
Recombination produces an opsin array that causes color vision deficiencies. A) Misalignment of the opsin gene arrays on the two X-chromosomes in a female allows a crossover in the region between the L and M genes in one array and the homologous region downstream of the M gene in the other array. This produces two new X-chromosome opsin gene arrays. One array has an L gene and two M genes and will confer normal color vision. The second array has a single opsin gene, an L gene, and produces the color vision defect, deuteranopia, when inherited by a male. B) Misalignment of the opsin gene arrays on the two X-chromosomes in a female allows a crossover between the L gene on one X chromosome and the M gene on the other X-chromosome. This produces two new arrays that differ in gene number from the parental arrays. A gene that derives part of its sequence from a parental L gene and part from a parental M gene encodes a pigment whose spectral sensitivity is primarily determined by the parental origin of exon 5. The array with one gene confers protanopia because the single gene derives exon 5 from the parental M gene. The array with three genes will cause a deutan color vision defect. The second gene in this array encodes an L-class pigment because it derives exon 5 from the parental L opsin gene. The severity of the deutan defect depends on the amino acid differences at the spectral tuning sites in the two L-class pigments. If there are no differences at the spectral tuning sites, deuteranopia will result; if there are differences, a male that inherits this array will be deuteranomalous. C) Recombination between an array with 3 opsin genes and another with 2 opsin genes is expected to produce arrays that cause color vision defects at a high frequency because the mismatch in gene number on the two arrays means there is no perfect alignment. Misalignment that results in a crossover between an L gene on one X-chromosome and the M gene on the other will produce two new arrays that cause color vision defects if inherited by a male. One array will have two genes, both of which encode opsins that form M-class pigments because the first gene in the array derives exon 5 from the parental M gene. Males inheriting such an array will have a protan defect, the severity of which is determined by amino acid differences specified at the spectral tuning sites. The second array produced has two L genes followed by an M gene. It will cause a male to have a deutan defect, the severity of which is determined by the spectral differences encoded by the genes encoding opsins that will form L-class pigments.
Because the L and M genes are adjacent to each other on the X-chromosome and they are nearly identical, an L gene from the paternal chromosome can align with the M gene from the maternal chromosome, as shown in Figure 2B. When X-chromosomes misalign, a crossover within the L gene on one X-chromosome and an M gene on the other X-chromosome produces two new arrays, each of which will cause a color vision defect when inherited by a male. One will cause a protan defect, the other will cause a deutan defect. In the protan-causing array the one remaining opsin gene is a hybrid between the parental L and M genes. As long as the hybrid has exon 5 from a parental M gene, the encoded photopigment will fall into the M class (Figure 1B). A male with a normal S-pigment and one X-chomosome pigment gene encoding an M pigment is an obligate protanope. The array associated with deutan color vision deficiency has a parental L gene as the first gene in the array. The second gene is a hybrid while the third gene is a parental M opsin gene. This array structure is the one most commonly found in deuteranomlous males, and it represented one of the most unexpected findings that has come from examining the molecular genetics associated with color vision deficiencies (Drummond-Borg et al., 1989, Nathans et al., 1986a, Neitz et al., 1996, Shevell et al., 1998). An initial explanation offered for the presence of apparently normal, intact M opsin genes in a color vision defect characterized by the absence of functional M cone contribution to vision was that the deutan hybrid gene and the normal M gene were co-expressed in the same cone, thereby shifting the spectral sensitivity of the “M-cone” toward that of the L cone. It was further hypothesized that the specific nature of the hybrid gene was important in determining the severity of the color vision defect (Nathans et al., 1986a). Understanding why a gene array like that illustrated Figure 2B confers a color vision defect when both normal L and M genes required for normal color vision are present requires information about the mechanism responsible for expression of the X-chromosome opsin genes. The key to understanding the color vision genotype in this case has come from studies demonstrating that, except in rare cases (Sjoberg, Neitz, Balding & Neitz, 1998), only the first two genes in the array are expressed (Bollinger, Sjoberg, Neitz & Neitz, 2004, Hayashi, Motulsky & Deeb, 1999, Neitz, Bollinger & Neitz, 2003, Winderickx, Battisti, Motulsky & Deeb, 1992a, Yamaguchi, Motulsky & Deeb, 1997).
Research relating genotypes to phenotypes uncovered another unexpected finding--in humans there has been a tremendous amount of intermixing of L and M opsin genes. In the classical view of color blindness genetics, a deuteranomalous allele of the M pigment gene would have arisen by a single event in which a normal M gene was mutated. Thus, 94% of the males were envisioned to have a normal M gene and 6% had mutants. Similarly, 98% of males would have the normal L gene and 2% had mutants. However, the homology of photopigment genes and their arrangement in a tandem array allows for a high mutation rate. Thus, instead of being produced by a single mutation, many of the genotypes associated with color vision deficiencies are the result of a series of mutational steps away from ancestral gene arrays, with one normal L and one normal M gene. The generation of a gene array associated with protanomaly, as illustrated in Figure 2C, is an example. In order to have the basis for protanomalous trichromacy as opposed to protanopia, protanomalous individuals must have at least two M genes encoding opsins that form two different pigments of the M class (Figure 1), but lack an L gene (as in Figure 2C). There is no way to produce an array with multiple M and no L genes from parental arrays with one L and one M. One parent must have at least two M genes, as produced by the rearrangement in Figure 2A. A subsequent crossover (Figure 2C) can produce a gene array with multiple genes with different spectral sensitivities falling in the M class.
We assume that the present variety of opsin genes arose from an ancestral arrangement with one normal L and one normal M gene. Presumably, the misalignment required for a gene rearrangement involving an unequal crossover is a very rare event and, initially, the rate of mutations producing genetic variety would have been relatively low. However, once variety in gene copy number was introduced by early rearrangements, the generation of females heterozygous for copy number would have greatly accelerated the rate of rearrangement. As shown in Figure 2C, for a female with one array with two genes and a second array with one gene, there is no perfect alignment. One of the two possible alignments pair an L with an M gene and if a crossover occurs when the genes are aligned in this way a gene rearrangement will occur. Thus, in females with X-chromosomes with unequal opsin gene numbers, there would be misalignments in 50% of the meioses that would produce a gene rearrangement if a crossover occurs within the gene array.
There is evidence that a remarkable amount of rearrangement has occurred in the L and M opsin genes over the course of human history. The two most obvious indicators are the variability in copy number among the L and M genes and the polymorphism at position 180 in which nearly half of normal males in some samples have A180, the amino acid presumed to be characteristic of the primordial M gene. In addition to position 180, variants at other spectral tuning sites in exons 2 and 4 have also been identified among males with normal color vision. It appears that all possible spectral types within the L class and within the M class shown in Figure 1 occur as normal variants. This is in striking contrast to the classic view, in which people with protan defects were viewed to have “the” normal M and a spectrally shifted “anomalous” L pigment and those with deutan color vision defects were believed to have “the” normal L and a spectrally shifted “anomalous” M pigment and the severity of the color vision deficiency was believed to be determined solely by the nature of anomalous pigment.
Assuming that, at one time, the majority of human ancestors had the typical Old World primate arrangement of one L and one M opsin gene on the X-chromosome, a cladistic method of classifying species into groups (clades) based on gene sequences can be used to determine the most likely amino acid sequences encoded by a single ancestral L and a single ancestral M opsin gene. Comparison of the deduced amino acid sequences of L and M opsin genes, for example among Caucasian males with normal color vision, reveals that relatively few individuals have genes encoding the ancestral opsins. The majority of males with normal color vision have L genes that encode some amino acids of the ancestral M opsin and M genes that encode some of the amino acids of the ancestral L opsins (Verrelli & Tishkoff, 2004). Furthermore, there is complete overlap between hybrid pigments mediating color vision in people with normal color vision and in people with red-green color vision deficiencies, although the frequencies of some hybrids are higher in color deficiency compared to normal trichromacy (Carroll et al., 2002, Crognale, Teller, Motulsky & Deeb, 1998, Neitz et al., 1996, Sharpe et al., 1998, Winderickx et al., 1993).
Prior to knowledge of the cone opsin gene sequences and the molecular mechanism responsible for spectral tuning, a key to understanding color anomaly appeared to be the characterization of the anomalous pigments (DeMarco, Pokorny & Smith, 1992), which, together with the putative normal L and M cone sensitivity functions, would comprise the characterization of the photopigment basis of anomalous trichromacy. The variability in normal pigments makes understanding the photopigment complement of color anomalous individuals more complicated; however, the discovery that the absorption spectra of primate photoreceptors assume a common shape when plotted on a normalized wave number axis (wave number divided by wave number of maximum sensitivity) (Baylor et al., 1987, Lamb, 1995, Mansfield, 1985), has greatly simplified the characterization of variant cone pigments. The implication is that all primate photopigments have a common shape that can be used to completely characterize the spectral properties of any human photopigment variant just from knowing its wavelength maximum. A comparison of the single pigment template curve (from Carroll, Bialozynski, Summerfelt, Neitz & Neitz, 2000) to human cone fundamentals derived from color matching functions (Stockman, MacLeod & Johnson, 1993) is shown in Figure 3, and there is generally good agreement over the entire spectrum when values of 420 nm, 530 nm and 557.5 nm are used for the spectral peaks of the S, M and L pigments, respectively. Over recent years, there has been the most uncertainty about the spectral peak of the S cone. However, the results in Figure 3 show that spectral peak obtained from human psychophysics agrees pretty well with those from MSP (419 nm) (Dartnall, Bowmaker & Mollon, 1983a) and from reconstituting the human S-pigment in vitro (Fasick, Lee & Oprian, 1999). We note that, theoretically, the human cone fundamentals derived from color matching functions should all have the identical shape when corrected for preretinal absorption and plotted on a log of wavenumber axis, and that shape should agree perfectly with the universal pigment template after appropriate corrections for optical density are made. The agreement is good, but not perfect. The discrepancies are likely due, in part, to the fact that the human cone fundamentals reflect average data from a number of humans who have different normal variants of M and L pigments. This might be expected to tend to broaden the cone fundamental curves. The fact that the human L fundamental is broader than either the human L or M curve is consistent with that idea, given that the normal human L pigments are the most variable.
Fig. 3.

Spectral sensitivities of human L, M and S cones plotted on a scale that is uniform in units of log of wave number. On this scale, all photopigments assume a common shape, described by a template curve (solid lines). The curve together with information about the spectral positions of the cone photopigments can be used to completely describe the photopigment basis for color vision in any individual. The template was derived by fitting an equation given at www.neitzvision.com to an amalgam of photopigment spectral sensitivity curves (Carroll, McMahon, Neitz & Neitz, 2000). A) The spectral peak of the template has been adjusted to fit cone fundamentals derived from color matching (Stockman & Brainard, 2010). B) All the curves from (A) have been shifted to a best fit, illustrating the close similarity between the shapes of the L, M and S spectral sensitivities and the template. The slight differences in psychophysically derived fundamentals may derive, in part, from variation in the normal cone pigments.
Taken together, the spectral tuning data from Figure 1 and the photopigment template curve of Figure 3, it should be possible to completely characterize the photopigments of any person just from examination of the sequences of the photopigment genes they express. There is very good agreement between the deduced photopigment complement and color vision capabilities of people with color vision defects (Barbur, Rodriguez-Carmona, Harlow, Mancuso, Neitz & Neitz, 2008, Neitz et al., 1996, Neitz et al., 2004, Shevell et al., 1998). An implication is that the technology is available to allow conventional color vision testing to be replaced by a genetic test.
The rest of this section summarizes what has been learned about genotype/phenotype relationships in red-green color vision deficiencies. Protan defects are characterized by the absence of an L cone contribution to vision. X-chromosome opsin gene arrays underlying the dichromatic form, protanopia, either have a single opsin gene that encodes an M pigment, or have multiple genes in which the first two encode opsins that produce M pigments that are identical in spectra (Jagla, Jägle, Hayashi, Sharpe & Deeb, 2002, Nathans et al., 1986a, Neitz, Neitz, He & Shevell, 1999, Neitz et al., 2004). The anomalous trichromatic form, protanomaly, is associated with opsin gene arrays in which the first two genes encode opsins that produce M pigments that differ in spectra. Most commonly, the spectral separation in the M pigments is the result of amino acid differences at spectral tuning sites that shift the peak sensitivity of one pigment relative to the other. Amino acid substitutions have a generally smaller effect on the spectral sensitivity of M pigments relative to L pigments (Figure 1), thus, individual protans are likely to have a smaller spectral separation between their two pigments drawn from the M class compared to deutans (described below), who draw two pigment from the L class. This can explain why, as a group, protanomalous observers have somewhat poorer color discrimination than deuteranomalous observers. In addition to the amino acids that shift the spectrum, protanomlous color vision may also arise from amino acid differences that do not shift the relative peak sensitivity of the underlying M pigments, but instead increase the optical density of one pigment relative to the other (Neitz et al., 1999). In the latter case, color vision becomes dichromatic under conditions that bleach the pigments and equalize their optical densities.
Deutan defects are characterized by the absence of an L cone contribution to vision. X-chromosome opsin gene arrays underlying the dichromatic form, deuteranopia, either have a single opsin gene that encodes an L pigment, or have multiple genes in which the first two encode opsins that produce L pigments that are identical in spectra (Neitz et al., 1996). The anomalous trichromatic form, deuteranomaly, is associated with arrays in which the first two genes encode opsins that produce L pigments that differ in spectral sensitivity. Among individuals with deuteranomalous color vision, there is variation in phenotypes. For example, on the American Optical Hardy Rand and Rittler pseudoisochromatic plate test, in which the stimuli have a range of difficulty giving a measure of severity, deuteranomalous performance ranges from nearly normal to nearly as poor as a dichromat. There is a very good correlation between deuteranomalous behavior in color vision tests and the magnitude of the spectral separation between the underlying L pigments; however, in some cases when people have very small spectral differences between their pigments, discrimination performance as measured by the range of anomaloscope settings accepted as a match is better than might be predicted from the spectral separation of the underlying pigments (Barbur et al., 2008, Neitz et al., 1996, Neitz et al., 2004, Shevell et al., 1998).
In recent years, it has been discovered that deleterious combinations of amino acids at the dimorphic positions can be produced as a consequence of intermixing of L and M opsin genes. One such combination at the exon 3 encoded amino acid positions is Leucine 153, Isoleucine 171, Alanine 174, Valine 178 and Alanine 180, abbreviated LIAVA (Carroll, Neitz, Hofer, Neitz & Williams, 2004, Crognale, Fry, Highsmith, Haegerstrom-Portnoy, Neitz, Neitz & Webster, 2004, Mizrahi-Meissonnier, Merin, Banin & Sharon, 2010, Neitz et al., 2004). The LIAVA combination is associated with the absence of cone function when specified either by an L or an M opsin gene. Males whose L opsin contains this deleterious combination are protanopes, males whose M opsin contains this combination are deuteranopes, males who have a single opsin gene on the X-chromosome that specifies this combination or for whom the first two opsin genes in the array specify this combination are blue cone monochromats. There is a perfect correlation between the opsin gene specifying this combination and color vision phenotype, despite the fact that the arrays differ dramatically in the number of opsin genes, and in the identity of the gene that specifies the LIAVA combination. This, together with the observations that the LIAVA combination has never been found in a person without vision problems and it has been found on a variety of genetic backgrounds, indicates that it is the causative mutation and rules out the possibility that the phenotype is caused by an unidentified mutation in linkage disequilibrium. Whether color vision defects caused by this deleterious combination are congenital or whether they develop over time as the photoreceptor becomes progressively non-functional is an open question.
A less frequent cause of inherited red-green color vision deficiencies has arisen through conventional mutational mechanisms that produce relatively rare, random mutations that cause the corresponding gene to encode an opsin that fails to form a functional photopigment or that prevents the gene itself from being transcribed (Bollinger, Bialozynski, Neitz & Neitz, 1999, Carroll, Rossi, Porter, Neitz, Roorda, Williams & Neitz, 2010, Jagla et al., 2002, Neitz et al., 2004, Winderickx, Sanocki, Lindsey, Teller, Motulsky & Deeb, 1992c). The most common such mutation is the replacement of the cysteine residue at position 203 with the amino acid arginine.
Inherited tritan color vision deficiencies are rare in comparison to the inherited red-green color vision defects and they have an autosomal dominant inheritance pattern (Kalmus, 1955). In addition, the inheritance pattern shows incomplete penetrance meaning that some people who have the causative gene do not have the phenotype. Weitz and coworkers (Weitz, Miyake, Shinzato, Montag, Zrenner, Went & Nathans, 1992a, Weitz, Went & Nathans, 1992b) showed that inherited tritan defects were caused by mutations in the S opsin gene that produce amino acid substitutions. They identified three amino acid substitutions in the S opsin gene as causing tritan defects: arginine substituted for glycine at position 79, proline substituted for serine at position 209, and serine substituted for proline at position 264. More recently, two new mutations--replacement of arginine at position 283 with glutamine, and replacement of leucine at position 56 with proline--have been found as causes of tritan defects (Baraas, Carroll, Gunther, Chung, Williams, Roster & Neitz, 2007, Gunther, Neitz & Neitz, 2003, Gunther, Neitz & Neitz, 2006).
The genetic underpinnings of two more rare forms of inherited color vision loss have been elucidated. One, termed blue cone monochromacy or incomplete achromatopsia, results from mutations that prevent proper transcription of the X-chromosome cone opsin genes, or renders the encoded opsins non-functional, as will be discussed in detail below. The second is complete achromatopsia and is caused by mutations in genes that are normally expressed in all three cone types (Kohl, Jägle, Sharpe & Wissinger, 2009, Thiadens, Somervuo, van den Born, Roosing, van Schooneveld, Kuijpers, van Moll-Ramirez, Cremers, Hoyng & Klaver, 2010). The most common mutations interfere with normal function of the cone photoreceptor cyclic GMP gated ion channel, which has two subunits, encoded by separate genes (CNGA3 and CNGB3). Mutations in the genes encoding the phototransduction proteins cone transducin (GNAT2 gene) and phosphodiesterace 6C (PDE6C) have also been found in association with achromatopsia. The most common cause of achromatopsia in humans is mutations in the CNGB3 gene, and a recent study using gene therapy in a canine model of a CNGB3 defect showed rescue of cone photoreceptor function (Komáromy, Alexander, SRowlan, Garcia, Chiodo, Kaya, Tanaka, Acland, Hauswirth & Aguirre, 2010).
Genes and the Cone Photoreceptor Mosaic
The question of what governs the organization of the three cone types in the retinal cone mosaic is central to understanding the circuitry for color vision and evolutionary strategies for optimizing the mosaic organization for extracting visual information. Over the last 25 years, several developmental studies in humans and other Old World primates have examined this question (Bumsted & Hendrickson, 1999, Bumsted, Jasoni, Szél & Hendrickson, 1997, Curcio, Sloan, Kalina & Hendrickson, 1990, Curcio, Sloan, Packer, Hendrickson & Kalina, 1987, Wikler & Rakic, 1991, Xiao & Hendrickson, 2000). In humans, immunohistochemistry has shown that S opsin is first detected in the fovea at about fetal week 10.9 and S cones cover the entire retina by about fetal week 19 (Bumsted & Hendrickson, 1999, Xiao & Hendrickson, 2000). The ability to detect L and M opsin immunologically occurs quite a bit later, with L/M cones first detected in the central retina at about fetal week 21.5, and extending over the whole retina by fetal weeks 34 to 37 (Xiao & Hendrickson, 2000). Because the L and M opsins are more than 96% identical in amino acid sequence, antibodies that recognize one, recognize both. In situ hybridization experiments in fetal human retinas using nucleic acid probes to label S and LM opsin mRNA give similar results with mRNA being detectable just shortly before opsin protein is detectable. As for antibody labeling, in situ hybridization methods are incapable of distinguishing between L and M cones. The significant lag in the appearance of LM versus S opsin protein and mRNA during development suggests that differentiation of S cones is independently controlled from LM cones; however, because these methods do not distinguish between L and M cones, they shed no light on whether L and M cones are independently controlled.
Insight into the mechanism of differentiation of L versus M cones has come from studies of New World primates in which trichromatic color vision evolved through allelic diversity of a single X-chromosome cone opsin gene locus rather than a gene duplication that placed two cone opsin genes on the same X-chromosome (Jacobs, Neitz & Neitz, 1993). For example, in squirrel monkeys, there are three alleles of the X-chromosome cone opsin gene. One encodes an opsin that forms a pigment that is similar in spectral peak to the human L pigment, another is similar to the human M pigment, and a third has a spectral peak that is intermediate between human L and M. All males of the species are dichromatic, having only one X-chromosome, and thus having S cones and a single cone type that absorbs in the middle-to-long wavelengths. Females have two X-chromosomes, and can either be homozygous or heterozygous for the X-chromosome opsin allele. If homozygous, they are dichromatic; however, females who carry both an allele for an L opsin and one for an M opsin have the equivalent of normal human color vision, having both L and M cones because the process of X-inactivation segregates the expression of the L and M opsin genes to separate populations of cones. The significance of this is that in trichromatic female squirrel monkeys, the difference between L and M cones is solely determined by the stochastic choice of which X-chromosome is retained as the active one. Variation in L:M cone ratio in female squirrel monkeys is similar to what is seen in human males with normal color vision. In squirrel monkeys the variation has been attributed to the stochastic process of X-inactivation, influenced by the number of cells present at the time of activation and other random factors in the inactivation process (Jacobs & Williams, 2006).
In humans, even though both L and M opsin genes reside on the X-chromosome, there is evidence that a stochastic mechanism also determines whether each individual cell expresses L versus M opsin. In their early work to investigate the genetic mechanisms of blue cone monochromacy, Nathans and colleagues discovered a DNA element upstream of the L opsin gene that is essential for transcription of the X-chromosome opsin genes (Nathans, Davenport, Maumenee, Lewis, Hejtmancik, Litt, Lovrien, Weleber, Bachynski, Zwas, Klingaman & Fishman, 1989). The DNA element was given the name Locus Control Region, abbreviated LCR, and it is an enhancer that mediates cell type specific expression of the X-chromosome opsin genes (Li, Timmers, Guy, Pang & Hauswirth, 2007, Wang, Macke, Merbs, Zack, Klaunberg, Bennett, Gearhart & Nathans, 1992). The LCR is highly conserved, and is present in all other mammalian species examined, the vast majority of which have a single X-chromosome opsin gene. Interactions between the opsin gene promoter and the LCR are thought to be required for opsin gene expression (Smallwood, Wang & Nathans, 2002).
The gene duplication event that ultimately led to there being both L and M opsin genes on the same X-chromosome duplicated an ∼ 40 kilobase pair segment that extends ∼450 basepairs upstream of the opsin gene and includes the promoter, and it extends about 18 kilobase pairs downstream of the coding sequence. The LCR was not included in the duplication, and so in humans and other Old World primates, the L and M opsin genes must share the same enhancer, meaning only one of the X-chromosome cone opsin genes can be expressed at a time. Epigenetic modification of the opsin gene locus may play an important role in opsin gene expression and in determination of the fate of a nascent cone photoreceptor as an L versus an M cone such that, during development, a competition between interactions that promote opsin gene expression and mechanisms that silence all genes that are not expressed as part of the cell's final differentiated phenotype ultimately leaves each L/M cone with one of the X-chromosome opsin genes actively transcribed, while all others are silenced (Johnston Jr. & Desplan, 2008).
Although there is no experimental data demonstrating a role for epigenetic silencing, the observation that the ratio of L:M pigment messenger RNA changes over the course of development (Knoblauch, Neitz & Neitz, 2006) has led us to suggest a model in which, during gestation, each cone cycles randomly between transcribing either the L or M opsin gene that can explain features of the topography of L and M cones in the retinal mosaic. According to the model, during development of the retina, at any given time a nascent cone is transcribing either an L or M opsin. At the time a gene is being expressed it is protected from silencing. This increases the probability that it will transcribe the same opsin in future cycles, while at the same time the non-transcribed genes are subject to epigenetic modifications that decrease their probabilities of being expressed in future cycles (Knoblauch et al., 2006). Probabilistic events result in one gene being expressed in each mature cone while all the others are silenced. Epigenetic changes are passed on to daughter cells when a cell divides. The gene silencing mechanism can explain why the L/M cone ratio increases from the center of the retina out to the periphery (Hagstrom, Neitz & Neitz, 1997, Neitz, Balding, McMahon, Sjoberg & Neitz, 2006). The cells in the peripheral retina are born later than those in the center (Bumsted et al., 1997, Xiao & Hendrickson, 2000); if the M pigment genes are more prone to silencing and the epigenetic modifications are ongoing as the retina develops, the probability of expressing an M gene would decrease in the periphery. The heritability of the epigenetic changes introduces a slight nonrandomness compared to the idea that each cone independently makes a completely random choice to express M or L opsin. Cells near one another are the product of related cell divisions and share some epigenetic memory, making them more likely than expected by chance to express the same opsin. This explains the slight clumpiness that has been observed for the human L/M cone mosaic (Hofer, Carroll, Neitz, Neitz & Williams, 2005). Genetic changes in non-coding DNA in the region of the opsin genes are expected to influence how prone the DNA is to silencing and polymorphisms upstream of the opsin gene array have been found to be associated with individual differences in L/M cone ratio (Gunther, Neitz & Neitz, 2008, Stamatoyannopoulos, Thurman, Noble, Kutyavin, Shafer, Dorschner & Deeb, 2005). DNA is packaged as loops of chromatin in cells, and the most likely explanation for only two of the X-chromosome opsin genes being expressed is that the loop structures inhibit access of the LCR to opsin genes that are downstream of the first two genes in the array. The rare cases in which more than two genes from the array have been demonstrated to be expressed may be the result of mutations that affect the looping structure. Proximity of the LCR to the first gene in the array has been proposed to explain the greater number of L than M cones in the retinas of some people. However, there is a huge range of L/M cone ratio and in some groups, having more M than L cones is common. Thus, it appears that the details of how the DNA is looped in the nucleus is a more important determinant of the relative access of L vs. M genes to the LCR, and thus the cone ratio, than the linear distances along the DNA (McMahon, Carroll, Awua, Neitz & Neitz, 2008).
A variety of methods ranging from immunohistochemistry in post-mortem eyes to adaptive optics and retinal densitometry in living eyes have been used to evaluate the relative number and distribution of S cones versus L and M cones (Bumsted & Hendrickson, 1999, Hofer et al., 2005, Roorda, Metha, Lennie & Williams, 2001). At 1 degree eccentric from the fovea, the average percentage of cones that were identified as S cones by adaptive optics was 5.72 percent, and the S cones are distributed in a relatively regular hexagonal array (Hofer et al., 2005). The relative ratio of L to M cones among individuals with normal color vision is highly variable. The first direct evidence for the relative distribution of L versus M cones came from adaptive optics and retinal densitometry (Hofer et al., 2005, Roorda & Williams, 1999), and the results confirm reports of variation in the L:M ratio among humans with normal color vision from studies that use indirect methods (Rushton & Baker, 1964, Carroll, Neitz, & Neitz, 2002, Kuchenbecker, Sahay, Tait, Neitz & Neitz, 2008, Mollon & Bowmaker, 1992, Neitz et al., 2006,). In summary, the development of S cones and their arrangement in the adult retina compared to L and M cones all point to the identity of S cones as being distinctly different from L and M cones, with S cones being non-randomly distributed. In contrast, the arrangement and ratio of L and M cones are consistent with their arrangements being determined by a stochastic process such that the L and M cones represent a single receptor population differing only by which X-chromosome opsin gene is expressed.
Typically, mammals, including most New World primates, are dichromatic with a single opsin gene on the X-chromosome, and all cones that are not S cones express the available X-chromosome opsin gene. Thus, human single gene dichromats are expected to express, by default, the one remaining X-chromosome opsin gene in all the cones that would be M or L in a normal trichromat. However, in the case of protan or deutan color vision defects caused by individual missense amino acid substitutions or by the LIAVA deleterious combination, it is expected that developing photoreceptors will transcribe and translate the mutant opsin gene. What affect this has on the mature adult cone mosaic has been investigated using adaptive optics and optical coherence tomography (OCT) for both the LIAVA mutation (Carroll et al., 2004) and the C203R mutation (Carroll, Baraas, Wagner-Schuman, Rha, Siebe, Sloan, Tait, Thompson, Morgan, Neitz & Neitz, 2009). The adaptive optics images of the retina from a male with deuteranopia whose M opsin gene encoded the LIAVA combination show a mottled appearance of the cone mosaic. There are large regions lacking visible photoreceptors interspersed with the visible photoreceptors. The outer nuclear layer thickness of the LIAVA retina by OCT was within the normal range suggesting the areas where no cones were visible by adaptive optics nonetheless contained cones, albeit nonfunctional ones that cannot waveguide light back into the camera and appear dark. In contrast, adaptive optics images of the retina from a color deficient male with the C203R mutation in his M opsin gene showed a cone mosaic that was relatively undisrupted in appearance compared to the LIAVA retina, although cone density was reduced compared to control retinas. These results suggest that, as for mutations at the corresponding cysteine residue in rhodopsin, the cones that express the C203R mutant opsin degenerate. The appearance of the cone mosaic suggests that the degeneration must occur prior to maturation and packing of the cones into the fovea, which occurs postnatally. This would account for the reduced cone density but relatively regular packing arrangement.
Results from psychophysics and from electroretinography suggested there is residual S cone function in some tritanopes. It also seemed possible that complete versus incomplete penetrance of inherited tritanopia was a function of whether the affected individual had mutations in both copies of the S opsin gene, or in just one copy (Weitz et al., 1992a, Weitz et al., 1992b). What has become clear more recently is that tritan color vision defects are analogous to retinitis pigmentosa caused by mutations in the gene encoding the rod pigment rhodopsin. Indeed, some of the mutations in the S opsin gene in tritan color vision deficiency occur at positions corresponding to positions in rhodopsin at which amino acid substitutions cause the rod photoreceptors to degenerate. In a recent study a father and his daughter, both of whom made tritan errors, were both shown to be heterozygous for a substitution of glutamine instead of the normally occurring arginine at position 283 of the S opsin. The father's tritan phenotype was severe, whereas the daughter's was relatively mild. Adaptive optics imaging revealed an absence of S cones in the father, but S cones were clearly present in the daughter. Taken together, the findings indicate that inherited tritan defects are associated with a progressive degeneration of the S cones, analogous to the degeneration of rods in retinitis pigmentosa (Baraas et al., 2007). This accounts for the apparent incomplete penetrance of the phenotype in that in early stages the S cones function and the tritan color vision defect does not manifest until S cones have reached a sufficient stage of degeneration.
It seemed equally possible that the rare disorder, blue cone monochromacy, which is associated with an absence of both L and M cone contribution to vision, could be due to mutations at the photoreceptor level or at a higher neural processing level until linkage mapping showed it to be linked to Xq28, the same chromosomal location as the L and M opsin genes (Lewis, Holcomb, Bromley, Wilscon, Roderick & Hejtmancik, 1987). Subsequent genetic analysis of affected individuals revealed a variety of different mutational mechanisms that give rise to BCM. One mechanism is the deletion of all but one opsin gene on the X-chromosome, with an inactivating mutation in the one gene remaining. Inactivating mutations similar to those found as rare causes of deutan and protan defects have been identified including deleterious combinations of normal polymorphisms, and rare random amino acid substitutions. A relatively common cause of BCM was identified as the deletion of the LCR (Nathans et al., 1989, Nathans, Maumenee, Zrenner, Sadowski, Sharpe, Lewis, Hansen, Rosenberg, Schwartz, Heckenlively, Trabousli, Klingaman, Bech-hansen, LaRouche, Pagon, Murphy & Weleber, 1993), which results in a complete absence of L and M cones because the cones cannot express any of the X-chromosome opsin genes. Retinas of female carriers of an LCR deletion have been imaged using adaptive optics (Carroll et al., 2010). Their retinal cone mosaics appear undisrupted and uniform in cone packing, but the cone density is greatly reduced and cone outer segments are much wider than normal suggesting that during foveal development, the surviving cones fill the available space taking on a morphology with a larger diameter.
Another rare disorder, enhanced S-cone syndrome, is autosomal recessive, and is characterized by an increase in the number of S-cones relative to L/M cones and rods. Mutations in the gene encoding a photoreceptor-specific nuclear receptor, NR2E3, have been identified in patients with this disorder (Kanda & Swaroop, 2009, Rocha-Sousa, Hayashi, Gomes, Penas, Brandão, Rocha, Urashima, Yamada, Tsuneoka & Falcão-Reis, in press).
Genes and the Circuitry For Color Vision
Background
Genetics play the central role in all of life's processes, including the circuitry for color vision. In the last decade, research in a number of areas has shed light on the process of how genes operate to give rise to circuitry for color vision. As discussed above, mutations and deletions of the photopigment genes give rise to color vision defects with reduced color vision capacities. More recent work, which is a topic in the following sections, has focused on understanding the converse--how the addition of an extra opsin gene in a dichromat can give rise to an expansion in color vision capacities.
At the level of the ganglion cells and the lateral geniculate nucleus (LGN), the circuits that carry color information are spectrally opponent, firing action potentials to some wavelengths and being inhibited by others. In early diagrams illustrating the putative first stages of color processing, e.g. (Wiesel & Hubel, 1966), post synaptic elements were drawn as having selective contacts to L, M and S cones with excitatory elements connecting exclusively to one cone class and inhibitory elements to a second class (Figure 4). The ideas that such selectivity in the wiring was necessary for color vision and was genetically specified were highlighted by proposals to explain inherited color vision deficiencies (Hurvich & Jameson, 1962).
Fig. 4.

Hubel and Wiesel's conception of cone photoreceptor contributions to circuitry responsible for red-green spectrally opponent cells recorded in the LGN. Excitatory connections were assumed to selectively connect to L cones, avoiding S and M cones. The inhibitory connections were assumed to selectively connect to M cones, avoiding S and M cones. Redrawn from Wiesel and Hubel (1966).
In contrast to the selective contacts hypothesis, just over a quarter of a century ago, the opposite idea was forwarded that spectrally opponent cells arise by completely random connections to cone photoreceptors (Paulus & Kroger-Paulus, 1983). The “random wiring hypotheses” held that, rather than requiring genetic instructions, the opponent properties of the wiring could, in part, be a consequence of the centers of midget ganglion cells receiving input from a single cone and from features of the topography of the cone mosaic (Jusuf, Martin & Grunert, 2006, Lennie, Haake & Williams, 1991).
Twenty-five years ago, there was very little evidence to distinguish between the genetically-specified cone-selective vs. random wiring dichotomy. This is an example of a genetics and circuitry issue that has largely been resolved over the last decade. The main conclusion is that the circuitry for color vision arose from the interplay between genetically specified cone-type-specific connectivity for S vs. L/M cones and random, or mostly random, e.g., (Field, Gauthier, Sher, Greschner, Machado, Jepson, Shlens, Gunning, Mathieson, Dabrowski, Paniinski, Litke & Chichilinsky, 2010) wiring for L vs. M connectivity. This difference in genetic specification of connections is the result of different evolutionary origins for the two types of connectivity.
Evolution of red-green color vision
All lower mammals are either monochromats or dichromats and Old World (OW) monkey and ape species are all trichromatic (Jacobs, 1981, Jacobs, 2004). Most New World (NW) monkeys have color vision that is an evolutionary intermediate between the dichromats and OW trichromats and their genetics and color vision has been a key discovery for answering questions about how new color vision capacities might arise from the addition of a photopigment gene.
Nothing evolves de novo. Everything new in biology arose by modification of some preexisting system. The major questions have been, from what preexisting circuitry did red-green color vision evolve and, what were the associated costs? The transition from dichromacy to trichromacy required the addition of a third cone class but what, if any, additional changes in the circuitry were required? Aspects of color vision in NW primates, as representatives of a transitional form of color vision between non-primates and the OW primates, have shed light on this question. NW monkey species that have what has been termed, “allelic trichromacy” include both individuals with trichromacy and with dichromacy as normal phenotypes (Boissinot, Tan, Shyue, Schneider, Sampaio, Neiswanger, Hewett-Emmett & Li, 1998, Jacobs, 1983, Mollon, Bowmaker & Jacobs, 1984). In these animals, the circuitry for their two normal forms of color vision, di- and trichromatic, must share the same genetic instructions. Consider the squirrel monkey (Saimiri sciureus); two thirds of females are trichromats; however, all males and one third of the females are dichromats (Jacobs & Neitz, 1987). Thus, a large majority, a full two-thirds of the individuals of this species, are dichromats.
The simplest idea is that, together with X-inactivation, a single mutation like the one at position 277 seen in prosimians (Jacobs, Deegan, Tan & Li, 2002) that blue-shifts the long wavelength pigment by about 12 nm, produced a mosaic of two subtypes of middle-to-long wavelength cone in the retinas of heterozygous female squirrel monkeys that, in turn, exploited preexisting circuitry, to provide a new dimension of color vision. A few subsequent amino acid changes in the opsin to increase the spectral separation produced a subset of females with cone complements like modern human trichromats. The idea that the presence of three cone types alone provided rewards of trichromacy without additional subsequent genetic changes seems likely based on the principle that each individual genetic change must, in the words of Darwin (1859) be “useful to its possessor.” Having each individual mutation provide an advantage is the only way to overcome the compounded improbabilities of multiple highly unlikely mutations occurring in one animal (or one genetic line of animals). Moreover, a nascent mutation that subsequently benefited trichromats by refining the circuitry for red-green color vision would not have increased in frequency in the species if it proved to be a disadvantage to the larger dichromatic subpopulation. Thus, the most straightforward hypothesis is that full red-green color vision, similar to that experienced by human trichromats, emerged as the result of the evolution of three cone types in the retina without any further genetically coded modifications to the circuitry.
A related issue is whether there could be a balance between the relative selective advantages of dichromacy vs. trichromacy in the natural world. For example, such a balance has been offered as an explanation for the high frequency of color defective humans (Regan, Julliot, Simmen, Vienot, Charles-Dominique & Mollon, 2001). However, in earlier sections we presented evidence that the high frequency of color defective humans is the result of an extraordinarily high mutation rate of those genes. NW primates are a different case; it appears that dichromacy is maintained in the majority of squirrel monkeys and related species simply because there are no males with the genes required for trichromacy (Jacobs & Neitz, 1985). In the absence of trichromatic genetic variants among the males, selection cannot act to increase the frequency of trichromacy in that gender. In contrast, natural selection can act on the allele frequencies in females to affect the relative number of dichromats vs. trichromats. In cebid monkeys in which there 3 equally spaced spectral types of photopigments, for females, a balancing selection produced by the heterozygous advantage of trichromacy has produced three alleles at near equal frequency, maximizing the number of trichromats (Jacobs & Neitz, 1985, Mollon et al., 1984). The fact that selective pressure has produced the maximum number of trichromatic squirrel monkeys allowed by their genetic repertoire indicates that trichromacy in these monkeys has a strong selective advantage over dichromacy. One counterexample to the advantages of tri- over dichromacy is that human dichromats can perform better than trichromats in detecting luminance edges in the presence of strong masking chromatic contours (Morgan, Adam & Mollon, 1992). The hidden digit designs of the Ishihara plates are a demonstration of this. This small subset of plates is intended to have a visible sign for colorblind people, but not be seen by normal trichromats. Dichromats seeing things that are invisible to trichromats is an attractive idea, especially to people with color vision defects; however, the masking effect is weak, and these particular plates are relatively ineffective in distinguishing people with color vision defects from normal. The lesson learned is that trichromacy provides powerful advantages with virtually no offsetting disadvantages.
Except for the extra opsin gene, there is no evidence for any differences between the dichromat and the trichromat. It is hard to reconcile this with the idea that there could be separate circuitry dedicated entirely to red-green color vision. Mollon (1989) popularized the now favored view that phylogenetically ancient neural machinery serves blue-yellow color vision, whereas a newly evolved subsystem serves red-green color vision. Certainly, red-green color vision depends on recently evolved subtypes of X-encoded cone opsins, and blue-yellow color vision is evolutionarily ancient based on its ubiquity across mammals; however, this does not necessarily imply the evolution of new circuitry dedicated to red-green color vision. Red-green color vision might represent an improvement in the function of circuitry that was already present in the dichromat. This might be analogous to the evolution of flight. A recent study suggests that the wings of the ancestor to modern birds, the archaeopteryx, could have been used for gliding but not flapping wing flight (Su, Luo, Terakita, Shichida, Liao, Kazmi, Sakmar & Yau, 2006, Xu, Zhou, Wang, Kuang, Zhang & Du, 2003). Flapping wings evolved as an improvement of wings used for gliding, not by the creation of a separate system. Similarly, trichromacy may represent an enhancement in the functions of pre-existing circuits. The emergent trichromats may have been able to make use of red-green opponent signals that were added to pre-existing circuitry with the addition of a third cone population rather than creating a separate system for red-green color vision.
Besides the opsin genes, there are no known differences in the genetic code between dichromats and trichromats. However, for other systems, during development, large differences in wiring of the visual system can result from differences in visual experience. Even though the circuitry for di- and trichromatic color vision share the same genetic instructions, this does not rule out the possibility that the differences in neural activity during development imposed by having different cone complements could be responsible for very different neural wiring in dichromats and trichromats including specialized circuitry for red-green color vision.
In the last decade, new genetic technology has been applied to questions about the neural plasticity of color vision. For the past 50 years, deprivation and ablation studies were the main tools for studying neural plasticity. However, recently, molecular genetics methods have made it possible to add inputs. In one series of experiments, genetic manipulations were used to generate trichromatic mice (Smallwood, Olveczky, Williams, Jacobs, Reese, Meister & Nathans, 2003). “Knock-in” mice had the endogenous M-cone (spectral peak = 511 nm) opsin gene replaced (Jacobs, Neitz & Deegan, 1991) with one encoding a human L-opsin (spectral peak = 555.5 nm). The knock-in mice were mated to wild-type mice to produce heterozygous females in which X-inactivation produced two middle-to-long wavelength cone submosaics, and some of the heterozygous mice gained red-green color vision capacities (Jacobs, Williams, Cahill & Nathans, 2007). Any color opponent signals in the genetically altered mice would have to arise by random connections to center-surround receptive fields in which, unlike primates, both centers and surrounds contain several cones. Some ganglion cells in the transgenic mice would carry red-green, spectrally opponent signals because of random differences in the relative numbers of L and M cones in the center vs. the surround; however, the number of ganglion cells with strong opponency would be small, and as expected from this, red-green color vision in the mice was relatively weak. Nonetheless, this demonstrates that in a species lacking red-green color vision it is possible to get trichromatic behavior just by adding a new cone subpopulation. These experiments do not, however, address the question of how much the ability to extract red-green color vision depends on visual experience during development. That question was addressed by experiments in which gene therapy was used to add a third cone population in adult animals.
Gene therapy was performed on adult squirrel monkeys that were missing the L-opsin gene and were colorblind since birth (Mancuso, Hauswirth, Li, Connor, Kuchenbecker, Mauck, Neitz & Neitz, 2009). L-opsin was added to a subset of M cones, providing the receptoral basis for trichromatic color vision. The addition of a third opsin in adult red–green color deficient primates was sufficient to produce trichromatic color vision behavior, demonstrating that trichromacy can arise from a single addition of a third cone class and it does not require a developmental process. The treated animals discriminated colors in the red and green range from each other and as different from gray. They could also discriminate among other color combinations that dichromats find impossible to tell apart. The ability to make new color discriminations was closely timed with the appearance of robust expression of the introduced opsin indicating that no rewiring or new circuitry was associated with the acquisition of red-green color vision.
Most midget ganglion cells in macaques are red-green spectrally opponent and a popular idea has been that the circuitry for red-green color vision evolved in an ancestral primate from the precursor to the midget ganglion cells that had been previously responsible for luminance contrast-based achromatic spatial vision. However, a new and different explanation has been offered for the newly acquired color vision in the treated squirrel monkeys--that they might be taking advantage of preexisting blue-yellow color vision circuits (Mancuso et al., 2009, Mancuso, Neitz, Hauswirth, Li, Connor, Kuchenbecker, Mauck & Neitz, 2010b, Shapley, 2009). The treated monkeys were protanopes and after treatment, a subset of the cones co-expressed L-pigment with the native M pigment. Shapley (2009) explains that if some of the receptive fields remained wild type S vs M like the untreated protanope and other receptive fields opposed S cones to the newly introduced L-like cones making them essentially S vs L, the animal would have two blue-yellow systems with different spectral response properties that could be responsible for the observed trichromatic behavior. We agree with Shapley's idea and have taken it one step farther suggesting that, not only does red-green color perception in the treated monkeys, in part, take advantage of the pre-existing blue-yellow system, the hue components of red-green color vision in all primates might represent an improvement in the function of the pre-existing blue-yellow system that was split in two by the addition of the third cone population (Mancuso et al., 2009). Understanding of the genetics and evolution of blue-yellow spectral opponency has been transformed over the last 25 years. In light of the recent suggestions of a relationship between blue-yellow and red-green hue systems in gene therapy treated monkeys, understanding the blue-yellow system may be critical to understanding the circuitry of hue mechanisms in general.
Evolution of blue-yellow opponency and S- and L/M cone specific connections
In terms of cone selectivity, primates can be considered to have two major cone classes: S and L/M. It is now abundantly clear that S and L/M cones in primates have separate circuitry, each with highly cone-type selective connections (Crook, Davenport, Peterson, Packer, Detwiler & Dacey, 2009, Dacey & Lee, 1994, Dacey, Lee, Stafford, Pokorny & Smith, 1996, Mariani, 1984, Martin & Grunert, 1999, Packer, Verweij, Li, Schnapf & Dacey, 2010). The major types of bipolar, horizontal and ganglion cell types likely to be relevant to color vision in primates are illustrated in Figure 5. Evolution is conservative, with all vertebrates sharing many common features. Receptors homologous to human S cones, L/M cones and rods are nearly universal features of vertebrate visual systems and the evolutionary roots of the photoreceptor cell-type specific circuitry seen in primates likely predates vertebrates. The ancestors to human S vs. L/M opsins predated the appearance of eyes (Neitz, Carroll & Neitz, 2001). A billion years before photopigments and photoreceptors served the function of vision, a primitive form of blue-yellow color vision was already in place driving circadian rhythms and vertical migration in one-celled organisms that predated bacteria (Lamb, Collin & Pugh, 2007, Spudich & Spudich, 2008). UV light triggered archaebacteria to descend away from the damaging UV rays of midday, and the gentle orange light of dusk resulted in upward migration to collect long wavelengths for a primitive form of photosynthesis. Emerging millions of years later in evolution, the hagfish ‘eye’ continued to function as a circadian organ. These primitive, jawless, eel-shaped marine chordates have ganglion cells that project predominantly to the hypothalamus (Fritzsch & Collin, 1990) just as their likely mammalian homologues, the melanopsin-containing retinal ganglion cells (Berson, 2003, Koyanagi, Kubokawa, Tsukamoto, Shichida & Terakita, 2005, Provencio, Jiang, WDeGrip, Hayes & Rollag, 1998, Provencio, Rodriguez, Jiang, Hayes, Moreira & Rollag, 2000, Rollag, Berson & Provencio, 2003). Thus, a form of “blue-yellow” chromatic opponency may be one of the oldest sensory capacities, having originally evolved to signal the large spectral changes in the sky at dawn and dusk. These chromatic signals precisely mark the phase of the day-night cycle and provide a powerful cue for circadian entrainment. Since blue-yellow opponency is a characteristic of the primitive receptor systems responsible for circadian entrainment extending from archaebacteria to the parietal eye of reptiles, (Solessio & Engbretson, 1993, Su et al., 2006) it is not surprising then that melanopsin ganglion cells in modern primates are blue-yellow spectrally opponent (Dacey, Liao, Peterson, Robinson, Smitch, Pokorny, Yau & Gamlin, 2005). We assume that the circuitry for blue-yellow color vision evolved by adapting preexisting components of the spectrally opponent functions of the circadian organs to new functions. The fact that biological mechanisms in which short- and long-wavelength lights have had opposing actions have been around since the emergence of animal life on earth makes it understandable why highly cone-type specific circuitry involving S vs. L/M cones would be inherent to the retina.
Fig. 5.

Primate retinal ganglion cells that receive cone selective connections and have the potential for playing a role in color vision. Upper and lower panels show how the addition of a third cone type changes the chromatic inputs to different ganglion cells in the primate. The cell bodies of ganglion cells are drawn as diamonds. Bipolar cells have circular cell bodies. Horizontal cells bodies are hexagonal. For the trichromat (lower panel), four ON/OFF pairs of midget ganglion cells are drawn. Different combinations of cone connectivity distinguish the four ganglion cell pairs. Two ON/OFF pairs of ganglion cells, one pair with an L cone center and one with an M cone center, receive input from cones that make contacts with H2 horizontal cells, which contact nearby S cones. Two other ganglion cell pairs (an L center and an M center) receive input from cones that do not have the potential for significant S cone input from the surround. Assuming that a small subset of ganglion cells receive S cone input from the surround, the M cones with S in the surround give rise to an OFF center ganglion cell with (S+L)-M opponency and an ON center ganglion cell with M-(S+L) providing the potential retinal basis for a red and green, respectively, hue pathway. L cones with S in the surround give rise to an OFF center ganglion cell with (S+M)-L opponency and an ON center ganglion cell with L-(S+M) providing the potential retinal basis for a blue and yellow, respectively, hue pathway. ON midget ganglion cells with no S cone input to the surround have L-M opponency when the center cone is L and M-L opponency when the center cone is M. M cones provide the center of one spectrally opponent ganglion cell but the surround of neighboring ganglion cells. If neighboring L-M and M-L are indiscriminately combined in the cortex, chromatic opponency for diffuse spots of light will cancel. This would make L-M and M-L ganglion cells the substrate for edge detectors that would also signal chromatic borders. Thus, the four pairs of midget ganglion cells could provide the retinal basis to serve red, green, blue and yellow hue perception and luminance/chromatic edge detection. One S cone bipolar cell is illustrated. It connects specifically to an S cone. It provides the S-ON input to the small bistratified ganglion cell, which is drawn showing dendritic arbors in both the ON and OFF sublamina (labeled OFF-CENTER and ON-CENTER). The single S cone bipolar cell also provides an S-OFF input via an inhibitory interneuron to the melanopsin ganglion cell (drawn in yellow with an “X” shaped dendritic arbor). Both the melanopsin ganglion cell and the small bistratified cells have large receptive fields so the ON component of the melanopsin and the OFF component of the small bistratified cell have M+L cone inputs, giving them (L+M)-S and S-(L+M) spectral opponency, respectively. A comparison of the “DICHROMAT” top panel with the “TRICHROMAT” bottom panel shows how the spectral opponent properties of each of the 10 ganglion cells illustrated change when the retina is transformed from having two cone types to having three. Those midget cells that are capable of only transmitting luminance information in the dichromat become L vs. M opponent in the trichromat. Putative midget ganglion cells with S vs. L inputs that could serve blue color vision are transformed into two pairs to serve blue-yellow and red-green color vision in the trichromat. An attempt was made to preserve some of the anatomical details of the retina in the cartoon. Cones and bipolar cells are shown with ribbon synapses. ON bipolars make connections to the ribbon and terminate in the ON lamina. OFF bipolar cells are shown making more lateral connections representing flat contacts and they terminate in the OFF sublamina. The inset illustrates that horizontal cells make reciprocal synapses.
The discovery of the melanopsin ganglion cells is directly part of the legacy of Nathans' original cloning of the opsin genes; it has been celebrated for its impact on circadian biology; however, it has been equally revolutionary for the field of color vision. Dermal melanophores migrate to the periphery of the pigment cells of frog skin (Xenopus laevis) in response to illumination and in an effort to identify the opsin responsible, Provenceo et al. (Provencio et al., 1998) screened a melanophore cDNA library for opsin-like nucleotide sequences. The opsin identified was termed melanopsin. Subsequently, Provencio et al. identified the human homolog and showed that, in mammals, melanopsin expression is restricted to the retina (Provencio et al., 2000). Melanopsin-positive ganglion cells matched the anatomical characteristics of cells known to project to the primary circadian pacemaker of the hypothalamus (Gooley, Lu, Chou, Scammell & Saper, 2001, Hattar, Liao, Takao, Berson & Yau, 2002). The surprise for those of us studying color vision was that Dacey and colleagues (Dacey et al., 2005) determined that the melanopsin containing ganglion cells are the S-OFF spectrally opponent cells of the primate retina. Over many years of recording, no anatomically identified (M+L) –S cells have been identified in the macaque retina other than the melanopsin ganglion cells. Remarkably, application of the ON-pathway agonist, 2-amino-4-phosphonobutyric acid (AP-4), completely blocks the S-OFF light response in the primate melanopsin ganglion cells (Dacey, Peterson & Robinson, 2002). This suggests the S-OFF signals are transmitted to the inner retina via the ON S cone bipolar cell, and that a sign inversion of the ON signal in the inner retinal circuitry is critical for generation of the S-OFF response (Dacey et al., 2002). Flat contacts on to cones are associated with OFF bipolar cells, and in electron micrographs, occasional flat contacts are observed on to S-cone terminals (Calkins, 2001, Klug, Herr, Ngo, Sterling & Schein, 2003). However, at the ganglion cell level the only physiologically identified (M+L) –S cells, the melanopsin ganglion cells, get their S-OFF response by sign inverting an input from S-cone ON bipolar cells. The flat contacts on to S-cones observed anatomically appear to be capricious since no major physiological function resulting from them has been identified.
The discovery of the S-OFF melanopsin ganglion cell reveals a major role for S-cone ON bipolar cells to serve an ancient spectrally opponent system that is not related to hue perception. The melanopsin-expressing ganglion cells have the largest dendritic tree diameters of any primate retinal ganglion cell (Dacey et al., 2005). The only way cells with such large dendritic trees could have cone-opponent circuitry is through genetically-specified cone-specific connections fed through two sets of circuitry with opposing polarities, L/M ON from L/M cone specific bipolars, and S-OFF through a sign reversing synapse from S cone specific bipolars (see Figure 5). The S-OFF melanopsin ganglion cells have an S-ON counterpart, the small bistratified ganglion cells, with which they share input from the S-ON bipolar cell (Dacey, 1993, Dacey & Lee, 1994). As shown in Figure 5, the S-ON small bistratified and S-OFF melanopsin ganglion cells appear to be partners in an ancient circuit sharing the same S-cone bipolar. The fact that the S-OFF component of this pair, the melanopsin ganglion cells, are not likely candidates for serving the yellow component of the blue-yellow hue system might raise doubt that S-ON small bistratified cells could be directly responsible for the blue hue component. There are several other reasons that might eliminate S-ON ganglion cells as candidates for blue-yellow hue perception. If the firing of action potentials by the small bistratified cells were the neural correlate for the percept of blue, then we would expect to perceive blue whenever the small bistratified cell fires. This is not the case; unlike the midget ganglion cells that give relatively sustained ON or OFF responses, depending on the type, the small bistratified cells produce equally robust responses to the onset of short wavelength light and to the offset of long wavelength lights presented in the dark (Crook et al., 2009). In contrast, short wavelength lights in the dark elicit blue percepts, but the offset of long wavelength lights do not. A yellow light can produce a blue afterimage, however, one must look at a white stimulus after the offset of yellow; there is no blue sensation with the offset of a yellow light in the dark. A common percept associated with the offset of a bright yellow light in the dark is a yellow afterimage. Another major disconnect between the physiological properties of small bistratified cells and blue-yellow perception is that the small bistratified cells have S-(M+L) cone inputs. They fire action potentials to S cone stimuli and they are inhibited by stimuli that selectively activate M or L cones. This does not match human blue-yellow perceptions for lights presented to the central retina. Percepts of blueness are elicited rather than inhibited by activation of M cones (DeValois & DeValois, 1993, Drum, 1989, Neitz & Neitz, 2008, Stockman & Brainard, 2010), so with respect to M-cone stimuli, the small bistratified cells behave opposite of what would be predicted if they served blue-yellow hue perception.
Genetics provide the final and most definitive test of the hypothesis that the S-ON small bistratified ganglion cell and the S-OFF melanopsin ganglion cell system are the substrate for blue-yellow color vision. Both of these ganglion cell types receive S cone input from S cone ON-bipolar cells via the metabotropic glutamate receptor, mGluR6. The hypothesis that these ganglion cells are the physiological substrates for blue-yellow perception predicts that people who have genetic mutations that render mGluR6 receptors nonfunctional will be unable to perform color discrimination and detection tasks based on S cones, resulting in deficits in blue-yellow color perception; however, people with genetic defects that eliminate function of mGluR6 receptors have none of these problems (Dryja, McGee, Berson, Fishman, Sandberg, Alexander, Derlacki & Rajagopalan, 2005).
Probably the major reason that the anatomically distinct S -(L+M) and (L+M)-S ganglion cell pathways have been thought to be substrates for blue-yellow color vision in the face of contradicting evidence is a perceived lack of other candidates. However, there have been reports (de Monasterio, 1979, Derrington, Krauskopf & Lennie, 1984, Tailby, Solomon & Lennie, 2008) of a set of underappreciated cells early in the visual pathway that are perfect candidates for providing hue perception. Our blue-yellow hue perception differs from that expected from S-(L+M) and (L+M)-S ganglion cells in that the direction of maximum activation of the perceptual hue mechanisms are “rotated in color space” (Webster, Miyahara, Malkoc & Raker, 2000) relying on (S+M)- L and L-(S+M) circuitry in which M cone responses are added to S rather than being differenced from them. Similarly, our red-green hue perception is based on (S+L)- M and M-(S+L) mechanisms which are also “rotated” in color space compared to that expected from ganglion cells in which L and M cones are opposed. de Monasterio and colleagues (de Monasterio, Gouras & Tolhurst, 1975) were the first to document the physiology of ganglion cells in the retina in which the L cone mechanism is opposed to the S and M cones, as expected for cells serving perceptual blue-yellow color vision. He also identified ganglion cells in which S and L cones are opposed to M as required to explain red-green hue perception. A characteristic of our hue perception is that the circuitry for all four unique hues, red, green, blue and yellow relies on input from all three cones while a preponderance of ganglion and LGN cells receive input from only two cone types--L and M. de Monasterio commented that the finding of a “cortical trichromatic organization and of a subcortical dichromatic organization could indicate a central reorganization of the processing of colour information”. However, his ganglion cell recordings showed “that many colour-opponent ganglion cells in the rhesus monkey retina receive input from all three cone mechanisms, indicating that trichromatic interactions begin in the retina.” Modern multistage color models have continued to propose central reorganization of the processing of color information to explain the (S+M) vs. L and (S+L) vs. M inputs to perceptual hue channels (DeValois & DeValois, 1993, Stockman & Brainard, 2010). However, de Monasterio's conclusions from a third of a century ago, that (S+M) vs. L and (S+L) vs. M pathways originate in the retina (as shown in Figure 5) seem worth revisiting in the light of genetic results and the constraints imposed by evolution, as reviewed here.
Molecular genetics, physiology and anatomy have converged to indicate that in primates, there is a single bipolar cell type that is highly S cone specific and there are others that are highly L/M cone specific (Figure 5) (Dacey, 1996, Li & DeVries, 2006, Martin & Grunert, 1999). The S vs. L/M cone specific connectivity severely restricts the possible ways in which S cone input can be combined with L and M signals at early stages of color processing. Thus, cells like the small bistratified cells and the melanopsin ganglion cells that receive input from large numbers of L and M cones and combine S and L/M inputs via circuitry in the inner retina are constrained to have only S vs. (L+M) connectivity. From the known anatomical cone-type selective connections shown in Figure 5, the only way to get retinal ganglion cells that combine inputs from all three cone types in the (S+L) vs. M and (S+M) vs. L combinations relevant to hue perception is via S cone input to the surrounds of midget ganglion cells via H2 horizontal cells (Figure 5). H2 horizontal cells (Dacey et al., 1996) receive a strong input from S cones and a weaker input from L- and M-cones and they provide the only known anatomical pathway for building ganglion cells that combine S with L or S with M cones, i.e., by providing S cone input to the surrounds of midget bipolar cells so that the cone type in the receptive field center will be opposed to S plus the other cone type in the midget bipolar cell's receptive field surround.
In summary, 1) S cone based hue perception in people whose S cone bipolar cells are nonfunctional is indistinguishable from normal in color vision tests. 2) S vs. L/M cone specificity ensures that ganglion cell-types receiving direct S signals via S cone bipolar cells will have S vs. (L+M) circuitry and 3) (S+L) vs. M, (S+M) vs. L and S vs. (L+M) cell types have all been reported early in the retinostriate visual pathway. Examples of the different chromatic cell types are evident in the results of Tailby et al. (Tailby et al., 2008) which have been reproduced in Figure 6. In primates, the midget ganglion cells are the major mediators of conscious perception making up more than 95% of the ganglion cells in the fovea (Dacey, 1994). The midget bipolar cells do not contact S cones (with the exception of the far periphery (Field et al., 2010); thus, the great majority of midget bipolar cells do not carry S cone signals (Sun, Smithson, Zaidi & Lee, 2006). H2 horizontal cells contact both S cones, which represent 5% of the total cone population, and L/M cones, which represent 95% of the cone population. Only the small subset of L and M cones that have strong H2 input and are in close proximity to S cones could have access to S cone signals that would be significant in the surrounds of midget ganglion cells. Thus, for midget ganglion cells in the central retina, if a small number have S cone input, it would always be mixed with L or with M cone signals in the surround, opposed to the single L or M cone in the receptive field center (Figure 7). This would provide the (S+L) vs. M and (S+M) vs. L combinations necessary for hue perception.
Fig. 6.

Cone weights of LGN cells recorded by (Tailby et al., 2008). Each dot represents the response properties of one LGN neuron. Normalized weights assigned to each cone type by the cells are plotted. The weight attached to M-cone input is plotted against that for the L-cone. The distance from the diagonals reflects the magnitude of S cone input. Thus, a cell plotted at the origin would have only S cone input and one on a diagonal line would have no S cone input. Only those neurons that were determined to have significant S cone input are represented. LGN neurons were found that represent all the spectrally opponent ganglion cells shown in Figure 5. The cells have been color coded to represent their putative role in hue perception. The cells that plot in the upper left and lower right triangles have L vs. M opponency and either excitatory or inhibitory S cone inputs as required to match the cone input to human hue perception. LGN cells with all the correct cone inputs to account for human hue perception were identified: M-(S+L) for green, (S+M)-L for blue, L-(S+M) for yellow and (S+L)-M for red; however, only one cell with the correct cone inputs to account for red percepts, (S+L)-M, was recorded from. A threshold was used to decide which LGN cells to include as receiving S cone input, it may be that other (S+L)-M cells were present but fell below the threshold. This seems reasonable because only 5% S cone contribution is required to account for normal hue perception. Redrawn from (Tailby et al., 2008).
Fig. 7.

A) In a dichromat, midget ganglion cells with an S cone in the surround could provide the basis for blue-yellow color vision with yellow percepts being mediated by ON ganglion cells and blue percepts mediated by OFF ganglion cells. Spectral response properties of each of the two spectrally opponent cells types are plotted. B) The addition of a third cone type to the retina transforms the former blue and yellow pathways. What was a single S vs. L receptive field type is transformed into two different receptive fields, one with an L cone center and one with an M cone center. ON and OFF pathways split the L center receptive fields into L-(S+M) and (S+M)-L and the M center pathways into M-(S+L) and (S+L)-M. The spectrally opponent response properties of each of the four trichromatic ganglion cell types is shown. The cells responsible for red, green, blue and yellow are all derived from a blue-yellow ancestor, but they all differ significantly from the preexisting blue yellow system.
For the midget system (or its predecessor), occasional S cone signals could come via horizontal cells to an M/L cone pedicle that, in turn, is served by an ON and an OFF bipolar cell (Figure 7). As has been described by Mancuso et al. (Mancuso et al., 2010b) this could provide an S-ON signal to mediate the percept of blueness (via the OFF bipolar) and an S-OFF signal to mediate the precept of yellowness (via the ON bipolar), thus, providing parallel and complementary blue-yellow circuitry in which there is one S-ON for every S-OFF, and the pathways would have equal and opposite opponency. For the midget ganglion cells, since S cone input would come from the surround, the OFF-bipolar cells could carry the S-ON signals. These would be unaffected by mutations in the mGluR6 gene that prevent ON bipolar signaling from cones, explaining why S cone based vision, as assayed by standard color vision tests, is unaffected in patents who lack mGluR6-mediated signaling. S-OFF bipolar cells would be functional in patients lacking functional mGluR6 receptors but S-ON midget cells would not. However, ON and OFF pathways have push-pull interactions mediated by crossover inhibition (Molnar & Werblin, 2007) at higher levels of the pathway; thus, as long as the S-ON side is intact, ON-OFF opponent interactions at higher levels would provide input to the S-OFF pathway.
A rationale in favor the H2 horizontal cell/midget ganglion cell based blue-yellow system hypothesis is that it provides the explanation for how red-green color vision could have arisen from dichromacy in evolution solely by the addition of a third cone population as we have proposed (Mancuso et al., 2010b) and is as illustrated in Figure 7. It also explains how gene therapy can produce a full, extra dimension of color vision in adults without a developmental process. As discussed above, Shapley has proposed the logical solution that the addition of a third cone type in gene therapy treated monkeys may have split the pre-existing blue-yellow hue system in two. The small bistratified cells receive input from several dozen L/M cones. Thus, the addition of a randomly placed third cone type would produce a relatively homogenous population of small bistratified cells with respect to mixed L/M cone contribution and there wouldn't be two populations. However, if blue-yellow color vision is based on a subset of midgets with S-cone input in the surrounds as proposed by Mancuso et al. (Mancuso et al., 2010b), the addition of the third cone type would break the blue-yellow system cleanly into two subpopulations depending on whether the cone center is M or L, explaining both the trichromacy of the monkeys treated with gene therapy and the evolution of trichromacy in primates (Figure 7). According to this idea, red-green hue perception represents an improvement in the preexisting blue-yellow system in which the hue dimensions are enhanced. Nothing is lost or compromised in this split because visual detection and discrimination based on S cones becomes distributed across the two newly formed hue systems. Since both are hybrids of the dichromatic blue-yellow circuit, it would explain why S cones play an important role in both red-green and blue-yellow hue systems.
Trichromacy resulting from splitting the blue–yellow pathway is a new idea stimulated by the gene therapy results. Similarly, the report that mice gained trichromatic vision after having a new cone introduced stimulated ideas about the circuitry responsible. Makous (2007) proposed an idea much more in keeping with conventional thinking that the observed red-green color vision behavior in the mice could be the result of taking advantage of existing circuits responsible for extracting luminance contrast for spatial vision. As illustrated in Figure 5, in the case of primates, there can be no doubt that preexisting circuits responsible for spatial vision in dichromats carry L/M opponent signals after the addition of a third cone type. In dichromatic primate ancestors, if midgets comprised greater than 90% of the ganglion cells in the central retina, as they do in modern monkeys, they must have provided the major source for luminance contrast form vision. The addition of a third cone type would have made the majority of formerly achromatic midget cells in the central retina L vs M spectrally opponent in addition to being responsive to achromatic luminance contrast edges. If these L-M opponent ganglion cells are the basis for detecting red-green chromatic boundaries in the absence of luminance contrast at edges, it could represent an improvement in the preexisting circuits for spatial vision enhancing their edge detecting capabilities to include pure red-green chromatic borders. For red-green opponent signals added in trichromats, it seems likely that both schools of thought are correct, L vs M signals could be used in both the preexisting blue-yellow color vision system (e.g., as suggested by Shapely) and the achromatic luminance contrast spatial vision system (e.g., as suggested by Makous.) for complementary functions that represents enhancements to each.
Makous (2007) suggested that the treated mice might “see” their own L/M cone mosaic. However, visualizing the stabilized L/M mosaic seems unlikely to be a problem for systems with emergent red-green color vision. A major transform for spatial vision in primary visual cortex is the properties of receptive fields. While most cells in the LGN respond well to small spots of light, cortical cells respond to moving oriented edges and bars. Responses to a stabilized L/M cone mosaic would be filtered out at this stage. The details of how the LGN receptive fields are transformed in the cortex is not completely clear; however, it is apparent that it involves combining the outputs from neighboring cells in the retina. Figure 8 illustrates how this would happen in the classic feedforward model of Hubel and Wiesel (Hubel & Wiesel, 1962). Combining adjacent ON receptive fields indiscriminately adds together L-M and M-L receptive fields, canceling opponent responses to diffuse colored lights while producing cortical cells that respond well to luminance contrast edges. However, receptive fields constructed of red-green opponent ganglion cells would be expected to maintain responsiveness to moving red-green equiluminant edges. If so, such cortical cells could respond both to achromatic luminance and to red-green chromatic edges. This is the character of many cells observed in area VI. They respond to dark-light edges but not diffuse colored lights; however, they will respond to edges of one color opposed to another at all relative intensities (Conway, Hubel & Livingston, 2002, Gouras & Kruger, 1979, Hubel & Livingston, 1990, Johnson, Hawken & Shapley, 2008, Johnson, Hawken & Shapley, 2001). Such cells respond to borders defined by luminance contrast, but they also allow the visual system to extract form information when objects are differentiated from their backgrounds only by chromatic borders. Thus, when trichromacy evolved, the addition of the red-green opponent signals to the preexisting edge detector system responsible for form vision would represent an enhancement in function. The only potential cost is that red-green chromatic edges could mask luminance edges. As mentioned above, this is the only consistently documented advantage of dichromacy over trichromacy.
Fig. 8.

A possible scheme for explaining the chromatic properties of cortical cell receptive fields adapted from Hubel and Wiesel (Hubel & Wiesel, 1962). A number of LGN cells, of which four are illustrated, project upon a single cortical cell. The synapses are presumed to be excitatory in this “feed forward” model. In this model, a number of the inputs must be active at the same time in order to exceed the threshold of the cortical cell. Indiscriminate connectivity to L vs. M opponent cells in which L-M cells are always nearby M-L cells cancels opponent responses to diffuse colored lights, but responses to luminance edges are enhanced.
The role in color vision for cells that respond to red-green equiluminant edges and luminance contrast has been uncertain. It may be unlikely that they contribute to hue perception because they appear to signal the presence of a chromatic edge, but not the difference between green-red as opposed to red-green boundaries (Conway et al., 2002, Hubel & Wiesel, 2005). If red-green isoluminant edge detection is handled by a high-resolution spatial contour detection system that was enhanced by the addition of L vs. M opponency to midget ganglion cells, the problem that the system does not provide hue information might be solved if hue perception is mediated by separate circuitry operating at lower spatial resolution that represents a subdivision of the preexisting blue-yellow system. L vs. M opponency of the contour detectors does correspond to one of the cardinal directions that characterize threshold data obtained in color discrimination experiments (Krauskopf, Williams & Heeley, 1982). In the past, the different spectral signatures obtained for psychophysical hue vs. detection tasks has been thought to represent two different stages of one color vision system (Stockman & Brainard, 2010). However, red-green opponent information may be carried in two parallel pathways and may represent improvements to two preexisting systems, one for spatial vision and the other for blue-yellow color vision rather than the creation of single hierarchical color vision system.
There is agreement that explaining all aspects of human color perception requires the activities of the cones to be processed through multiple stages, including transformations at the level of the ganglion cells and additional cortical stages. Multistage, color models, include those of Judd (1966), De Valois & De Valois (1993), and Stockman and Brainard (2010). From the De Valois and De Valois model as a starting point we have suggested that instead of being carried by a single pathway solely concerned with red-green color vision, red-green opponent signals in trichromats may be carried by two parallel pathways that preexisted in ancestral dichromatic primates, one for extracting contours relevant to spatial form and one that preexisted for blue-yellow hue perception (Mancuso, Mauck, Kuchenbecker, Neitz & Neitz, 2010a). This can reconcile the different ideas recently proposed for how red-green color vision might arise from preexisting circuits in genetically modified animals (Makous, 2007, Shapley, 2009). Evolution is purely opportunistic; by making use of different aspects of existing circuitry in which L vs. M opponent signals were added, evolution could have taken advantage of an opportunity to expand sensory capacities by a single genetic change at the level of the photopigments.
Finally, older multistage color models are primarily concerned with explaining our perceptual responses to isolated colored lights presented against dark backgrounds. As described by Hubel and Wiesel (Hubel & Wiesel, 2005), cells that respond to one part of the spectrum in their center and to a different part of the spectrum in their surround, cannot explain color contrast or color constancy. Thus, the (S+M) vs. L cells, described above, that we propose could account for blue-yellow hue perception would require an additional transformation at a higher processing stage to make them double opponent accounting for color constancy. Hubel and Wiesel (Hubel & Wiesel, 2005) have suggested that type I LGN cells, which presumably represent output of S-ON small bistratified ganglion cells, feed into double opponent cells of cortical V1. Since, as discussed above, early stages of blue-yellow hue circuitry could be based on midget ganglion cells with S cone input from the surround, S-ON small bistratified ganglion cells could have the role suggested by Hubel and Wiesel. S+M vs. L cell inputs from layer IV could be combined with an inhibitory input from small bistratified cell input to layers 2/3 (Chatterjee & Callaway, 2003) to produce double-opponent cells. An attractive feature of the midget ganglion cell/H2 horizontal input cells being the early basis of hue opponency is that it leaves open an important function for the small bistratified cells in forming the second half of double opponent cells.
Acknowledgments
JN is the Bishop Professor and MN is the Hill professor in Ophthalmology at the University of Washington. The work from our laboratories discussed here was funded by Research to Prevent Blindness and by NEI grants EY09620, EY09303, EY016861, and Core Grants for Vision Research (EY01931, EY01730). We thank K. Mancuso for critical reading of the manuscript and discussions, and J. Kuchenbecker for discussions and assistance with the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Asenjo AB, Rim J, Oprian DD. Molecular determinants of human red/green color discrimination. Neuron. 1994;12:1131–1138. doi: 10.1016/0896-6273(94)90320-4. [DOI] [PubMed] [Google Scholar]
- Baraas RC, Carroll J, Gunther KL, Chung M, Williams DR, Roster DH, Neitz M. Adaptive optics retinal imaging reveals S-cone dystrophy in tritan color vision deficiency. Journal of the Optical Society of America A. 2007;24:1438–1447. doi: 10.1364/josaa.24.001438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbur JL, Rodriguez-Carmona M, Harlow JA, Mancuso K, Neitz J, Neitz M. A study of unusual Rayleigh matches in deutan deficiency. Visual Neuroscience. 2008;25:507–516. doi: 10.1017/S0952523808080619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor DA, Nunn BJ, Schnapf JL. Spectral sensitivity of cones of the monkey Macaca fascicularis. Journal of Physiology. 1987;390:145–160. doi: 10.1113/jphysiol.1987.sp016691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson DM. Strange vision: Ganglion cells as circadian photoreceptors. Trends in Neurosciences. 2003;26(6):314–320. doi: 10.1016/S0166-2236(03)00130-9. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Tan Y, Shyue SK, Schneider H, Sampaio I, Neiswanger K, Hewett-Emmett D, Li WH. Origins and antiquity of X-linked triallelic color vision systems in New World monkeys. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13749–13754. doi: 10.1073/pnas.95.23.13749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger K, Bialozynski C, Neitz J, Neitz M. The importance of deleterious mutations in M pigment genes as a cause of color vision defects. Investigative Ophthalmology & Visual Science (Supplement) 1999;40(4):S354. [Google Scholar]
- Bollinger K, Sjoberg S, Neitz M, Neitz J. Topographical cone photopigment gene expression in deutan-type red-green color vision defects. Vision Research. 2004;34:135–145. doi: 10.1016/j.visres.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Bosten JM, Robinson JD, Jordan G, Mollon JD. Multidimensional scaling reveals a color dimension unique to ‘color-deficient’ observers. Current Biology. 2005;15(23):R950–R952. doi: 10.1016/j.cub.2005.11.031. [DOI] [PubMed] [Google Scholar]
- Bumsted K, Hendrickson A. Distribution and development of short-wavelength cones differ between Macaca monkey and human fovea. The Journal of Comparative Neurology. 1999;403:502–516. [PubMed] [Google Scholar]
- Bumsted K, Jasoni C, Szél Á, Hendrickson A. Spatial and temporal expression of cone opsins during monkey retinal development. The Journal of Comparative Neurology. 1997;378:117–134. [PubMed] [Google Scholar]
- Calkins DJ. Seeing with S cones. Progress in Retinal and Eye Research. 2001;20:255–287. doi: 10.1016/s1350-9462(00)00026-4. [DOI] [PubMed] [Google Scholar]
- Carroll J, Baraas RC, Wagner-Schuman M, Rha J, Siebe CA, Sloan C, Tait DM, Thompson S, Morgan JIW, Neitz J, Neitz M. Cone photoreceptor mosaic disruption associated with Cys203Arg mutation in the M-cone opsin. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20948–20953. doi: 10.1073/pnas.0910128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Bialozynski C, Summerfelt P, Neitz M, Neitz J. Estimates of L:M cone ratios from ERG flicker photometry and genetics. Investigative Ophthalmology & Visual Science (Supplement) 2000;41(4):S808. [Google Scholar]
- Carroll J, McMahon C, Neitz M, Neitz J. Flicker-photometric electroretinogram estimates of L : M cone photoreceptor ratio in men with photopigment spectra derived from genetics. Journal of the Optical Society of America A. 2000;17(3):499–509. doi: 10.1364/josaa.17.000499. [DOI] [PubMed] [Google Scholar]
- Carroll J, Neitz M, Hofer H, Neitz J, Williams DR. Functional photoreceptor loss revealed with adaptive optics: An alternate cause of color blindness. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(22):8461–8466. doi: 10.1073/pnas.0401440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Neitz M, Neitz J. Estimates of L:M cone ratio from ERG flicker photometry and genetics. Journal of Vision. 2002;2:531–542. doi: 10.1167/2.8.1. [DOI] [PubMed] [Google Scholar]
- Carroll J, Rossi E, Porter J, Neitz J, Roorda A, Williams D, Neitz M. Deletion of the X-linked opsin gene array locus control region (LCR) results in disruption of the cone mosaic. Proc Natl Acad Sci USA. 2010 doi: 10.1016/j.visres.2010.07.009. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Callaway E. Parallel colour-opponent pathways to primary visual cortex. Nature. 2003;426:668–671. doi: 10.1038/nature02167. [DOI] [PubMed] [Google Scholar]
- Chen J, Nakamura T, Ebry TG, Ok H, Konno K, Derguini F, Nakanishi K, Honig B. Wavelength regulation in iodopsin, a cone pigment. Biophysical Journal. 1989;55:725–729. doi: 10.1016/S0006-3495(89)82871-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway B, Hubel D, Livingston M. Color contrast in macaque V1. Cerebral Cortex. 2002;12:915–925. doi: 10.1093/cercor/12.9.915. [DOI] [PubMed] [Google Scholar]
- Crognale MA, Fry M, Highsmith J, Haegerstrom-Portnoy G, Neitz J, Neitz M, Webster MA. Characterization of a novel form of X-linked incomplete achromatopsia. Visual Neuroscience. 2004;21:197–204. doi: 10.1017/s0952523804213384. [DOI] [PubMed] [Google Scholar]
- Crognale MA, Teller DY, Motulsky AG, Deeb SS. Severity of color vision defects: electroretinographic (ERG), molecular and behavioral studies. Vision Research. 1998;38(21):3377–3385. doi: 10.1016/s0042-6989(97)00425-2. [DOI] [PubMed] [Google Scholar]
- Crook JD, Davenport CM, Peterson BB, Packer OS, Detwiler PB, Dacey DM. Parallel ON and OFF cone bipolar inputs establish spatially coextensive receptive field structure of blue-yellow ganglion cells in primate retina. Journal of Neuroscience. 2009;29:8372–8387. doi: 10.1523/JNEUROSCI.1218-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. The Journal of Comparative Neurology. 1990;292:497–523. doi: 10.1002/cne.902920402. [DOI] [PubMed] [Google Scholar]
- Curcio CA, Sloan KR, Packer O, Hendrickson AE, Kalina RE. Distribution of cones in human and monkey retina: Individual variability and radial asymmetry. Science. 1987;236:579–582. doi: 10.1126/science.3576186. [DOI] [PubMed] [Google Scholar]
- Dacey DM. Morphology of a small-field bistratified ganglion cell type in the macaque and human retina. Visual Neuroscience. 1993;10(6):1081–1098. doi: 10.1017/s0952523800010191. [DOI] [PubMed] [Google Scholar]
- Dacey DM. Physiology, morphology and spatial densities of identified ganglion cell types in primate retina. Ciba Foundation Symposia. 1994;184:12–28. doi: 10.1002/9780470514610.ch2. discussion 28-34, 63-70. [DOI] [PubMed] [Google Scholar]
- Dacey DM. Circuitry for color coding in the primate retina. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(2):582–588. doi: 10.1073/pnas.93.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacey DM, Lee BB. The blue-ON opponent pathway in primate retina originates from a distinct bistratified ganglion cell type. Nature. 1994;367:731–735. doi: 10.1038/367731a0. [DOI] [PubMed] [Google Scholar]
- Dacey DM, Lee BB, Stafford DK, Pokorny J, Smith VC. Horizontal cells of the primate retina: cone specificity without spectral opponency. Science. 1996;271:656–659. doi: 10.1126/science.271.5249.656. [DOI] [PubMed] [Google Scholar]
- Dacey DM, Liao HW, Peterson BB, Robinson FR, Smith VC, Pokorny J, Yau KW, Gamlin PD. Melanopsin-expressing ganglion cells in primate retina signal colour and irradiance and project to the LGN. Nature. 2005;433(7027):749–754. doi: 10.1038/nature03387. [DOI] [PubMed] [Google Scholar]
- Dacey DM, Peterson BB, Robinson FR. Identification of an S-cone opponent OFF pathway in the Macaque monkey retina: morphology, physiology and posible circuitry. Investigative Ophthalmology & Visual Science. 2002;43 E-abstract 2983. [Google Scholar]
- Dartnall HJA, Bowmaker JK, Mollon JD. Human visual pigments: microspectrophotometric results from the eye of seven persons. Proceedings of the Royal Society of London - Series B: Biological Sciences. 1983a;220:115–130. doi: 10.1098/rspb.1983.0091. [DOI] [PubMed] [Google Scholar]
- Dartnall HJA, Bowmaker JK, Mollon JD. Microspectrophotometry of human photoreceptors. In: Mollon J, Sharpe L, editors. Colour Vision: Physiology and Psychophysics. New York: Academic Press; 1983b. pp. 69–80. [Google Scholar]
- Darwin C. On the origin of the species by means of natural selection, or, The preservation of favoured races in the struggle for life. London: J. Murray; 1859. [PMC free article] [PubMed] [Google Scholar]
- de Monasterio FM. Signals from blue cones in “red-green” opponent-colour ganglion cells of the macaque retina. Vision Research. 1979;19:441–449. doi: 10.1016/0042-6989(79)90111-1. [DOI] [PubMed] [Google Scholar]
- de Monasterio FM, Gouras P, Tolhurst DJ. Trichromatic colour opponency in ganglion cells of the rhesus monkey retina. J Physiology. 1975;251:197–216. doi: 10.1113/jphysiol.1975.sp011087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarco P, Pokorny J, Smith VC. Full spectrum cone sensitivity functions for X-chromosome-linked anomalous trichromats. Journal of the Optical Society of America A. 1992;9:1465–1476. doi: 10.1364/josaa.9.001465. [DOI] [PubMed] [Google Scholar]
- Derrington AM, Krauskopf J, Lennie P. Chromatic mechanisms in lateral geniculate nucleus of macaque. Journal of Physiology. 1984;357:241–265. doi: 10.1113/jphysiol.1984.sp015499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeValois RL, DeValois KK. A multi-stage color model. Vision Research. 1993;33(8):1053–1065. doi: 10.1016/0042-6989(93)90240-w. [DOI] [PubMed] [Google Scholar]
- Drum B. Hue signals from short- and medium-wavelength sensitive cones. Journal of the Optical Society of America A. 1989;6(1):153–156. doi: 10.1364/josaa.6.000153. [DOI] [PubMed] [Google Scholar]
- Drummond-Borg M, Deeb SS, Motulsky AG. Molecular patterns of X-chromosome-linked color genes among 134 men of European ancestry. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:983–987. doi: 10.1073/pnas.86.3.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Berson EL, Fishman GA, Sandberg MA, Alexander KR, Derlacki DJ, Rajagopalan AS. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(13):4884–4889. doi: 10.1073/pnas.0501233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasick JI, Lee N, Oprian DD. Spectral tuning in the human blue cone pigment. Biochemistry. 1999;38:11593–11596. doi: 10.1021/bi991600h. [DOI] [PubMed] [Google Scholar]
- Field G, Gauthier J, Sher A, Greschner M, Machado T, Jepson J, Shlens J, Gunning D, Mathieson K, Dabrowski W, Paniinski L, Litke A, Chichilinsky E. Functional connectivity in the retina at the resolution of photoreceptors. Nature. 2010;467:673–677. doi: 10.1038/nature09424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Collin S. Dendritic distribution of two populations of ganglion cells and the retinopetal fibers in the retina of the silver almprey (Ichthyomyzon unicuspis) Visual Neuroscience. 1990;4:533–545. doi: 10.1017/s0952523800005745. [DOI] [PubMed] [Google Scholar]
- Gooley JJ, Lu J, Chou TC, Scammell TE, Saper CB. Melanopsin in cells of origin of the retinohypothalamic tract. Nature Neuroscience. 2001;4(12):1165. doi: 10.1038/nn768. [DOI] [PubMed] [Google Scholar]
- Gouras P, Kruger J. Responses of cells in foveal visual cortex of the monkey to pure colo contrast. J Neurophysiology. 1979;42:850–860. doi: 10.1152/jn.1979.42.3.850. [DOI] [PubMed] [Google Scholar]
- Gunther K, Neitz J, Neitz M. Nucleotide polymorphisms upstream of the X-chromosome opsin gene array tune L:M cone ratio. Visual Neuroscience. 2008;25:265–271. doi: 10.1017/S0952523808080280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther KL, Neitz J, Neitz M. A novel missense mutation in the S cone photopigment in a male who made Tritan errors on the Neitz Test of Color Vision. Investigative Ophthalmology & Visual Science (Supplement) 2003;44:B803–B803. [Google Scholar]
- Gunther KL, Neitz J, Neitz M. A novel mutation in the short-wavelength sensitive cone pigment gene associated with a tritan color vision defect. Visual Neuroscience. 2006;23:403–409. doi: 10.1017/S0952523806233169. [DOI] [PubMed] [Google Scholar]
- Hagstrom SA, Neitz J, Neitz M. Ratio of M/L pigment gene expression decreases with retinal eccentricity. In: Cavonius CR, editor. Colour Vision Deficiencies XIII. Dordrecht: Kluwer Academic Publishers; 1997. pp. 59–66. [Google Scholar]
- Hattar S, Liao HW, Takao M, Berson DM, Yau KW. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295(5557):1065–1070. doi: 10.1126/science.1069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Motulsky AG, Deeb SS. Position of a ‘green-red’ hybrid gene in the visual pigment array determines colour-vision phenotype. Nature Genetics. 1999 May;22:90–93. doi: 10.1038/8798. [DOI] [PubMed] [Google Scholar]
- Hofer H, Carroll J, Neitz J, Neitz M, Williams DR. Organization of the human trichromatic cone mosaic. Journal of Neuroscience. 2005;25(42):9669–9679. doi: 10.1523/JNEUROSCI.2414-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel D, Livingston M. Color and contrast sensitivity in the lateral geniculate body and primary visual cortex of the macaque monkey. J Neuroscience. 1990;10:2223–2237. doi: 10.1523/JNEUROSCI.10-07-02223.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel D, Wiesel T. Receptive fields, binocular interaction and functional architecture in the cat's visual cortex. J Physiology. 1962;160:106–154. doi: 10.1113/jphysiol.1962.sp006837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel D, Wiesel TN. Brain and Visual Perception: The story of a 25 year collaboration. New York: Oxford University Press; 2005. [Google Scholar]
- Hurvich L, Jameson D. Color theory and abnormal red-green vision. Documenta Ophthalmologica. 1962;16:409–442. doi: 10.1007/BF00146738. [DOI] [PubMed] [Google Scholar]
- Jacobs GH. Comparative Color Vision. New York: Academic Press; 1981. [Google Scholar]
- Jacobs GH. Within-species variations in visual capacity among squirrel monkeys (Saimiri sciureus): Sensitivity differences. Vision Research. 1983;23(3):239–248. doi: 10.1016/0042-6989(83)90112-8. [DOI] [PubMed] [Google Scholar]
- Jacobs GH. Comparative color vision. In: Chalupa L, Werner JS, editors. The Visual Neurosciences. Vol. 2. Cambridge, MA: MIT Press; 2004. pp. 962–973. [Google Scholar]
- Jacobs GH. Primate color vision: A comparative perspective. Visual Neuroscience. 2008;25:619–633. doi: 10.1017/S0952523808080760. [DOI] [PubMed] [Google Scholar]
- Jacobs GH, Deegan JF, 2nd, Tan Y, Li WH. Opsin gene and photopigment polymorphism in a prosimian primate. Vision Research. 2002;42(1):11–18. doi: 10.1016/s0042-6989(01)00264-4. [DOI] [PubMed] [Google Scholar]
- Jacobs GH, Neitz J. Color vision in squirrel monkeys: sex-related differences suggest the mode of inheritance. Vision Research. 1985;25:141–143. doi: 10.1016/0042-6989(85)90088-4. [DOI] [PubMed] [Google Scholar]
- Jacobs GH, Neitz J. Inheritance of color vision in a New World monkey (Saimiri sciureus) Proceedings of the National Academy of Sciences of the United States of America. 1987;84:2545–2549. doi: 10.1073/pnas.84.8.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs GH, Neitz J, Deegan JF., II Retinal receptors in rodents maximally sensitive to ultraviolet light. Nature. 1991;353:655–656. doi: 10.1038/353655a0. [DOI] [PubMed] [Google Scholar]
- Jacobs GH, Neitz J, Neitz M. Genetic basis of polymorphism in the color vision of platyrrhine monkeys. Vision Research. 1993;33:269–274. doi: 10.1016/0042-6989(93)90083-9. [DOI] [PubMed] [Google Scholar]
- Jacobs GH, Williams GA. L and M cone proportions in polymorphic New World monkeys. Visual Neuroscience. 2006;23:365–370. doi: 10.1017/S0952523806233066. [DOI] [PubMed] [Google Scholar]
- Jacobs GH, Williams GA, Cahill H, Nathans J. Emergence of Novel Color Vision in Mice Engineered to Express a Human Cone Photopigment. Science. 2007;315:1723–1725. doi: 10.1126/science.1138838. [DOI] [PubMed] [Google Scholar]
- Jagla WM, Jägle H, Hayashi T, Sharpe LT, Deeb SS. The molecular basis of dichromatic color vision in males with multiple red and green visual pigment genes. Human Molecular Genetics. 2002;11:23–32. doi: 10.1093/hmg/11.1.23. [DOI] [PubMed] [Google Scholar]
- Johnson E, Hawken M, Shapley R. The Orientation Selectivity of Color-Responsive Neurons in Macaque V1. J Neuroscience. 2008;28:8096–8106. doi: 10.1523/JNEUROSCI.1404-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Hawken M, Shapley R. The spatial transformation of color in the primary visual cortex of the macaque monkey. Nature Neuroscience. 2001;4:409–416. doi: 10.1038/86061. [DOI] [PubMed] [Google Scholar]
- Johnston RJ, Jr, Desplan C. Stochastic neuronal cell fate choices. Current Opinion in Neurobiology. 2008;18:20–27. doi: 10.1016/j.conb.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan G, Deeb S, Bosten J, Mollon JD. The dimensionality of color vision in carriers of anomalous trichromacy. J Vision. 2010;10:12. doi: 10.1167/10.8.12. [DOI] [PubMed] [Google Scholar]
- Jordan G, Mollon JD. A study of women heterozygous for colour deficiencies. Vision Research. 1993;33:1495–1508. doi: 10.1016/0042-6989(93)90143-k. [DOI] [PubMed] [Google Scholar]
- Jordan G, Mollon JD. Sons and mothers: classification of colour-deficient and heterozygous subjects by counterphase modulation photometry. In: Cavonius CR, editor. Colour Vision Deficiencies XIII. Dordrecht: Kluwer Academic Publishers; 1997. pp. 385–392. [Google Scholar]
- Jusuf P, Martin P, Grunert U. Random wiring in the midget pathway of primate retina. J Neuroscience. 2006;26:3908–3917. doi: 10.1523/JNEUROSCI.4891-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmus H. The familial distribution of congenital tritanopia. Annals of Human Genetics. 1955;20:39–56. doi: 10.1111/j.1469-1809.1955.tb01277.x. [DOI] [PubMed] [Google Scholar]
- Kanda A, Swaroop A. A comprehensive analysis of sequence variants and putative disease-causing mutations in photoreceptor-specific nuclear receptor NR2E3. Molecular Vision. 2009;15:2174–2184. [PMC free article] [PubMed] [Google Scholar]
- Klug K, Herr S, Ngo I, Sterling P, Schein SJ. Macaque retina contains an S-cone OFF midget pathway. J Neuroscience. 2003;39:9881–9887. doi: 10.1523/JNEUROSCI.23-30-09881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoblauch K, Neitz M, Neitz J. An urn model of the development of L/M cone ratios in human and macaque retina. Visual Neuroscience. 2006;23(3-4):591–596. doi: 10.1017/S0952523806233157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl S, Jägle H, Sharpe L, Wissinger B. GeneReviews [internet]1993-2004 Jun 24 [updated 2010 September 10] University of Washington; 2009. Achromatopsia. [Google Scholar]
- Komáromy A, Alexander J, SRowlan J, Garcia M, Chiodo V, Kaya A, Tanaka J, Acland G, Hauswirth W, Aguirre G. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010;19:2581–2593. doi: 10.1093/hmg/ddq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosower EM. Assignment of groups responsible for the “opsin shift” and light absorption of rhodopsin and red, green, and blue iodopsins (cone pigments) Proceedings of the National Academy of Sciences of the United States of America. 1988;85:1076–1080. doi: 10.1073/pnas.85.4.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi M, Kubokawa K, Tsukamoto H, Shichida Y, Terakita A. Cephalochordate melanopsin: evolutionary linkage between invertebrate visual cells and vertebrate photosensitive retinal ganglion cells. Current Biology. 2005;15(11):1065–1069. doi: 10.1016/j.cub.2005.04.063. [DOI] [PubMed] [Google Scholar]
- Kraft TW, Neitz J, Neitz M. Spectra of human L cones. Vision Research. 1998;38:3663–3670. doi: 10.1016/s0042-6989(97)00371-4. [DOI] [PubMed] [Google Scholar]
- Krauskopf J, Williams D, Heeley D. Cardinal Directions of Color Space. Vision Research. 1982;22:1123–1131. doi: 10.1016/0042-6989(82)90077-3. [DOI] [PubMed] [Google Scholar]
- Kuchenbecker J, Sahay M, Tait DM, Neitz M, Neitz J. Topography of the long- to middle-wavelength sensitive cone ratio in the human retina assessed with a wide-field color multifocal electroretinogram. Visual Neuroscience. 2008;25:301–306. doi: 10.1017/S0952523808080474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb T, Collin S, Pugh ENJ. Evolution of the vertebrate eye:opsins, photoreceptors, retina and eye cup. Nature Reviews Neuroscience. 2007;8:960–976. doi: 10.1038/nrn2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb TD. Photoreceptor spectral sensitivities: common shape in the long-wavelength region. Vision Research. 1995;35(22):3083–3091. doi: 10.1016/0042-6989(95)00114-f. [DOI] [PubMed] [Google Scholar]
- Lennie P, Haake PW, Williams DR. The design of chromatically opponent receptive fields. In: Landy MS, Movshon JA, editors. Computational Models of Visual Processing. Cambridge: MIT Press; 1991. pp. 71–82. [Google Scholar]
- Lewis RA, Holcomb JD, Bromley WC, Wilscon MC, Roderick TH, Hejtmancik F. Mappinx X-linked ophthalmic diseases III. Provisional assignment of the locus for blue cone monochromacy to Xq28. Archives of Ophthalmology. 1987;105:1055–1059. doi: 10.1001/archopht.1987.01060080057028. [DOI] [PubMed] [Google Scholar]
- Li Q, Timmers AM, Guy J, Pang J, Hauswirth WW. Cone-specific expression using a human red opsin promoter in recombinant AAV. Vision Research. 2007;48 doi: 10.1016/j.visres.2007.07.026. [DOI] [PubMed] [Google Scholar]
- Li W, DeVries SH. Bipolar cell pathways for color and luminance vision in a dichromatic mammalian retina. Nature Neuroscience. 2006;9(5):669–675. doi: 10.1038/nn1686. [DOI] [PubMed] [Google Scholar]
- Macke JP, Nathans J. Individual variation in the size of the human red and green visual pigment gene array. Investigative Ophthalmology & Visual Science. 1997;38(5):1040–1043. [PubMed] [Google Scholar]
- Makous W. Comment on “Emergence of Novel Color Vision in Mice Engineered to Express a Human Cone Photopigment”. Science. 2007;318:196. doi: 10.1126/science.1146084. [DOI] [PubMed] [Google Scholar]
- Mancuso K, Hauswirth WW, Li Q, Connor TB, Kuchenbecker JA, Mauck MC, Neitz J, Neitz M. Gene therapy for red-green colour blindness in adult primates. Nature. 2009;461:784–787. doi: 10.1038/nature08401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso K, Mauck MC, Kuchenbecker JA, Neitz M, Neitz J. A multi-stage color model revisited: Implications for a gene therapy cure for red-green colorblindness. Advances in Experimental Medicine and Biology. 2010a;664:631–638. doi: 10.1007/978-1-4419-1399-9_72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso K, Neitz M, Hauswirth WW, Li Q, Connor T, Kuchenbecker J, Mauck MC, Neitz J. Longer-term results of gene therapy for red-green color blindness in monkeys. IOVS. 2010b;51 doi: 10.1038/nature08401. e-abstract 6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield RJW. Primate photopigments and cone mechanisms. In: Fein A, Levine JS, editors. The Visual System. New York: A. R. Liss; 1985. pp. 89–106. [Google Scholar]
- Mariani AP. Bipolar cells in monkey retina selective for the cones likely to be blue-sensitive. Nature. 1984;308:184–186. doi: 10.1038/308184a0. [DOI] [PubMed] [Google Scholar]
- Martin PR, Grunert U. Analysis of the short wavelength-sensitive (“blue”) cone mosaic in the primate retina: Comparison of New World and Old World monkeys. The Journal of Comparative Neurology. 1999;406:1–14. doi: 10.1002/(sici)1096-9861(19990329)406:1<1::aid-cne1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- McMahon C, Carroll J, Awua S, Neitz J, Neitz M. The L:M cone ratio in males of African descent with normal color vision. Journal of Vision. 2008;8:1–9. doi: 10.1167/8.2.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merbs SL, Nathans J. Absorption spectra of the hybrid pigments responsible for anomalous color vision. Science. 1992;258:464–466. doi: 10.1126/science.1411542. [DOI] [PubMed] [Google Scholar]
- Merbs SL, Nathans J. Role of hydroxyl-bearing amino acids in differentially tuning the absorption spectra of the human red and green cone pigments. Photochemistry and Photobiology. 1993;58:706–710. doi: 10.1111/j.1751-1097.1993.tb04956.x. [DOI] [PubMed] [Google Scholar]
- Merbs SL, N J. Absorption spectra of human cone pigments. Nature. 1992;356:433–435. doi: 10.1038/356433a0. [DOI] [PubMed] [Google Scholar]
- Mizrahi-Meissonnier L, Merin S, Banin E, Sharon D. Variable retinal phenotypes caused by mutations in the X-linked photopigment gene array. Investigative Ophthalmology and Visual Science. 2010 doi: 10.1167/iovs.09-4592. [DOI] [PubMed] [Google Scholar]
- Mollon JD. “Tho' she kneel'd in that Place where they grew…”. The uses and origins of primate colour vision. Journal of Experimental Biology. 1989;146:21–38. doi: 10.1242/jeb.146.1.21. [DOI] [PubMed] [Google Scholar]
- Mollon JD, Bowmaker JK. The spatial arrangement of cones in the primate fovea. Nature. 1992;360:677–679. doi: 10.1038/360677a0. [DOI] [PubMed] [Google Scholar]
- Mollon JD, Bowmaker JK, Jacobs GH. Variations of colour vision in a New World primate can be explained by polymorphism of retinal photopigments. Proceedings of the Royal Society of London - Series B: Biological Sciences. 1984;222(1228):373–399. doi: 10.1098/rspb.1984.0071. [DOI] [PubMed] [Google Scholar]
- Molnar A, Werblin F. Inhibitory feedback shapes bipolar cell responses in the rabbit retina. J Neurophysiology. 2007;98:3423–3435. doi: 10.1152/jn.00838.2007. [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Adam A, Mollon JD. Dichromats detect colour-camouflaged objects that are not detected by trichromats. Proceedings of the Royal Society of London - Series B: Biological Sciences. 1992;248:291–295. doi: 10.1098/rspb.1992.0074. [DOI] [PubMed] [Google Scholar]
- Nagy A, MacLeod D, Heyneman N, Eisner A. Four cone pigments in women heterozygous for color deficiency. Journal of the Optical Society of America. 1981;71:719–799. doi: 10.1364/josa.71.000719. [DOI] [PubMed] [Google Scholar]
- Nathans J, Davenport CM, Maumenee IH, Lewis RA, Hejtmancik JF, Litt M, Lovrien E, Weleber R, Bachynski B, Zwas F, Klingaman R, Fishman G. Molecular genetics of human blue cone monochromacy. Science. 1989;245:831–838. doi: 10.1126/science.2788922. [DOI] [PubMed] [Google Scholar]
- Nathans J, Maumenee IA, Zrenner E, Sadowski B, Sharpe LT, Lewis RA, Hansen E, Rosenberg P, Schwartz M, Heckenlively JR, Trabousli E, Klingaman R, Bech-hansen NT, LaRouche GR, Pagon RA, Murphy WH, Weleber RG. Genetic heterogeneity among blue-cone monochromats. American Journal of Human Genetics. 1993;53:987–1000. [PMC free article] [PubMed] [Google Scholar]
- Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS. Molecular genetics of inherited variation in human color vision. Science. 1986a;232:203–210. doi: 10.1126/science.3485310. [DOI] [PubMed] [Google Scholar]
- Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. 1986b;232:193–202. doi: 10.1126/science.2937147. [DOI] [PubMed] [Google Scholar]
- Neitz J, Carroll J, Neitz M. Color vision: Almost reason enough for having eyes. Optics & Photonics News. 2001;12:26–33. [Google Scholar]
- Neitz J, Jacobs GH. Polymorphism of the long-wavelength cone in normal human color vision. Nature. 1986;323:623–625. doi: 10.1038/323623a0. [DOI] [PubMed] [Google Scholar]
- Neitz J, Neitz M. Colour vision: The wonder of hue. Current Biology. 2008;18:R700–R702. doi: 10.1016/j.cub.2008.06.062. [DOI] [PubMed] [Google Scholar]
- Neitz J, Neitz M, He JC, Shevell SK. Trichromatic color vision with only two spectrally distinct photopigments. Nature Neuroscience. 1999;2:884–888. doi: 10.1038/13185. [DOI] [PubMed] [Google Scholar]
- Neitz J, Neitz M, Jacobs GH. More than three different cone pigments among people with normal color vision. Vision Research. 1993;33:117–122. doi: 10.1016/0042-6989(93)90064-4. [DOI] [PubMed] [Google Scholar]
- Neitz J, Neitz M, Kainz PM. Visual pigment gene structure and the severity of human color vision defects. Science. 1996;274:801–804. doi: 10.1126/science.274.5288.801. [DOI] [PubMed] [Google Scholar]
- Neitz M, Balding SD, McMahon C, Sjoberg SA, Neitz J. Topography of long- and middle-wavelength sensitive cone opsin gene expression in human and Old World monkey retina. Visual Neuroscience. 2006;23(3-4):379–385. doi: 10.1017/S095252380623325X. [DOI] [PubMed] [Google Scholar]
- Neitz M, Bollinger K, Neitz J. Middle wavelength sensitive photopigment gene expression is absent in deuteranomalous colour vision. In: Mollon JD, Knoblauch K, Pokorny J, editors. Normal and Defective Colour Vision. Oxford, UK: Oxford University Press; 2003. pp. 318–327. [Google Scholar]
- Neitz M, Carroll J, Renner A, Knau H, Werner JS, Neitz J. Variety of genotypes in males diagnosed as dichromatic on a conventional clinical anomaloscope. Visual Neuroscience. 2004;21:205–216. doi: 10.1017/s0952523804213293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitz M, Kraft TW, Neitz J. Expression of L cone pigment gene subtypes in females. Vision Research. 1998;38:3221–3225. doi: 10.1016/s0042-6989(98)00076-5. [DOI] [PubMed] [Google Scholar]
- Neitz M, Neitz J. Numbers and ratios of visual pigment genes for normal red-green color vision. Science. 1995;267:1013–1016. doi: 10.1126/science.7863325. [DOI] [PubMed] [Google Scholar]
- Neitz M, Neitz J, Grishok A. Polymorphism in the number of genes encoding long-wavelength sensitive cone pigments among males with normal color vision. Vision Research. 1995a;35:2395–2407. [PubMed] [Google Scholar]
- Neitz M, Neitz J, Jacobs GH. Spectral tuning of pigments underlying red-green color vision. Science. 1991;252:971–974. doi: 10.1126/science.1903559. [DOI] [PubMed] [Google Scholar]
- Neitz M, Neitz J, Jacobs GH. Genetic basis of photopigment variations in human dichromats. Vision Research. 1995b;35:2095–2103. doi: 10.1016/0042-6989(94)00306-8. [DOI] [PubMed] [Google Scholar]
- Packer OS, Verweij J, Li PH, Schnapf JL, Dacey DM. Blue-yellow opponency in primate S cone photoreceptors. Journal of Neuroscience. 2010;30(2):568–572. doi: 10.1523/JNEUROSCI.4738-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus W, Kroger-Paulus A. A new concept of retinal colour coding. Vision Research. 1983;23(5):529–540. doi: 10.1016/0042-6989(83)90128-1. [DOI] [PubMed] [Google Scholar]
- Piantanida T. A replacement model of X-linked recessive colour vision defects. Annals of Human Genetics. 1974;37:393–404. doi: 10.1111/j.1469-1809.1974.tb01845.x. [DOI] [PubMed] [Google Scholar]
- Piantanida TP, Gille J. Methodology-specific Rayleigh-match distributions. Vision Research. 1992;32:2375–2377. doi: 10.1016/0042-6989(92)90100-w. [DOI] [PubMed] [Google Scholar]
- Pokorny J, Smith VC. Evaluation of a single pigment shift model of anomalous trichromacy. Journal of the Optical Society of America. 1977;67:1196–1209. doi: 10.1364/josa.67.001196. [DOI] [PubMed] [Google Scholar]
- Provencio I, Jiang G, WDeGrip W, Hayes W, Rollag M. Melanopsin: An opsin in melanophores, brain, and eye. Proc Natl Acad Sci USA. 1998;95:340–345. doi: 10.1073/pnas.95.1.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provencio I, Rodriguez I, Jiang G, Hayes W, Moreira E, Rollag M. A Novel Human Opsin in the Inner Retina. Journal of Neuroscience. 2000;20:600–605. doi: 10.1523/JNEUROSCI.20-02-00600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan BC, Julliot C, Simmen B, Vienot F, Charles-Dominique P, Mollon JD. Fruits, foliage and the evolution of primate colour vision. Philosophical Transactions of the Royal Society B: Biological Sciences. 2001;356:229–283. doi: 10.1098/rstb.2000.0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha-Sousa A, Hayashi T, Gomes N, Penas S, Brandão E, Rocha P, Urashima M, Yamada H, Tsuneoka H, Falcão-Reis F. A novel mutation (Cys83Tyr) in the second zinc finger of NR2E3 in enhanced S-cone syndrome. Graefes Archive for Clinical and Experimental Ophthalmology. doi: 10.1007/s00417-010-1482-y. in press. [DOI] [PubMed] [Google Scholar]
- Rollag MD, Berson DM, Provencio I. Melanopsin, ganglion-cell photoreceptors, and mammalian photoentrainment. Journal of Biological Rhythms. 2003;18(3):227–234. doi: 10.1177/0748730403018003005. [DOI] [PubMed] [Google Scholar]
- Roorda A, Metha A, Lennie P, Williams DR. Packing arrangement of the three cone classes in primate retina. Vision Research. 2001;41:1291–1306. doi: 10.1016/s0042-6989(01)00043-8. [DOI] [PubMed] [Google Scholar]
- Roorda A, Williams DR. The arrangement of the three cone classes in the living human eye. Nature. 1999;397:520–522. doi: 10.1038/17383. [DOI] [PubMed] [Google Scholar]
- Rowe M, Jacobs G. Naturalistic color discriminations in polymorphic platyrrhine monkeys: effects of stimulus luminance and duration examined with functional substitution. Visual Neuroscience. 2007;24:17–23. doi: 10.1017/S0952523807230159. [DOI] [PubMed] [Google Scholar]
- Rushton WAH, Baker HD. Red/green sensitivity in normal vision. Vision Research. 1964;4:75–85. doi: 10.1016/0042-6989(64)90034-3. [DOI] [PubMed] [Google Scholar]
- Sanocki E, Shevell SK, Winderickx J. Serine/alanine amino acid polymorphism of the L cone photopigment assessed by dual Rayleigh-type color matches. Vision Research. 1993;34:377–382. doi: 10.1016/0042-6989(94)90096-5. [DOI] [PubMed] [Google Scholar]
- Schnapf JL, Kraft TW, Baylor DA. Spectral sensitivity of human cone photoreceptors. Nature. 1987;325:439–441. doi: 10.1038/325439a0. [DOI] [PubMed] [Google Scholar]
- Shapley R. Vision: Gene therapy in colour. Nature. 2009;461:737–739. doi: 10.1038/461737a. [DOI] [PubMed] [Google Scholar]
- Sharpe LT, Stockman A, Jägle H, Knau H, Klausen G, Reitner A, Nathans J. Red, green, and red-green hybrid pigments in the human retina: correlations between deduced protein sequences and psychophysically measured spectral sensitivities. Journal of Neuroscience. 1998;18:10053–10069. doi: 10.1523/JNEUROSCI.18-23-10053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevell SK, He JC, Kainz PM, Neitz J, Neitz M. Relating color discrimination to photopigment genes in deutan observers. Vision Research. 1998;38:3371–3376. doi: 10.1016/s0042-6989(97)00434-3. [DOI] [PubMed] [Google Scholar]
- Sjoberg SA, Neitz M, Balding SD, Neitz J. L-cone pigment genes expressed in normal colour vision. Vision Research. 1998;38:3213–3219. doi: 10.1016/s0042-6989(97)00367-2. [DOI] [PubMed] [Google Scholar]
- Smallwood PM, Olveczky BP, Williams GL, Jacobs GH, Reese BE, Meister M, Nathans J. Genetically engineered mice with an additional class of cone photoreceptors: implications for the evolution of color vision. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(20):11706–11711. doi: 10.1073/pnas.1934712100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood PM, Wang Y, Nathans J. Role of a locus control region in the mutually exclusive expression of human red and green cone pigment genes. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:1008–1011. doi: 10.1073/pnas.022629799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solessio E, Engbretson GA. Antagonistic chromatic mechanisms in photoreceptors of the parietal eye of lizards. Nature. 1993;364(6436):442–445. doi: 10.1038/364442a0. [DOI] [PubMed] [Google Scholar]
- Solomon SG, Lennie P. The machinery of colour vision. Nature Reviews Neuroscience. 2007;8(4):276–286. doi: 10.1038/nrn2094. [DOI] [PubMed] [Google Scholar]
- Spudich J, Spudich E. The simplest eyes: rhodopsin-mediated phototaxis reception in microorganisms. In: Tsonis P, editor. Animal Models in Eye Research. Amsterdam: Elsevier; 2008. pp. 6–14. [Google Scholar]
- Stamatoyannopoulos J, Thurman R, Noble W, Kutyavin T, Shafer A, Dorschner M, Deeb SS. American Society of Human Genetics. Salt Lake City, Utah: American Society of Human Genetics; 2005. Quantitative phenotype variation in long- and middle-wave cone photoreceptors due to polymorphism in an upstream insulator element. p Abstract #273. [Google Scholar]
- Stockman A, Brainard D. Color vision mechanisms. In: Bass M, editor. OSA Handbook of Optics. New York: McGraw-Hill; 2010. pp. 11.11–11.104. [Google Scholar]
- Stockman A, MacLeod DIA, Johnson NE. Spectral sensitivities of the human cones. Journal of the Optical Society of America A. 1993;10(12):2491–2520. doi: 10.1364/josaa.10.002491. [DOI] [PubMed] [Google Scholar]
- Stockman A, Sharpe LT, Merbs S, Nathans J. Spectral sensitivities of human cone visual pigments determined in vivo and in vitro. Methods in Enzymology. 2000;316:626–650. doi: 10.1016/s0076-6879(00)16754-0. [DOI] [PubMed] [Google Scholar]
- Su C, Luo D, Terakita A, Shichida Y, Liao H, Kazmi M, Sakmar T, Yau KW. Parietal-eye phototransduction components and their potential evolutionary implications. Science. 2006;311:1617–1627. doi: 10.1126/science.1123802. [DOI] [PubMed] [Google Scholar]
- Sun H, Smithson HE, Zaidi Q, Lee BB. Specificity of cone inputs to Macaque retinal ganglion cells. Journal of Neurophysiology. 2006;95:837–849. doi: 10.1152/jn.00714.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailby C, Solomon SG, Lennie P. Functional asymmetries in visual pathways carrying S-cone signals in macaque. Journal of Neuroscience. 2008;28(15):4078–4087. doi: 10.1523/JNEUROSCI.5338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiadens A, Somervuo V, van den Born L, Roosing S, van Schooneveld M, Kuijpers R, van Moll-Ramirez N, Cremers F, Hoyng C, Klaver C. Progressive loss of cones in achromatopsia. An imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2010;51:2852–2859. doi: 10.1167/iovs.10-5680. [DOI] [PubMed] [Google Scholar]
- Ueyama H, Hayashi S, Tanabe S, Tanaka Y, Hayashi T, Deeb SS, Yamade S, Ohkubo I. Number and arrangement of the red and green visual pigment genes in color-normal Japanese males. Color Research & Application. 2001;26:S84–S88. [Google Scholar]
- Verrelli BC, Tishkoff SA. Signatures of selection and gene conversion associated with human color vision variation. American Journal of Human Genetics. 2004;75:363–375. doi: 10.1086/423287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollrath D, Nathans J, Davis RW. Tandem array of human visual pigment genes at Xq28. Science. 1988;240:1669–1672. doi: 10.1126/science.2837827. [DOI] [PubMed] [Google Scholar]
- Waaler GHM. Heredity of two normal types of colour vision. Nature. 1968;218:688–689. doi: 10.1038/218688a0. [DOI] [PubMed] [Google Scholar]
- Wald G. The molecular basis of visual excitation. Nature. 1967;219:800–807. doi: 10.1038/219800a0. [DOI] [PubMed] [Google Scholar]
- Wald G. The molecular basis of visual excitation. Science. 1968;162:230–239. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- Wang Y, Macke JP, Merbs SL, Zack DJ, Klaunberg B, Bennett J, Gearhart J, Nathans J. A locus control region adjacent to the human red and green visual pigment genes. Neuron. 1992;9:429–440. doi: 10.1016/0896-6273(92)90181-c. [DOI] [PubMed] [Google Scholar]
- Webster MA, Miyahara E, Malkoc G, Raker VE. Variations in normal color vision I. Cone-opponent axes. Journal of the Optical Society of America A. 2000;17(9):1535–1544. doi: 10.1364/josaa.17.001535. [DOI] [PubMed] [Google Scholar]
- Weitz CJ, Miyake Y, Shinzato K, Montag E, Zrenner E, Went LN, Nathans J. Human tritanopia associated with two amino acid substitutions in the blue sensitive opsin. American Journal of Human Genetics. 1992a;50:498–507. [PMC free article] [PubMed] [Google Scholar]
- Weitz CJ, Went LN, Nathans J. Human tritanopia associated with a third amino acid substitution in the blue sensitive visual pigment. American Journal of Human Genetics. 1992b;51:444–446. [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH. Spatial and chromatic interactions in the lateral geniculate body of the rhesus monkey. Journal of Neurophysiology. 1966;29(6):1115–1156. doi: 10.1152/jn.1966.29.6.1115. [DOI] [PubMed] [Google Scholar]
- Wikler KC, Rakic P. Relation of an array of early-differentiating cones to the photoreceptor mosaic in the primate retina. Nature. 1991;351:397–400. doi: 10.1038/351397a0. [DOI] [PubMed] [Google Scholar]
- Winderickx J, Battisti L, Hibibya Y, Motulsky AG, Deeb SS. Haplotype diversity in the human red and green opsin genes: evidence for frequent sequence exchange in exon 3. Human Molecular Genetics. 1993;2:1413–1421. doi: 10.1093/hmg/2.9.1413. [DOI] [PubMed] [Google Scholar]
- Winderickx J, Battisti L, Motulsky AG, Deeb SS. Selective expression of human X chromosome-linked green opsin genes. Proceedings of the National Academy of Sciences of the United States of America. 1992a;89:9710–9714. doi: 10.1073/pnas.89.20.9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winderickx J, Lindsey DT, Sanocki E, Teller DY, Motulsky AG, Deeb SS. Polymorphism in red photopigment underlies variation in colour matching. Nature. 1992b;356:431–433. doi: 10.1038/356431a0. [DOI] [PubMed] [Google Scholar]
- Winderickx J, Sanocki E, Lindsey DT, Teller DY, Motulsky AG, Deeb SS. Defective colour vision associated with a missense mutation in the human green visual pigment gene. Nature Genetics. 1992c;1:251–256. doi: 10.1038/ng0792-251. [DOI] [PubMed] [Google Scholar]
- Xiao M, Hendrickson A. Spatial and temporal expression of short, long/medium or both opsins in human fetal cones. The Journal of Comparative Neurology. 2000;425:545–559. [PubMed] [Google Scholar]
- Xu X, Zhou Z, Wang X, Kuang X, Zhang F, Du X. Four-winged dinosaurs from China. Nature. 2003;421:335–340. doi: 10.1038/nature01342. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Motulsky AG, Deeb SS. Visual pigment gene structure and expression in the human retinae. Human Molecular Genetics. 1997;6(7):981–990. doi: 10.1093/hmg/6.7.981. [DOI] [PubMed] [Google Scholar]