

Abstract
Although much current research focuses on developing new boron reagents and identifying robust catalytic systems for the cross-coupling of these reagents, the fundamental preparations of the nucleophilic partners (i.e., boronic acids and derivatives) has been studied to a lesser extent. Most current methods to access boronic acids are indirect and require harsh conditions or expensive reagents. A simple and efficient palladium-catalyzed, direct synthesis of arylboronic acids from the corresponding aryl chlorides using an underutilized reagent, tetrahydroxydiboron B2(OH)4, is reported. To insure preservation of the carbon-boron bond, the boronic acids were efficiently converted to the trifluoroborate derivatives in good to excellent yields without the use of a work-up or isolation. Further, the intermediate boronic acids can be easily converted to a wide range of useful boronates. Finally, a two-step, one-pot method was developed to couple two aryl chlorides efficiently in a Suzuki-Miyaura-type reaction.
The Suzuki–Miyaura cross-coupling reaction has become of immense utility and thus importance,1,2 and yet there does not exist a simple, robust, catalytic, and functional group tolerant method that provides direct access to the boronic acid, with straightforward conversion to related derivatives. Thus, arylboronic acids are normally accessed by hydrolysis of boronate esters, and consequently many other boronic acid derivatives (e.g., organotrifluoroborates) are derived ultimately from these same boronate esters. The boronate esters can be accessed in a number of ways, including iridium-catalyzed C–H activation with bis(pinacolato)diboron3 and transmetalation from reactive organometallics.4 The harsh reaction conditions used in the latter approach severely limit functional group incorporation into the boronic acid derivatives.5 Another common method makes use of bis(pinacolato)diboron or related reagents to access arylboronates catalytically according to Miyaura's widely used protocol.6–12 When boronic acids are the targets required in any of these protocols, the boronate esters must be converted by way of oxidation,13 hydrolysis,14 reduction,15 or transesterification.16 These methods are inherently inefficient both in terms of atom economy and step economy, employ harsh reaction conditions (NaIO4, aq HCl, LAH, diethanolamine, BBr3), or require the use of excess polymer-supported boronic acid.16,17 Further, the transformation of pinacol boronate esters to the analogous trifluoroborates often suffers from difficulties in purification, rendering these protocols exceedingly tedious.18,19
In this communication, the development of the first direct synthesis of boronic acids via palladium-catalyzed cross-coupling of aryl chlorides with tetrahydroxydiboron B2(OH)4 (1) is outlined.20–23 This protocol allows the direct synthesis and isolation of arylboronic acids and also permits the preparation of trifluoroborates and diverse boronate analogs without the use of highly reactive organometallic species or through manipulation of the arylboronates. Additionally, the method permits the coupling of two aryl chlorides in a two-step one-pot reaction, representing an efficient manner in which to perform Suzuki-Miyaura type coupling reactions. Further, all reagents are bench stable, obviating the use of a glove box, and the method does not require the use of anhydrous solvents.
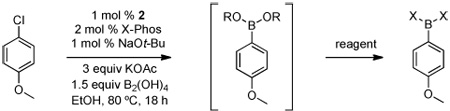
Microscale high-throughput experimentation (HTE) techniques24 revealed that the use of Pd(OAc)2 with X-Phos (3:1 ligand:palladium), KOAc (3 equiv), and EtOH at 80 °C for 18 h was optimal to convert 4-chloroanisole to the corresponding borylated product. Attempts to translate this lead into a robust scale-up procedure revealed that reaction success was highly dependent upon how the active catalyst was formed. During the HTE studies, the catalyst and ligand were dosed to the sample plates in appropriate solvents based on component solubility, and then the dosing solvents were removed under high vacuum prior to addition of the remaining reagents.
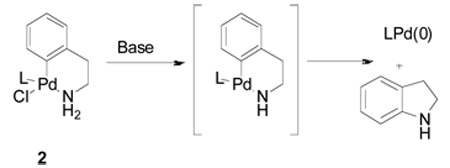
For initial synthetic studies and scale-up in the laboratory, the protocol was different: all of the components were mixed, and thus tetrahydroxydiboron 1 (eq 1) was exposed to the non-ligated Pd(II) precatalyst. The use of 11B NMR to monitor stability demonstrated that 1 decomposed completely after 1 h of stirring at rt in EtOH in the presence of various Pd(II) species {e.g., Pd[(allyl)Cl]2 and Pd(OAc)2}. However, if the Pd(II) source and the ligand were mixed together for 1 h at 65 °C in EtOH prior to the addition of 1, decomposition of 1 was greatly reduced. It thus appeared that in the HTE experiments, the solvent removal process allowed the Pd(OAc)2 to become efficiently complexed to the ligand, thereby minimizing the decomposition of 1. To eliminate the need to activate the catalyst before adding the B2(OH)4 and substrate, we found that the use of a recently reported preformed catalyst 2 performs as advertised in undergoing rapid transformation to the active Pd(0) species (eq 2), allowing the cross-coupling to occur in a more efficient and simplified manner without extensive decomposition of the tetrahydroxy diboron.25 As mentioned, the use of alcohol solvents, most notably ethanol, was optimal for solubility and reactivity. Under these conditions the tetrahydroxy diboron is in equilibrium with various ethyl esters, providing some similarity to bispinacol boronate (eq 1). In the HTE experiments the ligand to palladium ratio (3:1) and concentration (0.1 M) were also found to be essential to achieve high yields.
 |
(1) |
 |
(2) |
Utilizing this convenient protocol (Table 1), diverse boronate derivatives, including pinacol boronates (entry 1), MIDA boronates (entry 3), and pinanediol boronates (entry 4) were synthesized. The boronic acid can also be isolated in excellent yield after aqueous work-up by simply washing the crude solid with hexane (Table 1 entry 5).
Table 1.
Palladium-Catalyzed Direct Synthesis of a Boronic Acid and Derivatives
 | |||
|---|---|---|---|
| entry | reagent | product | isolated yield (%) |
| 1 | 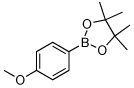 |
90 | |
| 2 |  |
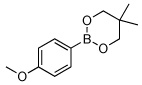 |
87 |
| 3 | 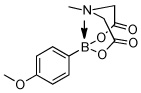 |
85 | |
| 4 | 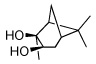 |
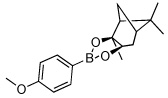 |
75 |
| 5 | hexanes wash | 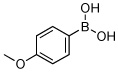 |
82 |
| 6 |  |
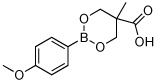 |
72 |
Of greatest interest was the direct conversion of the intermediates to the more robust aryltrifluoroborates (Table 2). Avoiding the work-up, treatment with KHF2 using an operationally simple and efficient protocol affords the 'protected' form of the boronic acid in higher yields. Thus all reagents were first weighed on the bench (including the aryl chloride if it was a solid) into a vessel capable of being sealed with a Teflon cap (commonly used for microwave reaction vials). The reactions were sealed and then purged with nitrogen. Degassed ethanol was added via syringe (followed by the aryl chloride if it was a liquid) and the reaction was heated to 80 °C for the required time (4–18 h). The reaction was cooled to rt, then filtered through a pad of Celite, rinsing with EtOAc. After concentrating the reaction, it was taken up in MeOH, cooled to 0 °C, and then aqueous KHF2 was added dropwise. The bath was removed, and the reaction was allowed to stir at rt until conversion to the trifluoroborate was complete (~20 min) as monitored by 11B NMR. The reaction was concentrated, dried, and purified by continuous Soxhlet extraction with acetone. Once the extraction was complete, the acetone was removed and the trifluoroborate was obtained by precipitation with ether. A similar, user-friendly procedure was used for the synthesis of the boronate esters (see Supporting Information for details).
Table 2.
Boronic Acid Synthesis and Direct Conversion to Trifluoroborates
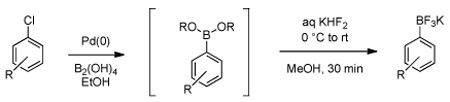 | |||||
|---|---|---|---|---|---|
| entry | product | isolated yield (%) | entry | product | isolated yield (%) |
| 1 | 90a (92)b | 8 | 75c | ||
| 2 | 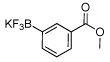 |
76a | 9 | 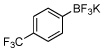 |
82c |
| 3 | 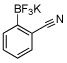 |
24a | 10 | 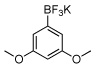 |
92a |
| 4 | 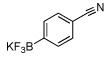 |
57c | 11 | 84a | |
| 5 | 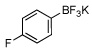 |
80a | 12 | 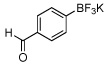 |
80c |
| 6 | 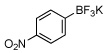 |
58c | 13 | 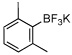 |
50c |
| 7 | 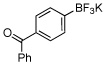 |
90c | 14 | 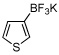 |
65c,d |
General conditions: 1 mol % of 2, 2 mol % of X-Phos, 1 mol % of NaOt-Bu, 3 equiv of KOAc, 1.5 equiv of 1, EtOH (0.1 M), 80 °C, 18 h.
6 mmol scale reaction.
3.0 equiv of 1,
3 mol % of 2, 50 °C, 24 h.
In this manner the trifluoroborate salts were obtained in good to excellent yields after their conversion from the corresponding boronic acid (Table 2). The method tolerates a wide range of substituents, including nitro, cyano, ester, and aldehyde groups. In general, aryl rings substituted with electron-rich functional groups (entries 10 and 11) fared better than those with electron withdrawing groups (entries 2, 3, 4 and 6). Systems with ortho substituted groups led to good yields (entry 8), and most impressively highly hindered 2,6-disubstituted aryl chlorides also cross-couple very effectively (entry 13). The latter compares very favorably to the only other related Miyaura-type coupling of such systems, which employed aryl bromides or iodides with 5–10 mol % catalyst loading and B2pin2 or pinacolborane.17b Importantly, the reaction works equally well on gram scale (entry 1). Aryl bromides were not effective under this set of conditions, owing to homocoupling, and research continues to find a suitable set of conditions for these substrates.
Heteroaryls generally require higher catalyst loading and longer reaction times (entry 14), and the reactions must be performed at reduced temperatures to eliminate homocoupling of the formed boronic acid with the starting chloride, which occurs rapidly at temperatures over 80 °C even at low catalyst loads. Initial optimization of the heteroaryl borylation reaction was carried out with 3-chlorothiophene and was not found to be general across different heteroaromatic systems. Further development of a general method utilizing heteroaryl chlorides is currently ongoing in our laboratory.
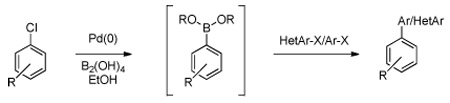
The method can also be extended to a 'one-pot' Suzuki-Miyaura reaction (Table 3).8 In this simple procedure, two aryl chlorides can be coupled to one another without the isolation of the intermediate boronic acid, which is the common practice. The first aryl chloride is used to synthesize the boronic acid in a manner similar to the methods described above. Upon the addition of a second aryl chloride and 3 equivalents of aqueous K2CO3 (which appears to decompose the excess tetrahydroxydiboron), the reaction was stirred overnight at 80 °C. Following an aqueous work-up, the unsymmetrically coupled compounds were obtained in good to excellent yield after column chromatography.
Table 3.
One-Pot Palladium-Catalyzed Cross-Coupling Reactions
 | ||||
|---|---|---|---|---|
| entry | first chloride | second chloride | product | isolated yield (%) |
| 1 | 85 | |||
| 2 | 90 | |||
| 3 |  |
 |
63 | |
| 4 |  |
 |
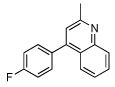 |
60 |
| 5 | 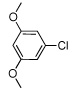 |
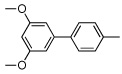 |
55 | |
General conditions: 2.5 mol % of 2, 5 mol % of X-Phos, 2.5 mol % of NaOt-Bu, 3 equiv of KOAc, 3 equiv of 1, EtOH (0.1 M), 80 °C, 2 h. 1 equiv of the second aryl or heteroaryl chloride, 3 equiv of 1.8 M K2CO3, 80 °C, 15 h.
In summary, we have reported the first palladium-catalyzed, direct synthesis of boronic acids with a wide range of aryl chlorides utilizing a preformed catalyst (2) and tetrahydroxydiboron (1). All of the reagents utilized are air stable so that the method does not require the use of a glove box or anhydrous solvents. The method tolerates a wide range of functional groups and allows easy and direct access to a diverse set of boronic acids, boronate esters, and trifluoroborates. In this process, the use of expensive pinacol is avoided, which also alleviates the need to hydrolyze and remove this unnecessary additive. Additionally, an efficient one-pot cross-coupling of two aryl- or heteroaryl chlorides can be accomplished in good to excellent yield over two steps with low catalyst loads. Efforts to extend the method to a more diverse set of aryl bromides and heteroaryls are currently ongoing.
Supplementary Material
Acknowledgements
We thank the NIGMS (R01 GM035249) and NSF GOALI program (CHE-0848460) for their financial support of this research. SLJT thanks past and present members of the Merck Research Laboratories West Point Medicinal Chemistry Department for their support and Merck & Co., Inc. for funding this PhD research. Additionally, Dr. Corneliu Stanciu (University of Pennsylvania) is acknowledged for his many insightful conversations, and Dr. Rakesh Kohli (University of Pennsylvania) for his help in obtaining HRMS data.
Footnotes
Supporting Information available: Experimental details and spectral data for all compounds synthesized. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Bellina F, Carpita A, Rossi R. Synthesis. 2004:2419–2440. [Google Scholar]
- 2.Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633–9695. [Google Scholar]
- 3.(a) Cho J–Y, Iverson CN, Smith MR., III J. Am. Chem. Soc. 2000;122:12868–12869. [Google Scholar]; (b) Shimada S, Batsanov AS, Howard JAK, Marder TB. Angew. Chem. Int. Ed. 2001;40:2168–2171. doi: 10.1002/1521-3773(20010601)40:11<2168::AID-ANIE2168>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; (c) Tse MK, Cho J–Y, Smith MR., III Org. Lett. 2001;3:2831–2833. doi: 10.1021/ol0162668. [DOI] [PubMed] [Google Scholar]; (d) Cho J–Y, Tse MK, Holmes D, Maleczka RE., Jr Science. 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; (e) Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J. Am. Chem. Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]; (f) Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew. Chem. Int. Ed. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]; (g) Maleczka RE, Jr, Shi F, Holmes D, Smith MR., III J. Am. Chem. Soc. 2003;125:7792–7793. doi: 10.1021/ja0349857. [DOI] [PubMed] [Google Scholar]; (h) Chotana GA, Rak MA, Smith MR., III J. Am. Chem. Soc. 2005;127:10539–10544. doi: 10.1021/ja0428309. [DOI] [PubMed] [Google Scholar]; (i) Paul S, Chotana GA, Holmes D, Reichle RC, Maleczka RA, Jr, Smith MR., III J. Am. Chem. Soc. 2006;128:15552–15553. doi: 10.1021/ja0631652. [DOI] [PubMed] [Google Scholar]; (j) Murphy JM, Tzschucke CC, Hartwig JF. Org. Lett. 2007;9:757–760. doi: 10.1021/ol062903o. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki A, Brown HC. Organic Syntheses via Boranes. Vol. 3. Milwakee: Aldrich Chemical Company; 2003. [Google Scholar]
- 5.Li W, Nelson D, Jensen M, Hoerrner S, Cai D, Larsen R, Reider P. J. Org. Chem. 2002;67:5394–5397. doi: 10.1021/jo025792p. [DOI] [PubMed] [Google Scholar]
- 6.Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508–7510. [Google Scholar]
- 7.Billingsley KL, Buchwald SL. J. Org. Chem. 2008;73:5589–5591. doi: 10.1021/jo800727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Billingsley K, Barder T, Buchwald S. Angew. Chem. Int. Ed. 2007;46:5359–5363. doi: 10.1002/anie.200701551. [DOI] [PubMed] [Google Scholar]
- 9.Rosen B, Huang C, Percec V. Org. Lett. 2008;10:2597–2600. doi: 10.1021/ol800832n. [DOI] [PubMed] [Google Scholar]
- 10.Moldoveanu C, Wilson D, Wilson C, Leowananwat P, Resmerita A, Lui C, Rosen B, Percec V. J. Org. Chem. 2010;75:5438–5452. doi: 10.1021/jo101023t. [DOI] [PubMed] [Google Scholar]
- 11.Wilson D, Wilson C, Moldoveanu C, Resmerita A, Corcoran P, Hoang L, Rosen B, Percec V. J. Am. Chem. Soc. 2010;132:1800–1801. doi: 10.1021/ja910808x. [DOI] [PubMed] [Google Scholar]
- 12.Moldoveanu C, Wilson D, Wilson C, Corcoran P, Rosen B, Percec V. Org. Lett. 2009;11:4974–4977. doi: 10.1021/ol902155e. [DOI] [PubMed] [Google Scholar]
- 13.(a) Nakamura H, Fujiwara M, Yamamoto Y. J. Org. Chem. 1998;63:7529–7530. doi: 10.1021/jo980818r. [DOI] [PubMed] [Google Scholar]; (b) Falck JR, Bondlela M, Venkaraman SK. J. Org. Chem. 2001;66:7148–7150. doi: 10.1021/jo015838z. [DOI] [PubMed] [Google Scholar]; (c) Yu S, Saenz J, Srirangam JK. J. Org. Chem. 2002;67:1699–1170. doi: 10.1021/jo016131f. [DOI] [PubMed] [Google Scholar]; (d) Deng J-H, Liu K, Kuntz KW, Snapper ML, Hoveyda AH. J. Am. Chem. Soc. 2003;125:9032–9034. doi: 10.1021/ja030249r. [DOI] [PubMed] [Google Scholar]
- 14.(a) Song YL, Morin C. Synlett. 2001:266–268. [Google Scholar]; (b) Jung ME, Lazarova TI. J. Org. Chem. 1999;64:2976–2977. doi: 10.1021/jo9902751. [DOI] [PubMed] [Google Scholar]; (c) Ma D, Wu Q. Tetrahedron Lett. 2001;42:5279–5281. [Google Scholar]; (d) Zaidlewicz M, Wolan A. J. Organomet. Chem. 2002;657:129–135. [Google Scholar]
- 15.Yang W, He H, Drueckhammer DG. Angew. Chem. Int. Ed. 2001;40:1714–1718. [PubMed] [Google Scholar]
- 16.Pennington T, Kardiman C, Hutton CA. Tetrahedron Lett. 2004;45:6657–6660. [Google Scholar]
- 17.(a) Malan C, Morin C. J. Org. Chem. 1998;63:8019–8020. [Google Scholar]; (b) Diemer V, Chaumeil H, Defoin A, Carre C. Tetrahedron. 2010;66:918–929. [Google Scholar]
- 18.Yuen A, Hutton C. Tetrahedron. Lett. 2005;46:7899–7903. [Google Scholar]
- 19.Bagutski V, Ros A, Aggarwal VK. Tetrahedron. 2009;65:9956–9960. [Google Scholar]
- 20.Sebelius S, Olsson V, Szabo K. J. Am. Chem. Soc. 2005;127:10478–10479. doi: 10.1021/ja052885q. [DOI] [PubMed] [Google Scholar]
- 21.Selander N, Kipke A, Sebelius S, Szabo K. J. Am. Chem. Soc. 2007;129 doi: 10.1021/ja074917a. 13723-12731. [DOI] [PubMed] [Google Scholar]
- 22.Selander N, Szabo K. Chem. Commun. 2008:3420–3422. doi: 10.1039/b804920c. [DOI] [PubMed] [Google Scholar]
- 23.Marcuccio S. U.S. Patent 9912940. 1999
- 24.Dreher SD, Dormer PG, Sandrock DL, Molander GA. J. Am. Chem. Soc. 2008;130:9257–9259. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biscoe MR, Fors BP, Buchwald SL. J. Am. Chem. Soc. 2008;130:6686–6687. doi: 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.