

Abstract
Type I interferon (IFN-α/β) is comprised of a family of highly related molecules that exert potent antiviral activity by interfering with virus replication and spread. IFN-α/β secretion is tightly regulated through pathogen sensing pathways that are operative in most somatic cells. However, specialized antigen-presenting plasmacytoid dendritic cells are uniquely equipped with the capacity to secrete extremely high levels of IFN-α/β, suggesting a key role for this cytokine in priming adaptive T-cell responses. Recent studies in both mice and humans have demonstrated a role for IFN-α/β in directly influencing the fate of both CD4+ and CD8+ T cells during the initial phases of antigen recognition. As such, IFN-α/β, among other innate cytokines, is considered an important ‘third signal’ that shapes the effector and memory T-cell pool. Moreover, IFN-α/β also serves as a counter-regulator of T helper type 2 and type 17 responses, which may be important in the treatment of atopy and autoimmunity, and in the development of novel vaccine adjuvants.
Keywords: CD4/CD8 T cells, cytokine, interferon, memory
Type I interferon – the oldest cytokine
Since the discovery of interferon-α/β (IFN-α/β) over 50 years ago, this family of cytokines has proven to be a critical regulator of innate immunity via its pleiotropic actions on virtually all somatic cell types. Interferon-α/β was first reported in 1957 by Isaacs and Lindenmann as an activity that ‘interfered’ with influenza A infection.1,2 Type I interferon is a family of highly related monomeric secreted proteins.3 In humans, there are approximately 20 IFN-α subtype genes in addition to individual genes encoding IFN-β, -κ, -ε and -ω. In mice, there exists an additional region of gene duplications resulting in approximately 20 genes encoding IFN-ζ isoforms (also known as ‘limitin’).4
Interferon-α/β programmes a state of resistance to intracellular pathogens and serves to alarm cells of both innate and adaptive immunity to the threat of infections. As such, IFN-α has been used therapeutically for over 25 years to treat hepatitis B and chronic hepatitis C as well as other viral infections.5 The antiviral effects of IFN-α/β have been appreciated since its discovery but many other unique biological properties of IFN-α/β have been revealed and harnessed for the treatment of multiple sclerosis and a variety of cancers. However, in these cases, it is not clear what specific immunological processes are being modulated by IFN-α/β to mediate these disparate effects.
Considering the numbers of IFN-α/β subtype genes, remarkably only one IFN-α/β receptor (IFNAR) has been identified, which is ubiquitously and constitutively expressed.3 All IFN-α/β isoforms tested can bind the IFNAR, albeit with varying affinities. However, IFN-α/β gene products bind the IFNAR in a species-specific fashion. Only one subtype of human IFN-α [recombinant hIFN-α (A/D)] has been shown to cross-react with the murine IFNAR and can activate both human and mouse cells. Although there is divergence in the structure and sequence of type I interferons and their receptor across species, many biological activities are shared.
The IFNAR is a heterodimeric complex composed of two type I transmembrane subunits designated R1 and R2. Both the human and mouse IFNARs are constitutively associated with the janus kinases (JAKs) Jak1 and Tyk2 (reviewed in ref. 3). Before cytokine activation, the N-terminus of signal transducer and activator of transcription 2 (STAT2) mediates an interaction with the cytoplasmic tail of the IFNAR2.6 Pre-association of STAT2 with the IFNAR is a required step for IFN-α/β signal transduction, and we will discuss the role of STAT N-domains in more depth later in this review. Upon receptor activation by IFN-α/β, the two receptor subunits co-ligate and promote activation of the JAKs that phosphorylate tyrosine (Y) residues within the cytoplasmic domains of the IFNAR1/2 chains.7,8 STAT2 becomes phosphorylated on Y-690 located just distal to the SH2 domain. Unlike STAT2, STAT1 is recruited to the receptor complex indirectly by docking to phosphorylated Y-690 on STAT2.8 The STAT1–STAT2 heterodimer then associates with interferon regulatory factor-9 to form the interferon-sensitive gene factor-3 (ISGF3). The ISGF3 regulates expression of the majority of interferon-sensitive genes (ISGs) by directly transactivating interferon-sensitive response elements found within their promoters.
Interferon-α/β is primarily induced by viral infection but can also be secreted in response to a variety of biological stresses including bacterial infection, UV irradiation, inflammation and heat shock.9,10 Virtually all cells have the inherent capacity to secrete some level of IFN-α/β in response to certain viral infections. However, professional antigen-presenting cells, particularly plasmacytoid dendritic cells (pDCs), are a key source of IFN-α/β. Plasmacytoid DCs are a specialized subset of DCs whose maturation is guided by innate cytokines [interleukin-3 (IL-3), Flt2 ligand, granulocyte–macrophage colony-stimulating factor and IL-4] and signalling through pattern recognition receptors during infections.11,12 These signals promote the secretion of a variety of innate cytokines, notably IL-12, IL-18, and importantly, IFN-α/β.11,13,14 Although these cells are not as efficient at activating CD4+ T cells as monocyte-derived DCs because of their lower expression of MHC-II, pDCs play a significant role in promoting T helper priming through cytokine secretion.15,16 In this review, we will survey recent advances in delineating the direct from the indirect effects of IFN-α/β in regulating the development of T-cell effector responses and its novel role in promoting T-cell memory.
IFN-α/β signalling and regulation of effector and memory CD4+ Th1 cells
Since the discovery of CD4+ T-cell subsets, a major quest in T-cell biology has been to understand the signals that control the differentiation of these subpopulations. One of the first signals identified was found to control T helper type 1 (Th1) differentiation, with IL-12 being the key cytokine governing this pathway.17–19 Binding of IL-12 to its receptor (IL-12R) on CD4+ cells triggers the activation of the JAKs Jak2 and Tyk2,20 leading to the phosphorylation and activation of STAT4.21,22 Phosphorylated STAT4 plays a critical role during Th1 commitment by promoting expression of T-bet,23–26 and recent studies have defined unique roles for both STAT4 and T-bet in regulating IFN-γ gene expression within committed Th1 cells.27 Finally, IFN-γ enhances both T-bet and IL-12Rβ2 expression, reinforcing IL-12-mediated Th1 commitment.28,29 Hence, in both mice and humans, IL-12 signalling through STAT4 and T-bet was established as a key pathway to IFN-γ production and the Th1 phenotype.
In parallel studies, the role of IFN-α/β in Th1 development was examined with seemingly conflicting results. In mouse, STAT4 activation was not detected in response to IFN-α/β compared with IL-12,22 yet studies with human cells reported just the opposite, suggesting a species difference in IFN-α/β-mediated STAT4 phosphorylation.30–32 However, as new and more specific reagents became available, low levels of phosphorylated STAT4 could be detected in mouse cells in response to IFN-α/β.33 The apparent species difference in STAT4 activation was found to involve STAT2.32 Like the IFNAR, STAT2 is also highly divergent across species, and the mouse sequence harbours a unique minisatellite sequence in the C-terminus that is not found in any other species. Moreover, the human STAT2 C-terminus was found to be critical in regulating IFN-α/β-dependent STAT4 activation in human cells, as the mouse STAT2 C-terminus failed to reconnect STAT4 recruitment to the human IFNAR.34,35
In an effort to determine the significance of the species-specific difference in STAT2, a knock-in mouse was generated in which the C-terminus of murine STAT2 was replaced with the human sequence, resulting in a chimeric mouse/human STAT2 molecule.36 Interferon-α/β treatment of STAT2 knock-in CD4+ T cells led to normal ISGF3 formation and ISG expression. However, IFN-α/β did not promote STAT4 phosphorylation or IFN-γ expression in CD4+ T cells expressing the chimeric STAT2 molecule. Hence, although the C-terminus of human STAT2 was required in human cells to promote efficient STAT4 phosphorylation in response to IFN-α/β, it was not sufficient to restore this pathway in mouse cells. Indeed, recent studies have highlighted the importance of STAT N-terminal domains in coordinating additional contacts with cytokine receptors that form the pre-assembled complexes necessary for cytokine-driven STAT activation.6,37,38 Specifically, the STAT4 N-terminus was found to be critical for IFN-α/β-dependent STAT4 activation through specific contacts made with the human, but not mouse, IFNAR2 subunit.39 These studies have revealed additional levels of complexity of cytokine receptors and their underlying molecular interactions that coordinate STAT activation.
Although the biochemical nature of STAT4 tyrosine phosphorylation differed quantitatively between mouse and human, there still remained the issue regarding the function of IFN-α/β-dependent STAT4 activation during Th1 commitment. Given the pronounced role of IL-12 signalling through STAT4 to drive Th1 commitment, these early studies assumed that any signalling pathway that activated STAT4 would promote Th1 development. Recent studies have challenged this assumption. Virtually all receptors that signal via the JAK/STAT pathway promote STAT tyrosine phosphorylation within minutes following receptor engagement. However, the duration of signalling varies between receptors and among STAT family members. Hilkens and colleagues40 first demonstrated a clear difference in the duration of STAT4 tyrosine phosphorylation between IL-12 and IFN-α/β signalling in human CD4+ T cells, with IL-12 promoting sustained STAT4 activation compared with IFN-α/β signalling. The inability of IFN-α/β to maintain STAT4 activation was correlated with a marked deficit in IFN-α/β-dependent Th1 development. Further kinetic comparisons of IL-12 and IFN-α/β clearly demonstrated that while IL-12 promoted STAT4 phosphorylation up to 24 hr, STAT4 was rapidly dephosphorylated within 6 hr of IFN-α/β stimulation.26 As a result, only cells treated with IL-12 expressed sustained levels of T-bet sufficient for IFN-γ secretion and Th1 commitment. Hence, while IFN-α/β may be more efficient at promoting acute STAT4 phosphorylation in human cells than in mouse, IFN-α/β cannot sustain STAT4 phosphorylation and is therefore not sufficient to drive Th1 commitment in either species.33,36,41,42
The in vitro differentiation studies described above can only address issues of sufficiency for a cytokine to regulate the development of specific phenotypes. However, when assessed in vivo, IFN-α/β signalling seemed to contribute to Th1 development.43,44 Likewise, mice deficient in IL-12 were still able to generate Th1 cells in response to murine hepatitis virus infection, demonstrating that multiple pathways were involved and may be required for Th1 development.45 One possible pathway involves IL-18, which was shown to synergize with IFN-α/β to activate STAT4 in the absence of IL-12.46,47 Interferon-α/β also promotes the expression of IL-21 and the IL-21R in T cells.48 As IL-21 induces Th1-associated genes, possibly in synergy with IL-18, this may represent another pathway by which IFN-α/β contributes to Th1 development.49,50 Taken together, these studies suggest that while IFN-α/β is not sufficient to drive Th1 commitment via direct and sustained STAT4 activation, it contributes to Th1 responses in vivo by collaborating with other cytokines that are differentially induced in response to various classes of pathogens.
Finally, IFN-α/β may play a broader role in CD4+ T-cell functions by regulating the development and stability of long-lived memory cells. Although IFN-α/β may promote cell cycle arrest and, in some cases, apoptosis in certain cell types, CD4+ T cells respond quite differently depending upon their activation status. Marrack et al.51 demonstrated that IFN-α/β protected cells from undergoing acute activation-induced cell death. Though not directly driving proliferation, IFN-α/β seemed to block apoptosis following antigen stimulation in vitro, which may be related to the development of long-lived central memory cells. As central memory cells were first described as having decreased effector capabilities, they display enhanced recall proliferation coincident with elevated secretion of IL-2.52 Recently, Davis et al.53 demonstrated a direct role for IFN-α/β in promoting the development of human central memory-like CD4+ T cells and preserving elevated IL-2 expression preferentially within these cells versus their effector cell counterparts. Hence, IFN-α/β acts to prevent terminal differentiation of effector CD4+ T cells by selectively regulating IL-2 expression at the expense of driving inflammatory cytokine secretion.
Negative regulation of Th2 and Th17 differentiation
As IFN-α/β is induced during Th1-dominant antiviral immune responses, IFN-α/β production may act to suppress the development of other subsets and their associated effector functions. Indeed, a growing body of literature has highlighted the role of IFN-α/β in cross-regulating the differentiation and stability of both Th2 and Th17 cells. These two subsets are guided by distinct signals, with Th2 cells controlled by IL-4, and Th17 cells responding to transforming growth factor-β, IL-6 and IL-1β. The counter-regulation of these varied signals by IFN-α/β is of clinical interest and is currently under investigation.
T helper type 2 development can be influenced by such cytokines as IL-33 and thymic stromal lymphopoietin,54,55 but IL-4 remains the primary signal that drives Th2 commitment from naive precursors.55,56 The Th2 differentiation involves the integration of signals both from the T-cell receptor and from IL-4 signalling via STAT6, which culminates in the induction of the GATA3 transcription factor. GATA3 subsequently promotes transcription at the Th2 cytokine locus containing the IL-4, IL-5 and IL-13 genes. This pathway also acts acutely to inhibit expression of the IL-12Rβ2 subunit.57 Consequently, induction of GATA3 serves to block Th1 development while positively regulating Th2 commitment. Moreover, while there seems to be some level of plasticity in Th2 cells,58 GATA3 is involved in an autoregulatory feedback loop that maintains Th2 commitment even in the absence of further IL-4 signalling.59,60 Hence, autoregulation by GATA3 represents an important stabilizing mechanism for Th2 commitment. However, early reports demonstrated that IFN-α/β could inhibit IL-5 secretion and eosinophil migration during allergic responses.61,62 Furthermore, IFN-α/β treatment of bulk CD4+ T cells during acute stimulation seemed to inhibit IL-5, but not IL-4 or IL-13. This was somewhat curious considering the dominant role played by IL-4 and GATA3 in Th2 effector function. Yet, despite these and other similar studies, one central question remained: can IFN-α/β regulate the ability of IL-4 to drive Th2 differentiation?
Recently, Huber et al.63 found that unlike the Th1-promoting cytokines IL-12 and IFN-γ, IFN-α/β potently and specifically inhibited the ability of IL-4 to drive Th2 differentiation of human cells but not murine cells. Moreover, IFN-α/β destabilized pre-committed Th2 cells and blocked Th2 cytokine expression. Interferon-α/β also reduced expression of the Th2 marker, CRTH2. It appears to do this, at least in part, by suppressing mRNA and protein levels of GATA3, which is critical for expression of CRTH2 as well as Th2-associated cytokines. While the underlying mechanism of GATA3 suppression is not yet clear, there are a few clues. First, as neither IL-12 nor IFN-γ inhibits Th2 commitment, the effect is not likely to be mediated by STAT4 or STAT1. Furthermore, the inhibition of Th2 cells by IFN-α/β paralleled recent studies demonstrating that type-III interferon (IFN-λ) can also suppress Th2 responses.64 Since both IFN-α/β and IFN-λ activate STAT2 and drive ISGF3 complex formation,65 STAT2 may play a crucial role in suppressing human Th2 development.
In addition to Th2 cells, there is increasing evidence that Th17 cells contribute to a variety of inflammatory processes involved in autoimmunity and allergic diseases.66 The Th17 cells are regulated by combined signalling via transforming growth factor-β, IL-6, IL-23 and IL-1β, culminating in the induction of the transcription factor retinoic acid-related orphan receptor γT.67 T helper type 17 cells mediate a variety of inflammatory reactions through their selective secretion of IL-17A, IL-17F and IL-22.68 However, unlike IL-4-mediated Th2 development, a variety of signals can block Th17 commitment including IFN-γ, IL-4 and IL-12. Interferon-α/β was also demonstrated to negatively regulate Th17 development in mice,69 and the suppression of Th17 development by IFN-α/β has recently been extended to human Th17 cells.70 Consequently, Th17 cells represent a more flexible developmental programme that can be counter-regulated by various signals, particularly by IFN-α/β. Given the use of IFN-β clinically for the treatment of multiple sclerosis, a disease associated with increased inflammation and IL-17 levels in the central nervous system,71 the ability of IFN-α/β to limit Th17 cells may explain the effectiveness of this treatment.72 Furthermore, the ability of IFN-α/β to inhibit Th2 and Th17 cells suggests that it may play a key role in controlling allergic responses.
The importance of IFN-α/β-mediated suppression of allergic T cell subsets is underscored by studies demonstrating that pDCs from asthma patients secrete less IFN-α/β than healthy donor pDCs in response to viral infections and toll-like receptor (TLR) ligands.73–75 Likewise, Gill et al.76 compared the induction of IFN-α by influenza virus in pDCs isolated from patients with asthma or healthy subjects and found that influenza virus infection promoted significantly less IFN-α secretion by pDCs from patients with asthma patients. Considering recent observations that IFN-α blocks Th2 development and stability,63 we propose that the defect in IFN-α production in pDCs from patients with asthma may skew T-cell priming toward Th2 development. It has been suggested that the reduction in IFN-α/β secretion during upper respiratory viral infections may lead to exacerbated lung pathology in those with asthma because of the inability of innate secretion of IFN-α/β to control viral replication in the lungs.75 While this is possible, asthma exacerbation by viruses may also be attributed to the lack of counter-regulation normally provided by IFN-α/β. Given that respiratory viral infections, such as RSV, have been linked to the induction of asthma, it is possible that the inflammation accompanying these infections supports priming of bystander allergen-specific Th2 cells. Furthermore, as people with asthma encounter recurrent infections, the lack of IFN-α secretion may allow additional Th2 priming.
Although pDCs are a significant source of IFN-α/β secretion during viral infections, these cells also express relatively elevated levels of the high-affinity IgE receptor FcεRI. Although it is not clear what specific role pDCs may play in allergen-induced asthma via IgE-mediated activation, Liu and colleagues77 recently demonstrated a reciprocal regulation of TLR9 and FcεRI upon receptor–ligand engagement. Here, IgE cross-linking significantly reduced TLR9 expression, resulting in decreased IFN-α production in response to CpG DNA. These results are intriguing because they suggest that sensitization with allergens may block IFN-α secretion during viral infections. Moreover, Gill et al.76 demonstrated that IgE, but not IgG, cross-linking significantly reduced IFN-α secretion from pDCs in response to both influenza A and B virus infection. Collectively, these results demonstrate that pDCs from patients with asthma secrete significantly less IFN-α, and IgE cross-linking blocks IFN-α secretion even in pDCs from healthy controls in response to influenza virus, suggesting both an intrinsic and extrinsic mechanism for IFN-α suppression. Hence, IFN-α/β seems to be a key focal point of reciprocal antagonism by antiviral and allergic responses. As mentioned earlier, IFN-α/β promotes IL-21 secretion, which is reported to negatively regulate both IgE production and allergic rhinitis.78–80 These findings are supported by early studies demonstrating that IFN-α/β can suppress IgE class switching during B-cell priming.81,82 In summary, IFN-α/β may prove to be a potent cross-regulatory signal to block Th2/Th17 development as well as IgE production, which underscores its potential therapeutic use in atopic diseases. The role of IFN-α/β in modulating CD4+ Th responses is summarized in Fig. 1.
Figure 1.
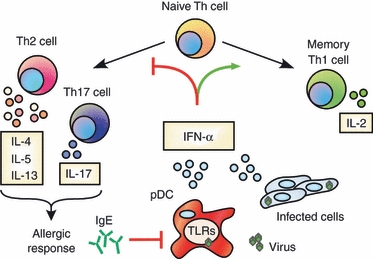
Type I interferon (IFN) regulates CD4+ T helper cell development. IFN-α/β contributes to various functions of T helper type 1 (Th1) cells, particularly the secretion of interleukin-2 (IL-2) by memory cells. Conversely, IFN-α/β restricts the development of alternative populations such as Th2 and Th17. pDCs, plasmacytoid dendritic cells; TLR, toll-like receptor.
Regulation of effector and memory CD8+ T cells
In CD4+ T cells, IL-12 dominates as a unique signal driving effector Th1 commitment in both mice and humans.26,40,41 Although IFN-α/β may play ancillary roles in effector Th1 commitment, the two signals are not redundant. However, this division of labour may not be so distinct in CD8+ T cells, particularly in the mouse. Both IL-12 and IFN-α/β have been reported to enhance CD8+ T-cell effector activity. One of the first studies examining the role of IL-12 in CD8+ T-cell effector function concluded that neither IFN-γ secretion nor cytolytic activity was regulated by IL-12.83 This study also demonstrated that STAT4 knock-out CD8+ T cells could become functional effector cells, albeit to a lesser extent than wild-type cells. However, Mescher and colleagues84–87 have recently proposed that both IL-12 and IFN-α/β can act as a ‘third signal’ to promote both IFN-γ secretion and expression of perforin and granzymes in murine CD8+ cells. Furthermore, both IL-12 and IFN-α/β were found to markedly enhance cytolytic activity, and these effects were dependent upon STAT4.86 Based on these observations, it was concluded that IL-12 and IFN-α/β shared redundant roles in the regulation of CD8+ development and effector function.
Interferon-α/β can play a significant role in priming effector responses and maintaining pools of memory cells via indirect actions through other cytokines and by enhancing antigen presentation. For example, IFN-α/β can act indirectly on innate cells to elicit IL-15 secretion, and perhaps IL-15 alone or in combination with IFN-α/β can drive homeostatic proliferation and maintenance of memory CD8+ T cells in vivo.88,89 Moreover, both IFN-α/β and IFN-γ can promote homeostatic maintenance of CD8+ memory cells, but with differing requirements for additional cytokines like IL-12, IL-18 and IL-15.90 A major component of IFN-α/β-driven antiviral properties is the marked induction of genes involved in antigen processing and presentation, particularly expression of class I genes and associated endocytic proteins involved in proteolysis and peptide loading. By engaging this pathway in an in vivo model of antigen cross-priming, Tough and colleagues91,92 demonstrated that IFN-α/β enhanced CD8+ T-cell expansion as well as cytolytic activity, which may explain the strong adjuvant effect of IFN-α/β on protein vaccination strategies.
While the individual roles of IL-12 and IFN-α/β can be assessed in isolation in vitro, in vivo studies have revealed unique roles for IFN-α/β and IL-12 that depend upon priming conditions and the class of pathogen. Initial studies demonstrated that the induction of IFN-α/β by CpG stimulation led to antigen-presenting cell-dependent T-cell proliferation, which required IFN-α/β signalling within the responding T cells.93 These early studies did not directly compare IFN-α/β with the powerful inflammatory effects of IL-12. However, comparing primary CD8+ responses with various pathogens, Murali-Krishna and colleagues94 demonstrated that IFN-α/β signals were required for CD8+ expansion in response to lymphocytic choriomeningitis virus (LCMV), but less so in response to vaccinia virus or Listeria monocytogenes infections.44 Based on this observation, it was postulated that antigenic load may contribute to CD8+ dependence on IFN-α/β for full expansion, as LCMV viral titres are much higher during the peak of the infection than vaccinia virus titres. Furthermore, a recent study demonstrated that CD8+ responses to Trypanosoma cruzi were completely independent of IFN-α/β signalling.95 This is somewhat surprising given the dependence on IFN-α/β during cross-priming reported by Tough and colleagues. Nonetheless, all of these reports highlight the potential for IL-12 and IFN-α/β to significantly regulate CD8+ effector responses, which were originally reported to be IL-12- and STAT4-independent.
Interleukin-12 and IFN-α/β may also play distinct roles in regulating CD8+ T-cell memory development. First, although IL-12 has been reported to play a positive role in generating CD8+ effector cells, it seems to have an inverse role in generating memory cells. Pearce et al.96 recently demonstrated that the kinetics and magnitude of the CD8+ memory response to L. monocytogenes were significantly enhanced in IL-12Rβ2−/− cells. This observation correlated with enhanced CD8+ memory in T-bet knockout mice, as IL-12 has been reported to positively regulate T-bet expression.97,98 Moreover, as cells expand in response to antigen stimulation, the enhanced expression of T-bet driven by IL-12 generates populations of terminally differentiated cytotoxic effector cells.99,100 Conversely, Murali-Krishna and colleagues94 demonstrated a severe block in CD8+ memory in IFNAR−/− CD8+ T cells during LCMV infections, perhaps because the cells failed to expand during the primary response. The mechanism for this defect has not been described. If IL-12 negatively regulates memory cell development while IFN-α/β positively regulates this process, it remains puzzling how memory cells develop when both of these cytokines are secreted during intracellular pathogen infections.
In mice, both IL-12 and IFN-α/β are sufficient to promote effector function in CD8+ T cells when activated in vitro, albeit IFN-α/β is not quite as potent as IL-12 in regulating cytokine expression.86,101 However, there seems to be less redundancy between these two cytokine pathways in driving human CD8+ T-cell effectors. Recently, Ramos et al.102 compared the ability of IL-12 and IFN-α to promote cytokine secretion and lytic activity in primary naive human CD8+ T cells. In contrast to mouse, IL-12 induced robust lytic activity and secretion of IFN-γ and tumour necrosis factor-α, but treatment with IFN-α alone had little effect on these activities compared with cells activated under neutralizing conditions. Two recent studies claim that IFN-α enhances IFN-γ production103 and granzyme expression104 in human CD8+ T cells, but those reports only compared IFN-α to neutralizing conditions. Indeed, IFN-α does marginally increase IFN-γ production over the baseline control, but this level is still 10-fold less than the magnitude of production induced by IL-12.102 Consequently, IL-12 appears to be the main signal driving the expression of effector cytokines. However, while IFN-α failed to regulate effector cell development, IFN-α enhanced the development of CD8+ central memory (TCM) cells.102 This activity was unique to IFN-α, because IL-12 promoted only effector cell (TEM) but not TCM development. These cells lack immediate effector function but rapidly acquire these responses following secondary stimulation, hence representing a functional memory population. Interestingly, when naive cells receive signals from both IL-12 and IFN-α, both TEM and TCM cells develop simultaneously, and they are derived from subpopulations of cells that differentially progress through cell division. The IL-12 programmes TEM phenotypes in actively dividing cells, whereas IFN-α induces TCM development by limiting proliferation and terminal differentiation in a subset of cells. These points are summarized in Fig. 2.
Figure 2.
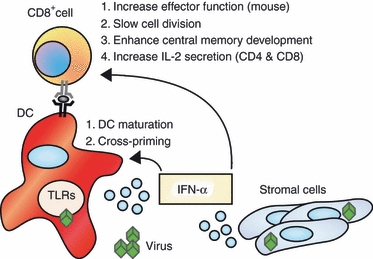
Interferon-α/β (IFN-α/β) supports CD8+ T-cell priming through direct effects on both the antigen-presenting cells and the T cells. IFN-α/β signalling in antigen-presenting cells enhances their ability to activate CD8+ T cells. IFN-α/β also acts directly on CD8+ T cells to support the development of effector and memory populations. DC, dendritic cell; IL-2, interleukin-2; TLR, toll-like receptor.
Regarding the mechanism of this developmental programme, Ramos et al.102 demonstrated that the development of distinct effector and memory phenotypes of human CD8+ T cells occurred through the reciprocal regulation of their respective cytokine receptors. Development of TCM was regulated by marked induction of the IFNAR with low expression of the IL-12R, whereas effector cells rapidly divided and progressively lost IFNAR while gaining IL-12R expression. Differential sensitivity to IL-12 and IFN-α therefore dictates the fate of cells that commit to TEM and TCM phenotypes. This study demonstrates for the first time that IL-12 and IFN-α are not redundant signals in the development of human CD8+ T-cell responses, instead creating a system for concomitant development of effector and memory human CD8+ T cells that is directly influenced by cytokine signalling. These observations offer an important leap forward in the understanding of human CD8+ T-cell development and indicate a new model for the role of innate cytokines in the genesis of memory and effector responses during infection.
Conclusion
In summary, our understanding of the role of type I IFN in T-cell development has historically been complicated by numerous differences between mice and humans. Nevertheless, the emerging picture shows that IFN-α/β plays an important and multifaceted part in regulating adaptive responses through both direct and indirect effects. Interferon-α/β directly enhances the development of CD4+ and CD8+ T cells with TCM characteristics, while also contributing to TEM development via collaboration with other cytokines or feedback by antigen-presenting cells. In addition, IFN-α/β ensures the proper differentiation of Th1 cells by restricting the development of alternative subsets like Th2 and Th17. This novel function is immunologically important for appropriate antiviral responses, and also suggests new therapeutic uses for IFN-α/β.
Acknowledgments
J.P.H and J.D.F. are supported by grants and fellowships from the National Institutes of Health and the National Institute of Allergy and Infectious Diseases. We thank Fatema Z. Chowdhury and Sarah R. Gonzales for critically reviewing the manuscript.
Disclosures
The authors have no conflicts of interest.
References
- 1.Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci. 1957;147:268–73. [PubMed] [Google Scholar]
- 2.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–67. [PubMed] [Google Scholar]
- 3.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–64. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 4.Hardy MP, Owczarek CM, Jermiin LS, Ejdeback M, Hertzog PJ. Characterization of the type I interferon locus and identification of novel genes. Genomics. 2004;84:331–45. doi: 10.1016/j.ygeno.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Leung S, Kerr IM, Stark GR. Functional subdomains of STAT2 required for preassociation with the alpha interferon receptor and for signaling. Mol Cell Biol. 1997;17:2048–56. doi: 10.1128/mcb.17.4.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan H, Krishnan K, Greenlund AC, Gupta S, Lim JT, Schreiber RD, Schindler CW, Krolewski JJ. Phosphorylated interferon-alpha receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. EMBO J. 1996;15:1064–74. [PMC free article] [PubMed] [Google Scholar]
- 8.Qureshi SA, Leung S, Kerr IM, Stark GR, Darnell JE., Jr Function of Stat2 protein in transcriptional activation by alpha interferon. Mol Cell Biol. 1996;16:288–93. doi: 10.1128/mcb.16.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doly J, Civas A, Navarro S, Uze G. Type I interferons: expression and signalization. Cell Mol Life Sci. 1998;54:1109–21. doi: 10.1007/s000180050240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Maeyer E, De Maeyer-Guignard J. Type I interferons. Int Rev Immunol. 1998;17:53–73. doi: 10.3109/08830189809084487. [DOI] [PubMed] [Google Scholar]
- 11.Naik SH, Sathe P, Park HY, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. 2007;8:1217–26. doi: 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 12.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 13.Ito T, Amakawa R, Kaisho T, et al. Interferon-alpha and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J Exp Med. 2002;195:1507–12. doi: 10.1084/jem.20020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iho S. Type I IFN synthesis in plasmacytoid dendritic cells. J Immunol. 2003;171:2767. doi: 10.4049/jimmunol.171.6.2767. [DOI] [PubMed] [Google Scholar]
- 15.Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. 2000;1:305–10. doi: 10.1038/79747. [DOI] [PubMed] [Google Scholar]
- 16.Krug A, Veeraswamy R, Pekosz A, Kanagawa O, Unanue ER, Colonna M, Cella M. Interferon-producing cells fail to induce proliferation of naive T cells but can promote expansion and T helper 1 differentiation of antigen-experienced unpolarized T cells. J Exp Med. 2003;197:899–906. doi: 10.1084/jem.20021091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–9. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 18.Macatonia SE, Hsieh CS, Murphy KM, O'Garra A. Dendritic cells and macrophages are required for Th1 development of CD4+ T cells from alpha beta TCR transgenic mice: IL-12 substitution for macrophages to stimulate IFN-gamma production is IFN-gamma-dependent. Int Immunol. 1993;5:1119–28. doi: 10.1093/intimm/5.9.1119. [DOI] [PubMed] [Google Scholar]
- 19.Macatonia SE, Hosken NA, Litton M, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–9. [PubMed] [Google Scholar]
- 20.Bacon CM, McVicar DW, Ortaldo JR, Rees RC, O'Shea JJ, Johnston JA. Interleukin 12 (IL-12) induces tyrosine phosphorylation of JAK2 and TYK2: differential use of Janus family tyrosine kinases by IL-2 and IL-12. J Exp Med. 1995;181:399–404. doi: 10.1084/jem.181.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacon CM, Petricoin EF, 3rd, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O'Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci U S A. 1995;92:7307–11. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, Murphy KM. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181:1755–62. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 24.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–42. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 25.Placek K, Gasparian S, Coffre M, Maiella S, Sechet E, Bianchi E, Rogge L. Integration of distinct intracellular signaling pathways at distal regulatory elements directs T-bet expression in human CD4+ T cells. J Immunol. 2009;183:7743–51. doi: 10.4049/jimmunol.0803812. [DOI] [PubMed] [Google Scholar]
- 26.Ramos HJ, Davis AM, George TC, Farrar JD. IFN-alpha is not sufficient to drive Th1 development due to lack of stable T-bet expression. J Immunol. 2007;179:3792–803. doi: 10.4049/jimmunol.179.6.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thieu VT, Yu Q, Chang HC, Yeh N, Nguyen ET, Sehra S, Kaplan MH. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity. 2008;29:679–90. doi: 10.1016/j.immuni.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–24. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–57. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 30.Cho SS, Bacon CM, Sudarshan C, Rees RC, Finbloom D, Pine R, O'Shea JJ. Activation of STAT4 by IL-12 and IFN-alpha: evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol. 1996;157:4781–9. [PubMed] [Google Scholar]
- 31.Rogge L, D'Ambrosio D, Biffi M, Penna G, Minetti LJ, Presky DH, Adorini L, Sinigaglia F. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J Immunol. 1998;161:6567–74. [PubMed] [Google Scholar]
- 32.Farrar JD, Smith JD, Murphy TL, Murphy KM. Recruitment of Stat4 to the human interferon-alpha/beta receptor requires activated Stat2. J Biol Chem. 2000;275:2693–7. doi: 10.1074/jbc.275.4.2693. [DOI] [PubMed] [Google Scholar]
- 33.Berenson LS, Farrar JD, Murphy TL, Murphy KM. Frontline: absence of functional STAT4 activation despite detectable tyrosine phosphorylation induced by murine IFN-alpha. Eur J Immunol. 2004;34:2365–74. doi: 10.1002/eji.200324829. [DOI] [PubMed] [Google Scholar]
- 34.Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–9. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- 35.Farrar JD, Murphy KM. Type I interferons and T helper development. Immunol Today. 2000;21:484–9. doi: 10.1016/s0167-5699(00)01710-2. [DOI] [PubMed] [Google Scholar]
- 36.Persky ME, Murphy KM, Farrar JD. IL-12, but not IFN-alpha, promotes STAT4 activation and Th1 development in murine CD4+ T cells expressing a chimeric murine/human Stat2 gene. J Immunol. 2005;174:294–301. doi: 10.4049/jimmunol.174.1.294. [DOI] [PubMed] [Google Scholar]
- 37.Ota N, Brett TJ, Murphy TL, Fremont DH, Murphy KM. N-domain-dependent nonphosphorylated STAT4 dimers required for cytokine-driven activation. Nat Immunol. 2004;5:208–15. doi: 10.1038/ni1032. [DOI] [PubMed] [Google Scholar]
- 38.Murphy TL, Geissal ED, Farrar JD, Murphy KM. Role of the Stat4 N domain in receptor proximal tyrosine phosphorylation. Mol Cell Biol. 2000;20:7121–31. doi: 10.1128/mcb.20.19.7121-7131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tyler DR, Persky ME, Matthews LA, Chan S, Farrar JD. Pre-assembly of STAT4 with the human IFN-alpha/beta receptor-2 subunit is mediated by the STAT4 N-domain. Mol Immunol. 2007;44:1864–72. doi: 10.1016/j.molimm.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. J Immunol. 2004;172:61–9. doi: 10.4049/jimmunol.172.1.61. [DOI] [PubMed] [Google Scholar]
- 41.Berenson LS, Gavrieli M, Farrar JD, Murphy TL, Murphy KM. Distinct characteristics of murine STAT4 activation in response to IL-12 and IFN-{alpha} J Immunol. 2006;177:5195–203. doi: 10.4049/jimmunol.177.8.5195. [DOI] [PubMed] [Google Scholar]
- 42.Davis AM, Hagan KA, Matthews LA, Bajwa G, Gill MA, Gale M, Jr, Farrar JD. Blockade of virus infection by human CD4+ T cells via a cytokine relay network. J Immunol. 2008;180:6923–32. doi: 10.4049/jimmunol.180.10.6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176:3315–9. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- 44.Way SS, Havenar-Daughton C, Kolumam GA, Orgun NN, Murali-Krishna K. IL-12 and type-I IFN synergize for IFN-gamma production by CD4 T cells, whereas neither are required for IFN-gamma production by CD8 T cells after Listeria monocytogenes infection. J Immunol. 2007;178:4498–505. doi: 10.4049/jimmunol.178.7.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schijns VE, Haagmans BL, Wierda CM, Kruithof B, Heijnen IA, Alber G, Horzinek MC. Mice lacking IL-12 develop polarized Th1 cells during viral infection. J Immunol. 1998;160:3958–64. [PubMed] [Google Scholar]
- 46.Sareneva T, Julkunen I, Matikainen S. IFN-alpha and IL-12 induce IL-18 receptor gene expression in human NK and T cells. J Immunol. 2000;165:1933–8. doi: 10.4049/jimmunol.165.4.1933. [DOI] [PubMed] [Google Scholar]
- 47.Matikainen S, Paananen A, Miettinen M, Kurimoto M, Timonen T, Julkunen I, Sareneva T. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur J Immunol. 2001;31:2236–45. [PubMed] [Google Scholar]
- 48.Strengell M, Julkunen I, Matikainen S. IFN-alpha regulates IL-21 and IL-21R expression in human NK and T cells. J Leukoc Biol. 2004;76:416–22. doi: 10.1189/jlb.1003488. [DOI] [PubMed] [Google Scholar]
- 49.Strengell M, Matikainen S, Siren J, Lehtonen A, Foster D, Julkunen I, Sareneva T. IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J Immunol. 2003;170:5464–9. doi: 10.4049/jimmunol.170.11.5464. [DOI] [PubMed] [Google Scholar]
- 50.Strengell M, Sareneva T, Foster D, Julkunen I, Matikainen S. IL-21 up-regulates the expression of genes associated with innate immunity and Th1 response. J Immunol. 2002;169:3600–5. doi: 10.4049/jimmunol.169.7.3600. [DOI] [PubMed] [Google Scholar]
- 51.Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. 1999;189:521–30. doi: 10.1084/jem.189.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 53.Davis AM, Ramos HJ, Davis LS, Farrar JD. Cutting edge: a T-bet-independent role for IFN-alpha/beta in regulating IL-2 secretion in human CD4+ central memory T cells. J Immunol. 2008;181:8204–8. doi: 10.4049/jimmunol.181.12.8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ziegler SF. The role of thymic stromal lymphopoietin (TSLP) in allergic disorders. Curr Opin Immunol. 2010;22:795–9. doi: 10.1016/j.coi.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–35. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, Afkarian M, Murphy TL. Signaling and transcription in T helper development. Annu Rev Immunol. 2000;18:451–94. doi: 10.1146/annurev.immunol.18.1.451. [DOI] [PubMed] [Google Scholar]
- 57.Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, Murphy KM. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–55. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- 58.Hegazy AN, Peine M, Helmstetter C, et al. Interferons direct Th2 cell reprogramming to generate a stable GATA-3+ T-bet+ cell subset with combined Th2 and Th1 cell functions. Immunity. 2010;32:116–28. doi: 10.1016/j.immuni.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, Murphy KM. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/s1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- 60.Farrar JD, Ouyang W, Lohning M, Assenmacher M, Radbruch A, Kanagawa O, Murphy KM. An instructive component in T helper cell type 2 (Th2) development mediated by GATA-3. J Exp Med. 2001;193:643–50. doi: 10.1084/jem.193.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zielinski RM, Lawrence WD. Interferon-alpha for the hypereosinophilic syndrome. Ann Intern Med. 1990;113:716–8. doi: 10.7326/0003-4819-113-9-716. [DOI] [PubMed] [Google Scholar]
- 62.Schandene L, Del Prete GF, Cogan E, Stordeur P, Crusiaux A, Kennes B, Romagnani S, Goldman M. Recombinant interferon-alpha selectively inhibits the production of interleukin-5 by human CD4+ T cells. J Clin Invest. 1996;97:309–15. doi: 10.1172/JCI118417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huber JP, Ramos HJ, Gill MA, Farrar JD. Cutting edge: type I IFN reverses human Th2 commitment and stability by suppressing GATA3. J Immunol. 2010;185:813–7. doi: 10.4049/jimmunol.1000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dai J, Megjugorac NJ, Gallagher GE, Yu RY, Gallagher G. IFN-lambda1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood. 2009;113:5829–38. doi: 10.1182/blood-2008-09-179507. [DOI] [PubMed] [Google Scholar]
- 65.Dumoutier L, Tounsi A, Michiels T, Sommereyns C, Kotenko SV, Renauld JC. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem. 2004;279:32269–74. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- 66.Ciprandi G, De Amici M, Murdaca G, Fenoglio D, Ricciardolo F, Marseglia G, Tosca M. Serum interleukin-17 levels are related to clinical severity in allergic rhinitis. Allergy. 2009;64:1375–8. doi: 10.1111/j.1398-9995.2009.02010.x. [DOI] [PubMed] [Google Scholar]
- 67.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 68.Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21:274–80. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 69.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 70.Moschen AR, Geiger S, Krehan I, Kaser A, Tilg H. Interferon-alpha controls IL-17 expression in vitro and in vivo. Immunobiology. 2008;213:779–87. doi: 10.1016/j.imbio.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 71.Bennett JL, Stuve O. Update on inflammation, neurodegeneration, and immunoregulation in multiple sclerosis: therapeutic implications. Clin Neuropharmacol. 2009;32:121–32. doi: 10.1097/WNF.0b013e3181880359. [DOI] [PubMed] [Google Scholar]
- 72.Chen M, Chen G, Nie H, et al. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4+ T cells in MS. Eur J Immunol. 2009;39:2525–36. doi: 10.1002/eji.200838879. [DOI] [PubMed] [Google Scholar]
- 73.Tversky JR, Le TV, Bieneman AP, Chichester KL, Hamilton RG, Schroeder JT. Human blood dendritic cells from allergic subjects have impaired capacity to produce interferon-alpha via Toll-like receptor 9. Clin Exp Allergy. 2008;38:781–8. doi: 10.1111/j.1365-2222.2008.02954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bufe A, Gehlhar K, Grage-Griebenow E, Ernst M. Atopic phenotype in children is associated with decreased virus-induced interferon-alpha release. Int Arch Allergy Immunol. 2002;127:82–8. doi: 10.1159/000048173. [DOI] [PubMed] [Google Scholar]
- 75.Gehlhar K, Bilitewski C, Reinitz-Rademacher K, Rohde G, Bufe A. Impaired virus-induced interferon-alpha2 release in adult asthmatic patients. Clin Exp Allergy. 2006;36:331–7. doi: 10.1111/j.1365-2222.2006.02450.x. [DOI] [PubMed] [Google Scholar]
- 76.Gill MA, Bajwa G, George TA, Dong CC, Dougherty II, Jiang N, Gan VN, Gruchalla RS. Counterregulation between the FcepsilonRI pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol. 2010;184:5999–6006. doi: 10.4049/jimmunol.0901194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schroeder JT, Bieneman AP, Xiao H, Chichester KL, Vasagar K, Saini S, Liu MC. TLR9- and FcepsilonRI-mediated responses oppose one another in plasmacytoid dendritic cells by down-regulating receptor expression. J Immunol. 2005;175:5724–31. doi: 10.4049/jimmunol.175.9.5724. [DOI] [PubMed] [Google Scholar]
- 78.Ozaki K, Spolski R, Feng CG, et al. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–4. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- 79.Shang XZ, Ma KY, Radewonuk J, Li J, Song XY, Griswold DE, Emmell E, Li L. IgE isotype switch and IgE production are enhanced in IL-21-deficient but not IFN-gamma-deficient mice in a Th2-biased response. Cell Immunol. 2006;241:66–74. doi: 10.1016/j.cellimm.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 80.Hiromura Y, Kishida T, Nakano H, Hama T, Imanishi J, Hisa Y, Mazda O. IL-21 administration into the nostril alleviates murine allergic rhinitis. J Immunol. 2007;179:7157–65. doi: 10.4049/jimmunol.179.10.7157. [DOI] [PubMed] [Google Scholar]
- 81.Finkelman FD, Svetic A, Gresser I, Snapper C, Holmes J, Trotta PP, Katona IM, Gause WC. Regulation by interferon alpha of immunoglobulin isotype selection and lymphokine production in mice. J Exp Med. 1991;174:1179–88. doi: 10.1084/jem.174.5.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pene J, Rousset F, Briere F, et al. IgE production by normal human lymphocytes is induced by interleukin 4 and suppressed by interferons gamma and alpha and prostaglandin E2. Proc Natl Acad Sci U S A. 1988;85:6880–4. doi: 10.1073/pnas.85.18.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carter LL, Murphy KM. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon gamma production from CD4+ versus CD8+ T cells. J Exp Med. 1999;189:1355–60. doi: 10.1084/jem.189.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–71. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 85.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–51. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–9. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 87.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, Popescu F, Xiao Z. Signals required for programming effector and memory development by CD8+ T cells. Immunol Rev. 2006;211:81–92. doi: 10.1111/j.0105-2896.2006.00382.x. [DOI] [PubMed] [Google Scholar]
- 88.Berard M, Brandt K, Bulfone-Paus S, Tough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol. 2003;170:5018–26. doi: 10.4049/jimmunol.170.10.5018. [DOI] [PubMed] [Google Scholar]
- 89.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001;167:1179–87. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 90.Tough DF, Zhang X, Sprent J. An IFN-gamma-dependent pathway controls stimulation of memory phenotype CD8+ T cell turnover in vivo by IL-12, IL-18, and IFN-gamma. J Immunol. 2001;166:6007–11. doi: 10.4049/jimmunol.166.10.6007. [DOI] [PubMed] [Google Scholar]
- 91.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–15. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 92.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, Tough DF. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol. 2006;176:4682–9. doi: 10.4049/jimmunol.176.8.4682. [DOI] [PubMed] [Google Scholar]
- 93.Sun S, Zhang X, Tough DF, Sprent J. Type I interferon-mediated stimulation of T cells by CpG DNA. J Exp Med. 1998;188:2335–42. doi: 10.1084/jem.188.12.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–50. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martin DL, Murali-Krishna K, Tarleton RL. Generation of Trypanosoma cruzi-specific CD8+ T-cell immunity is unaffected by the absence of type I interferon signaling. Infect Immun. 2010;78:3154–9. doi: 10.1128/IAI.00275-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J Immunol. 2007;179:2074–81. doi: 10.4049/jimmunol.179.4.2074. [DOI] [PubMed] [Google Scholar]
- 97.Intlekofer AM, Takemoto N, Wherry EJ, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–44. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 98.Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol. 2006;177:7515–9. doi: 10.4049/jimmunol.177.11.7515. [DOI] [PubMed] [Google Scholar]
- 99.Cui W, Joshi NS, Jiang A, Kaech SM. Effects of signal 3 during CD8 T cell priming: bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine. 2009;27:2177–87. doi: 10.1016/j.vaccine.2009.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–95. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. Programming for CD8 T cell memory development requires IL-12 or type I IFN. J Immunol. 2009;182:2786–94. doi: 10.4049/jimmunol.0803484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood. 2009;113:5516–25. doi: 10.1182/blood-2008-11-188458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hervas-Stubbs S, Riezu-Boj JI, Gonzalez I, et al. Effects of IFN-alpha as a signal-3 cytokine on human naive and antigen-experienced CD8+ T cells. Eur J Immunol. 2010;40:3389–402. doi: 10.1002/eji.201040664. [DOI] [PubMed] [Google Scholar]
- 104.Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, Woodland DL. Type I interferons regulate cytolytic activity of memory CD8+ T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. doi: 10.1016/j.immuni.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]