

Abstract
The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 are involved in variety of inflammatory disorders including multiple sclerosis, rheumatoid arthritis, psoriasis and sarcoidosis. Two alternatively spliced variants of the human CXCR3-A receptor have been described, termed CXCR3-B and CXCR3-alt. Human CXCR3-B binds CXCL9, CXCL10, CXCL11 as well as an additional ligand CXCL4. In contrast, CXCR3-alt only binds CXCL11. We report that CXCL4 induces intracellular calcium mobilization as well as Akt and p44/p42 extracellular signal-regulated kinase phosphorylation, in activated human T lymphocytes. These responses have similar concentration dependence and time–courses to those induced by established CXCR3 agonists. Moreover, phosphorylation of Akt and p44/p42 is inhibited by pertussis toxin, suggesting coupling to Gαi protein. Surprisingly, and in contrast with the other CXCR3 agonists, stimulation of T lymphocytes with CXCL4 failed to elicit migratory responses and did not lead to loss of surface CXCR3 expression. Taken together, our findings show that, although CXCL4 is coupled to downstream biochemical machinery, its role in T cells is probably distinct from that of CXCR3-A agonists.
Keywords: chemokine receptors, inflammation, signal transduction, T cells
Introduction
Members of the chemokine (chemotactic cytokine) superfamily and their receptors play a major role in the trafficking of immune cells under homeostatic and inflammatory conditions. They have also been implicated in distinct biological processes such as angiogenesis, proliferation and apoptosis.1–5 The interferon-γ (IFN-γ) -inducible chemokines CXCL9 (monokine induced by human IFN-γ/Mig), CXCL10 [IFN-γ-inducible 10 000 molecular weight (MW) protein/IP10] and CXCL11 (IFN-γ-inducible T-cell α chemoattractant/ITAC) bind to their receptor CXCR3.6–8 Recent studies have shown that different CXCR3 ligands exhibit unique temporal and spatial expression patterns, suggesting that they have non-redundant functions in vivo. Moreover, the CXCR3 ligands share low sequence homology (around 40% amino acid identity) and exhibit differences in their potencies and efficacies at CXCR3.9–12 CXCL11 is believed to be the dominant CXCR3 agonist, as it is more potent and efficacious than CXCL10 or CXCL9 as a chemoattractant and in stimulating calcium flux and receptor desensitization, whereas CXCL9 and CXCL10 exhibit lower affinities for human CXCR3 in comparison with CXCL11.6
CXCR3 is preferentially expressed on T helper type 1 (Th1) rather than Th2 cells, and is thought to play a key role in the recruitment of Th1 cells to sites of inflammation.13–15 It is also expressed on activated CD8+ cells as well as natural killer cells, malignant B lymphocytes, endothelial cells and thymocytes.13–15 In addition, functional CXCR3 is expressed on B cells,16–18 mast cells,19–21 vascular pericytes, and several other cell types.22–24 Binding of agonists to the CXCR3 receptor results in cellular responses such as integrin activation, actin reorganization and directional migration. In T lymphocytes, the stimulation of CXCR3 by its agonists leads to elevation of intracellular calcium25 as well as activation of phosphoinositide 3-kinase (PI3K)/Akt-dependent signalling and p44/p42 extracellular signal-regulated kinase (ERK) pathways.26 CXCR3 activation has also been shown to induce rapid tyrosine phosphorylation of several proteins including zeta-associated protein of 70 000 MW (ZAP-70), linker for the activation of T cells (LAT) and phospholipase-C-γ1 (PLCγ1).27
Expression of CXCR3-binding chemokines dramatically increases during inflammation and tissue injury, most likely as a consequence of increased IFN-γ secretion. These chemokines are crucial in directing activated T cells to the sites of inflammation and CXCR3 and its agonists have been implicated in the induction and perpetuation of several human inflammatory disorders15 including atherosclerosis,28 autoimmune diseases,29 transplant rejection30,31 and viral infections32 making this receptor an attractive target for new anti-inflammatory drugs. In recent years, two variants of the CXCR3 receptor have been identified, namely CXCR3-B33 and CXCR3-alt.34 Both variants are generated via alternative splicing of mRNA encoding the original CXCR3 receptor (henceforth referred to as CXCR3-A). In the case of CXCR3-B, alternative splicing results in extension of the N terminus by 52 amino acids and this form of the receptor has been shown to bind CXCL4 (platelet factor 4, PF4) in addition to the three established CXCR3 agonists.33 Agonist–receptor interactions between CXCR3-B and CXCL4 were shown to induce apoptotic signals in microvascular endothelial cells; however, neither a chemotactic response towards a CXCL4 gradient nor calcium flux was observed in CXCR3-B transfectants.33 In contrast, CXCR3-alt is a 101-amino-acid-truncated version of CXCR3 that consequently exhibits a dramatically altered C terminus and a predicted 4–5 transmembrane domain structure.34 Despite this drastically modified structure, CXCR3-alt has been shown to bind to CXCL11 and elicit biochemical signals, yet curiously it does not signal in response to CXCL9 or CXCL10.34 In addition, CXCL11 (but not other CXCR3 agonists) binds to CXCR7, a receptor that has been associated with increased adhesiveness, invasiveness and reduced apoptosis of human umbilical vein endothelial cells and tumour cells.35,36
In this study we examined the expression of CXCR3 variants on activated human T lymphocytes and used CXCL4 as a tool to investigate CXCR3-B-mediated responses in T cells. We observed that CXCL4 induced biochemical responses (intracellular calcium mobilization as well as phosphorylation of Akt and p44/42 ERK) in T lymphocytes. However, despite being comparable in magnitude with signals elicited by CXCR3 agonists, the CXCL4-induced signals are insufficient to elicit a migratory response.
Materials and methods
Reagents
Recombinant human CXCL9, CXCL10, CXCL11 and CXCL4 were obtained from PeproTech (London, UK). Phycoerythrin (PE) and fluorescein isothiocyanate (FITC) -conjugated mouse IgG1, anti-human CXCR3 monoclonal antibody (clone 49801), IgG2A isotype control, FITC-conjugated mouse anti-human CD8 (isotype IgG2B, clone 37006) and appropriate isotype were obtained from R&D Systems (Abingdon, UK). The FITC-conjugated mouse anti-human CD3 (isotype IgG1κ, clone WT31), CD4 (IgG1κ, clone 11830) antibodies and IgG1κ isotype control were purchased from BD Biosciences (Oxford, UK). Polyclonal anti-phospho-p44/p42 Erk (Thr202/Tyr204) antibody and polyclonal anti-phospho-Akt (Ser473) antibody, both produced in rabbit, were obtained from Cell Signaling Technology, New England Biolabs (Hitchin, UK). Akt1 and Erk1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary anti-rabbit horseradish peroxidase (HRP) and anti-goat HRP antibodies were from Dako Ltd (Cambridge, UK). Bordetella pertussis toxin was purchased from Calbiochem (Leicestershire, UK). The CXCR3 antagonists T487 (AMG487), and NBI-74330 were synthesized and supplied to S.G.W. for research purposes only by UCB (Cambridge, UK).
Isolation and in vitro expansion of T cells
Procedures for the use of human blood were carried out under University and Departmental safety and ethical guidelines for the use of human tissue. Peripheral blood-derived mononuclear cells (PBMCs) were isolated from heparinized peripheral blood obtained from healthy volunteers and isolated as detailed previously.37 Briefly, whole blood was mixed 1 : 1 with RPMI-1640 medium and separated by differential centrifugation using Lymphoprep (Axis-Shield, Cambridgeshire, UK). The PBMC layer was diluted in RPMI-1640, washed three times and resuspended in RPMI-1640 complete medium (RPMI-1640 supplemented with 10% fetal calf serum and 50 U/ml penicillin plus 50 μg/ml streptomycin). The PBMCs were stimulated for 3 days using 10 μg/ml Staphylococcal enterotoxin B (SEB; Sigma-Aldrich, Poole, UK) and cultured at 37° in a humidified 5% CO2 environment. On day 3, cells were washed from SEB, and kept in culture in RPMI-1640 complete medium supplemented every 2–3 days with interleukin-2 (IL-2; 20 ng/ml) (PeproTech). Cells were maintained up to a maximum of 12 days, and used 9–12 days after isolation and activation. Our method of activated peripheral blood-derived T-lymphocyte generation consistently yielded an almost pure T-lymphocyte population that was approximately 80% CD4+ at days 5 and 12 post-isolation.37
Reverse transcription-PCR, primers
Total RNA was purified from cultured T cells isolated from the blood obtained from different donors using TRIzol® reagent (Invitrogen, Paisley, UK) according to the manufacturer's instructions. The cDNA was prepared by reverse transcription with oligo-dT using the Omniscript RT kit (Qiagen, Crawley, UK) according to the manufacturer's protocol and used as a template for amplification by PCR with primers specific to the CXCR3-A, CXCR3-B and CXCR3-alt genes. The PCR were performed for CXCR3-A and CXCR3-alt (accession number X95876), CXCR3-B (accession no: AF469635) specific primers (CXCR3-A 5′ primer: CCAAGTGCTAAATGACGCCG; CXCR3-A 3′-primer: CAAAGGCCACCACGACCACCACCA which yield products of 770 bp;34 CXCR3-B 5′ primers ATGGAGTTGAGGAAGTACGGCCCTGGAAG; CXCR3-B 3′ primers: AAGTTGATGTTGAAGAGGGCACCTGCCAC, which yield 545-bp products; CXCR3-alt 5′ primers CCAAGTGCTAAATGACGCCG, CXCR3-alt 3′ primers CTCCCGGAACTTGACCCCTGTG which yield 622-bp products. This primer was designed to combine the CXCR3-alt-specific sequence that arises from joining bases on positions 695 and 1033.34β-Actin primers were used as loading controls. Products were separated by electrophoresis on a 1·2% agarose gel and visualized by UV transillumination.
Generation of CXCR3 constructs
To generate the CXCR3 variant constructs, the CXCR3-A, -B and -alt open-reading frames were amplified by PCR from human cDNA (Clontech, Saint-Germain-en-Laye, France). HindIII and KpnI sites were introduced to the PCR products which were then subcloned into the pEGFP vector (Clontech) allowing fusion with enhanced green fluorescent protein at the C termini of CXCR3 variants. Fragments were then cloned into pcDNA3.1 vector (Invitrogen) via HindIII and NotI sites. Plasmids were then transformed into TOP10 or DH5α bacteria. Positive colonies were screened by colony PCR and confirmed by sequencing. The HEK293 human embryonic kidney cells maintained in RPMI-1640 complete medium were transfected using LT1-Trans transfection reagent (Mirus; Cambridge BioScience, Cambridge, UK) following the manufacturer's protocol. Mock transfections were carried in the same fashion with an empty pcDNA3.1 vector.
Cell stimulation and immunoblotting
To analyse biochemical signalling through the CXCR3 receptor, days 9–12 human T cells were washed three times in RPMI-1640 medium and re-suspended to a concentration of 1 × 106/500 μl. Cells were then incubated for 30 min at 37° in the absence or presence of antagonists. Cells were stimulated with CXCR3 agonists diluted in RPMI-1640, then centrifuged and lysed by addition of 100 μl solubilization buffer (50 mm Tris–HCl pH 7·5, 150 mm NaCl, 1% Nonidet P40, 5 mm EDTA, 1 mm sodium vanadate, 1 mM sodium molybdate, 10 mm sodium fluoride, 40 μg/ml PMSF, 0·7 μg/ml pepstatin A, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 μg/ml soyabean trypsin inhibitor). The samples were mixed and gently rotated at 4° for 20 min and then centrifuged at 600 g for 10 min. The supernatant was transferred to fresh tubes and diluted 1 : 1 with 10% SDS containing 1 × sample buffer. Before loading into the gel, samples were boiled for 5 min at 100°. The samples were separated by electrophoresis in 10% SDS–PAGE. Proteins were then electro-transferred onto nitrocellulose membrane, blocked in 5% milk and incubated with rabbit phosphospecific anti-p44/p42 Erk (1/1000), and anti-Akt/PKB serine 473 (1/1000) as primary antibody and anti-rabbit HRP (1/10 000) as secondary antibody. Immune complexes were visualized using enhanced chemiluminescence (ECL Western blotting system; Amersham Bioscience, Little Chalfont, UK). To confirm equal loading, membranes were stripped by incubation in stripping buffer (100 mm 2-mercaptoethanol, 2% SDS, 62·5 nm Tris–HCl pH 6·7) at 60° for 20 min and re-probed with Akt1 or Erk1 antibodies.
Flow cytometry
Day 9–12 SEB-activated and IL-2-maintained T cells (1 × 106 per sample) were washed, re-suspended in ice-cold FACS buffer (PBS containing 5% fetal calf serum) and incubated with PE-conjugated mouse monoclonal anti-CXCR3 antibody or mouse IgG1 isotype control, on ice. After 30 min, cells were washed twice in FACS buffer and analysed using a FACSCanto™ flow cytometer (BD Biosciences) and diva software.
CXCR3 surface expression assay
CXCR3-expressing T cells were incubated with various concentrations of CXCL9, CXCL10, CXCL11 or CXCL4 for different times as indicated. In some experiments, T cells were pre-incubated for 30 min with different concentrations of CXCR3 antagonists before the addition of chemokines. At the end of these experiments, ice-cold FACS buffer was added, and cells were studied for cell surface expression of CXCR3 as described above. Mean fluorescence intensity (MFI) values were obtained by subtracting the MFI of the isotype control from the MFI of the positively stained sample. Decrease in CXCR3 surface expression was expressed as a percentage of baseline expression using the formula: MFI of stimulated cells/MFI of untreated cells × 100.
In vitro chemotaxis assay
Chemotaxis assays were performed using a 96-well plate-based ChemoTx® system as described previously.38 Briefly, cells were washed and resuspended at 3·2 × 106 cells/ml in RPMI-1640 medium containing 0·1% BSA. Wells were loaded with 29 μl chemokine (lower chamber) and overlaid with a 5 μm pore-size filter. The cell suspension (25 μl) was placed on the hydrophobic surface surrounding each well on the filter (upper chamber), and the plate was incubated in a humidified incubator at 37°, 5% CO2 for 3 hr. Cells migrating to the bottom chamber were counted using flow cytometry as previously described.39
Intracellular calcium mobilization
Previously activated T cells were washed twice in RPMI-1640 medium, resuspended at the concentration 1 × 106 cells/ml in the same medium and loaded with 5 μm Fluo-4AM (Invitrogen, Renfrew, UK). Cells were then washed twice, resuspended in assay buffer (147 mm NaCl, 2 mm KCl, 10 mm HEPES, 12 mm glucose, 1 mm MgCl2, 2 mm CaCl2, pH 7·3 with NaOH) and aliquots were placed in a 96-well plate at 1 × 105 cells/well and treated as described in the figure legends. Real-time fluorescence was recorded (excitation 485 nm, emission 520 nm) every 3–10 seconds using a multimode platereader (Fluostar Optima; BMG Labtech, Aylesbury, UK) before and after stimulation with chemokines. Data were expressed as a change in fluorescence units or fold basal response.
Results
Determining expression of CXCR3 in human-blood-derived T lymphocytes
Freshly isolated human T lymphocytes express low levels of CXCR3 on their surface, but expression was markedly up-regulated following T-cell activation with the superantigen SEB (which provides an intense polyclonal immune response without the need of antigen-specific recognition) and subsequent maintenance in IL-2 (Fig. 1a). The commercially available anti-CXCR3 antibodies are unable to distinguish between CXCR3-A, CXCR3-B and CXCR3-alt, although selective CXCR3-B antibodies are not widely available and there are currently no reported CXCR3-alt specific antibodies. Therefore, we investigated the presence of mRNA expression for each variant of CXCR3 (Fig. 1b). Our data show that at the mRNA level, CXCR3-A, CXCR3-B and CXCR3-alt are all expressed on SEB/IL-2-activated T lymphocytes.
Figure 1.
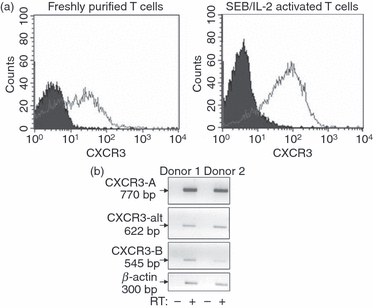
CXCR3 receptor is highly expressed on activated human peripheral blood-derived T lymphocytes. (a) Freshly isolated (left histogram) and Staphylococcus enterotoxin B (SEB)/interleukin-2 (IL-2) activated (day 9) T lymphocytes (right histogram) were stained with anti-CXCR3 monoclonal antibody (open histograms) or with isotype-matched immunoglobulin controls (filled histograms) as detailed in Materials and methods. (b) Messenger RNA from 5 × 106 SEB/IL-2-activated T cells was extracted and incubated in the presence (+) or the absence (−) of reverse transcriptase to ensure that samples were not contaminated with genomic DNA. Reverse transcription-PCR was performed using primers to specifically amplify CXCR3-A, CXCR3-B, and CXCR3-alt cDNA and products were separated on agarose and visualized under UV light. Data from two donors are shown, representative of results from three other different donors.
CXCR3 agonists and CXCL4 stimulate intracellular calcium elevation and PI3K/Akt and p44/p42 ERK signalling
Given the limitations of existing CXCR3-B antibodies, we assessed responsiveness to CXCL4 as an indirect estimate to establish whether functional CXCR3-B was expressed on T cells. This chemokine was initially reported to activate CXCR3-B but not other CXCR3 isoforms.33 However, further studies by Mueller et al.40 suggested that CXCL4 binds to and activates both A and B isoforms of CXCR3. For further examination of the role of CXCL4 and its receptor interaction in T cells, we tested the ability of CXCL4 to induce the elevation of intracellular free calcium and compared this response with those induced by other CXCR3 agonists. All chemokines examined, increased intracellular free calcium levels in T cells previously activated with SEB (Fig. 2). However, CXCL4 elicited a less robust response compared with other CXCR3 agonists and a high (micromolar) concentration of CXCL4 was required to induce intracellular calcium elevation to levels comparable with the responses induced by nanomolar amounts of CXCL9, CXCL10 or CXCL11 (Fig. 2). CXCL11 was also able to stimulate higher maximal responses than the other chemokines examined.
Figure 2.
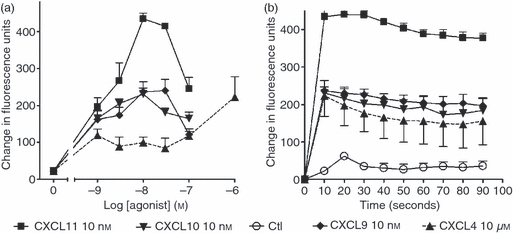
CXCR3 agonists and CXCL4 induce intracellular calcium flux in T cells. Staphylococcus enterotoxin B (SEB)/interleukin-2 (IL-2) activated T cells (day 9–12) were loaded with Fluo-4 AM at 37° for 30 min. Cells were then washed twice in RPMI-1640 medium and aliquoted at 1 × 105 cells/well as described in the Materials and methods and treated with the indicated concentrations of each agonist (a) or with 10 nm of each agonist for the times indicated (b). Changes in fluorescence were measured using multimode plate reader (Fluostar Optima). Peak responses from each stimulation were taken to create a concentration–response curve. Error bars represent mean ± SEM of four experiments using cells from different donors.
To study signalling, the effect of each chemokine on PI3K/Akt and p44/p42 ERK pathways was assessed in previously activated T lymphocytes. CXCL9, CXCL10, CXCL11 and CXCL4 stimulated PI3K/Akt-dependent signalling, as measured by phosphorylation of Akt/PKB at Ser473. In addition, all agonists stimulated p44/p42 phosphorylation at Thr202 and Tyr204 (Fig. 3). Curiously, the responses to CXCL4 lacked a conventional concentration-dependent profile and the significance of this is discussed later. The Akt and p44/p42 phosphorylation responses to all agonists occurred rapidly and transiently with complete attenuation of responses after 10 min stimulation (Fig. 4a), although CXCL11-stimulated phosphorylation was detectable earlier (within 30 seconds) and was more sustained in comparison with the other agonist responses.
Figure 3.
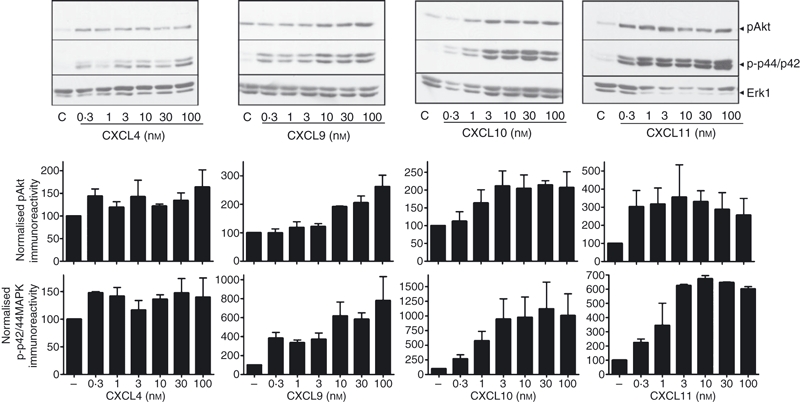
CXCR3 agonists and CXCL4 induce phosphorylation of p44/p42 extracellular signal-regulated kinase (ERK) and protein kinase B (PKB)/Akt. Aliquots of activated T cells (1 × 106 cells/500 μl) were left untreated or stimulated in parallel at 37° with 0.3–100 nm CXCR3 agonist for 2 min. Cells were lysed by the addition of 1 × sample buffer. Cell lysates were resolved by SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with a phospho-specific p44/42 ERK or Akt antibody with affinity for the active Ser473-phosphorylated form of Akt and proteins were visualized with enhanced chemiluminescence. The blots were stripped and reprobed with anti-Erk1 antibody to verify equal loading and efficiency of protein transfer (lower panel). p44/p42 ERK and Akt phosphorylation (mean from four experiments, ±SEM) was quantified by densitometry and corrected for total Erk1 expression on stripped blots. The immnoblots shown are derived from a single experiment representative of at least four others using cells from different donors.
Figure 4.
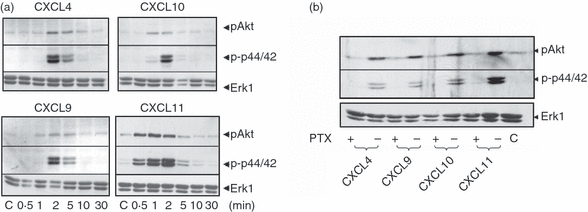
CXCR3 agonists and CXCL4 stimulate time-dependent phosphorylation of p44/p42 extracellular signal-regulated kinase (ERK) and protein kinase B (PKB)/Akt. Aliquots of activated T cells (1 × 106 cells/500 μl) were left untreated or stimulated in parallel at 37° with 1 nm CXCR3 agonist for the times indicated (a) or pre-treated with 10 ng/ml pertussis toxin for 16 hr before stimulation with CXCR3 agonists (1 nm) for 2 min (b). Cells were lysed by the addition of 1× sample buffer. Cell lysates were resolved by SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with a phospho-specific Erk or Akt antibody with affinity for the active Ser473-phosphorylated form of Akt and proteins were visualized with enhanced chemiluminescence. The blots were stripped and reprobed with anti-Erk1 antibody to verify equal loading and efficiency of protein transfer (lower panel). The data shown are derived from a single experiment performed three times using cells from different donors.
Sensitivity of CXCR3 agonist responses to pertussis toxin
The CXCR3 receptor has been previously shown to be coupled to Gi protein, yet the CXCR3-B variant has been reported to be pertussis toxin-insensitive and possibly Gs coupled.33 Therefore, for further investigation of signalling through CXCR3-A and CXCR3-B, T lymphocytes were pre-treated for 16 hr with 10 ng/ml pertussis toxin and its effect on Akt and p44/42 ERK phosphorylation was determined. As expected, pre-treatment with pertussis toxin completely inhibited CXCL9-, CXCL10- and CXCL11-induced phosphorylation of both Akt and p44/p42 MAP kinases (Fig. 4b). Previously, CXCR3-B-mediated responses to CXCL4 in other systems were reported to be pertussis toxin-resistant.33 It was surprising therefore that in our experimental system, CXCL4-induced phosphorylation of Akt and p44/p42 was inhibited by pertussis toxin (Fig. 4b).
Migratory responses to CXCL9, CXCL10, CXCL11 and CXCL4
Having established pertussis toxin-sensitive biochemical responsiveness to CXCL4 we next explored whether it could elicit functional responsiveness by assessing cell migration. The SEB-activated T lymphocytes mounted migratory responses to increasing concentrations of CXCL9, CXCL10 and CXCL11. Commensurate with the more robust activation of biochemical signals stimulated by CXCL11, this agonist elicited migratory responses greater than either CXCL9 or CXCL10 (Fig. 5). In contrast, we were unable to detect any migratory response towards CXCL4 (Fig. 5) over the same concentration range and which had previously been demonstrated to elicit biochemical responses (Figs 3 and 4).
Figure 5.
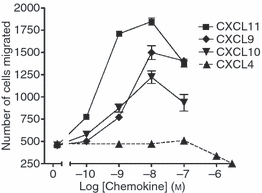
CXCL9, CXCL10 and CXCL11 but not CXCL4 stimulate migratory responses in human activated T lymphocytes. Staphylococcus enterotoxin B (SEB)/interleukin-2 (IL-2) activated T lymphocytes (3·2 × 106 cells/ml) were placed on the upper membrane of a 96-well chemotaxis plate above a lower chamber containing CXCR3 agonists at the indicated concentrations as described in the Materials and methods. Cell migration across a 5-μm pore size membrane was determined after a 3 hr incubation at 37° in 5% CO2. Cells migrating to the bottom chamber were counted using flow cytometry as described in the Materials and methods. Data shown are mean ± SEM of triplicate values from a single experiment and are representative of three other experiments using cells from different donors.
Agonist-induced down-regulation of CXCR3 surface expression in response to CXCL9, CXCL10, CXCL11 and CXCL4
A feature of chemokine interaction with the G-protein-coupled receptor (GPCR) is rapid receptor internalization, which leads to signal attenuation and desensitization, providing a regulatory mechanism for intracellular responses.41,42 We used the loss of surface expression of CXCR3 as an indirect measurement of internalization in response to agonist binding. T lymphocytes were exposed to various concentrations of agonists for 30 min or alternatively were incubated with 100 nm of each agonist for different periods of time. CXCR3 surface expression was then detected by flow cytometry using a mouse monoclonal anti-human CXCR3 antibody. Because the antibody does not distinguish between CXCR3 variants we cannot distinguish between effects on CXCR3-A, CXCR3-B and CXCR3-alt and could only interpret our results as decreased total surface CXCR3. Accordingly, CXCL9, CXCL10 and CXCL11 all induced concentration-dependent and time-dependent decreases in total CXCR3 surface expression (Fig. 6a,b), with CXCL11 being the most efficient stimulus for loss of cell surface CXCR3. Stimulation with CXCL11 (100 nm) reduced surface expression of CXCR3 by about 85% of control level after 1 hr incubation (Fig. 6b). In contrast, the maximum losses of surface expression detected in response to the same concentrations of CXCL9 or CXCL10 were around 30% and 50% of basal level, respectively. Importantly, incubation with CXCL4 did not result in any detectable internalization of CXCR3 at comparable time-points and concentrations used for the other CXCR3 agonists.
Figure 6.
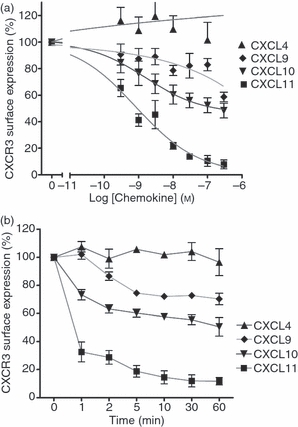
Stimulation with CXCL9, CXCL10 or CXCL11, but not CXCL4, induces down-regulation of CXCR3 surface expression. Previously activated T cells (day 9–12, 1 × 106/500 μl) were stimulated with increasing concentrations of CXCL4, CXCL9, CXCL10 or CXCL11 over 30 min (a), or with 100 nm of each agonist for up 1–60 min (b). Agonists were then washed off and cells were incubated with anti-CXCR3 antibody or isotype control at 4° followed by analysis FACSCanto™ flow cytometer as described in the Materials and methods. Decreases in CXCR3 surface expression is expressed as a percentage of CXCR3 levels detected on unstimulated cells. Presented data represent mean ± SEM of at least three independent experiments using cells from different donors.
Effect of small CXCR3 antagonists on CXCR3-mediated responses
To investigate further whether the biochemical signalling elicited by CXCL4 was indeed mediated by a CXCR3 isoform, we used the small non-peptide CXCR3 antagonists, T487 and its analogue NBI-74330.43,44 First, we verified that these inhibitors targeted CXCR3 by examining their effect on CXCL11-stimulated migration, loss of receptor expression and biochemical signalling, given that it exhibited higher potency and efficacy in our experiments in comparison with CXCL9 and CXCL10. Both T487 and NBI-74330 inhibited directional migration to CXCL11 in a concentration-dependent manner with IC50 values of 69 and 2·3 nm, respectively (Fig. 7a), which are comparable with those reported elsewhere.43,44 Further, loss of surface expression of CXCR3 in response to CXCL11 was inhibited after treatment with T487 and NBI-74330 (Fig. 7b) with IC50 values of about 140 and 0·2 nm, respectively. However, these antagonists did not completely inhibit the loss of surface expression, perhaps because of low levels of constitutive CXCR3 internalization occurring independently of agonist binding. Third, both compounds inhibited CXCL11-stimulated phosphorylation of Akt/PKB and p44/p42 ERK (Fig. 7c). Inhibition of signalling was seen over a concentration range similar to that observed in our chemotaxis and internalization experiments. Migratory and biochemical responses to CXCL9 and CXCL10 were also inhibited by both T487 and NBI-74330 at concentrations known to elicit near-maximum inhibition of CXCL11 responses (Figs 7d and 8). Neither T487 nor NBI-74330 had any effect on responses to the CXCR4 agonist CXCL12, which was used as negative control (Fig. 7d). As CXCL4 was not able to induce migratory responses in human T cells, we examined the effect of CXCR3 antagonists against CXCL4-stimulated Akt/PKB and p42/p44 ERK phosphorylation. We found that neither Akt/PKB nor p42/p44 phosphorylation induced by CXCL4 was sensitive to these CXCR3 inhibitors (Fig. 8).
Figure 7.
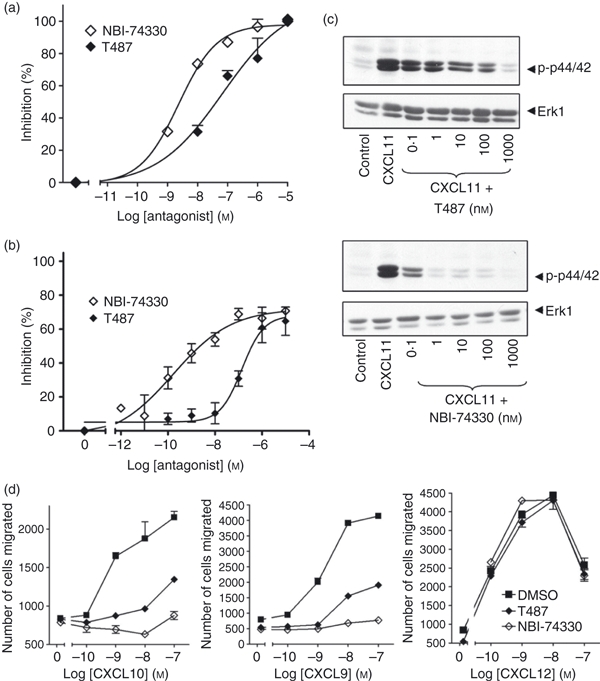
CXCR3 antagonists inhibit CXCL11-mediated responses in human T cells. Staphylococcus enterotoxin B (SEB)/interleukin-2 (IL-2) activated T lymphocytes (3·2 × 106 cells/ml; day 9–12 post-activation) were incubated for 30 min with the CXCR3 antagonists T487 or NBI-74330 at concentrations indicated (a, b, c) or 1 μm and 100 nm, respectively (d). Cells were then used in migration (a,d), biochemical (c) or receptor surface expression assays (b). For migration assays, cells placed on the upper membrane of a 96-well chemotaxis plate above a lower chamber containing 1 nm CXCL11 (a) or concentrations indicated (d). Cell migration across a 5-μm pore size membrane was determined as described in the Materials and methods. For receptor surface expression assays (b), T cells were stimulated with 30 nm of CXCL11 for 5 min. Agonist was then washed off and cells were incubated with anti-CXCR3 antibody or isotype control at 4° followed by analysis FACSCanto™ flow cytometer as described in the Materials and methods. Data (a, b, d) represent mean ± SEM of at least three independent experiments using cells from different donors. (c) Aliquots of activated T cells (1 × 106 cells/500 μl) were left untreated or stimulated at 37° with 1 nm CXCL11 for 2 min in the absence or presence of 30 min pre-treatment with T487 or NB-74330 at the concentrations indicated. Cell lysates were resolved by SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with a phospho-specific Erk or Akt antibody with affinity for the active Ser473-phosphorylated form of Akt and proteins were visualized with enhanced chemiluminescence. The blots were stripped and reprobed with anti-Erk-1 antibody to verify equal loading and efficiency of protein transfer (lower panel). The data are derived from a single experiment representative of at least three others using cells from different donors.
Figure 8.
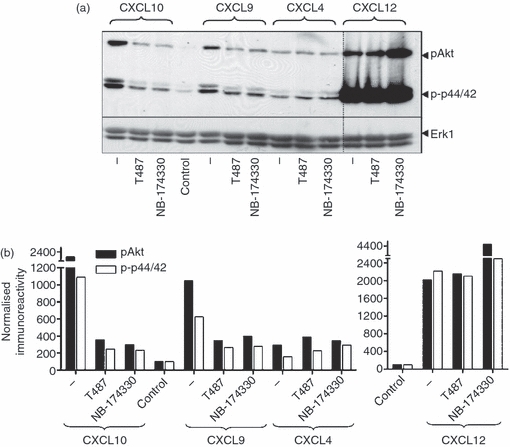
CXCR3 antagonists inhibit CXCR3-mediated phosphorylation of p44/p42 extracellular signal-regulated kinase (ERK) and protein kinase B (PKB)/Akt in human T cells. Aliquots of activated T cells (1 × 106 cells/500 μl) were left untreated or stimulated at 37° with 1 nm CXCL11 or CXCL12 for 2 min in the absence or presence of 30 min pre-treatment with T487 or NB-74330 at 1 μm and 100 nm, respectively. Cell lysates were resolved by SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with a phospho-specific Erk or Akt antibody with affinity for the active Ser473-phosphorylated form of Akt and proteins were visualized with enhanced chemiluminescence (a). The blots were stripped and reprobed with anti-Erk-1 antibody to verify equal loading and efficiency of protein transfer (lower panel). Phosphorylation of Akt and p44/p42 ERK was quantified by densitometry and corrected for total Erk 1 expression on stripped blots (b). The data are derived from a single experiment representative of at least three others using cells from different donors.
CXCR3-B expressing HEK293 cells are responsive to CXCL4
For further investigation of the responses mediated by identified CXCR3 isoforms, we transiently expressed cDNA constructs expressing CXCR3-A, CXCR3-B and CXCR3-alt in HEK293 cells (Fig. 9a). As reported previously for other cell types, such as the pre-B-cell line L1.2 and also in endothelial cells,33,40 consistently lower levels of surface expression were obtained for CXCR3-B transfectants making direct comparison difficult. In addition, apparent expression of CXCR3-alt was even lower than that observed for CXCR3-B. Differences in expression levels may also be explained by different affinities of antibody binding of CXCR3-B and CXCR3-alt. We then went on to assess the ability of CXCL11 and CXCL4 to induce intracellular calcium elevation in HEK293 expressing each isoform of CXCR3. CXCL11 (30 nm) induced responses in HEK293 cells expressing all variants of CXCR3. Responses detected in CXCR3-A transfectants were more robust than those observed in CXCR3-B- and CXCR3-alt-expressing cells. These observations are supported by previous findings that CXCL11 was shown to have higher affinity for CXCR3-A than other isoforms,33,34 but may also reflect different levels of receptor expression. CXCL4 (300 nm) induced calcium elevation in cells expressing CXCR3-A or CXCR3-B. This was surprising because it has been previously reported that CXCL4 exhibits the highest affinity for the CXCR3-B isoform,33,40 although the more robust responses detected in CXCR3-A-expressing cells might be explained by high surface expression of CXCR3-A. Elevations in intracellular free calcium were not observed in cells transfected with an empty vector (Fig. 9b), indicating that responses were specific for the receptor transfected.
Figure 9.
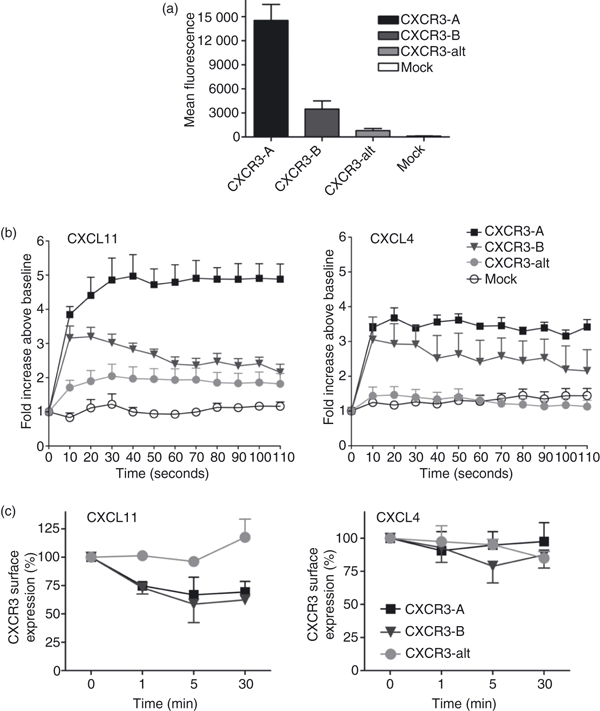
CXCR3-A and CXCR3-B but not CXCR3-alt transfectants are responsive to CXCL4. (a) HEK293 cells were transfected with plasmids encoding CXCR3-A, CXCR3-B and CXCR3-alt. Surface expression of each variant of CXCR3 is expressed as mean fluorescence intensity, measured by flow cytometry after staining with phycoerythrin-conjugated anti-CXCR3 antibody. Staining with an appropriate isotype control was subtracted. (b) HEK293 cells expressing CXCR3-A, CXCR3-B or CXCR-alt were loaded with Fluo-4 at 37° for 30 min. Cells were then washed twice in assay buffer and aliquoted at 1 × 105 cells/well as described in the Materials and methods and treated with indicated concentrations of each agonist. Changes in fluorescence were measured using a multimode plate reader (Fluostar Optima). Data shown are the mean of two separate experiments performed in duplicate. (c) HEK293 cells expressing CXCR3 variants were stimulated with 100 nm CXCL4 or CXCL11 over 30 min. Agonists were then washed off and cells were incubated with anti-CXCR3 antibody or isotype control at 4° followed by analysis on a FACSCanto™ flow cytometer as described in the Materials and methods. Decrease in CXCR3 surface expression is expressed as a percentage of CXCR3 levels detected on unstimulated cells. Presented data represent mean ± SEM of three independent experiments.
Despite the relatively low surface expression of CXCR3-B and CXCR3-alt isoforms in transfected HEK293 cells, we investigated the effect of CXCL11 and CXCL4 on detected surface receptor expression (Fig. 9c). We found that CXCL11 induced down-regulation of the CXCR3-B receptor to about 60% of basal expression, comparable with the down-regulation of CXCR3-A. In contrast, upon stimulation with CXCL11, surface expression of CXCR3-alt increased by around 25%, and a further 75% increase was observed after extending the incubation time to 120 min (data not shown). Treatment of the cells with CXCL4 led to a modest decrease (around 25%) of CXCR3-B surface expression but no effect on CXCR3-A was detected. Similarly, the (low basal) level of CXCR3-alt surface expression remained unchanged.
Discussion
In this study we have demonstrated the expression of CXCR3 as well as of two of its known variants, CXCR3-B and CXCR3-alt, in ex vivo activated human peripheral blood-derived T lymphocytes. The established CXCR3 agonists CXCL9, CXCL10, CXCL11 all elicited elevation of intracellular calcium and stimulate pertussis toxin-sensitive phosphorylation of p44/p42 ERK and PI3K/Akt in these cells. Similar responses of comparable magnitude were also observed following treatment with CXCL4, which has previously been reported as an agonist for CXCR3-B.33 Remarkably, while CXCL9, CXCL10 and CXCL11 all stimulate the loss of surface expression of CXCR3 and elicited directional migration responses of T lymphocytes, no such responses were observed following CXCL4 stimulation of these cells. In addition, CXCL4-stimulated phosphorylation of Akt and p44/42 ERK was resistant to two non-competitive CXCR3 antagonists at concentrations that inhibited the phosphorylation of this signalling molecule in response to the three established CXCR3 agonists.
All of the CXCR3 agonists elicited transient p44/42 ERK and Akt phosphorylation responses, which is consistent with receptor desensitization and internalization. However, CXCL11 induced the most robust biochemical and functional responses consistent with previous observations.42 In addition to binding CXCR3, CXCL11 (unlike CXCL9 and CXCL10) is also reported to bind CXCR7. It seems unlikely that the more robust responses to CXCL11 are the result of additional signalling via CXCR7 because previous studies indicate that CXCL11 is unable to induce calcium signalling, or p44/42 or Akt phosphorylation, through CXCR7.45 It is interesting to note however that while chemotaxis and calcium responses to all three established CXCR3 agonists were dependent on the C terminus and the DRY sequence, distinct domains are required for internalization.7 Hence, CXCL11 predominantly induces internalization via a C-terminal independent pathway whereas CXCL9- and CXCL10-stimulated internalization occurs via a C-terminus-dependent pathway.7
The existence of binding sites or receptors for CXCL4 on T lymphocytes has previously been suggested because this ligand can suppress T-cell function by decreasing proliferation and IL-2 mRNA expression, as well as inhibiting the release of IL-2 and IFN-γ.46 In addition, CXCL4 induces proliferation of regulatory T cells (CD4+ CD25+) and regulates Th1/Th2 polarization via differential regulation of transcription factors T-bet and GATA-3.47,48 The significance of T-cell regulation by CXCL4 is unclear though it has been postulated that the deposition of micromolar concentrations of CXCL4 by activated platelets onto the endothelium may result in the recruitment of activated T lymphocytes, leading to their infiltration into the tissues and the amplification of inflammation.49,50 Consistent with our data, CXCL4 has previously been demonstrated to stimulate elevation of intracellular calcium in activated T lymphocytes.40 Mueller et al. have reported that CXCL4 induces migration of activated T lymphocytes in a pertussis toxin-sensitive manner,40 although we were unable to detect this response. One reason for the discrepancy between our data and those of Mueller et al. may be that the previous study used T cells that had been activated with concanavalin A, whereas we used SEB as our initial activating stimulus. This may lead to many discrepancies between the two model systems including differences in the overall CD4+/CD8+ ratio in the two studies. Interestingly, T cells activated with alternative stimuli such as anti-CD3/CD28 antibody-coated beads, also did not migrate to CXCL4 (data not shown), suggesting that our observations are not restricted to cells that have been activated with SEB.
It has previously been reported that in addition to binding the established CXCR3 agonists, CXCR3-B also binds CXCL4 with high affinity.33 Therefore, one interpretation of our observations relating to the biochemical responses initiated by CXCL4 is that these reflect expression and activation of CXCR3-B. Indeed, we can detect CXCR3-B in activated T cells at the mRNA level. However, several lines of evidence are inconsistent with the CXCL4 responses being mediated via CXCR3-B. First, CXCL4-stimulated Akt phosphorylation and p44/p42 ERK is pertussis toxin sensitive, yet CXCL4-mediated calcium fluxes and inhibitory effects on proliferation in CXCR3-B-transfected endothelial cell models are pertussis toxin-resistant.33 This may simply reflect differential coupling of CXCR3-B to the pertussis toxin-sensitive Gαi/Go subunits and the pertussis toxin-insensitive Gαq subunits in different cell types. Second, the biochemical responses to CXCL4 were resistant to the CXCR3 inhibitor T487 and its analogue NBI-74330. The selectivity of these compounds for CXCR3-A versus CXCR3-B is not known. CXCR3-B differs only in the N-terminus region and given the non-competitive nature of these compounds, one might predict that they will inhibit both variants with similar potencies. A related CXCR3 antagonist is reported to inhibit CXCL4-induced T-cell migration responses,40 but is unable to displace radiolabelled CXCL4 binding to CXCR3-B-expressing Chinese hamster ovary (CHO) cells. The relevance of findings made in CXCR3-B-transfected CHO cells to observations made using T cells, is questionable. It is clear though that the interactions of CXCR3 with CXCL4 in T cells seem quite distinct from its interactions with other ligands as CXCL4 is unable to displace CXCL10 from activated T lymphocytes.40 The lack of a conventional concentration–response relationship with regard to CXCL4 effects on Akt and p44/42 ERK phosphorylation in our studies, underlines this conclusion.
The failure of CXCL4 to elicit loss of CXCR3 surface expression, may account for the lack of migratory responses because receptor internalization has been reported to be necessary for chemotactic responses following activation of CXCR2 in some cell models.51–55 In this regard, β-arrestins are known to play a key role in the internalization of ligand-activated chemokine receptors.56 The internalized GPCR–β-arrestin complex can form a signalosome that activates signalling proteins such as ERK1/2, p38 and Jun N-terminal kinase and act as a scaffold that connects the GPCR to tyrosine kinases, c-Src, PI3K/Akt and nuclear factor-κB pathways.56 CXCL4 does not cause internalization (as assessed by surface expression) and is therefore unlikely to lead to the formation/activation of GPCR–β-arrestin-dependent signalosomes, this may explain the lack of migratory responses. Although we observed little difference in either kinetics or magnitude of Akt and p44/p42 ERK phosphorylation, the CXCL4-stimulated calcium responses were markedly weaker compared with the other CXCR3 agonists. It is also important to stress that our assays measure total cellular biochemical responses and the lack of internalization/GPCR–β-arrestin-dependent signalosome formation may impair the correct compartmentalization and localization of biochemical events that are necessary to provide the stimulus for migration. However, the requirement for receptor internalization in migratory responses to chemokines varies according to cell and receptor type and there are several examples of migration occurring in the absence of receptor internalization.55,57,58 Indeed, mutation of the DRY sequence can ablate CXCL11-induced calcium mobilization, p44/42 phosphorylation and chemotaxis with no effect on CXCR3 internalization.7
It is now apparent that GPCRs can possess multiple binding sites and are able to modulate multiple signalling pathways. The concept that different ligands selectively stabilize different ‘active’ conformations of GPCRs and, as such, might be able to selectively regulate different pathways is well established.59,60 There is considerable interest in assessing whether such ligands may offer therapeutic advantage if they regulate a signal associated with a beneficial outcome without activating a second pathway that may be contraindicated. Our results show that although CXCL4 does not share functional properties with CXCR3 agonists, it is still able to induce intracellular signalling that is likely to be independent of the classical CXCR3. This suggests a distinct role in T-cell biology and regulation of human immune response that merits further investigation with the possibility of revealing a new target for manipulation of T-cell activity during inflammatory or autoimmune diseases. This is underlined by reports that it exerts opposite effects on production of Th1 and Th2 cytokines to those observed with CXCL10.47 Importantly, the design of CXCR3 antagonists with a view to combat Th1-associated diseases would seem unlikely to have effects on the Th2-associated effects of CXCL4.
Disclosures
The authors have no financial conflicts of interest related to this study.
References
- 1.Luster AD. Chemokines – chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–45. doi: 10.1056/NEJM199802123380706. [DOI] [PubMed] [Google Scholar]
- 2.Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. 2001;2:123–8. doi: 10.1038/84219. [DOI] [PubMed] [Google Scholar]
- 3.Sallusto F, Mackay CR, Lanzavecchia A. The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol. 2000;18:593–620. doi: 10.1146/annurev.immunol.18.1.593. [DOI] [PubMed] [Google Scholar]
- 4.von Andrian UH, Mackay CR. Advances in immunology: T-cell function and migration – two sides of the same coin. N Engl J Med. 2000;343:1020–33. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 5.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 6.Cole KE, Strick CA, Paradis TJ, et al. Interferon-inducible T cell α chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009–21. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colvin RA, Campanella GSV, Sun JT, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. 2004;279:30219–27. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- 8.Cox MA, Jenh CH, Gonsiorek W, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell α chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59:707–15. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- 9.Amichay D, Gazzinelli RT, Karupiah G, Moench TR, Sher A, Farber JM. Genes for chemokines MuMig and Crg-2 induced in protozoan and viral infections in response to IFN-γ with patterns of tissue expression that suggest nonredundant roles in vivo. J Immunol. 1996;157:4511–20. [PubMed] [Google Scholar]
- 10.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–204. doi: 10.4049/jimmunol.168.7.3195. [DOI] [PubMed] [Google Scholar]
- 11.Khan IA, MacLean JA, Lee FS, et al. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12:483–94. doi: 10.1016/s1074-7613(00)80200-9. [DOI] [PubMed] [Google Scholar]
- 12.Widney DP, Xia YR, Lusis AJ, Smith JB. The murine chemokine CXCL11 (IFN-inducible T cell α chemoattractant) is an IFN-γ- and lipopolysaccharide-inducible glucocorticoid-attenuated response gene expressed in lung and other tissues during endotoxemia. J Immunol. 2000;164:6322–31. doi: 10.4049/jimmunol.164.12.6322. [DOI] [PubMed] [Google Scholar]
- 13.Loetscher M, Gerber B, Loetscher P, et al. Chemokine receptor specific for IP10 and Mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–9. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 15.Qin SX, Rottman JB, Myers P, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–54. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones D, Benjamin RJ, Shahsafaei A, Dorfman DM. The chemokine receptor CXCR3 is expressed in a subset of B-cell lymphomas and is a marker of B-cell chronic lymphocytic leukemia. Blood. 2000;95:627–32. [PubMed] [Google Scholar]
- 17.Muehlinghaus G, Cigliano L, Huehn S, et al. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood. 2005;105:3965–71. doi: 10.1182/blood-2004-08-2992. [DOI] [PubMed] [Google Scholar]
- 18.Trentin L, Agostini C, Facco M, et al. The chemokine receptor CXCR3 is expressed on malignant B cells and mediates chemotaxis. J Clin Invest. 1999;104:115–21. doi: 10.1172/JCI7335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brightling CE, Ammit AJ, Kaur D, et al. The CXCL10/CXCR3 axis mediates human lung mast cell migration to asthmatic airway smooth muscle. Am J Respir Crit Care Med. 2005;171:1103–8. doi: 10.1164/rccm.200409-1220OC. [DOI] [PubMed] [Google Scholar]
- 20.Brightling CE, Kaur D, Berger P, Morgan AJ, Wardlaw AJ, Bradding P. Differential expression of CCR3 and CXCR3 by human lung and bone marrow-derived mast cells: implications for tissue mast cell migration. J Leukoc Biol. 2005;77:759–66. doi: 10.1189/jlb.0904511. [DOI] [PubMed] [Google Scholar]
- 21.Jinquan T, Anting L, Jacobi HH, et al. CXCR3 expression on CD34+ hemopoietic progenitors induced by granulocyte-macrophage colony-stimulating factor: II. Signaling pathways involved. J Immunol. 2001;167:4405–13. doi: 10.4049/jimmunol.167.8.4405. [DOI] [PubMed] [Google Scholar]
- 22.Bonacchi A, Romagnani P, Romanelli RG, et al. Signal transduction by the chemokine receptor CXCR3 – activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276:9945–54. doi: 10.1074/jbc.M010303200. [DOI] [PubMed] [Google Scholar]
- 23.Palmqvist C, Wardlaw AJ, Bradding P. Chemokines and their receptors as potential targets for the treatment of asthma. Br J Pharmacol. 2007;151:725–36. doi: 10.1038/sj.bjp.0707263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang XK, Yue TL, Ohlstein EH, Sung CP, Feuerstein GZ. Interferon-inducible protein-10 involves vascular smooth muscle cell migration, proliferation, and inflammatory response. J Biol Chem. 1996;271:24286–93. doi: 10.1074/jbc.271.39.24286. [DOI] [PubMed] [Google Scholar]
- 25.Rabin RL, Park MK, Liao F, Swofford R, Stephany D, Farber JM. Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J Immunol. 1999;162:3840–50. [PubMed] [Google Scholar]
- 26.Smit MJ, Verdijk P, van der Raaij-Helmer E, et al. CXCR3-mediated chemotaxis of human T cells is regulated by a G(i)- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood. 2003;102:1959–65. doi: 10.1182/blood-2002-12-3945. [DOI] [PubMed] [Google Scholar]
- 27.Dar WA, Knechtle SJ. CXCR3-mediated T-cell chemotaxis involves ZAP-70 and is regulated by signalling through the T-cell receptor. Immunology. 2007;120:467–85. doi: 10.1111/j.1365-2567.2006.02534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mach F, Sauty A, Iarossi AS, et al. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. 1999;104:1041–50. doi: 10.1172/JCI6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sorensen TL, Tani M, Jensen J, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103:807–15. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hancock WW, Lu B, Gao W, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–9. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD. Donor-derived IP-10 initiates development of acute allograft rejection. J Exp Med. 2001;193:975–80. doi: 10.1084/jem.193.8.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu MT, Chen BP, Oertel P, et al. Cutting edge: the T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J Immunol. 2000;165:2327–30. doi: 10.4049/jimmunol.165.5.2327. [DOI] [PubMed] [Google Scholar]
- 33.Lasagni L, Francalanci M, Annunziato F, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197:1537–49. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ehlert JE, Addison CA, Burdick MD, Kunkel SL, Strieter RM. Identification and partial characterization of a variant of human CXCR3 generated by posttranscriptional exon skipping. J Immunol. 2004;173:6234–40. doi: 10.4049/jimmunol.173.10.6234. [DOI] [PubMed] [Google Scholar]
- 35.Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–13. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balabanian K, Lagane B, Infantino S, et al. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem. 2005;280:35760–6. doi: 10.1074/jbc.M508234200. [DOI] [PubMed] [Google Scholar]
- 37.Coopman K, Smith L, Wright KL, Ward SG. Temporal variation in CB2R levels following T lymphocyte activation: evidence that cannabinoids modulate CXCL12-induced chemotaxis. Int Immunopharmacol. 2007;7:360–71. doi: 10.1016/j.intimp.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Smith LD, Hickman ES, Parry RV, Westwick J, Ward SG. PI3K γ is the dominant isoform involved in migratory responses of human T lymphocytes: effects of ex vivo maintenance and limitations of non-viral delivery of siRNA. Cell Signal. 2007;19:2528–39. doi: 10.1016/j.cellsig.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Webb A, Johnson A, Fortunato M, et al. Evidence for PI-3K-dependent migration of Th17-polarized cells in response to CCR2 and CCR6 agonists. J Leukoc Biol. 2008;84:1202–12. doi: 10.1189/jlb.0408234. [DOI] [PubMed] [Google Scholar]
- 40.Mueller A, Meiser A, McDonagh EM, et al. CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J Leukoc Biol. 2008;83:875–82. doi: 10.1189/jlb.1006645. [DOI] [PubMed] [Google Scholar]
- 41.Aragay AM, Ruiz-Gomez A, Penela P, et al. G protein-coupled receptor kinase 2 (GRK2): mechanisms of regulation and physiological functions. FEBS Lett. 1998;430:37–40. doi: 10.1016/s0014-5793(98)00495-5. [DOI] [PubMed] [Google Scholar]
- 42.Sauty A, Colvin RA, Wagner L, Rochat S, Spertini F, Luster AD. CXCR3 internalization following T cell-endothelial cell contact: preferential role of IFN-Inducible T cell α chemoattractant (CXCL11) J Immunol. 2001;167:7084–93. doi: 10.4049/jimmunol.167.12.7084. [DOI] [PubMed] [Google Scholar]
- 43.Heise CE, Pahuja A, Hudson SC, et al. Pharmacological characterization of CXC chemokine receptor 3 (CXCR3) ligands and a small-molecule antagonist. FASEB J. 2005;19:A1415. doi: 10.1124/jpet.105.083683. [DOI] [PubMed] [Google Scholar]
- 44.Johnson M, Li AR, Liu JW, et al. Discovery and optimization of a series of quinazolinone-derived antagonists of CXCR3. Bioorg Med Chem Lett. 2007;17:3339–43. doi: 10.1016/j.bmcl.2007.03.106. [DOI] [PubMed] [Google Scholar]
- 45.Proost P, Mortier A, Loos T, et al. Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood. 2007;110:37–44. doi: 10.1182/blood-2006-10-049072. [DOI] [PubMed] [Google Scholar]
- 46.Fleischer J, Grage-Griebenow E, Kasper B, et al. Platelet factor 4 inhibits proliferation and cytokine release of activated human T cells. J Immunol. 2002;169:770–7. doi: 10.4049/jimmunol.169.2.770. [DOI] [PubMed] [Google Scholar]
- 47.Romagnani P, Maggi L, Mazzinghi B, et al. CXCR3-mediated opposite effects of CXCL10 and CXCL4 on Th1 or Th2 cytokine production. J Allergy Clin Immunol. 2005;116:1372–9. doi: 10.1016/j.jaci.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 48.Liu CY, Battaglia M, Lee SH, Sun QH, Aster RH, Visentin GP. Platelet factor 4 differentially modulates CD4+ CD25+ (regulatory) versus CD4+ CD25– (nonregulatory) T cells. J Immunol. 2005;174:2680–6. doi: 10.4049/jimmunol.174.5.2680. [DOI] [PubMed] [Google Scholar]
- 49.Sachais BS, Turrentine T, Mckenna JMD, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57BI/6 and apoE–/– mice. Thromb Haemost. 2007;98:1108–13. [PubMed] [Google Scholar]
- 50.Pitsilos S, Hunt J, Mohler ER, et al. Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb Haemost. 2003;90:1112–20. doi: 10.1160/TH03-02-0069. [DOI] [PubMed] [Google Scholar]
- 51.Richardson RM, Pridgen BC, Haribabu B, Ali H, Snyderman R. Differential cross-regulation of the human chemokine receptors CXCR1 and CXCR2 – evidence for time-dependent signal generation. J Biol Chem. 1998;273:23830–6. doi: 10.1074/jbc.273.37.23830. [DOI] [PubMed] [Google Scholar]
- 52.Fan GH, Yang W, Wang XJ, Qian QH, Richmond A. Identification of a motif in the carboxyl terminus of CXCR2 that is involved in adaptin 2 binding and receptor internalization. Biochemistry. 2001;40:791–800. doi: 10.1021/bi001661b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benbaruch A, Bengali KM, Biragyn A, et al. Interleukin-8 receptor-β– the role of the carboxyl-terminus in signal-transduction. J Biol Chem. 1995;270:9121–8. doi: 10.1074/jbc.270.16.9121. [DOI] [PubMed] [Google Scholar]
- 54.Yang W, Wang DZ, Richmond A. Role of clathrin-mediated endocytosis in CXCR2 sequestration, resensitization, and signal transduction. J Biol Chem. 1999;274:11328–33. doi: 10.1074/jbc.274.16.11328. [DOI] [PubMed] [Google Scholar]
- 55.Richardson RM, Marjoram RJ, Barak LS, Snyderman R. Role of the cytoplasmic tails of CXCR1 and CXCR2 in mediating leukocyte migration, activation, and regulation. J Immunol. 2003;170:2904–11. doi: 10.4049/jimmunol.170.6.2904. [DOI] [PubMed] [Google Scholar]
- 56.Vroon A, Heijnen CJ, Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J Leukoc Biol. 2006;80:1214–21. doi: 10.1189/jlb.0606373. [DOI] [PubMed] [Google Scholar]
- 57.Cole SW, Jamieson BD, Zack JA. cAMP up-regulates cell surface expression of lymphocyte CXCR4: implications for chemotaxis and HIV-1 infection. J Immunol. 1999;162:1392–400. [PubMed] [Google Scholar]
- 58.Arai H, Monteclaro FS, Tsou CL, Franci C, Charo IF. Dissociation of chemotaxis from agonist-induced receptor internalization in a lymphocyte cell line transfected with CCR2B – evidence that directed migration does not require rapid modulation of signaling at the receptor level. J Biol Chem. 1997;272:25037–42. doi: 10.1074/jbc.272.40.25037. [DOI] [PubMed] [Google Scholar]
- 59.Kenakin T. Collateral efficacy in drug discovery: taking advantage of the good (allosteric) nature of 7TM receptors. Trends Pharmacol Sci. 2007;28:407–15. doi: 10.1016/j.tips.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 60.Baker JG, Hill SJ. Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci. 2007;28:374–81. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]