

Abstract
Interleukin-10 (IL-10) is an immunosuppressive cytokine that inhibits inflammatory gene expression. Phosphatidylinositol 3-kinase (PI3K) -mediated signalling regulates inflammatory responses and can induce IL-10 production, but a role for PI3K signalling in cellular responses to IL-10 is not known. In this study we investigated the involvement of the PI3K-Akt-GSK3 signalling pathway in IL-10-induced gene expression and IL-10-mediated suppression of Toll-like receptor-induced gene expression in primary human macrophages. A combination of loss and gain of function approaches using kinase inhibitors, expression of constitutively active Akt, and RNA interference in primary human macrophages showed that expression of a subset of IL-10-inducible genes was dependent on PI3K-Akt signalling. The effects of PI3K-Akt signalling on IL-10 responses were mediated at least in part by glycogen synthase kinase 3 (GSK3). In accordance with a functional role for PI3K pathways in contributing to the suppressive actions of IL-10, PI3K signalling augmented IL-10-mediated inhibition of lipopolysaccharide-induced IL-1, IL-8 and cyclo-oxygenase-2 expression. The PI3K signalling selectively modulated IL-10 responses, as it was not required for inhibition of tumour necrosis factor expression or for induction of certain IL-10-inducible genes such as SOCS3. These findings identify a new mechanism by which PI3K-mediated signalling can suppress inflammation by regulating IL-10-mediated gene induction and anti-inflammatory function.
Keywords: glycogen synthase kinase 3, human, interleukin-10, macrophages, phosphatidylinositol 3-kinase
Introduction
Interleukin-10 (IL-10) is a major immunosuppressive and anti-inflammatory factor that is indispensable for the homeostatic control of infection and inflammation.1 Key IL-10 functions include deactivation of myeloid lineage cells and suppression of inflammatory cytokine production. Interleukin-10 activates the transcription factor STAT3 (signal transducer and activator of transcription 3), which is required for the anti-inflammatory effects of IL-10, including inhibition of expression of pro-inflammatory genes.2–4 Emerging evidence indicates that STAT3 does not directly inhibit inflammatory gene expression. Instead, STAT3 induces the expression of genes and transcriptional repressors that in turn mediate the repression of cytokine and inflammatory gene promoters.3,5 Despite intensive study and genome-wide characterization of IL-10-inducible gene expression, the identity and mechanisms of action of IL-10-induced inhibitors of cytokine production are not well understood. A hallmark action of IL-10 is suppression of cell activation and gene induction by inflammatory factors such as Toll-like receptor (TLR) ligands, and it has been proposed that IL-10 can inhibit TLR signalling. However, inhibition of TLR signalling by IL-10 has not been consistently detected in numerous studies and cannot explain gene-specific inhibitory effects, i.e. why IL-10 inhibits the expression of only approximately 25% of TLR-inducible genes.6
Phosphatidylinositol 3-kinase (PI3K) is activated by numerous cytokines and inflammatory factors and signalling via the PI3K-Akt-glycogen synthase kinase 3 (GSK3) pathway has been implicated in macrophage survival, differentiation, activation and cytokine production.7 The PI3K-Akt signalling is strongly activated by the key macrophage survival/differentiation factor macrophage colony-stimulating factor (M-CSF) and, similar to other systems,8 Akt plays a key role in promoting macrophage survival. Both IL-4 and IL-13 also strongly activate PI3K-Akt signalling, which has been implicated in alternative macrophage differentiation promoted by these cytokines. Induction of PI3K-Akt signalling by activating receptors such as TLRs has been shown to act as a feedback inhibitory mechanism that limits inflammatory cytokine production.9 Early work showed that TLR-induced PI3K-Akt signalling could attenuate mitogen-activated protein kinase activation, and also contributed to the induction of IL-10.10 More recent work has implicated the GSK3 kinase downstream of Akt as an inhibitor of IL-10 expression.11–13 Akt-mediated phosphorylation and inactivation of GSK3 releases the IL10 gene from GSK3-mediated repression and results in increased IL-10 production. Although PI3K-Akt-GSK3 signalling has clearly been implicated in regulating IL-10 production, little is known about the role of this pathway in regulating macrophage responses to IL-10.
We previously reported that IL-10-induced expression of subsets of IL-10 target genes is attenuated in macrophages obtained from patients with inflammatory arthritis or pretreated with interferon-γ (IFN-γ).14,15 This altered pattern of IL-10-induced gene expression in IFN-γ-activated macrophages correlated with attenuated IL-10 anti-inflammatory function and ability to suppress TLR-induced cytokine production. We and others have shown that IFN-γ suppresses Akt phosphorylation and increases GSK3 activity in macrophages,12,16 which suggests the possibility that IFN-γ may modulate IL-10 responses via regulation of the PI3K-Akt-GSK3 pathway. In the present study we explored the involvement of PI3K, Akt and GSK3 in IL-10-induced gene expression and IL-10-mediated suppression of TLR-induced cytokine expression. We found that in human primary macrophages expression of a subset of IL-10-inducible genes was dependent on PI3K-Akt signalling. This PI3K-Akt signalling also contributed to the ability of IL-10 to suppress a subset of TLR-inducible cytokine genes. The effects of PI3K-Akt signalling on IL-10 responses were mediated at least in part by GSK3. These results implicate PI3K-Akt-GSK3 signalling in the regulation of IL-10 responses and IL-10-mediated suppression of inflammation.
Materials and methods
Cells and reagents
Cytokines were purchased from R & D Systems (Minneapolis, MN). Pharmacological inhibitors were acquired from Calbiochem (La Jolla, CA). Human peripheral blood mononuclear cells were isolated from the blood of healthy donors by centrifugation on a Ficoll density gradient (Gibco-BRL, Gaithersburg, MD). Monocytes were obtained from the peripheral blood mononuclear cells using anti-CD14 magnetic beads (Miltenyi Biotech, Auburn, CA) as previously described,14 and were > 96% pure as verified by flow cytometry. Monocytes were differentiated into macrophages by culturing for 3 days in RPMI-1640 medium (Gibco-RBL) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) and M-CSF (20 ng/ml). Cells were stimulated with IL-10 (20 ng/ml) or with IFN-γ (100 U/ml) for 3 hr or as specified. The viability and purity of macrophages were comparable in all conditions. Samples were processed according to a protocol approved by the Institutional Review Board of the Hospital for Special Surgery.
Real-time quantitative PCR
Real-time PCR was used to examine changes in gene expression. Briefly, 0·5 μg total RNA was reverse-transcribed using a first strand cDNA synthesis kit (Fermentas, Hanover, MD). Real-time PCR was performed using the iCycler (BioRad, Hercules, CA) and SYBR Green PCR Core Reagents kit (Applied Biosystems, Foster City, CA). Amounts of mRNA were normalized relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. The sequences of primers used for quantitative PCR are available in Supporting information, Table S1.
Immunoblotting
Whole cell extracts were obtained by lysis in buffer containing 20 mm HEPES (pH 7·0), 300 mm NaCl, 10 mm KCl, 1 mm MgCl2, 0·1% Triton X-100, 0·5 mm dithiothreitol, 20% glycerol and 1 × proteinase inhibitor cocktail (Roche, Basel, Switzerland), as previously described.12 The protein levels of extracts were quantified using the Bradford assay (BioRad).
For immunoblotting, 10 μg of cell lysates was fractionated on 7·5% or 10% polyacrylamide gels using SDS–PAGE, transferred to PVDF membranes (Millipore, Billerica, MA), incubated with specific antibodies in PBS-T supplemented with 5% BSA (Sigma, St. Louis, MO), and enhanced chemiluminescence (Amersham, Piscataway, NJ) was used for detection.
ELISA
Supernatants were collected and assayed by sandwich ELISA. Paired IL-1β capture and detection and antibody were used according to the manufacturer's protocol (R&D Systems).
Adenoviral transduction
Adenoviral transduction was performed as described before.17 Recombinant replication-deficient adenoviral particles encoding a constitutively active membrane-targeted mutant of Akt1 (Ad5-cytomegalovirus-Akt1) were purchased from Vector Biolabs (Philadelphia, PA), and adenoviral particles encoding green fluorescent protein (Ad5-cytomegalovirus-GFP) were purchased from the Vector Development Laboratory (Houston, TX). For adenoviral transduction, human monocytes were incubated for 5 days in complete RPMI-1640 medium supplemented with human M-CSF (40 ng/ml). Then, cells were incubated in low-serum medium containing 0·5% (vol/vol) fetal bovine seurm, supplemented with human M-CSF (20 ng/ml), for 1 hr, followed by transduction with adenoviral particles (50 particles per cell). After 12 hr of incubation, the culture was supplemented with an equal volume RPMI-1640 medium containing 10% (vol/vol) fetal bovine serum. Transduction efficiency was monitored by the fluorescence of GFP and was typically more than 87%.
RNA interference
RNA interference (RNAi) was performed using SMARTpool RNA duplexes that target human GSK3α, GSK3β or control duplexes (Dharmacon, Chicago, IL) as described before,18 using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) and the manufacturer's protocol optimized for primary cells. In brief, on the day of isolation primary human monocytes were incubated with 200 pmol small interfering RNA oligonucleotides and Lipofectamine RNAiMAX reagent (20 μl/ml). The transfection was repeated the next day and cells were collected after an additional 24 hr of culture.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP-IT Express kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions. Chromatin was prepared from 2 × 107 primary human monocytes per condition and was sheared to an average size of 500 bp using a Missonics Sonicator 3000. Rabbit polyclonal antibodies against STAT3, STAT1 or control rabbit IgG (Santa Cruz Biotechnologies, Santa Cruz, CA) were used for immunoprecipitation; each immunoprecipitation contained chromatin derived from 2 × 106 monocytes. Immunoprecipitates were subjected to PCR with primers that span sequences upstream of the transcription start sites of the human Autotaxin gene.
Statistical analysis
Paired Student's t-test was used to analyse data. A probability level of P < 0·05 was considered significant.
Results
Expression of a subset of IL-10 inducible genes is dependent on PI3K signalling
The PI3K-Akt pathway in macrophages is activated by M-CSF and serum growth factors and may be further transiently augmented after IL-10 stimulation.19 We tested the effects of inhibiting PI3K on IL-10-mediated induction of genes that we previously found were highly induced by IL-10 in primary human macrophages using microarray analysis.15,20 Expression of seven out of nine of the most highly IL-10-inducible genes was nearly completely abrogated in macrophages pretreated with two different PI3K inhibitors, LY294002 and wortmannin (Figs 1 and 3 and Supporting information, Table S2). In contrast, expression of highly inducible SOCS3 and KIAA0963 genes was not affected by PI3K inhibitors (Fig. 1 and Supporting information, Fig. S1 and data not shown), indicating specificity of inhibition for selective IL-10 target genes. The effect of LY294002 on gene expression was dose dependent (Supporting information, Fig. S2). These results suggest that PI3K is essential for the expression of a subset of IL-10-inducible genes, but does not globally regulate IL-10-induced gene expression.
Figure 1.

Inhibitors of phosphatidylinositol 3-kinase (PI3K) pathway selectively suppress interleukin-10 (IL-10) -induced gene expression. Human primary macrophages were pre-treated with LY294002 (20 μm) or Wortmannin (0·5 μm) for 1 or 3 hr, correspondingly, before stimulation with IL-10 (20 ng/ml) for 3 hr. Amounts of mRNA were measured using quantitative real-time PCR and normalized relative to GAPDH. Representative data of at least five independent blood donors are shown (mean ± SD of triplicate determinations). P-values for cumulative data from multiple blood donors were calculated by paired Student's t-test. P < 0·02 for differences in IL-10 versus LY294002 + IL-10, n = 14 (DKFZp586O0118, Autotaxin), n = 10 (ARNT2), n = 11 (SOCS3); P < 0·05 for differences in IL-10 versus Wortmannin + IL-10, n = 5 (DKFZp586O0118, Autotaxin, ARNT2, SOCS3). See also Supporting information, Fig. S1.
Figure 3.

Phosphatidylinositol 3-kinase (PI3K) -dependence of direct interleukin-10 (IL-10) target genes. Human primary macrophages were pretreated with cycloheximide (15 μg/ml) or LY294002 (20 μm) for 1 hr, and then stimulated with IL-10 (20 ng/ml) for 3 hr. Amounts of mRNA were measured using quantitative real-time PCR and normalized relative to GAPDH. Representative data of at least three independent blood donors are shown (mean ± SD of triplicate determinations). P-values for cumulative data were calculated by paired Student's t-test. P < 0·05 for differences in control versus IL-10, cycloheximide (CHX) versus CHX + IL-10, and IL-10 versus LY294002 + IL-10 for EGR2, Caspase 5, Autotaxin and SLAM (n = 3).
To further address the selective role of PI3K signalling in cytokine-induced gene expression, we tested the effects of PI3K inhibitors on IFN-γ-induced gene expression. Whereas IL-10 uses primarily STAT3 for gene induction, induction of many IFN-γ target genes is mediated primarily by STAT1. In contrast to IL-10-induced gene expression, LY294002 did not alter the expression of various STAT1-dependent IFN-γ-inducible genes that were tested (Fig. 2 and data not shown). Hence, IL-10-induced gene expression appeared more dependent on PI3K than IFN-γ-induced gene expression. We next wished to test if PI3K signalling was required for expression of direct IL-10 target genes, i.e. primary response genes that are induced by IL-10 rapidly and independently of new protein synthesis. Screening of our panel of highly IL-10-inducible genes showed that IL-10 was able to induce the expression of Autotaxin, Egr2, Caspase 5 and the signaling lymphocytic activation molecule (SLAM) in the presence of the protein synthesis inhibitor cycloheximide, indicating that these are primary response genes. The IL-10-induced expression of Autotaxin, Egr2, Caspase 5 and SLAM was potently inhibited by LY294002 (Fig. 3). These results exclude the possibility that PI3K solely regulates secondary response genes, i.e. those genes whose induction by IL-10 requires prior synthesis of IL-10-induced PI3K-dependent transcription factors. Instead, PI3K can regulate the expression of genes directly activated by IL-10 signalling.
Figure 2.

Expression of interferon-γ (IFN-γ) -inducible genes is not dependent on phosphatidylinositol 3-kinase (PI3K). Human primary macrophages were pretreated with LY294002 (20 μm) for 1 hr, and then stimulated with IFN-γ (100 U/ml) for 3 hr. Amounts of mRNA were measured using quantitative real-time PCR and normalized relative to GAPDH. Representative data of at least three independent blood donors are shown (mean ± SD of triplicate determinations). P-values for cumulative data were calculated by paired Student's t-test. The differences in genes induction by IFN-γ versus LY294002 + IFN-γ were not statistically significant (n = 3).
IL-10-induced STAT3 activation and promoter recruitment are minimally dependent on PI3K signalling
STAT3 is an obligatory component of IL-10 signalling and mediates the direct activation of IL-10 target genes.3 We next tested whether IL-10-induced STAT3 activation is dependent on PI3K. Interleukin-10 induced robust tyrosine phosphorylation of STAT3, and, consistent with previous reports,1,14 weak tyrosine phosphorylation of STAT1 (Fig. 4). In most experiments, IL-10-induced STAT3 tyrosine phosphorylation was not affected when PI3K was inhibited (Fig. 4), although a modest decrease in STAT3 tyrosine phosphorylation was detected in some donors (see Supporting information, Fig. S3). Hence, the nearly complete abrogation of IL-10-induced gene expression that was observed (Fig. 1) could not be explained based on inhibition of STAT3 tyrosine phosphorylation. Indeed, preserved STAT3 tyrosine phosphorylation explains the intact induction of SOCS3 expression even when PI3K was inhibited (Fig. 1). The weak activation of STAT1 by IL-10 was not dependent on PI3K signalling (Fig. 4). We did not detect serine phosphorylation of STAT3, which can modulate its transcriptional activity, in our experimental system (data not shown). These results show that proximal IL-10 signalling via STATs is minimally dependent on PI3K in primary human macrophages.
Figure 4.

Interleukin-10 (IL-10) -induced tyrosine phosphorylation of signal transducer and activator of transcription 3 (STAT3) and STAT1 is minimally dependent on phosphatidylinositol 3-kinase (PI3K). Human primary macrophages were left untreated or were treated with or LY294002 (20 μm) for 1 hr, and then stimulated with IL-10 (20 ng/ml) for 5 or 15 min. Whole-cell lysates were analysed by immunoblotting. The figure is representative of at least three independent blood donors.
To address the question of whether IL-10-induced binding of STATs to promoters of endogenous genes requires PI3K activity we used ChIP assays. As individual genes that are obligatory for IL-10 anti-inflammatory function have not been definitively identified,5 we examined Autotaxin, which is among the most highly inducible IL-10 target genes and whose induction was strongly dependent on PI3K signalling. Autotaxin expression was also induced by IFN-γ, and so we analysed occupancy of the Autotaxin promoter by STAT3 and STAT1 before and after IL-10 or IFN-γ stimulation of primary human macrophages. The IL-10 induced recruitment of STAT3, and to a lesser extent of STAT1, to the Auotaxin promoter (Fig. 5a,b). Specificity controls using irrelevant IgG for immunoprecipitation or amplification of STAT immunoprecipitation using primers specific for the Autotaxin 3′ untranslated region revealed a low background signal (Fig. 5a,c). As expected, IFN-γ induced less STAT3 recruitment, and more STAT1 recruitment, than did IL-10 (Fig. 5a,b). Inhibition of PI3K had a modest effect on IL-10-induced STAT3 recruitment and no discernable effect on IL-10-induced STAT1 recruitment to the Autotaxin promoter (Fig. 5a,b); this stands in stark contrast to the near-complete abrogation of IL-10-induced Autotaxin expression by PI3K inhibitors (Fig. 1). Hence, suppression of IL-10-induced Autotaxin expression by PI3K inhibitors is unlikely to be solely explained on the basis of diminished STAT recruitment to the promoter.
Figure 5.
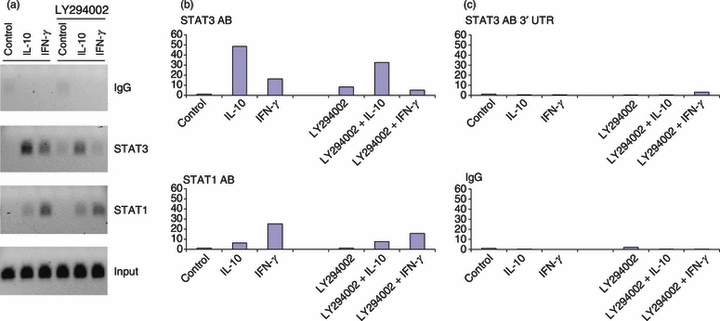
Effect of phosphatidylinositol 3-kinase (PI3K) pathway inhibition on the recruitment of signal transducer and activator of transcription (STAT) proteins to the Autotaxin promoter after interleukin-10 (IL-10) or interferon-γ (IFN-γ) stimulation. Human primary macrophages were left untreated or treated with or LY294002 (20 μm) for 1 hr, and then stimulated with IL-10 (20 ng/ml) for 60 min. (a) Binding of STAT3 and STAT1 proteins was examined by chromatin immunoprecipitation (ChIP) and analysed by PCR with primers specific for the Autotaxin promoter and gel electrophoresis of amplification products. (b) Recruitment of STAT3 and STAT1 to the Autotaxin promoter was evaluated by ChIP and quantitative real-time PCR. (c) Recruitment of STAT3 to 3′ untranslated region (UTR) of Autotaxin (top panel) and control immunoprecipitation using irrelevant IgG were performed to confirm specificity of the ChIP experiment. Data are representative of two independent blood donors. P-values were calculated by paired Student's t-test. P < 0·05 for IL-10, IFN-γ, LY294002 + IL-10, or LY294002 + IFN-γ versus untreated control for STAT3 and STAT1 immunoprecipitations; P = 0·50 IL-10 versus LY294002 + IL-10 (STAT3 IP), P = 0·43 IL-10 versus LY294002 + IL-10 (STAT1 IP), P = 0·29 IFN-γ versus LY294002 + IFN-γ (STAT3 IP), P = 0·75 IFN-γ versus LY294002 + IFN-γ (STAT1 IP).
Akt-GSK3 signalling regulates IL-10-induced gene expression
The PI3K activates the serine-threonine protein kinase Akt,7 which in turn phosphorylates and thereby inactivates GSK3, a kinase implicated in suppression of gene expression in other systems.11,21 We next tested the role of Akt-GSK3 signalling in IL-10-inducible gene expression in primary human macrophages. First, we transduced human macrophages with adenoviral particles encoding a membrane-targeted mutant of Akt1 (Ad-Akt1-CA) that contains a membrane localization sequence. This increases Akt activation by basal PI3K activity by bringing Akt1 into closer approximation with PI3K. Nevertheless, the mutant Akt1 can lose its activity after incubation with LY294002 as the result of dephosphorylation in the absence of ongoing phosphorylation by basal PI3K activity.22 This mutant is commonly termed ‘constitutive active’ (CA), because it is active after expression in most cell types without requiring a stimulus to increase PI3K activity over baseline. Akt1-CA super-induced the expression of PI3K-dependent IL-10 target genes DKFZp586O0118, ARNT2 and Caspase 5 (Fig. 6). In contrast, IL-10 target genes that were not dependent on PI3K, SOCS3 and KIAA0963, were not superinduced by constitutively active Akt1 (Fig. 6 and data not shown). In addition, CA-Akt1 partially but substantially overcame LY294002-mediated inhibition of IL-10-induced gene expression (Fig. 6, compare columns 4 and 8), further supporting a role for Akt downstream of PI3K in augmenting IL-10-induced gene expression. Consistent with a positive role for Akt in IL-10 responses, two different Akt inhibitors effectively suppressed IL-10-induced gene expression (see Supporting information, Fig. S4).
Figure 6.

Constitutively active Akt1 augments the induction of IL-10-responsive genes and partially reverses their suppression by phosphatidylinositol 3-kinase (PI3K) inhibitors. Human macrophages were transduced with adenoviral particles encoding constitutively active Akt1 (Ad-CMV-AktCA) or green fluorescent protein (Ad-CMV-GFP), as a control. After 48 hr, cells were treated with LY294002 (20 μm) for 1 hr, and then stimulated with IL-10 (20 ng/ml) for 3 hr. Amounts of mRNA were measured using quantitative real-time PCR and normalized relative to GAPDH. Representative data of three independent blood donors are shown (mean ± SD of triplicate determinations). P-values for cumulative data were calculated by paired Student's t-test. P < 0·05 for differences in IL-10 + GFP versus IL-10 + AktCA for DKFZp586O0118, ARNT2 and Caspase 5 (n = 3). The differences in KIAA0963 induction by IL-10 + GFP versus that by IL-10 + AktCA were not statistically significant (n = 3).
Next we investigated the role of GSK3 in IL-10-induced gene expression. Consistent with most cell types,11 GSK3 activity was detected at baseline, as assessed by measuring levels of its substrate β-catenin (data not shown). However, baseline serine phosphorylation of GSK3 was observed, indicating partial inhibition of GSK3 activity by signalling pathways basally active in macrophages (Fig. 4). The GSK3 phosphorylation disappeared when PI3K was inhibited, but was not affected by IL-10 stimulation (Fig. 4). These results place GSK3 downstream of PI3K, which is probably activated by M-CSF and serum growth factors in human macrophages and partially suppresses GSK3 activity. We used RNAi to knock down GSK3 expression in primary human macrophages and test the role of GSK3 in IL-10-induced gene expression. The inverse relationship between PI3K-Akt and GSK3 activity led us to predict that downregulation of GSK3 would have the opposite effect from PI3K and Akt inhibitors, namely increased expression of IL-10-inducible genes. Indeed, knockdown of the GSK3α isoform using RNAi enhanced induction of IL-10 target genes, but not of IFN-γ-inducible ISG54 (Fig. 7a,b). Similar results were obtained using GSK3 inhibitors (see Supporting information, Fig. S4). The RNAi-mediated knockdown of GSK3β had a lesser effect on IL-10-inducible gene expression than did knockdown of GSK3α (data not shown). As cAMP response element-binding protein (CREB) is one of the targets of GSK3,12,13 and GSK-3-mediated phosphorylation of CREB inhibits CREB transcriptional activity,23 we tested the involvement of CREB in IL-10-induced gene expression. However, down-regulation of CREB with RNAi did not have any apparent effect on the induction of IL-10-dependent genes (Fig. 7a,b). Collectively, these results implicate PI3K-Akt and GSK3 in the regulation of IL-10-induced gene expression, with Akt promoting gene expression by suppressing the repressive functions of GSK3.
Figure 7.

Down-regulation of glycogen synthase kinase 3α (GSK3α) expression by RNA interference (RNAi) correlates with enhanced induction of interleukin-10 (IL-10) -dependent genes. (a) Human primary macrophages were transfected with control, GSK3α, or cAMP response element-binding protein (CREB) short interfering RNAs for 48 hr, and then stimulated with IL-10 (20 ng/ml) for 3 hr. Amounts of mRNA were measured using quantitative real-time PCR and normalized relative to GAPDH. (b) mRNA and protein levels of GSK3α and CREB were measured using immunoblotting. Representative data of three independent blood donors are shown. P-values for cumulative data were calculated by paired Student's t-test. P < 0·05 for differences in Control RNAi versus GSK3α RNAi for IL-10 induction of DKFZp586O0118, ARNT2 and Caspase 5 (n = 3).
IL-10-mediated suppression of a subset of inflammatory genes is partially dependent on PI3K signalling
A key biological activity of IL-10 is to suppress the expression of pro-inflammatory genes.1,3 Accordingly, we examined the role of PI3K signalling in IL-10-mediated suppression of lipopolysaccharide (LPS) -induced inflammatory genes. As expected, IL-10 inhibited expression of the inflammatory genes for cyclo-oxygenase-2 (COX2), IL-1, IL-8 and tumour necrosis factor-α (TNF-α) induced by the TLR4 ligand LPS (Fig. 8, compare columns 3 and 4). Interleukin-10-mediated suppression of LPS-induced COX2 expression was essentially completely abrogated when PI3K (Fig. 8a, top left panel, compare columns 3–4 to 7–8) or Akt1/2 was inhibited (see Supporting information, Fig. S5). In addition, inhibition of PI3K partially reversed IL-10-mediated suppression of IL-8 and IL-1 expression (Fig. 8a). These results were confirmed by measuring IL-1β protein concentrations in culture supernatants. The IL-10 inhibited IL-1β production by 85% in control macrophages and 48% when PI3K was inhibited (Fig. 8b and see Supporting information, Fig. S6, P < 0·04). Similar observations were made when IL-1 was used instead of LPS to stimulate human macrophages: IL-10 more weakly suppressed IL-1-induced inflammatory gene expression in the presence of PI3K inhibitors (see Supporting information, Fig. S7). In contrast to the results with COX2, IL-8 and IL-1, suppression of TNF-α expression by IL-10 remained intact when PI3K was inhibited (Fig. 8a), showing that inhibition of PI3K did not globally abrogate IL-10 suppressive function. In summary, decreased expression of a subset of IL-10 target genes when PI3K signalling is inhibited correlated with impaired ability of IL-10 to suppress the expression of a subset of inflammatory genes. These results suggest that PI3K signalling selectively modulates IL-10 anti-inflammatory function.
Figure 8.

Inhibition of phosphatidylinositol 3-kinase (PI3K) attenuates interleukin-10 (IL-10)-mediated suppression of lipopolysaccharide (LPS)-induced cyclo-oxygenase 2 (COX2), IL-1β and IL-8 expression. Cells were pretreated with LY294002 (20 μm) for 1 hr, stimulated with IL-10 (20 ng/ml) for 30 min, and then LPS (1 ng/ml) was added for 3 hr. (a) Amounts of mRNA were measured using quantitative real-time PCR and normalized relative to GAPDH. Representative data of at least three independent blood donors are shown (mean ± SD of triplicate determinations). P-values for cumulative data were calculated by paired Student's t-test. P < 0·05 for differences in IL-10 + LPS versus LY294002 + IL-10 + LPS for COX2, IL-8 and IL-1 genes (n = 3). (b) IL-1β concentrations in collected supernatants were measured by ELISA after 5 hr of incubation of cells with LPS (2 ng/ml). Results obtained with one blood donor are shown; results obtained with an additional three donors are shown in Supporting information, Fig. S6.
Discussion
Similar to most signalling pathways, PI3K-mediated pathways can either augment or suppress inflammatory and immune responses depending on context.7,9 A suppressive role for the PI3K-Akt-GSK3 signalling module in TLR-induced macrophage and DC activation and cytokine production has been established, in part mediated by induction of IL-10 production.12,13 In the current study we present pharmacological and genetic evidence using primary human macrophages that PI3K-Akt-GSK3 signalling augments IL-10 responses by enhancing expression of a subset of IL-10-inducible genes and contributing to IL-10-mediated suppression of certain inflammatory genes, including COX2, IL-1β and IL-8. Hence, the PI3K-Akt-GSK3 pathway modulates both IL-10 production and macrophage responses to IL-10. These findings identify the enhancement of IL-10 responses as a new mechanism by which PI3K-mediated signalling can suppress inflammatory responses and highlight the interconnections between PI3K pathways and IL-10. Modulation of PI3K signalling represents an opportunity to fine tune and selectively regulate a subset of IL-10 responses in macrophages.
Basal PI3K signalling in macrophages is maintained by growth and survival factors such as M-CSF, to which macrophages are continuously exposed in vivo.24 Interestingly, the suppressive properties of M-CSF have been described, for example generation of suppressive tumour-associated macrophages,25 and our findings suggest that increased responsiveness to IL-10 is one mechanism by which M-CSF may promote a suppressive environment. In addition, PI3K-Akt activity can be acutely boosted (and GSK3 activity further suppressed) as part of feedback inhibition by inflammatory receptors such as TLRs,9,12,13 and by IL-4, IL-13 and IL-10 itself.19,26,27 Interleukin-4 and IL-13 promote generation of alternatively activated macrophages, one of whose functions is to regulate inflammatory responses.26,27 The PI3K signalling can contribute to differentiation of M2 alternatively activated macrophages,28 which are highly responsive to IL-10. In contrast, IFN-γ that activates macrophages and induces inflammatory M1 differentiation suppresses PI3K-Akt signalling and augments GSK3 activity.11,12,16 Interferon-γ selectively suppresses expression of a subset of IL-10-inducible genes and attenuates their anti-inflammatory activity without affecting STAT3 phosphorylation.14 These previous findings can potentially be explained by IFN-γ-mediated increases in GSK3 activity that selectively suppress a subset of IL-10-indcible genes, as described in the current study. Overall, inputs from multiple cytokines important for macrophage function will determine the level of PI3K-Akt signalling and GSK3 activity in macrophages and will indirectly affect IL-10 responses (Fig. 9).
Figure 9.

Phosphatidylinositol 3-kinase (PI3K)-Akt-glycogen synthase kinase 3 (GSK3) signalling is required for expression of a subset of interleukin-10 (IL-10) inducible genes. Effective induction of certain genes by IL-10 is dependent on PI3K-mediated signalling in human macrophages, which is activated by various cytokines and growth factors.
Our results suggest a model where the level of PI3K-Akt signalling generated in response to various environmental cues in turn modulates macrophage responses to IL-10 (Fig. 9). Importantly, the dependence of IL-10 target gene induction on PI3K signalling is gene-specific, which is consistent with the modest effects of inhibition of PI3K on proximal IL-10 signalling and STAT3 activation and promoter recruitment. A subset of IL-10 target genes that includes SOCS3 is induced independently of the status of PI3K-Akt signalling (Fig. 9, right, termed PI3K-independent genes). In contrast, the majority of highly IL-10-inducible genes analysed in this study require input from PI3K signalling for full activation (Fig. 9, left, termed PI3K-dependent genes). Our findings show that PI3K signalling augments IL-10-inducible gene expression at least in part by reversing the repressive effects of GSK3 (Fig. 9). However, it is possible that PI3K-Akt signalling also induces expression of positive regulators of IL-10-inducible genes that cooperate with IL-10-induced signals (Fig. 9, dashed line). It is tempting to speculate that some of the PI3K-dependent genes contribute to IL-10-mediated inhibition of COX2, IL-8 and IL-1β, which was also dependent on PI3K signalling. Identification of specific IL-10-inducible genes that mediate the suppression of inflammatory gene expression has represented a major challenge in understanding mechanisms of IL-10 action, and analysis of the functions of IL-10-inducible PI3K-dependent genes will be the subject of future work.
One previous report documented activation of PI3K by IL-10 in primary monocytes using in vitro lipid kinase assays and measuring phosphorylation of p70 S6 kinase; however, phosphorylation of PI3K or other substrates was not detected and phosphorylation of Akt or GSK3 was not measured.19 We did not observe IL-10-induced changes in Akt or GSK3 phosphorylation over baseline. The differences between the two studies are most likely explained by difficulty in measuring weak signals in Akt or GSK3 phosphorylation over baseline, although it is possible that the increase in lipid kinase activity that was detected19 did not translate into activation of downstream signalling via Akt. Although IL-10 can contribute to the level of PI3K-Akt signalling and regulate GSK3 activity in macrophages, our results suggest that this may represent a relatively minor contribution. A potentially more significant issue is the conclusion by the same group that PI3K does not play a role in the anti-inflammatory effects of IL-10.19,29 This conclusion was reached based solely on the analysis of regulation of TNF and TNF receptor expression. Our findings are concordant with these previous reports in that we also found that PI3K signalling did not affect IL-10-mediated regulation of TNF expression. In the current study, we undertook a more extensive analysis that took advantage of previous microarray data identifying highly IL-10-inducible genes and also examined COX2, IL-1 and IL-8 expression and found a selective and gene-specific role of PI3K signalling in IL-10 responses in human macrophages.
In our system, inhibition of PI3K only modestly affected IL-10-induced STAT3 tyrosine phosphorylation and recruitment to the Autotaxin promoter. Although this modest inhibition was insufficient to explain the dramatic changes in gene induction that we observed, in other contexts PI3K-Akt signalling and GSK3 can more substantially modulate Jak-STAT signalling. Mammalian target of rapamycin (mTOR), another downstream substrate of the PI3K-Akt pathway, was identified as a positive regulator of STAT3 signalling.30,31 The inhibitor of mTOR, rapamycin, suppressed STAT3 phosphorylation at late time points (3–6 hr) after LPS stimulation.30,31 However, this effect appears to be caused not by direct inhibition of STAT3 activation, but results from inhibition of IL-10 expression by rapamycin,30 or inhibition of a type I IFN-mediated autocrine loop that supports IL-10 expression and STAT3 activity.31 Rapamycin did not inhibit direct activation of gene expression by IL-10 in our system (T. Antoniv, unpublished data). In addition, interactions between GSK3 and STAT3 in astrocytes and microglia have been described.32 Overall, the findings suggest that PI3K-Akt-GSK3 signalling regulates IL-10 responses by several mechanisms, including mTOR-dependent regulation of IL-10 production,30,31 GSK3-mediated regulation of IL-10 production,12,13 GSK3–STAT interactions,11,32 and, as described in this study, GSK3-mediated modulation of IL-10-induced gene expression.
In conclusion, our findings expand the regulatory role of the PI3K-Akt-GSK3 pathway in anti-inflammatory responses by showing that this pathway regulates IL-10-induced gene expression and the ability of IL-10 to suppress a subset of inflammatory genes. Understanding of molecular mechanisms that modulate IL-10-induced gene expression may serve as the basis for novel therapeutic approaches to selectively target subsets of the IL-10 response and hence fine tune IL-10-mediated anti-inflammatory function.
Acknowledgments
We thank K. H. Parkmin and X. Hu for reading the manuscript. This work was supported by grants from the National Institutes of Health (to L.B.I.).
Glossary
Abbreviations
- ChIP
chromatin immunoprecipitation assay
- COX2
cyclo-oxygenase 2
- CREB
cAMP response element-binding protein
- GFP
green fluorescent protein
- GSK3
glycogen synthase kinase-3
- IFN
interferon
- IL-10
interleukin-10
- LPS
lipopolysaccharide
- M-CSF
macrophage colony-stimulating factor
- mTOR
mammalian target of rapamycin
- PI3K
phosphatidylinositol 3-kinase
- RNAi
RNA interference
- STAT
signal transducer and activator of transcription
- TLR
Toll-like receptor
- TNF
tumour necrosis factor
Disclosures
The authors declare no financial or commercial conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Inhibitor of phosphatidylinositol 3-kinase (PI3K) pathway selectively suppresses interleukin-10 (IL-10)-induced gene expression.
Figure S2. Concentration gradient of LY294002.
Figure S3. Phosphorylation of signal transducer and activator of transcription 3 (STAT3) by interleukin-10 (IL-10) was modestly affected by pre-treatment of macrophages with LY294002 in some donors.
Figure S4. Inhibitors of glycogen synthase kinase 3 (GSK3) and Akt have opposite effects on induction of interleukin-10 (IL-10)-dependent genes.
Figure S5. Inhibition of Akt1/2 attenuates interleukin-10 (IL-10)-mediated suppression of lipopolysaccharide (LPS)-induced cyclo-oxygenase 2 (COX2).
Figure S6. Suppression of interleukin-1β (IL-1β) expression by IL-10 was attenuated in human macrophages following phosphatidylinositol 3-kinase (PI3K) inhibition.
Figure S7. The capacity of interleukin-10 (IL-10) to suppress IL-1β-induced gene expression was attenuated in macrophages pre-treated with phosphatidylinositol 3-kinase (PI3K) inhibitor.
Table S1. Primers used for real-time quantitative PCR.
Table S2. Most highly induced genes after 3 hr stimulation of macrophages by interleukin-10.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than about missing material) should be directed to the corresponding author for the article.
References
- 1.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 2.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 3.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–9. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 4.Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172:567–76. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- 5.El Kasmi KC, Smith AM, Williams L, et al. Cutting edge: a transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215–9. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- 6.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–63. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 7.Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol. 2004;22:563–98. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- 8.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–63. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 10.Stumpo R, Kauer M, Martin S, Kolb H. IL-10 induces gene expression in macrophages: partial overlap with IL-5 but not with IL-4 induced genes. Cytokine. 2003;24:46–56. doi: 10.1016/s1043-4666(03)00270-9. [DOI] [PubMed] [Google Scholar]
- 11.Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3) Trends Immunol. 2009;31:24–31. doi: 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–74. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 13.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–84. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herrero C, Hu X, Li WP, Samuels S, Sharif MN, Kotenko S, Ivashkiv LB. Reprogramming of IL-10 activity and signaling by IFN-γ. J Immunol. 2003;171:5034–41. doi: 10.4049/jimmunol.171.10.5034. [DOI] [PubMed] [Google Scholar]
- 15.Antoniv T, Ivashkiv LB. Dysregulation of IL-10-dependent gene expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum. 2006;54:2711–21. doi: 10.1002/art.22055. [DOI] [PubMed] [Google Scholar]
- 16.Tsai CC, Kai JI, Huang WC, et al. Glycogen synthase kinase-3β facilitates IFN-γ-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J Immunol. 2009;183:856–64. doi: 10.4049/jimmunol.0804033. [DOI] [PubMed] [Google Scholar]
- 17.Yarilina A, Park-Min KH, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9:378–87. doi: 10.1038/ni1576. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Tassiulas I, Park-Min KH, et al. ‘Tuning’ of type I interferon-induced Jak-STAT1 signaling by calcium-dependent kinases in macrophages. Nat Immunol. 2008;9:186–93. doi: 10.1038/ni1548. [DOI] [PubMed] [Google Scholar]
- 19.Crawley JB, Williams LM, Mander T, Brennan FM, Foxwell BM. Interleukin-10 stimulation of phosphatidylinositol 3-kinase and p70 S6 kinase is required for the proliferative but not the antiinflammatory effects of the cytokine. J Biol Chem. 1996;271:16357–62. doi: 10.1074/jbc.271.27.16357. [DOI] [PubMed] [Google Scholar]
- 20.Antoniv TT, Park-Min KH, Ivashkiv LB. Kinetics of IL-10-induced gene expression in human macrophages. Immunobiology. 2005;210:87–95. doi: 10.1016/j.imbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Woodgett JR, Ohashi PS. GSK3: an in-Toll-erant protein kinase? Nat Immunol. 2005;6:751–2. doi: 10.1038/ni0805-751. [DOI] [PubMed] [Google Scholar]
- 22.Andjelkovic M, Alessi DR, Meier R, et al. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–24. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 23.Force T, Woodgett JR. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J Biol Chem. 2009;284:9643–7. doi: 10.1074/jbc.R800077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14:628–38. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 25.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–66. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 27.Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev. 2008;226:205–18. doi: 10.1111/j.1600-065X.2008.00706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rauh MJ, Ho V, Pereira C, et al. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–74. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Williams L, Lali F, Clarke C, Brennan F, Foxwell B. Interleukin 10 modulation of tumour necrosis factor receptors requires tyrosine kinases but not the PI 3-kinase/p70 S6 kinase pathway. Cytokine. 2000;12:934–43. doi: 10.1006/cyto.1999.0673. [DOI] [PubMed] [Google Scholar]
- 30.Ohtani M, Nagai S, Kondo S, et al. Mammalian target of rapamycin and glycogen synthase kinase 3 differentially regulate lipopolysaccharide-induced interleukin-12 production in dendritic cells. Blood. 2008;112:635–43. doi: 10.1182/blood-2008-02-137430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weichhart T, Costantino G, Poglitsch M, et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–77. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Beurel E, Jope RS. Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem. 2008;283 doi: 10.1074/jbc.M802481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.