

Abstract
Histidine decarboxylase (HDC), the unique enzyme responsible for histamine generation, is highly expressed in myeloid cells but its function is poorly understood. Here, we show that Hdc knockout mice exhibit a markedly increased rate of colon and skin carcinogenesis. Using Hdc-EGFP BAC transgenic mice, we demonstrate that Hdc is expressed primarily in CD11b+Ly6G+ immature myeloid cells (IMCs) that are recruited early on in chemical carcinogenesis. Transplant of Hdc-deficient bone marrow to wildtype recipients results in increased CD11b+Ly6G+ cell mobilization and reproduces the cancer susceptibility phenotype. In addition, IMCs from Hdc knockout mice promote the growth of cancer xenografts and colon cancer cells downregulate Hdc expression through promoter hypermethylation and inhibits myeloid cell maturation. Exogenous histamine induces the differentiation of IMCs and suppresses their ability to support the growth of xenografts. These data indicate key roles for Hdc and histamine in myeloid cell differentiation, and CD11b+Ly6G+ IMCs in early cancer development.
Keywords: histidine decarboxylase (HDC), histamine, immature myeloid cells (IMCs), tumor-associated neutrophils (TANs), inflammation, carcinogenesis, receptor for advanced glycation end product (RAGE)
Accumulating evidence suggests that myeloid cells are a major component of the tumor mass and play a pivotal role in promoting tumor progression 1–4. In the bone marrow, CD11b+Gr-1+ immature myeloid cells (IMCs) are thought to represent myeloid precursor cells5. However, the myeloid lineage includes both monocytic cells, such as tumor-associated macrophages (TAMs)4, as well as granulocytic cells. It is also increasingly recognized that tumorigenesis in mice is associated with the recruitment of CD11b+Ly6G+ tumor-associated neutrophils (TANs) that consist of two polarized phenotypes: N1 TANs that have hypersegmented or lobulated nuclei and are cytotoxic to tumor cells, and N2 TANs that exhibit circular or ovoid nuclei similar to immature neutrophils of bone marrow and are protumorigenic 6,7. N2 TANs support tumor growth by producing nitric oxide, angiogenic factors and matrix-degrading enzymes 8. TGF-β appears to promote N2 polarization6, and these cells then contribute to suppression of antitumor immune responses 9 and promotion of metastatic behavior 10.
Most TANs are CD11b+Ly6G+, and thus are classified as a subset of CD11b+Gr-1+ cells, which also include myeloid-derived suppressor cells (MDSCs), a heterogeneous population of myeloid cells in mice that are excessively produced in cancer. MDSCs can be detected in the circulation in cancer patients and accumulate in larger numbers in the spleen of cancer-bearing mice 11–13. MDSCs include both monocytic (Ly6G−Ly6Chigh) and granulocytic (Ly6G+Ly6Clow) cells 14, with the latter sharing cell surface markers similar to TANs. However, MDSCs are defined functionally by their ability to suppress T cell responses 15, and recent studies have suggested that the functional MDSCs are CD11b+CD49d+ and monocytic (CD11b+Ly6C+)16. CD11b+Gr-1+ immature myeloid cells can be detected in the bone marrow and occasionally in the spleen of cancer-free animals11, raising a number of questions regarding the origins of TANs and MDSCs, and the role of IMCs in early stages of tumorigenesis6,14.
Histamine is a biogenic amine that has well defined roles in allergic responses and gastric acid secretion, and has also been linked to the modulation of immune responses. For example, histamine has been shown to regulate T cells, enhancing Th1-type responses through the H1-receptor and downregulating both Th1 and Th2 type responses through the H2-receptor 17. Furthermore, histamine is believed to play an immune regulatory function in myeloid cells 18,19,20. Histamine can be taken up in the diet, but endogenous histamine is generated through conversion of L-histidine to histamine by the action of a unique enzyme, histidine decarboxylase (HDC). Although mast cells are known to store histamine, recent studies have suggested that other types of myeloid cells may be important sources of histamine production 21,22. In mouse models of atherosclerosis, Hdc-expressing myeloid cells are primarily bone marrow derived and appear more like monocytic precursors rather than mature macrophages 23. Knockout of the Hdc gene has been reported24,25,26, but little is known regarding the role of histamine in carcinogenesis. Although patients with atopic allergy and excessive histamine release are reported to have a reduced incidence of cancer, the mechanisms remain unclear27,28. Here, we report Hdc is primarily expressed in CD11b+Ly6G+ IMCs within the bone marrow, where it promotes myeloid cell differentiation and suppresses cancer formation.
RESULTS
Hdc−/− mice exhibit susceptibility to colon and skin carcinogenesis
Histamine–deficient Hdc knockout (Hdc−/−) and wildtype mice were injected intraperitoneally with one dose of a genotoxic colonic carcinogen, azoxymethane (AOM), followed by 10 days of a non-genotoxic carcinogen, dextran sodium sulfate (DSS) oral exposure29. While both sets of mice developed colonic tumors, Hdc−/− mice exhibited significantly greater colorectal tumor number and size compared to wildtype mice (Fig. 1a, b). Pathological analysis revealed that wildtype mice exhibited low-grade dysplasia and tubular adenoma with scattered goblet cells, whereas Hdc−/− mice showed high-grade dysplasia, intramucosal carcinoma and invasive adenocarcinoma with rare goblet cells (Fig. 1c and Supplementary Fig. S1a), along with a greater degree of inflammatory infiltrate and a significant increase in proinflammatory cytokines IL-1α, IL-1β, and IL-6 (Supplementary Fig. S1b, c). Tumors from the Hdc−/− mice showed abundant nuclear staining for beta-catenin, which was less often present in tumors from wildtype mice (Supplementary Fig. S1f).
Figure 1. Histamine-deficient Hdc−/− mice are highly susceptible to colorectal and skin carcinogenesis.
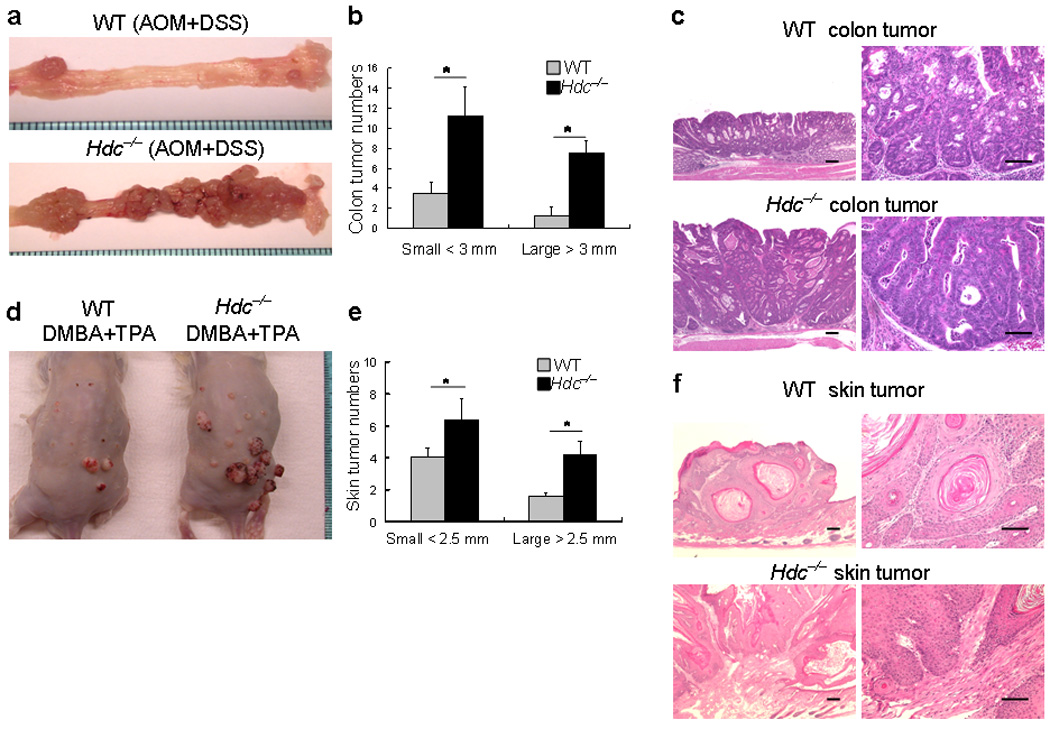
(a) Macroscopic appearance of colorectal tumors from wildtype and Hdc−/− mice. Hdc−/− mice and wildtype mice (both male Balb/c background) were injected with a single dose of AOM followed by DSS in the drinking water for 10 days. Fifteen weeks post DSS treatment, more large colonic tumors were observed in the distal colon and rectum of Hdc−/− mice.
(b) The number of large (diameter > 3mm) and small (diameter < 3mm) colorectal tumors in Hdc−/− mice versus wildtype mice (* P < 0.05; mean ± s.d. n =8, each group).
(c) Representative haematoxylin-eosin stained colonic tumor sections from wildtype and Hdc−/− mice (Scale bar, 50 µm).
(d) Macroscopic appearance of the skin tumors from wildtype and Hdc−/− mice. Wildtype and Hdc−/− mice were treated with the tumor initiator DMBA and then treated twice weekly with the tumor promoter TPA. Four months following exposure to DMBA + TPA treatment, a large number of visibly apparent papillomas were seen on the dorsum of Hdc−/− mice.
(e) The number of skin papillomas in Hdc−/− mice versus wildtype mice, including both large (diameter > 2.5mm) and small (diameter < 2.5mm) papillomas (* P < 0.05; mean ± s.d. n = 5, each group).
(f) Representative haematoxylin-eosin stained skin tumor sections from wildtype and Hdc−/− mice (Scale bar, 50 µm).
To extend these observations, we employed the 2-stage inflammation-associated skin carcinogenesis protocol using 9, 10-dimethyl-1, 2-benzanthracene (DMBA) and 12-O-tetradecanoylpherorbol 13-acetate (TPA). Hdc−/− mice first developed microscopic skin papillomas 10 weeks after TPA promotion, followed by macroscopic skin tumors 4 months later. The number, size and rate of the skin tumors in Hdc−/− mice were significantly greater than in wildtype mice (Fig. 1d, e). Histological analysis revealed that most of lesions were predominantly composed of benign papillomas; however, some of the skin tumors in Hdc−/− mice progressed to cancer (Fig. 1f). There was a greater number of infiltrating inflammatory cells and increased expression of proinflammatory cytokines IL-1α and IL-6 in Hdc−/− animals (Supplementary Fig. S1d, e). Taken together, these carcinogenesis studies suggest that Hdc and histamine likely play a protective role in inflammation-associated cancer.
Hdc is expressed primarily in CD11b+Ly6G+ immature myeloid cells
To determine the source of Hdc responsible for the protection against cancer, we generated an Hdc-EGFP BAC transgenic reporter mouse using a previously published EGFP-containing cassette30 (Supplementary Fig. S2a). EGFP fluorescence was observed in a variety of adult tissues, consistent with previous reports of endogenous Hdc expression 31–33. In the gastrointestinal tract, EGFP fluorescence was detectable in enteroendocrine cells at the base of the gastric oxyntic glands, while rare EGFP+ enteroendocrine cells were apparent in the small intestine and colon, confirmed by staining for E-cadherin and chromogranin A (Supplementary Fig. S3a). Scattered EGFP+ cells were seen in the dermis of the skin and connective tissue layer of the lung, and numerous EGFP+ cells were distributed in the red pulp of the spleen. Consistent with earlier literature, marked EGFP fluorescence was found in the ventral hypothalamus (Supplementary Fig. S2b).
Although for many years mast cells were postulated to be the major histamine-releasing cells in the intestine and other organs, immunostaining for tryptase confirmed that the majority of EGFP+ cells in the intestine, peritoneum and bone marrow were not mast cells (Supplementary Fig. S3c–f). Immunostaining also confirmed that the vast majority of c-Kit+ mast cells in the dermis of TPA treated Hdc-EGFP mice, and in the colonic tumor stroma of AOM + DSS treated Hdc-EGFP mice, showed no co-localization with EGFP+ cells (Supplementary Fig. S3g, h). Furthermore, FACS analysis demonstrated suppression of Hdc-EGFP expression in c-kit+FcεR+ mast cells of the spleen, peritoneum, and IL-3 dependent bone marrow-derived mast cells (Supplementary Fig. S4a, b). Instead, the most robust site of EGFP expression was found in bone marrow cells. FACS analysis revealed that 30–40% of bone marrow cells were EGFP+ and thus Hdc-expressing (Fig. 2a). Staining with antibody to Hdc and RT-PCR analysis (Supplementary Fig. S5a, b) confirmed Hdc expression in EGFP+ cells, whereas, no detectable Hdc expression was seen in EGFP− cells. Further FACS analysis showed that the vast majority of EGFP+ cells were of myeloid lineages, as nearly all of the cells expressed CD11b (~90 %) and Gr-1 (~87%), while they were generally negative for CD3, CD19, CD34, and Flk-1 (Supplementary Fig. S5c). FACS data also indicated that EGFP+ cells in the peritoneum were CD11b+ myeloid cells, rather than c-kit+FcεR+ mast cells (Fig. 2c). Taken together, these data would suggest that CD11b+Gr-1+ IMCs are the major source of Hdc expression within the bone marrow.
Figure 2. CD11b+Ly6G+ IMCs are the predominant source of Hdc-EGFP expression in the bone marrow.
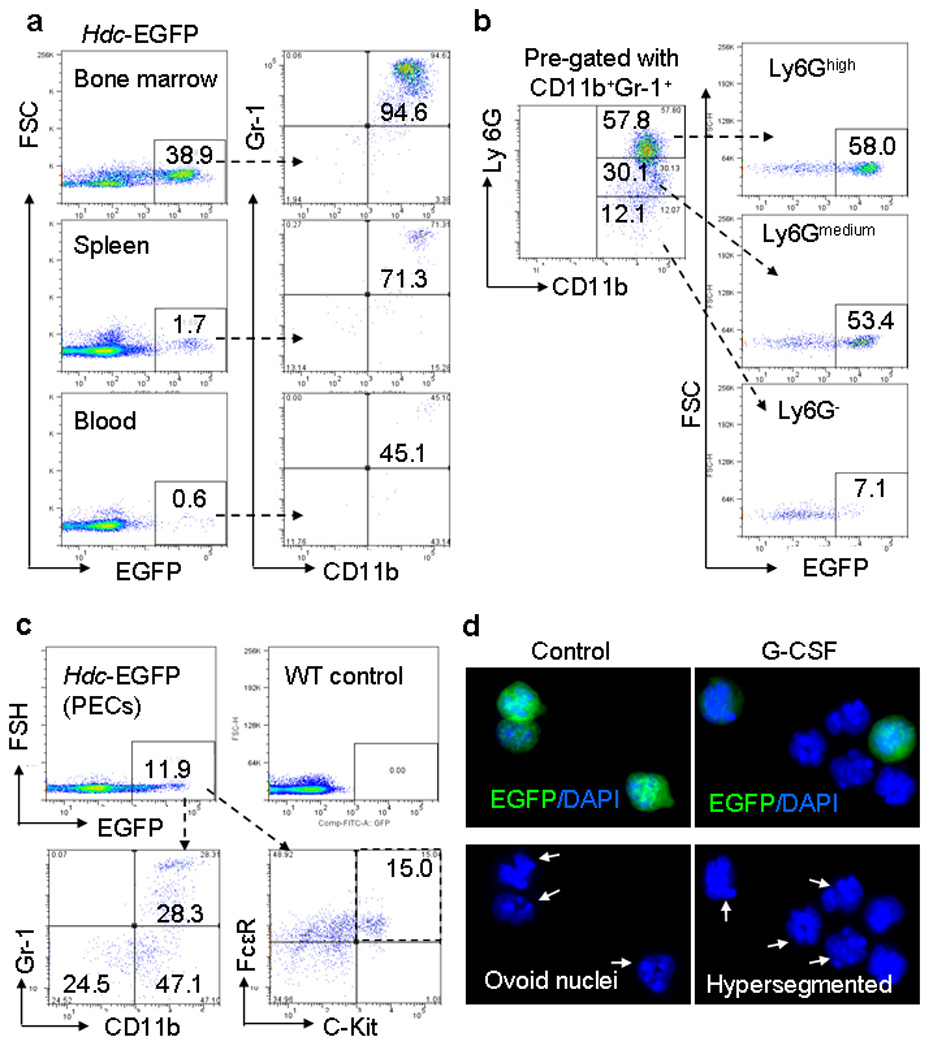
(a) The left panel shows the percentage of EGFP+ cells within the bone marrow (BM), spleen, and peripheral blood of Hdc-EGFP mice; the right panel shows the relative proportion of CD11b+ and Gr-1+ cells gated from the EGFP+ subset respectively.
(b) Expression of CD11b+Ly6G+ cells. CD11b+Gr-1+ myeloid cells were gated from the bone marrow of Hdc-EGFP mice. FACS analysis here indicates the proportion of EGFP+ cells that were CD11b+Gr-1+Ly6Ghigh, CD11b+Gr-1+Ly6Gmid, and CD11b+Gr-1+Ly6G− cells. This figure shows representative data from five experiments.
(c) Analysis of peritoneal exudative cells (PECs). Approximately 12% of EGFP+ were detected in the PEC by FACS. CD11b+Gr-1+ (28%) and CD11b+Gr-1− (47%) myeloid cells were the major EGFP+ cells. Approximately 15% of EGFP+ cells expressed mast cell markers: c-kit+FcεR+.
(d) Differentiation of splenic Hdc-EGFP+ IMCs. Immunofluorescence of DAPI stained EGFP+ CD11b+Ly6G+ IMCs treated with or without G-CSF for 48 hours showed suppression of EGFP expression with granulocyte differentiation (The pictures show representative data from three independent experiments).
CD11b+Gr-1+ IMCs comprise both the Ly6G+ granulocytic and Ly6C+ monocytic subsets. Using an antibody to Ly6G, we demonstrated that EGFP is highly expressed in ~60% of CD11b+Ly6Ghigh and ~50% of CD11b+Ly6Gmid subsets than in CD11b+Ly6G− subset (~10%) (Fig. 2b). Thus, Hdc-EGFP is expressed primarily in CD11b+Ly6G+ myeloid subset that by cell surface markers shows some similarity to CD11b+Ly6G+ tumor-associated neutrophils (TANs)6. In addition, we confirmed that Hdc-EGFP was mainly expressed in CD11b+CD49d− cells (Supplementary Fig. S6a–c), consistent with an immature, granulocytic phenotype previously shown to lack the ability to suppress T cell proliferation16. Interestingly, we found EGFP expression markedly decreased in differentiated granulocytes (neutrophils) derived from EGFP+ CD11b+Ly6G+ cells treated with granulocyte colony-stimulating factor (G-CSF) (Fig. 2d). Similarly, incubation of either EGFP+ or EGFP− CD11b+Gr-1+ IMCs with granulocyte-macrophage colony-stimulating factor (GM-CSF) resulted in differentiation into monocytes, and in the EGFP+ IMCs this resulted in repression of EGFP expression (Supplementary Fig. S7a). Real-time RT-PCR result confirmed that Hdc expression was significantly decreased in the differentiated monocytes/macrophages (Supplementary Fig. S7b). Taken together, the Hdc-EGFP+ myeloid lineage appears to comprise precursor cells for both the monocytic and granulocytic lineages, and expression of Hdc is suppressed during in vitro differentiation.
Histamine regulates the differentiation of CD11b+Gr-1+ IMCs
The expression of Hdc in IMCs, but not in mature granulocytes and macrophages, suggested the possibility of a role for histamine in the differentiation of CD11b+Gr-1+ myeloid subsets. Consequently, we examined the effects on myeloid subsets of histamine in Hdc−/− mice (Fig. 3a) and the irreversible Hdc inhibitor alpha-fluoromethylhistidine (α-FMH) in wildtype mice (Supplementary Fig. S8a). FACS analysis revealed that repression of Hdc function using these two approaches significantly increased the percentage of CD11b+Gr-1+ IMCs in the bone marrow, spleen and peripheral blood. Furthermore, the relative proportion of the CD11b+Ly6G+ subset was markedly increased in the peripheral blood of Hdc−/− mice, in particular in AOM + DSS induced colon tumor-bearing Hdc−/− mice (Fig. 3b). The increased proportion of granulocytic (CD11b+Ly6G+Ly6C−) and CD11b+Ly6G+Ly6C+ IMCs in the spleen and circulation of Hdc−/− mice, and the decreased proportion of CD11b+Ly6G−Ly6C+ (Fig. 3c), CD11b+Gr-1− (Supplementary Fig. S9a–c) or CD11b+CD49d+ (Supplementary Fig. S10a) monocytic cells, suggested that histamine-deficiency might inhibit the differentiation of monocytes. In addition, Hdc−/− mice also showed a reduced circulating level of mature neutrophils (CD11b−Gr-1+), suggesting histamine-deficiency might block the maturation of granulocytes (Supplementary Fig. S9a, b, d). In contrast, Complete Blood Count (CBC) of wildtype and Hdc−/− mice showed that numbers of neutrophils was increased, whereas monocytes exhibited no statistical change (Supplementary Fig. S8b–d). Thus, automated CBC analysis of neutrophils and monocytes is not able to distinguish between mature and immature myeloid cells, and the increase in neutrophils also reflects the increase in immature CD11b+Ly6G+ cells.
Figure 3. Hdc–deficiency upregulates CD11b+Gr-1+ and CD11b+Ly6G+ IMCs.
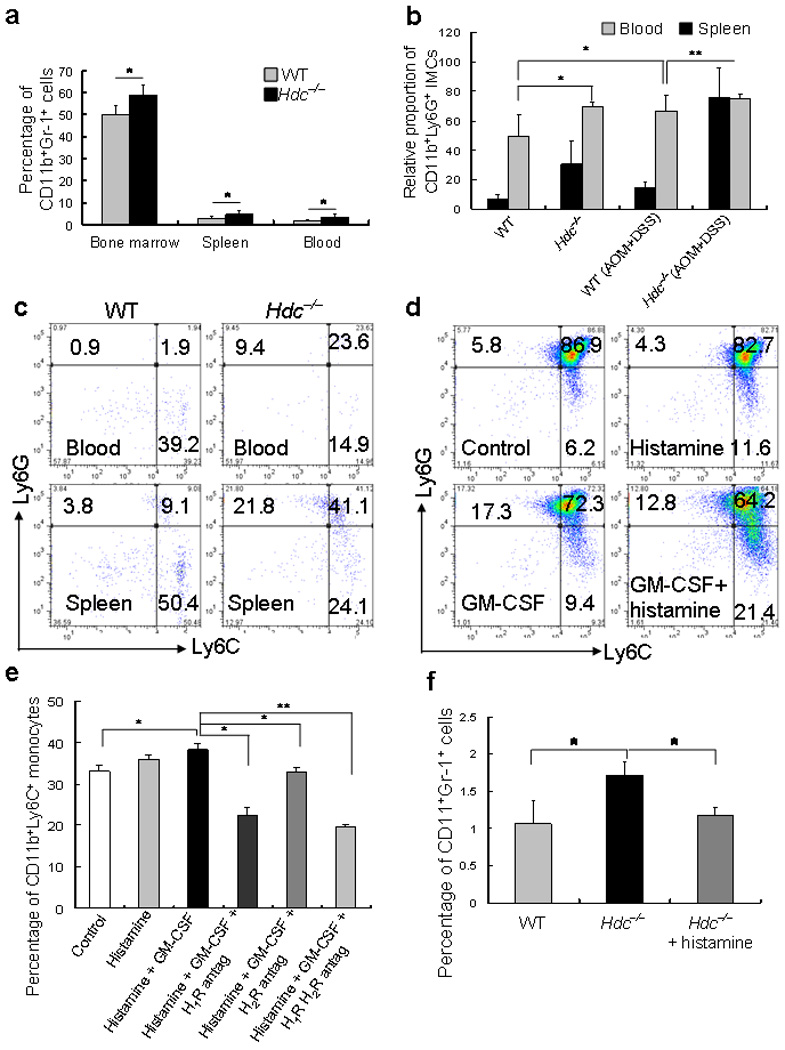
(a) The percentage of CD11b+Gr-1+ IMCs in the BM, spleen, and peripheral blood in wildtype and Hdc−/− mice were measured by FACS analysis (* P < 0.05; mean ± s.d. n = 10, each group).
(b) The relative proportion of CD11b+Ly6G+ cells in the peripheral blood and spleen of wildtype and Hdc−/− mice, with and without AOM + DSS-induced colon tumors, as measured by FACS analysis (* P < 0.05; ** P < 0.01; mean ± s.d. n = 5, each group).
(c) FACS analysis shows the abundance of the Ly6G and Ly6C subsets in CD11b+ cells in the peripheral blood (top) and spleen (bottom) of wildtype (left) and Hdc−/− (right) mice. The figure is representative of data from five mice of each group.
(d) Differentiation of IMCs. In vitro incubation of BM-derived EGFP+ CD11b+Gr-1+ IMCs with GM-CSF and histamine followed by FACS analysis for Ly6G and Ly6C surface expression. The figure is representative of data from three independent experiments.
(e) Histamine receptor blockade inhibits CD11b+Ly6C+ monocyte differentiation. CD11b+Gr-1+ IMCs sorted from the BM of Hdc−/− mice were incubated with histamine, GM-CSF, histamine and GM-CSF, histamine, GM-CSF, and H1R antagonist, histamine, GM-CSF, and H2R antagonist, or histamine, GM-CSF, H1R, and H2R antagonists, respectively. The proportion of CD11b+Ly6C+ monocytes was then assessed by FACS analysis (* P < 0.05; ** P < 0.01; Student’s t test. The figure shows the mean ± s.d. from three independent experiments).
(f) Effect of exogenous histamine on circulating CD11b+Gr-1+ IMCs in Hdc−/− mice. The percentage of CD11b+Gr-1+ IMCs by FACS in the peripheral blood of wildtype, Hdc−/−, and Hdc−/− mice treated with exogenous histamine (* P < 0.05; mean ± s.d. n = 5, each group).
To further characterize the effects of histamine, EGFP+ IMCs were isolated from the bone marrow of Hdc-EGFP mice, and analyzed by FACS following incubation with GM-CSF alone or GM-CSF plus histamine. GM-CSF incubation induced both CD11b+Ly6G+ granulocytic and CD11b+Ly6C+ monocytic differentiation, and exogenous histamine promoted the differentiation of the CD11b+Ly6C+ monocytic subset and decreased the proportion of the CD11b+Ly6G+ granulocytic subset (Fig. 3d). To address further the hypothesis that histamine regulates the maturation of CD11b+Ly6G+ granulocytes, we compared the morphologic appearance of splenic CD11b+Ly6G+ granulocytes from Hdc−/− and wildtype mice. The nuclear morphology of CD11b+Ly6G+ myeloid cells from Hdc−/− mice was characterized by an ovoid or circular nucleus (N2 phenotype), similar in some respects to TAN/N26, whereas CD11b+Ly6G+ cells from wildtype mice showed hypersegmented nucleus typical of mature neutrophils (N1 phenotype) (Supplementary Fig. S10b).
Hdc-deficiency leads to a marked reduction in histamine production and availability, and thus we postulated that within the bone marrow histamine acts in an autocrine or paracrine manner through defined histamine receptors (HRs). We examined HR expression in myeloid subsets within the bone marrow. Among the four histamine receptors (HR1–4), H1R and H2R were identified as the predominant receptors in CD11b+Gr-1+IMCs, and H1R was highly expressed in CD11b+Ly6C+ monocytic cells (Supplementary Fig. S10c). Staining with antibodies to H1R or H2R confirmed H1R and H2R protein expression in CD11b+Ly6G+ and CD11b+Ly6C+ IMCs, and also revealed increased H1R and H2R expression in weakly fluorescent EGFP+ cells (Supplementary Fig. S10d). In addition, H1R- and H2R- antagonists were both effective (although H1R more so) in inhibiting the monocytic differentiation of CD11b+Ly6C+ cells from IMCs after histamine plus GM-CSF treatment (Fig. 3e). Finally, administration of exogenous histamine in Hdc−/− mice significantly decreased circulating CD11b+Gr-1+ IMCs (Fig. 3f). Taken together, histamine promotes the differentiation of CD11b+Gr-1+ IMCs and CD11b+Ly6C+ monocytes through H1R > H2R autocrine and paracrine pathways.
Hdc+ IMCs are recruited to the inflamed tissue and colon tumor setting
To show that Hdc+ myeloid cells are recruited by carcinogenic stimuli, we examined EGFP expression in the AOM + DSS mouse model of colon cancer. Twenty weeks following AOM + DSS treatment, at a time when gross colonic tumors could be observed in most (80%) AOM + DSS treated Hdc-EGFP mice; there was marked infiltration of EGFP+ cells in the colonic adenomas compared to only rare submucosal EGFP+ cells in the colons of control mice (Fig. 4a and data not shown). In the AOM + DSS treated mice, we confirmed the presence of numerous CD11b+Gr-1+ myeloid cells in the stroma of tumors (Fig. 4a, bottom). Interestingly, at earlier time points (8 weeks) after AOM + DSS but prior to tumor development (20 weeks), there was an even greater number of Hdc-EGFP+ myeloid cells (Fig. 4b). Thus, there is a strong correlation between the early recruitment of EGFP+ IMCs and tumor development. Consistent with this notion, one or two doses of AOM resulted in no significant recruitment of EGFP+ cells to the colon and also no induction of cancer (data not shown), suggesting that the tumor promoter DSS induces cancer in part through IMC recruitment. Indeed, DSS treatment resulted in significant mobilization of CD11b+Gr-1+ IMCs in the peripheral blood within 4 days, with minimal changes in CD11b+Gr-1− monocytes or CD11b−Gr-1+ granulocytes (Supplementary Fig. S11a). These EGFP+ cells in the inflamed mucosa were positive for CD11b- and Gr-1-specific antibodies staining (Supplementary Fig. S11b).
Figure 4. EGFP+ IMCs are recruited to the inflamed tissue and increased in colonic carcinogenesis.
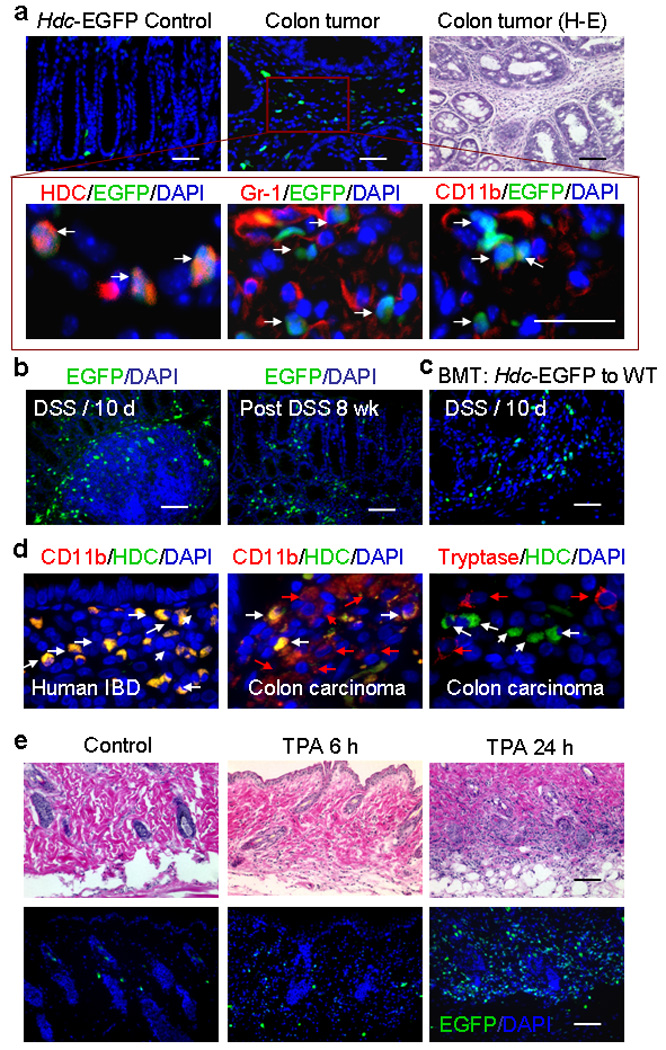
(a) Immunofluorescence and H&E sections from AOM + DSS–treated (20 weeks) and control Hdc-EGFP mice (n = 5). Top: Rare submucosal EGFP+ cells were observed in the colon of control mice, whereas, a large number of EGFP+ cells were observed in the colonic adenomas of AOM + DSS–treated mice. H&E staining of colonic tumors confirmed abundant inflammatory cells in the stroma. Bottom: Immunostaining of serial sections demonstrates that EGFP+ cells are Hdc+ and approximately 50% of CD11b+ and Gr-1+ cells are EGFP+.
(b) Left: Immunofluorescence image showing EGFP+ cells in the DAPI-stained colon of Hdc-EGFP mice treated with DSS (10 days). Right: Immunofluorescence image showing a greater number of EGFP+ cells in the DAPI-stained colon of Hdc-EGFP mice 8 weeks following AOM + DSS treatment. (i.e. compared to Fig. 4a, prior to tumor development)
(c) Immunofluorescence image of DAPI–stained colon from BM transplanted mice 10 days post DSS showing that infiltrating EGFP+ cells are BM–derived (donor: Hdc-EGFP mice; recipient: wildtype B6 mice).
(d) Immunofluorescence stained human colonic Inflammatory Bowel Disease (IBD) and colitis-associated carcinoma tissue sections showing HDC (green), CD11b (red) or tryptase (red). White arrows show HDC+ cells (which all co-localize with CD11b+ cells in IBD tissue); while red arrows show cells that express CD11b or tryptase alone. The figure is representative of data from analysis of five IBD and five cancer samples.
(e) Immunofluorescence and H&E stained sections show the presence of inflammatory cells and EGFP+ cells in the skin following a single TPA treatment. Top: H&E stained skin sections. Bottom: Immunofluorescence shows a large number of EGFP+ inflammatory cells in the dermis 24 hours following TPA treatment, compared to 6 hours following TPA treatment or acetone treated controls (n = 5).
To determine if HDC+ myeloid cells are also present in human colon cancer, we carried out an immunohistochemical study of human ulcerative colitis (UC, preneoplastic lesion) and colitis-associated colorectal cancer (CAC). Similar to our mouse models (Supplementary Fig. S11c), human UC was characterized by a marked infiltration of HDC+ cells that were largely restricted to CD11b+ cells. Furthermore, in CAC, CD11b+ cells were still extremely abundant in areas adjacent to the tumor, but showed marked downregulation of HDC and an immature morphology in tumor (Fig. 4d and Supplementary Fig. S11d). Consistent to the observation from mouse colon tumors (Supplementary Fig. S3h), HDC+ inflammatory cells in human CAC are not tryptase+ mast cells (Fig. 4d right).
It has previously been reported that a single application of TPA results in rapid recruitment of Gr-1+ inflammatory cells and increased Hdc activity in the skin 34,35. Following TPA treatment, a substantial number of Hdc-EGFP+ cells were recruited to the dermis and subcutaneous tissue, with a much greater number at 24 h compared to 6 h post treatment (Fig. 4e). FACS analysis of circulating cells in the peripheral blood confirmed that CD11b+Gr-1+ cells are increased ~3.5 fold one day following a single TPA application (data not shown). More than half of the CD45+ cells in the TPA-treated dermis expressed EGFP and the vast majority of EGFP+ cells expressed Gr-1 or CD11b (Supplementary Fig. S12a, b), but did not express tryptase (Supplementary Fig. S12c) and c-kit (Supplementary Fig. S3g). In addition, bone marrow transplant experiments showed that the EGFP+ cells that infiltrated the colon (Fig. 4c) and dermis of skin (Supplementary Fig. S12d) were bone marrow-derived.
We next examined gene expression patterns of bone marrow-derived CD11b+Gr-1+ IMCs and colon tumor associated CD11b+Gr-1+ MDSCs from wildtype and Hdc−/− mice using the Mouse Array directly after FACS sorting (Gene Expression Omnibus (GEO) database, Series Accession number is GSE23502). These studies revealed that at baseline Hdc−/− bone marrow-derived IMCs showed increased inflammatory cytokine (i.e. IL-1) and chemokine (i.e. CCR-2) expression compared to wildtype IMCs. More importantly, we found that tumor-associated MDSCs from both wildtype and Hdc−/− mice showed a much greater (5-fold) increase in the expression of IL-1 family ligands (IL-1α, IL-1β and IL-18) and IL-6 (Supplementary Table 1), along with overexpression of a number of chemokines and chemokine receptors, including CXCL-1, CXCL-2, and CCR-2 (Supplementary Table 2), and adhesion molecules like ICAM-1, VCAM-1, and CD44 (Supplementary Table 3).
IMCs from Hdc−/− mice accelerate tumor growth
In order to confirm that loss of Hdc expression specifically in bone marrow cells promotes tumor growth, we studied wildtype mice reconstituted with either wildtype or Hdc−/− bone marrow. Mice transplanted with Hdc−/− bone marrow showed greater susceptibility to carcinogenesis in both the AOM + DSS-induced colon carcinogenesis model (Fig. 5a) and the DMBA + TPA induced skin carcinogenesis model (data not shown), at a level closer to that found in Hdc−/− mice. Hdc−/− mice reconstituted with wildtype bone marrow exhibited reduced susceptibility to colon carcinogenesis compared with Hdc−/− mice (Fig. 5a).
Figure 5. Bone marrow derived IMCs from Hdc−/− mice accelerate tumor growth.
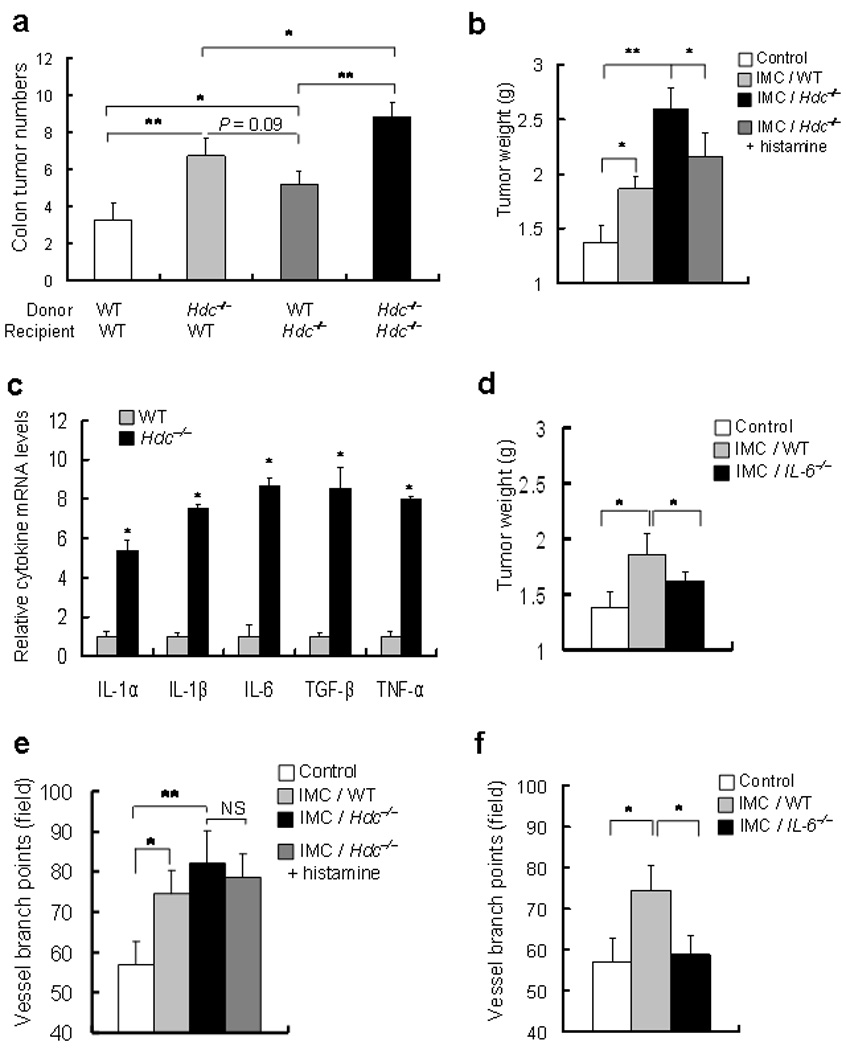
(a) Colonic tumor numbers in BM transplanted mice at 20 weeks following AOM + DSS. Wildtype and Hdc−/− mice were lethally irradiated and reconstituted with wildtype or Hdc−/− bone marrow (n = 6, * P < 0.05 compared to indicated control).
(b) Xenograft tumor weight. CT26 colon cancer cells were implanted in NOD-SCID mice alone (control) or with CD11b+Ly6G+ BM-derived IMCs from wildtype mice, or Hdc−/− mice, with or without exogenous histamine (800 µg kg−1 weight per day, I.P injection) (* P < 0.05; ** P < 0.01; mean ± s.d. n = 7, each group).
(c) Cytokine expression levels in BM-derived IMCs. Messenger RNA expression of IL-1α, IL-1β, IL-6, Tgf-β, and Tnf-α in CD11b+Ly6G+ IMCs from Hdc−/− mice compared to IMCs from wildtype mice were analyzed by Q-RT-PCR.
(d) Effect of IL-6 deficiency on induction of tumor xenograft growth by IMCs. CD11b+Ly6G+ bone marrow IMCs isolated from IL-6−/− mice exhibited suppression of CT26 cancer cells when implanted into NOD-SCID mice alone (control) or with CD11b+Ly6G+ bone marrow IMCs from wildtype or IL-6−/− mice. Tumor weight was measured (* P < 0.05; mean ± s.d. n = 7).
(e) and (f) The number of vessel branch points in xenograft tumors were counted in xenograft tumors derived from CT26 cancer cells injected with no CD11b+Ly6G+ IMCs (Control) or IMCs from wildtype mice, Hdc−/− mice, Hdc−/− cells + histamine, or IL-6−/− mice (* P < 0.05; mean ± s.d. n = 7, each group).
To demonstrate that Hdc-expressing CD11b+Gr-1+ or CD11b+Ly6G+ IMCs contribute directly to tumor growth, we used a xenograft tumor model in which mouse (CT26) colon cancer cells were injected subcutaneously into NOD-SCID mice with bone marrow-derived IMCs isolated from wildtype or Hdc−/− mice36. We found that CD11b+Ly6G+ IMCs from Hdc−/− mice more strongly promoted the growth of CT26 cancer cells compared to the IMCs of wildtype mice (Fig. 5b and Supplementary Fig. S14). CD11b+Gr-1+ IMCs from Hdc−/− mice also promoted greater growth of the xenograft tumors than wildtype CD11b+Gr-1+ IMCs (Supplementary Fig. S13a). Furthermore, we confirmed that bone marrow-derived Hdc-EGFP+ CD11b+Gr-1+ IMCs can strongly promote the growth of CT26 cancer cells (Supplementary Fig. S13b). In order to confirm that bone marrow-derived IMCs did not promote tumor growth as a result of immunosuppression, bone marrow-derived EGFP+ CD11b+Gr-1+ IMCs were incubated with splenic CD4+ T cells. In contrast to splenic CD11b+Gr-1+ MDSCs that suppressed IFN-γ from T cells, bone marrow-derived IMCs failed to have this effect (Fig.S13c). Taken together, these data indicate that bone marrow-derived CD11b+Gr-1+ and CD11b+Ly6G+ IMCs can directly promote tumor proliferation independent of effects on T cell suppression.
Indeed, the CD11b+Gr-1+ or CD11b+Ly6G+ IMCs from Hdc−/− mice showed much higher expression levels of pro-inflammatory cytokines, including an 8-fold increase in IL-6 (Fig. 5c, and Supplementary Table 1), compared to those from wildtype mice. Since previous studies have indicated an important role for IL-6 in colonic carcinogenesis 37,38, we examined CD11b+Ly6G+ IMCs from bone marrow of IL-6−/− mice. IL-6-deficiency resulted in significantly less stimulation of xenograft tumor growth compared with wildtype CD11b+Ly6G+ IMCs, indicating an important role for IL-6 in the promotion of tumor growth by IMCs (Fig. 5d). In addition, xenograft tumors that were co-implanted with the EGFP+ CD11b+Gr-1+ IMCs showed much greater neovascularization (Supplementary Fig. S13b), and the number of vessel branch points was significantly increased in xenograft tumors implanted with CD11b+Ly6G+ IMCs, an effect that was inhibited by deletion of the IL-6 gene (Fig. 5e, f and Supplementary Fig. S14, bottom). Finally, xenograft tumors with EGFP+ CD11b+Gr-1+ IMCs showed greater proliferation, and recruitment of α-SMA+ cancer-associated fibroblasts (Fig. 6a). Colonic adenomas from Hdc−/− mice also showed much greater α-SMA+ stromal cells compared to wildtype mice (Fig. 6b). Thus, these data suggest that CD11b+Gr-1+ or CD11b+Ly6G+ IMCs promote the growth of tumors in part through IL-6 expression and modulation of the tumor microenvironment.
Figure 6. Migration of circulating CD11b+Gr-1+ IMCs is RAGE-dependent and suppression of Hdc occurs through a methylation-dependent mechanism.
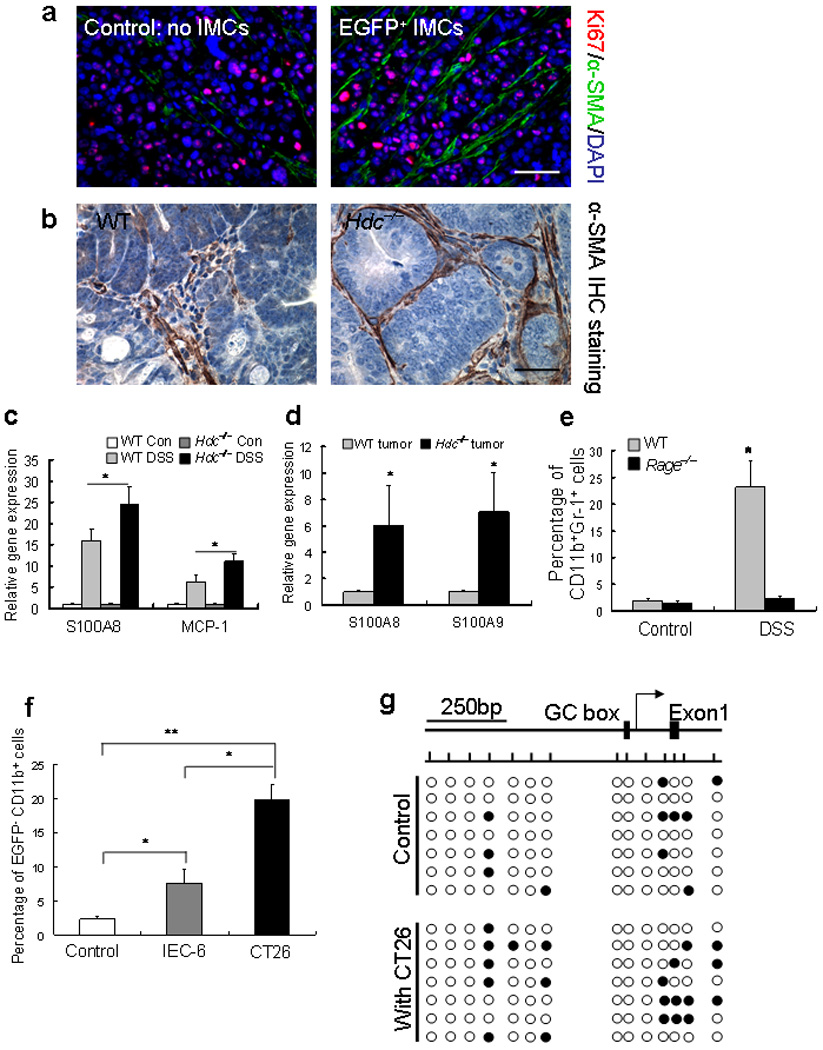
(a) Immunofluorescence staining of xenograft tumor sections with or without co-implanted EGFP+ CD11b+Gr-1+ IMCs. Increased expression of Ki67 and α-SMA in tumors that were co-implanted with EGFP+ IMCs.
(b) Colonic adenomas from Hdc−/− mice treated with AOM + DSS showed greater α-SMA+ myofibroblasts in the stroma compared to wildtype mice. (The pictures in panel a and b show representative data from five tumors in each group)
(c) Q-RT-PCR results showed a significant increase in the RAGE ligand S100A8 and MCP-1 expression in DSS–induced colitis tissue.
(d) Q-RT-PCR results showed a significant increase in the expression of RAGE ligands S100A8 and S100A9 in colonic tumors of Hdc−/− mice compared to wildtype mice treated with AOM + DSS.
(e) FACS analysis revealed a significant decrease in CD11b+Gr-1+ IMCs in the blood of Rage−/− mice following AOM + DSS treatment (* P < 0.05; mean ± s.d. n = 5, each group).
(f) CT26 cancer cells suppressed Hdc-EGFP expression in BM-derived CD11b+Ly6G+ IMCs to a greater extent than control IEC-6 rat intestinal epithelial cells (* P < 0.05; mean ± s.d. n = 3, each group).
(g) Hdc promoter methylation analysis by sodium bisulfate DNA sequencing. BM-derived Hdc-EGFP+ CD11b+Ly6G+ IMCs were co-cultured with CT26 cancer cells for 48 hours and DNA of IMCs was extracted for detection of DNA CpG methylation sites. Top: Bent arrow shows the transcription start site and boxes represent the GC box and exon 1. Short vertical lines across the horizontal line indicate CpG sites (−800 and +200 bp from the transcript start site). Bottom: Filled and open circles represent methylated and unmethylated cytosine residues respectively.
To further explore the mechanism of CD11b+Gr-1+ IMCs migration and recruitment in inflamed tissue, circulating EGFP+ CD11b+Gr-1+ IMCs were monitored in real-time using Intravital confocal microscopy. The ears of Hdc-EGFP and Hdc-EGFP/Hdc−/− mice were examined 6 h and 24 h following a single application of TPA. Six hours after TPA stimulation, a large number of EGFP+ inflammatory cells were observed in the dermis (Supplementary Fig. S15a) and ear vessels (Supplementary video), compared to rare EGFP+ cells in the circulation and tissue of control mice (data not shown).
The increased recruitment of IMCs in Hdc−/− mice could be related in part to the increased number of circulating cells or to increased migration in response to the incipient tumor site. Using immunofluorescence staining and FACS we found that when the same number of bone marrow-derived Hdc-EGFP+ CD11b+Gr-1+ IMCs from Hdc-EGFP or Hdc-EGFP/Hdc−/− mice were injected by tail vein into CT26 colon tumor-bearing NOD-SCID mice, approximately three times more tumor-infiltrating EGFP+CD11b+ cells, CD11b+Gr-1+ IMCs, and F4/80+ macrophages from Hdc−/− mice migrated to the tumor sites versus from wildtype mice (Supplementary Fig. S15b–d). Recently, a number of ligands for RAGE (receptor for advanced glycation end production) have been indicated in monocyte and macrophage recruitment39. To examine the expression of RAGE ligands in DSS-induced colitis, q-RT-PCR was performed and we detected a significant increase in the expression of the RAGE ligand S100A8 in colitis tissue (Fig. 6c) and tumors (Fig. 6d). When AOM + DSS treatment was applied to Rage−/− mice, we observed a significant reduction in circulating CD11b+Gr-1+ IMCs compared to wildtype mice (Fig. 6e). These data suggest that Hdc-deficiency increases the activation and recruitment of CD11b+Gr-1+ or CD11b+Ly6G+ IMCs in response to carcinogenic stimuli, and that bone marrow-derived IMCs constitute the majority of recruited EGFP+ inflammatory cells in carcinogenesis.
Given the downregulation of Hdc expression in colitis-associated cancer, we reasoned that cancer cells might be selectively modulating myeloid cells through repression of Hdc. First, we confirmed that when CD11b+Ly6G+ or CD11b+Ly6C+ IMCs isolated from Hdc-EGFP mice were co-cultured under differentiating conditions in vitro with CT26 cancer cells, there was greater downregulation of Hdc-EGFP expression (Supplementary Fig. S16a, b) along with a less differentiated nuclear morphology (ovoid or circular nuclei) (Supplementary Fig. S16c). This appeared to be cancer specific, since CT26 cancer cell suppressed Hdc-EGFP expression in IMCs to a much greater extent compared to IEC-6 nontransformed intestinal epithelial cells (Fig. 6f and Supplementary Fig. S16d). Furthermore, the addition of exogenous histamine to IMCs co-cultured in vitro with CT26 cancer cells was able to promote the differentiation of these cells (Supplementary Fig. S16c). This was confirmed through in vivo studies, where exogenous histamine given by i.p. injection inhibited the promotion of xenograft tumor growth by CD11b+Ly6G+ IMCs from Hdc−/− mice (Fig. 5b). We demonstrated that Hdc was also downregulated in our xenograft models, by decreased EGFP expression in the Hdc-EGFP+ CD11b+Gr-1+ IMCs two days following implantation with CT26 cancer cells in NOD-SCID mice. In contrast, CD11b+Gr-1+ bone marrow-derived IMCs from UBC-EGFP mice did not show such loss of EGFP expression when co-implanted with CT26 cancer cells (Supplementary Fig. S17). Previous reports have indicated that Hdc transcription is largely regulated by promoter CpG methylation40 41, and we found increased DNA CpG methylation sites in the Hdc promoter region of Hdc-EGFP+ CD11b+Ly6G+ cells following 48 hrs co-culture with CT26 cancer cells (Fig. 6f). In addition, microarray studies indicated the upregulation of a number of DNA methylation genes (e.g. Dnmt 3) in tumor-associated MDSCs compared to bone marrow-derived IMCs, supporting an induction of DNA methylation in IMCs by colonic tumors (Supplementary Table 4). Overall, these data are consistent with the notion that cancer cells down-regulate Hdc expression to prevent maturation of myeloid cells, and thus promote tumor growth.
DISCUSSION
We show here that the Hdc gene is expressed primarily in bone marrow CD11b+Gr-1+ IMCs, and that these IMCs contribute to the initiation and promotion of cancer. Histamine appears to be involved in the normal maturation and differentiation of myeloid cells, and a deficiency in Hdc leads to abnormal myeloid differentiation and the promotion of colonic and skin carcinogenesis. Cancers appear to mimic locally the Hdc knockout phenotype by downregulating Hdc expression in nearby myeloid cells through the induction of Hdc promoter hypermethylation, thereby inhibiting further myeloid differentiation. The increased carcinogenesis observed in the Hdc−/− mouse was due primarily to Hdc-deficiency in the bone marrow cells, and was associated with increased recruitment of IMCs to the cancer sites in a RAGE-dependent manner39. The induction of cancer by IMCs was mediated through an IL-6-dependent mechanism, since deletion of IL-6 in the IMCs resulted in inhibition of tumor growth and angiogenesis.
Previously, mast cells were considered the primary source of Hdc since they are known to store and release histamine. However, our study demonstrates that Hdc is not expressed by tryptase+ or c-kit+FcεR+ tissue mast cells, but rather by CD11b+Gr-1+ IMCs that constitute 30–40% of bone marrow hematopoietic cells. Hdc expression was largely localized to the CD11b+Ly6G+ granulocytic subset within the bone marrow where it appeared to contribute to myeloid differentiation in a H1R- and H2R- dependent manner. Deletion or inhibition (via α-FMH) of Hdc resulted in marked abnormalities in myeloid differentiation and decreased circulating mature granulocytes and monocytes and increased CD11b+Ly6G+ IMCs. Although these changes were not clearly reflected in automated (coulter) counting of peripheral leukocytes, the changes were confirmed by both FACS and manual counting of peripheral smears (not shown). Thus, Hdc+ CD11b+Gr-1+ cells might function as precursors for both mature neutrophils and monocytes.
In both our mouse and human studies, we observed that most CD11b+ cells were also HDC+ in preneoplastic tissues, while there was a much lower frequency of HDC+ in CD11b+ cells in established cancers. Hypermethylation of CpG promoter sites was associated with downregulation of Hdc expression in Hdc-EGFP+ IMCs co-cultured with colon cancer cells suggesting that this may be a tumor-induced phenomenon. Cancer cells appear to epigenetically reprogram myeloid progenitors which might account for the development of TANs or even MDSCs in the setting of cancer. Indeed, our microarray data showed substantial differences in gene expression between bone marrow-derived Hdc+ CD11b+Gr-1+ IMCs and tumor-associated MDSCs, including the upregulation of methylation genes associated with epigenetic reprogramming. Thus, while bone marrow-derived Hdc+ CD11b+Gr-1+ (mainly expressing CD11b+Ly6G+) IMCs resemble superficially MDSCs with respect to surface markers, they are CD11b+CD49d− and not able to suppress T cell function, and thus are functionally distinct from MDSCs16, and more closely resemble TANs.
In early stages of skin and colonic carcinogenesis, Hdc+ IMCs are rapidly recruited to the sites of carcinogenic stimuli exposure where they contribute to tumor development. Interestingly, we show here that tumor promoting agents, such as TPA in the skin or DSS in the colon, work in a large part by mobilizing and recruiting Hdc+ IMCs. Increased cancer susceptibility in Hdc−/− mice was a general phenomenon mediated largely by Hdc-deficiency in bone marrow cells; while we could not completely exclude a role for non-bone marrow derived Hdc in our transplants, the evidence for recipient Hdc was related to the incomplete (~70%) nature of the marrow transplants. The increased cancer was due to an increase in circulating IMCs, as well as a 3-fold more rapid migration and recruitment of Hdc−/− IMCs to the cancer site in a RAGE-dependent manner. Hdc−/− IMCs also expressed increased IL-6 upon LPS stimulation, and knockout of IL-6 in bone marrow IMCs reduced their ability to promote tumor growth and angiogenesis, consistent with previous studies 37,38. Our findings also point to a direct effect of IMCs on tumor promotion as we found that in the setting of histamine-deficiency, exogenous histamine partially reversed the effects. Lack of complete reversal by exogenous histamine is consistent with the previous findings that in immature mast cells exogenous histamine is unable to completely substitute for endogenous histamine42.
Growing evidence points to an important role for CD11b+Gr-1+ cells in the cancer promotion. While cancerous tissues have for many years been known to be histamine rich, our studies indicates that histamine is derived largely from Hdc+ CD11b+Ly6G+ IMCs. In previous studies, H2R antagonists have generally not been shown to be effective in the treatment of cancer 43–45; this may have been due, in part, to their inhibitory effects on the differentiation of IMCs. However, further studies are needed to assess the impact on cancer risk of HR antagonists, particularly H1R antagonists that have not been examined46. Interestingly, patients with atopic disease, associated with increased histamine secretion, appear to be less susceptible to carcinogenesis27,28,47,48. This has been attributed to enhanced immunosurveillance, but given our findings, could also be related to histamine-dependent effects on IMCs. Recently, a genetic mutation of HDC leading to loss of function has been described in association with Tourette’s syndrome, however, the cancer susceptibility of this family has not been reported26. Overall, our results suggest that the balance of histamine within the tumor microenvironment may be important, and that CD11b+Gr-1+ IMCs represent an important therapeutic target for the control of cancer growth.
Gene Expression Omnibus (GEO) database, Series Accession number is GSE23502.
METHODS
Generation of Hdc-EGFP BAC transgenic mice
The PKD4-NICD-EGFP plasmid was used as a PCR template to amplify a fragment containing EGFP and 5′ and 3′ ends homologous to sequences flanking the mouse Hdc ATG (PCR primers: forward 5′-GGTCCCTGTGCGCCCCGCCCGGCCCGCAGGCAGCCACCTGGCGAGTCTGACATGGTGAGCAAGGGCGAGGAGCT-3′, reverse 5′-GGCCGGACGCGAACGTGGAGAAGGACGGGAGCAGAGCGTCGCTGACAGCCATTCCTCCTTAGTTCCTATTCCGA-3′, with the underlined region homologous to the sequence flanking the Hdc translational start site). The BAC clone (# RP23-474H6 from CHORI) was introduced into the bacterial strain BL250. BL250 bacteria containing the BAC RP23-474H6 were made competent and transformed with the amplified fragment by electroporation according to previously described protocols 30. Clones in which the amplified fragment was inserted via homologous recombination were selected using chloramphenicol and Kanamycin resistance cassettes. Selected colonies were screened for correct recombination by amplification using EGFP- and Hdc-specific primers. The FRT-flanked KANA gene was excised from Hdc-EGFP-KANA BAC clones by expressing the FLP recombinase. CHEF gel electrophoresis and DNA sequencing confirmed correct transgene construction and integrity of the BAC flanking sequence (data not shown). The Hdc-EGFP BAC was then purified for pronuclear microinjection. Potential founder mice (B6/CBA mixed background) were genotyped by tail DNA amplification using primers specific for the EGFP coding sequence. The founder transgenic mice were then backcrossed with wildtype C57BL/6 mice.
In some experiments, Hdc-EGFP transgenic mice were crossed to Hdc−/− mice in which exon 5 of Hdc gene was replaced with a Neomycin cassette24. All the transgenic mice were housed in micro-isolator, solid-bottomed polycarbonate cages of a pathogen free condition. All experiments were approved by the Institutional Animal Care and Use Committees of Columbia University, College of Physicians and Surgeons.
Antibodies and immunohistochemical staining
The following antibodies were used in the current studies: Hdc-specific antiserum (EURO-DIAGNOSTICA); GFP and E-Cadherin-specific antibodies (Invitrogen); antibody to Chromogranin A (ImmunoStar); antibody to tryptase (abcam); PE-conjugated lineage specific antibodies (CD3, CD19, CD45, CD49d, and Ly6G), APC-conjugated antibody to CD11b, PercP-conjugated antibodies to Ly6C and Gr-1 (Ly6G and Ly-6c) (all from BD Pharmingen). PE-conjugated antibodies CD34, F4/80, and FcεRIα, Pe-cy7-conjugated antibody to CD117 (c-kit) (all from eBioscience); H1R and H2R-specific antibodies from Novus Biologicals. Texas Red-conjugated rabbit-specific secondary antibody and Texas red-conjugated rat-specific secondary antibody (Vector Laboratories) were used for immunohistochemistry analysis. Tissue samples from Hdc-EGFP transgenic mice were fixed with 4% Paraformaldehyde for 12 hours followed by 30% sucrose overnight. EGFP fluorescence expression was directly observed using an inverted fluorescence microscope (Nikon Eclipse TE2000-U). Images were analyzed with Adobe Photoshop.
RNA extraction and quantitative RT-PCR analysis
Total RNA was extracted from tissues of Hdc-EGFP transgenic mice and control mice using Trizol (Invitrogen). High fidelity cDNA was generated from each RNA samples with Superscript III cDNA amplification System (Invitrogen). Quantitative RT-PCR reaction samples were prepared as a mixture with Quantitect SYBR Green PCR kit (Qiagen) following the manufacturer’s instructions. Reactions were performed using an Applied Biosystems Prism 9700 PCR machine. The PCR conditions were as follows: 95 °C for 15 min followed by 45 cycles of 95 °C for 15 sec, 55 °C for 30 sec and 72 °C for 30 sec. The primer sequences used are as shown below: mHdc forward 5′-TGCCTGTGTTTGTCTGTGCAACG-3′; reverse 5′-ATCTGCCAATGCATGAAGTCCGTG-3′; IL-1α forward 5-GAGCGCTCAAGGAGAAGACCAG-3′; reverse 5′-CAGGTGCACCCGACTTTGTTCT-3′; I L-1β forward 5′-CAAGCAACGACAAAATACCTGTG-3′; reverse 5′-AGACAAACCGTTTTTCCATCTTCT-3′; I L-6 forward 5′-CCGGAGAGGAGACTTCACAGAG-3′; reverse 5′-CTGCAAGTGCATCATCGTTGTT-3′; TNF-α forward 5′-TGGCCCAGACCCTCACACTCAG-3′, reverse 5′-ACCCATCGGCTGGCACCACT-3′; T G F-β forward 5′-ACCGGAGAGCCCTGGATACCA-3′, reverse 5′-TATAGGGGCAGGGTCCCAGACA-3′; GAPDH forward 5′-CCACTCACGGCAAATTCAAC-3′; reverse 5′-GTAGACTCCACGACATACTCAG-3′.
Single-cell preparations for FACS analysis and sorting
Bone marrow-derived cells (BMDCs) from the femur and tibia of euthanized mice were flushed with ice-cold Hanks' Balanced Salt Solution (GIBCO) and depleted of red blood cells (RBCs) using RBC lysing buffer (BD Biosciences). Total nucleated cells in peripheral blood were isolated after erythrocyte lysis. Single-cell suspensions were made by filtrating cells through 40 µm strains. For FACS analysis, single-cell suspensions were stained with antibodies and evaluated by multi-color flow cytometry using a LSRII flow cytometer (BD Biosciences). Data were analyzed using FlowJo7 software (Tree Star). EGFP-expressing bone marrow cells and immature myeloid cells (CD11b+Gr-1+ and CD11b+Ly6G+) were sorted with a FACStarPlus flow cytometer (BD Biosciences). Similar protocols were used for EGFP+ IMCs from tumors. EGFP+ cells were isolated for in vitro co-culture experiments and co-implanted with tumor cells in xenograft models.
DSS induced colitis and AOM and DSS induced colonic carcinogenesis models
According to the protocol previously described, twenty 3-month-old Hdc-EGFP mice were treated with 3% non-genotoxic agent dextran sodium sulfate (DSS) (MP Biomedical, MW is 36,000 to 50,000) given in the drinking water29. For the acute colitis model, control mice were treated with distilled water. Colonic tissues and peripheral blood were collected at days 4, 10 and 15. In the AOM + DSS colonic carcinogenesis experiments, control groups included untreated Balb/C mice, mice injected with a single dose of the genotoxic colonic carcinogen, azoxymethane (AOM, 12.5 mg kg−1 weight), and mice treated with DSS alone (10 days); the AOM + DSS group were given one dose of AOM intraperitoneal injection followed by one cycle of DSS in drinking water for 10 days. 15 weeks (for Balb/C mice) and 20 weeks (for C57BL/6 mice) later mice were sacrificed; colonic tissue fixed with 4% PFA and 30% sucrose for frozen tissue or fixed in 10% formalin for paraffin embeddings. Each group had 8 mice.
TPA induced inflammatory response in the skin and DMBA and TPA induced skin carcinogenesis model
Eight 3-month-old Hdc-EGFP mice were treated with 12-O-tetradecanoylpherorbol 13-acetate (TPA) (8.5 nmol in 0.2 ml of acetone, P-8139 Sigma) which was painted on the shaved dorsal skin. Control mice were painted with 0.2 ml of acetone. Animals were sacrificed by cervical dislocation 6 hours and 24 hours after application of TPA. A piece of dorsal skin (2.0 × 2.0 cm) was used for histological analysis. In order to develop skin cancer, wildtype mice and Hdc−/− mice (Balb/C background, each group had 5 mice) were initiated once with 100 nmol of 9, 10-dimethyl-1, 2-benzanthracene (DMBA, Sigma), following twice weekly 8.5 nmol TPA treatment. Skin tissue were fixed with 4% PFA and 30% sucrose for frozen tissue or fixed in 10% formalin for paraffin embeddings.
Intravital microscopy study
Intravital microscopy of the cutaneous microvasculature of the ear was performed as described previously 49. Briefly, an acute inflammatory response was induced in the ears of EGFP-expressing wild type and Hdc−/− mice by application of a single dose of TPA (8.5 nmol, 20 µl per mouse). The number of EGFP-expressing cells that had emigrated from the blood into the surrounding tissue was determined at 6 and 24 hours after TPA stimulation. This was accomplished using an Intravital system consisting of a Zeiss Axiotech vario microscope with a 20 X water-immersion Olympus objective (LUMPlanFl, 0.5 NA), a Yokogawa CSU-22 spinning disk confocal scanner, a iXON EM camera, and a 488-nm laser line (Revolution XD, Andor Technology, South Windsor, CT) to detect EGFP-labeled cells. The extent of cell migration was measured by off-line analysis (ImagePro).
Tumor xenograft experiments
CT26 mouse colon cancer cells were grown in complete media. Once cells were 70–80% confluent, cells were washed with sterile PBS 3–4 hours before harvesting in order to remove dead and detached cells. Trypsin-EDTA was used to detach cells and cells were then centrifuged at 1200 rpm for 5 min followed by washing twice with PBS. Cells were then counted using a hemocytometer. Cells were prepared for injection by suspending cells in a volume so that 100 µl PBS contained 1.0 × 106 cells. Sorted EGFP+ and EGFPCD11b+ Gr-1+ or CD11b+Ly6G+ IMCs were also counted using a hemocytometer. A combination of 100 µl CT26 cells and 100 µl IMCs were then injected subcutaneously (2.0 × 106 cells) into the two-sides of the lower back of the mice. Exogenous histamine (800 µg kg−1 weight per day, I.P injection) was administered for 20 days to inhibit xenograft tumor growth. About 21–25 days later, tumors were harvested to measure tumor weight and size. Each group had 6 SCID mice. A similar protocol was used for mouse gastric cancer cells MNK45 and mouse skin cancer cells TGAC–43 experiment (data not shown).
Immature myeloid cell differentiation studies
EGFP+ and EGFP− IMCs were sorted from the bone marrow and spleen of Hdc-EGFP mice. Cells were resuspended and IMCs cultured in 6-well plates with 15% FBS RPMI 1640 media. Histamine (5 × 10−7M), H1R antagonist (1 µM pyrilamine maleate, sigma), H2R antagonist (50 µM cimetidine, sigma), Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and Granulocyte Colony-Stimulating Factor (G-CSF) were added (final concentration is 60 ng ml−1) to the media and differentiated myeloid cells harvested at various time points (24, 48 or 72 hours).
Microarray performance and statistical analysis
Differentially expressed genes of CD11b+Gr-1+ IMCs were examined by microarray analysis (Affymetrix Mouse 430.2 array). Total RNA of Hdc-expressing CD11b+Gr-1+ IMCs from the bone marrow was extracted from Hdc-EGFP and Hdc-EGFP/Hdc−/− mice (3 mice in each group). CD11b+Gr-1+ MDSCs of colonic tumors were sorted from 10–12 colon tumors of wildtype and Hdc−/− mice (5 mice in each group) and pooled to extract total RNA for Microarray studies. RNA concentration and integrity were measured using standard protocols; cRNA synthesis, labeling, microarray hybridization and scanning were performed by Columbia University Irving Cancer Center shared Resources, according to the protocol of Gene CHIP 3’ IVT EXPRESS KIT (AFFYMETRIX 901228). Pooled samples from each mixture were hybridized to two arrays. Raw CEL files were subjected to an established battery of quality tests using the Bioconductor package. Arrays were normalized using GCRMA algorithm. The microarray data were deposited in the GEO database (Gene Expression Omnibus) (Series Accession number is GSE23502). Differential expression of four comparisons was evaluated using LIMMA (LInear Models for MicroArrays). Both Limma and GCRMA are part of the Bioconductor suite which runs in the R statistical computing environment. The relative expression of an experimental condition (e.g. Hdc knockout) compared to a control condition (e.g. WT) was expressed as a Log2 ratio. A statistical significance cutoff of the Benjamini-Hochberg false discovery rate (FDR) ≤ 0.05 was used. Four sets of comparisons were performed to screen the upregulated or downregulated genes in the Hdc knockout CD11b+Gr-1+ IMCs or MDSCs (experiment group) versus the equivalent controls from the WT mice. Set 1: Hdc-expressing CD11b+Gr-1+ IMCs from the bone marrow of Hdc knockout mice compared to bone marrow IMCs from WT mice. Set 2: CD11b+Gr-1+ MDSCs in colonic tumors of Hdc knockout mice compared to WT mice. Set 3: CD11b+Gr-1+ MDSCs from colonic tumors of WT mice compared to IMCs from WT bone marrow cells. Set 4: CD11b+Gr-1+ MDSCs from colonic tumors of Hdc knockout mice compared to IMCs from bone marrow cells of Hdc knockout mice.
Measurement of cytokine levels by ELISA
CD11b+Gr-1+ IMCs in the bone marrow and spleen of colon tumor-bearing Hdc−/− and wildtype mice and CD4+ T cells in the spleen were sorted by FACS. 2.5 × 105 IMCs and 2.5 × 105 T cells were cultured in anti-CD3e pre-coated 24-well plates (with Concanavalin A at 5 µg ml−1 of culture medium). The levels of IFN-γ in the supernatant of cells cultured were determined using ELISA (cat 555138, BD Company, San Diego, CA). Absorbance was measured at 450 nm by a Multiscan MC reader, and the samples were analyzed by DELTA SOFT II software (BioMetallics, Inc., Princeton, NJ).
Sodium bisulfite treatment and sequencing analysis
Bone marrow-derived Hdc-EGFP-expressing CD11b+Ly6G+ IMCs were co-cultured with CT26 cancer cells for 48 hours. Genomic DNA of IMCs were extracted and subjected to sodium bisulfite modification (EZ DNA Methylation-Gold Kit, Zymo Research, Orange, CA) as described previously. After PCR amplification using primers (−800 and +200 bp from the transcript start site of mouse Hdc gene), the PCR products were cloned into the TOPO vector. The inserted PCR fragments of the individual clones obtained from each sample were sequenced with both M13 reverse and M13 forward primers using an ABI automated sequencer. The primer sequences used are as shown below: Primer 1 forward 5′-TAGGAGGAGTAATTTAAGGGAGATGA-3′; reverse 5′-AAAAAAAACACCACTAAAACCAAAC-3′; Primer 2 forward 5′-TTTGAGTTTAGTTTGGTTAGTTATGG-3′; reverse 5′-CAACTATCAACAAAACTTCTTAATC-3′; Primer 3 forward 5′-TTTTTAGTTTTGTTTGTTGTAGAAG-3′; reverse 5′-ATAAAATTACTCCTACCCTAACTTCTCTAT-3′; Primer 4 forward 5′-ATTTGATAAAGTGGTAATTTTTTTTT-3′; reverse 5′-AACACTTATTCTAAATTCTTTACCTC-3′.
Statistical Analysis
Data from at least three independent experiments or five mice per group are represented as mean ± s.d. Statistical difference between groups was evaluated by Student’s t test. A P value < 0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank S.P Tu, S.W. Wang, S. Takaishi, and H. Tomita for their helpful contributions. The authors thank K.S. Betz for her help with animal procedures. We are grateful to A. Leiter (University of Massachusetts) for the PKD4-NICD-EGFP plasmid. Receptor for advanced glycation end products (RAGE) knockout mice were kindly provided by A. M. Schmidt (Columbia University). This work was funded by the NIH grants RO1 DK52778 and U54 CA126513. S. Asfaha was supported by a Canadian Institutes for Health Research Clinician Scientist Award and Alberta Heritage Foundation for Medical Research Clinical Fellowship.
Footnotes
AUTHOUR CONTRIBUTIONS
X.D.Y. was involved in the study design, completion of experiments, data analysis and interpretation and manuscript preparation. W. A. constructed the Hdc-EGFP transgenic mouse and helped with examination of Hdc-EGFP expression. S. A. helped with the data interpretation and contributed to the manuscript preparation and revision. G. B. provided human colon specimen collection and did pathology assessment. R.A.F. carried out the microarray data analysis. G.C.J. helped with the colon cancer experiments and data analysis. H.P. helped with the skin carcinogenesis experiments and data analysis. B.S. performed histological analysis of the brain of Hdc-EGFP mice. T.G.D. carried out intravital microscopy studies. A.F. constructed the Hdc knockout mice and provided helpful suggestions for our study. T.C.W. designed the study, and contributed to the data analysis and writing of the manuscript. All authors discussed the results and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431:405–406. doi: 10.1038/431405a. [DOI] [PubMed] [Google Scholar]
- 2.Luo Y, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. The Journal of clinical investigation. 2006;116:2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shojaei F, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450:825–831. doi: 10.1038/nature06348. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in immunology. 2002;23:549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 5.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen L, et al. Inhibition of human neutrophil degranulation by transforming growth factor-beta1. Clinical and experimental immunology. 2007;149:155–161. doi: 10.1111/j.1365-2249.2007.03376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. The Journal of experimental medicine. 1995;181:435–440. doi: 10.1084/jem.181.1.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer research. 2001;61:4756–4760. [PubMed] [Google Scholar]
- 10.Tazawa H, et al. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. The American journal of pathology. 2003;163:2221–2232. doi: 10.1016/S0002-9440(10)63580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe S, et al. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J Immunol. 2008;181:3291–3300. doi: 10.4049/jimmunol.181.5.3291. [DOI] [PubMed] [Google Scholar]
- 13.Haile LA, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology. 2008;135:871–881. doi: 10.1053/j.gastro.2008.06.032. 881 e871–875. [DOI] [PubMed] [Google Scholar]
- 14.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu S, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haile LA, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J Immunol. 185:203–210. doi: 10.4049/jimmunol.0903573. [DOI] [PubMed] [Google Scholar]
- 17.Jutel M, et al. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature. 2001;413:420–425. doi: 10.1038/35096564. [DOI] [PubMed] [Google Scholar]
- 18.Elenkov IJ, et al. Histamine potently suppresses human IL-12 and stimulates IL-10 production via H2 receptors. J Immunol. 1998;161:2586–2593. [PubMed] [Google Scholar]
- 19.van der Pouw Kraan TC, et al. Histamine inhibits the production of interleukin-12 through interaction with H2 receptors. The Journal of clinical investigation. 1998;102:1866–1873. doi: 10.1172/JCI3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirasawa N, et al. Pharmacological analysis of the inflammatory exudate-induced histamine production in bone marrow cells. Immunopharmacology. 1997;36:87–94. doi: 10.1016/s0162-3109(96)00164-6. [DOI] [PubMed] [Google Scholar]
- 21.Higuchi S, et al. Effects of histamine and interleukin-4 synthesized in arterial intima on phagocytosis by monocytes/macrophages in relation to atherosclerosis. FEBS letters. 2001;505:217–222. doi: 10.1016/s0014-5793(01)02823-x. [DOI] [PubMed] [Google Scholar]
- 22.Zwadlo-Klarwasser G, et al. Generation and subcellular distribution of histamine in human blood monocytes and monocyte subsets. Inflamm Res. 1998;47:434–439. doi: 10.1007/s000110050357. [DOI] [PubMed] [Google Scholar]
- 23.Sasaguri Y, et al. Role of histamine produced by bone marrow-derived vascular cells in pathogenesis of atherosclerosis. Circulation research. 2005;96:974–981. doi: 10.1161/01.RES.0000166325.00383.ed. [DOI] [PubMed] [Google Scholar]
- 24.Ohtsu H, et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS letters. 2001;502:53–56. doi: 10.1016/s0014-5793(01)02663-1. [DOI] [PubMed] [Google Scholar]
- 25.Ohtsu H, Watanabe T. New functions of histamine found in histidine decarboxylase gene knockout mice. Biochemical and biophysical research communications. 2003;305:443–447. doi: 10.1016/s0006-291x(03)00696-x. [DOI] [PubMed] [Google Scholar]
- 26.Ercan-Sencicek AG, et al. L-histidine decarboxylase and Tourette's syndrome. The New England journal of medicine. 362:1901–1908. doi: 10.1056/NEJMoa0907006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prizment AE, et al. History of allergy and reduced incidence of colorectal cancer, Iowa Women's Health Study. Cancer Epidemiol Biomarkers Prev. 2007;16:2357–2362. doi: 10.1158/1055-9965.EPI-07-0468. [DOI] [PubMed] [Google Scholar]
- 28.Vajdic CM, et al. Atopic disease and risk of non-Hodgkin lymphoma: an InterLymph pooled analysis. Cancer research. 2009;69:6482–6489. doi: 10.1158/0008-5472.CAN-08-4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka T, et al. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer science. 2003;94:965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nature biotechnology. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]
- 31.Nissinen MJ, Panula P. Developmental patterns of histamine-like immunoreactivity in the mouse. J Histochem Cytochem. 1995;43:211–227. doi: 10.1177/43.2.7822777. [DOI] [PubMed] [Google Scholar]
- 32.Nissinen MJ, Karlstedt K, Castren E, Panula P. Expression of histidine decarboxylase and cellular histamine-like immunoreactivity in rat embryogenesis. J Histochem Cytochem. 1995;43:1241–1252. doi: 10.1177/43.12.8537641. [DOI] [PubMed] [Google Scholar]
- 33.Karlstedt K, Nissinen M, Michelsen KA, Panula P. Multiple sites of L-histidine decarboxylase expression in mouse suggest novel developmental functions for histamine. Dev Dyn. 2001;221:81–91. doi: 10.1002/dvdy.1127. [DOI] [PubMed] [Google Scholar]
- 34.Ikuta S, Edamatsu H, Li M, Hu L, Kataoka T. Crucial role of phospholipase C epsilon in skin inflammation induced by tumor-promoting phorbol ester. Cancer research. 2008;68:64–72. doi: 10.1158/0008-5472.CAN-07-3245. [DOI] [PubMed] [Google Scholar]
- 35.Kitamura Y, et al. Increase in histidine decarboxylase activity in mouse skin after application of tumor promoters. Princess Takamatsu symposia. 1983;14:327–334. [PubMed] [Google Scholar]
- 36.Rad FH, et al. VEGF kinoid vaccine, a therapeutic approach against tumor angiogenesis and metastases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2837–2842. doi: 10.1073/pnas.0611022104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grivennikov S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bollrath J, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Yan SF, Ramasamy R, Schmidt AM. Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nature clinical practice. 2008;4:285–293. doi: 10.1038/ncpendmet0786. [DOI] [PubMed] [Google Scholar]
- 40.Kuramasu A, Saito H, Suzuki S, Watanabe T, Ohtsu H. Mast cell-/basophil-specific transcriptional regulation of human L-histidine decarboxylase gene by CpG methylation in the promoter region. The Journal of biological chemistry. 1998;273:31607–31614. doi: 10.1074/jbc.273.47.31607. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki-Ishigaki S, et al. The mouse L-histidine decarboxylase gene: structure and transcriptional regulation by CpG methylation in the promoter region. Nucleic acids research. 2000;28:2627–2633. doi: 10.1093/nar/28.14.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiener Z, et al. Bone marrow-derived mast cell differentiation is strongly reduced in histidine decarboxylase knockout, histamine-free mice. Int Immunol. 2002;14:381–387. doi: 10.1093/intimm/14.4.381. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds JL, Akhter J, Adams WJ, Morris DL. Histamine content in colorectal cancer. Are there sufficient levels of histamine to affect lymphocyte function? Eur J Surg Oncol. 1997;23:224–227. doi: 10.1016/s0748-7983(97)92388-x. [DOI] [PubMed] [Google Scholar]
- 44.Bolton E, King J, Morris DL. H2-antagonists in the treatment of colon and breast cancer. Seminars in cancer biology. 2000;10:3–10. doi: 10.1006/scbi.2000.0301. [DOI] [PubMed] [Google Scholar]
- 45.Velicer CM, Dublin S, White E. Cimetidine use and the risk for prostate cancer: results from the VITAL cohort study. Annals of epidemiology. 2006;16:895–900. doi: 10.1016/j.annepidem.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 46.Robertson DJ, et al. Histamine receptor antagonists and incident colorectal adenomas. Alimentary pharmacology & therapeutics. 2005;22:123–128. doi: 10.1111/j.1365-2036.2005.02529.x. [DOI] [PubMed] [Google Scholar]
- 47.Wang H, Diepgen TL. Is atopy a protective or a risk factor for cancer? A review of epidemiological studies. Allergy. 2005;60:1098–1111. doi: 10.1111/j.1398-9995.2005.00813.x. [DOI] [PubMed] [Google Scholar]
- 48.Gandini S, Lowenfels AB, Jaffee EM, Armstrong TD, Maisonneuve P. Allergies and the risk of pancreatic cancer: a meta-analysis with review of epidemiology and biological mechanisms. Cancer Epidemiol Biomarkers Prev. 2005;14:1908–1916. doi: 10.1158/1055-9965.EPI-05-0119. [DOI] [PubMed] [Google Scholar]
- 49.Radeke HH, Ludwig RJ, Boehncke WH. Experimental approaches to lymphocyte migration in dermatology in vitro and in vivo. Experimental dermatology. 2005;14:641–666. doi: 10.1111/j.0906-6705.2005.00350.x. [DOI] [PubMed] [Google Scholar]
- 50.Salcedo R, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. The Journal of experimental medicine. 207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.