

Abstract
In Parkinson's disease (PD), loss of striatal dopaminergic (DA) terminals and degeneration of DA neurons in the substantia nigra (SN) are associated with glial reactions. Such inflammatory processes are commonly considered an epiphenomenon of neuronal degeneration. However, there is increasing recognition of the role of neuroinflammation as an initiation factor of DA neuron degeneration. To investigate this issue, we established a new model of brain inflammation by injecting the Toll-like receptor 3 (TLR-3) agonist polyinosinic:polycytidylic acid [poly(I:C)] in the SN of adult rats. Poly(I:C) injection induced a sustained inflammatory reaction in the SN and in the dorsolateral striatum. Significant changes were detected in proteins relevant to synaptic transmission and axonal transport. In addition, cytoplasmic mislocalization of neuronal TAR DNA binding protein TDP-43 was observed. Poly(I:C) injection increased the susceptibility of midbrain DA neurons to a subsequent neurotoxic trigger (low-dose 6-hydroxydopamine). Systemic delivery of interleukin-1 receptor antagonist protected SN DA neurons exposed to combined poly(I:C) induced inflammatory and neurotoxic oxidative stress.
These data indicate that viral-like neuroinflammation induces predegenerative changes in the DA system, which lowers the set point toward neuronal dysfunction and degeneration. New powerful neuroprotective therapies for PD might be considered by targeting critical inflammatory mechanisms, including cytokine-induced neurotoxicity.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disorder (Dauer and Przedborski, 2003). Most PD cases are sporadic, although many genetic mutations have been shown as causes or risk factors. The low penetrance of these mutations and the low disease concordance in relatives suggest that there must be interactions between multiple risk factors (Farrer, 2006). The risk factors for PD include genetic predisposition, age, and environmental triggers (Kasten et al., 2007). One such environmental stressor could be neuroinflammation. Activated microglia and high levels of proinflammatory molecules are present in the basal ganglia and brainstem of PD patients, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxicated patients, and in the basal ganglia in the 6-hydroxydopamine (6-OHDA), MPTP, and rotenone animal models of PD (McGeer et al., 1988; Langston et al., 1999; Liberatore et al., 1999; Cicchetti et al., 2002; Sherer et al., 2003). Although these data suggest that inflammation is involved in PD progression, whether such inflammatory processes are pathogenic or simply represent responses to neuronal degeneration is unclear. Recent evidence suggests that neuroinflammation can be involved early in such disease processes and can be coupled with predegenerative changes in the dopaminergic (DA) system (Koprich et al., 2008; Chung et al., 2009). Several environmental triggers, including bacterial and viral infections, may initiate neuroinflammatory processes. It has been hypothesized that viruses have an etiological role in the development of several neurodegenerative disorders, including PD (Mattson, 2004; Jang et al., 2009). Such a hypothesis is in line with the origin of postencephalitic parkinsonism (Economo Von, 1931; Dale et al., 2004). Neurotropic viruses (e.g., arboviruses, influenza viruses, herpes viruses, JC virus, rotaviruses) may cause fulminant encephalitis, sometimes resulting in lethal consequences. More commonly, viruses enter the CNS during subclinical infections, by the hematogenous route across the blood–brain barrier (BBB) or by the peripheral nerve route (Berth et al., 2009). Jang et al. (2009) showed that the intranasal administration of the neurotropic influenza virus H5N1 in mice triggers chronic microglia activation, phosphorylation and aggregation of α-synuclein (α-Syn), and a significant loss of dopamine in the substantia nigra (SN). However, it is not clear whether the inflammatory reaction that accompanies subclinical CNS infections can initiate a degenerative process or how inflammation can alter the normal neuronal function. Viral infections are indeed accompanied by a cytokine storm in the brain (Weller et al., 1991), and such inflammatory activation could be responsible for the CNS priming to degeneration and a possible aberrant response to subsequent neurotoxic triggers.
To investigate the role of neuroinflammatory processes induced by viral infections and the sensitization of DA neurons to neurotoxic triggers, we therefore developed a novel in vivo model of viral-like neuroinflammation. We injected the Toll-like receptor 3 (TLR-3) agonist polyinosinic:polycytidylic acid [poly(I:C)] into the SN of adult rats and characterized the cellular and molecular mechanisms underlying the resultant neuroinflammatory reaction. We then investigated whether these changes may hasten dysfunction of vulnerable nigral DA neurons, increasing their susceptibility to subsequent neurotoxic triggers.
Materials and Methods
Animals.
Female Sprague Dawley rats weighing ∼250 g (Charles River Laboratories) were used in all animal experiments. All animal procedures were performed in accordance with current National Institutes of Health guidelines and McLean Hospital/Harvard University Institutional Animal Care and Use Committee protocols.
Stereotaxic injection of poly(I:C) into SN.
All stereotaxic coordinates were derived from the rat atlas of Paxinos and Watson (1986). For each surgery animals were deeply anesthetized with ketamine and xylazine (60 and 3 mg/kg, i.m., respectively). Animals received a 4.0 μl stereotaxic injection of poly(I:C) (total amount, 10, 20, or 40 μg) (Axxora) delivered at a rate of 0.5 μl/min using microinfusion pumps (Stoelting) with a 5 min wait time after injection. All injections were made into the SN using the following anteroposterior (AP), mediolateral (ML), and dorsoventral (DV) coordinates: AP, −5.5 mm; ML, −2.0 mm; and DV, 7.5 mm relative to dura. Toothbar (TB) was set at −3.3 mm.
6-OHDA intra-striatal injection.
Animals received a single 3.5 μl stereotaxic injection of 6-OHDA (total dose, 5.0 μg of 6-OHDA prepared as free base; Sigma-Aldrich) delivered at a rate of 0.5 μl/min and a 5 min wait time after injection. Striatum injection coordinates were as follows: AP, +0.2 mm; ML, −3.0 mm; DV, −5.0 mm; and TB, −3.3 mm.
Interleukin-1 receptor antagonist administration.
Osmotic pumps (model 2ML1; Alzet) were filled with either interleukin-1 receptor antagonist (IL-1ra) (3.64 mg · kg−1 · h−1, s.c.; Anakinra; Amgen) or vehicle and implanted subcutaneously. Pumps were replaced weekly until completion of the study.
Immunohistochemistry.
Animals were terminally anesthetized by an intraperitoneal injection of sodium pentobarbital (100 mg/kg) and perfused intracardially with heparinized saline (0.1% heparin in 0.9% saline), followed by paraformaldehyde (4% in PBS). Immunohistochemical staining was performed as described previously (Bjorklund et al., 2002; Chung et al., 2009). The following primary antibodies were used: rabbit anti-Iba1 (1:500; Wako), rabbit anti-GAD67 (1:5000; Sigma-Aldrich), rabbit anti-glial fibrillary acidic protein (GFAP) (1:1000; Dako), mouse anti-major histocompatibility complex (MHC) class II RT1B (1:100; AbD Serotec), mouse anti-CD68 (1:100; AbD Serotec), mouse anti-CD11c (1:100; AbD Serotec), mouse anti-NMDA receptor 1 (NMDAR1) (1:250; BD Biosciences Transduction Laboratories), mouse anti-neuronal-specific nuclear protein (NeuN) (1:1000; Millipore Bioscience Research Reagents), mouse anti-postsynaptic density 95 (PSD-95) (1:500; BD Biosciences Transduction Laboratories), mouse anti-TLR3/CD28 (1:50; Imgenex), rabbit anti-TAR DNA binding protein 43 (TDP-43) (1:200; Abcam), and rabbit anti-tyrosine hydroxylase (TH) (1:1000; Pel Freez). Sections were counterstained with Hoechst.
Stereological procedures.
Quantification of TH-positive (TH+) neuronal number within the SN and ventral tegmental area (VTA) was performed by investigators blind to group treatments using an integrated Axioskop 2 microscope (Carl Zeiss) and Stereo Investigator software (MBF Bioscience) and stereological principles as described previously (Chung et al., 2007; Koprich et al., 2008). Briefly, SN and VTA were outlined by using the third cranial nerve as a landmark to define the vertical border between SN and VTA and by using the medial lemniscus as their dorsal border. The total number of TH-positive neurons was estimated from coded slides using the optical fractionator method. The number of sampling sites and other stereological parameters were experimentally determined to obtain a target coefficient of error (Gundersen's m = 1) of <0.10 for each case, thereby satisfying all criteria for objective stereological cell counts (Slomianka and West, 2005). Coexpression of Iba1/MHC-II was assessed in five to seven random fields in all available sections from one series.
Optical densitometry.
Optical densitometry on TH-positive fibers in the striatum was performed using the NIH ImageJ software version 1.3. For each animal, the average optical density of five separate sections through the striatum was determined. According to the atlas of Paxinos and Watson (1986), the approximate sections were as follows relative to bregma: +0.22 mm, 0 mm, −0.26 mm, −0.74 mm, and −1.22 mm. Optical densitometry from a region of cortex immediately adjacent to the corpus callosum was used to correct striatal optical densitometry measurements for nonspecific background.
Tissue preparation for multiplex ELISA and Western blot analysis.
Animals were terminally anesthetized the same way described above and perfused intracardially with heparinized saline (0.1% heparin in 0.9% saline). Striatum and SN were hand dissected on ice-cold surface, assisted by a tissue chopper.
Multiplex ELISA.
Tissue samples were homogenized in lysis buffer (TPER; Pierce) as described previously (Koprich et al., 2008). Halt protease and phosphatase inhibitor cocktail (1:100) was added fresh before cell lysis (Pierce). Samples were analyzed for the simultaneous detection of cytokines, chemokines, and chemokine receptors using a multiplex ELISA-based format and performed in duplicate (Pierce Thermo Fisher Scientific) (Koprich et al., 2008).
Western blot analysis.
Tissue samples were homogenized using hand-held Polytron homogenizer in ice-cold TEVP buffer, pH 7.4 [10 mm Tris-HCl, 5 mm NaF, 1 mm Na3VO4, 1 mm EDTA, 1 mm EGTA, 320 mm sucrose, and protease inhibitors (1:100; Sigma P8340)]. The homogenate was centrifuged at 800 × g to separate P1 (nuclear and debris) and supernatant (S1) fraction. S1 fraction was centrifuged at 9200 × g to separate P2 (membranous fraction) and S2 (cytosolic fraction). Ten to 20 μg of tissue proteins were used to run a gel. The respective volumes were mixed 1:1 with sample buffer and then boiled for 5 min. Samples were loaded into the Criterion precast 4–15, 10, or 12.5% SDS polyacrylamide gel system (Bio-Rad). Proteins were transferred from the gel to a polyvinylidene difluoride membrane electrically at 80 V for 1 h. Then, the membranes were washed in Tris-buffered saline with 0.1% Tween 20 (TBS-T). After at least 1 h of blocking in 5% nonfat dry milk, the membranes were incubated overnight at 4°C in various primary antibodies: CX3CR1 (1:500; Abcam), Iba1 (1:200; Santa Cruz Biotechnology), TLR-3 (1:200; Santa Cruz Biotechnology), α-Syn (1:500; BD Biosciences), SNAP-25 (1:2000; Millipore Bioscience Research Reagents), Rabphilin3A (1:2000; BD Biosciences Transduction Laboratories), RAB3B (1:2000; Affinity Bioreagents), AMPA receptor (glutamate receptors GluR2/GluR3/GluR4) (1:500; Cell Signaling Technology), PSD-95 (1:500; Neuromab), Drebrin (1:750; Genetex), NMDAR1 (1:400; BD Biosciences Transduction Laboratories), SAP102 (1:500; Cell Signaling Technology), syntaxin (1:1000; Millipore Bioscience Research Reagents), synaptophysin (1:200; Santa Cruz; Biotechnology), synaptotagmin (1:1000; BD Biosciences Transduction Laboratories), synapsin (1:5000; Millipore Bioscience Research Reagents), Munc-18 (1:7500; Affinity Bioreagents), KIF1A (clone 16; 1:2000; BD Biosciences Transduction Laboratories), KIF1B (1:2000; Novus Biologicals), KIF2A (1:10,000; Abcam), KIF3A (1:2000; Abcam), KIF5 (1:1000; Abcam), KIF17 (1:1000; Abcam), myosin Va (1:500; Sigma), dynein (clone 74.1; 1:1000; Millipore Bioscience Research Reagents), dynamitin (1:1000; Millipore Bioscience Research Reagents), TDP-43 (1:800; Abcam), α-tubulin (1:2000; Affinity Bioreagents), β-tubulin (1:5000; Covance), γ-tubulin (clone GTU-88; 1:500; Sigma), actin (clone C-2; 1:500; Santa Cruz Biotechnology), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5000; Millipore Bioscience Research Reagents). Then, the membranes were incubated in HRP-conjugated secondary antibodies for 1 h at room temperature. After washing with TBS-T, ECL or ECL plus (GE Healthcare) was used to probe for immunoreactive (IR) bands and exposed to film using the Hyblot film system (Denville Scientific). Optical density analysis (NIH ImageJ) was used to determine the relative abundance of protein in each sample.
Statistical analysis.
The numbers of TH-IR-positive neurons in the SN and VTA and optical density measurements of striatal TH fiber density were compared by one-way ANOVA, followed by post hoc testing using Tukey's honestly significant difference method. Unpaired two-tailed t test applied at the 95% confidence level (p < 0.05) was used to identify significantly different serum levels for each cytokine/chemokine and protein levels in rat SN and striatum, numbers of TH-IR neurons, and TH fiber density in IL1-ra experiment. Values of p < 0.05 were accepted as statistically significant. The Statistical Package GraphPad Prism version 4.00 (GraphPad Software) was used to analyze the data.
Results
The injection of the TLR-3 agonist poly(I:C) induces a long-lasting inflammatory reaction in the SN and dorsolateral striatum
To mimic the neuroinflammatory cascade elicited by a viral infection, we injected the TLR-3 agonist poly(I:C) into the substantia nigra pars compacta (SNc) of adult Sprague Dawley rats. We first determined the optimal dose of poly(I:C) that provided a neuroinflammatory response without inducing TH-IR cell loss. Sprague Dawley rats were injected with different doses of poly(I:C) (10, 20, and 40 μg) in the SNc. We examined the neuroinflammatory reaction by immunohistochemical analysis using Iba1 as a marker of microglia, and we quantified DA cell loss by TH-IR cell counting in the SNc at 12 d after injection. The intranigral injection of poly(I:C) induced microglial activation in a dose-dependent manner, as revealed by greater Iba1 immunostaining in animals injected with higher doses (data not shown). The injection of 40 μg of poly(I:C) produced significant TH-IR cell loss within the SNc (data not shown), whereas lower doses (10 and 20 μg) did not induce significant cell loss.
To examine the time course of poly(I:C)-induced neuroinflammatory reaction, animals were injected with 20 μg of poly(I:C) in the SNc, and the degree of microglial activation was determined by Iba1 immunostaining at 4, 12, and 33 d after the injection. It has been reported that the intracerebroventricular injection of poly(I:C) in mice induces robust microglia activation in the cerebral cortex via TLR-3 activation (Town et al., 2006). The intranigral injection of poly(I:C) induced microglial activation as shown by the presence of Iba1-immunoreactive cells with rod-like morphology as early as 4 d after the injection, which subsequently peaked at 12 d and persisted for 33 d (Fig. 1 A–D). Immunostaining for OX6 (a monomorphic determinant of the rat MHC class II antigen, expressed by activated microglial cells) confirmed the persistent microglial activation after the intranigral injection of poly(I:C) (Fig. 1 E–H; supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). We found that 25% of Iba1+ cells coexpressed MHC-II antigen 12 d after poly(I:C) injection (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Many MHC-II+ cells were found in the perivascular areas. We further characterized the microglial reaction using antibodies against the macrophage marker CD68 and dendritic cell marker CD11c. Poly(I:C) injection induced activation of perivascular and parenchymal CD68+ macrophages (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). Interestingly, many perivascular CD11c+/Iba1+ cells were detected after poly(I:C) injection, suggesting dendritic cell activation (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). A few parenchymal CD11c+ cells were also detected after poly(I:C) injection (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). We did not detect T-cell infiltration in brains of rats injected with poly(I:C). The neuroinflammatory response was quantified by Western blot analysis, which showed increased levels of Iba1, TLR-3, and the fractalkine receptor CX3CR1 in the SN at 12 d after poly(I:C) injection (Fig. 2 I). We next examined astrocytic activation by GFAP immunostaining in both groups of rats after administration of either poly(I:C) or vehicle. At 12 d after injection, the GFAP immunoreactivity was increased in the SN of animals injected with poly(I:C) when compared with control animals. Astrocytic activation was still evident in the SN 33 d after poly(I:C) injection (Fig. 1 I–L). Interestingly, the intranigral injection of poly(I:C) induced a neuroinflammatory response in the dorsolateral striatum. Starting at 12 d after poly(I:C) injection, Iba1-positive microglia in the dorsolateral striatum showed an activated phenotype as evident by their larger soma with short swollen processes compared with the elongated and needle-like shape in the control animals (Fig. 2 A–C).
Figure 1.
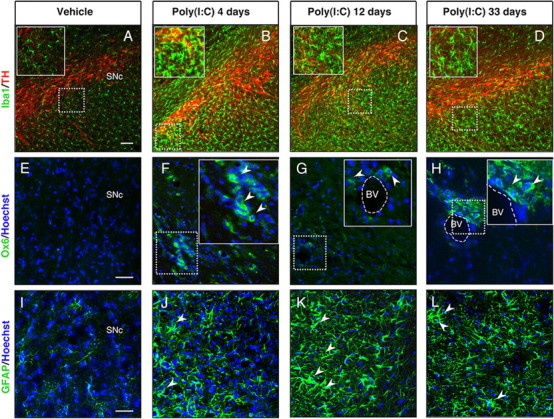
Intranigral injection of poly(I:C) induces microglial and astrocytic activation within the SN. Sprague Dawley rats were injected with vehicle or 20 μg of poly(I:C) into the SN. Brains were isolated at 4, 12, and 33 d after injection, and immunofluorescence microscopy was performed for TH (red), Iba1 (green), MHC class II antigen OX6 (green), and GFAP (green). A–D, Iba1 immunohistochemistry revealed resting highly ramified microglia within the SN in vehicle-injected animals (A). In contrast, animals injected with poly(I:C) at 4, 12, and 33 d after the injection showed activated microglia with shorter and thicker processes and larger cell bodies (insets) (B–D). E–H, MHC-II+ cells (arrowheads) were present in poly(I:C)-injected rats at 4,12 and 33 d after injection. I–L, Astrocytic reaction (arrowheads) was found in the SN at 4, 12, and 33 d after poly(I:C) injection. Cell nuclei were counterstained with Hoechst. Dashed lines in G and H indicate blood vessels. Scale bars: A–D, 100 μm; E–L, 50 μm; insets in B–D, 50 μm; insets in F–H, 25 μm. BV, Blood vessel.
Figure 2.

Intranigral injection of poly(I:C) induces microgliosis and astrocytic activation in the dorsolateral striatum. The dorsolateral striatum was immunostained for Iba1 (green) and GFAP (green) at 4, 12, and 33 d after the intranigral injection of vehicle or 20 μg of poly(I:C). Poly(I:C) promoted microglia activation (A–D) and astrocytic reaction (E–H) within the dorsolateral striatum starting at 12 d after the injection. At day 33, activated astrocytes were still present within the dorsolateral striatum (H), whereas microglial cells showed a resting phenotype (D). Cell nuclei were counterstained with Hoechst. Scale bars: A–H, 50 μm; insets in A–D, 25 μm. I, J, Western blots of SN and striatum extracts from vehicle and poly(I:C)-injected animals. Poly(I:C) injection induced an increase of the levels of Iba1, TLR3, and CX3CR1 in the SN. Levels of Iba1 were increased in the dorsolateral striatum. Vehicle, n = 9; poly(I:C), n = 10; *p < 0.05, **p < 0.01, unpaired t test.
Striatal microglial activation decreased over time and returned to baseline conditions 33 d after the injection (Fig. 2 D). Microglia activation was quantified by Western blot analysis, which revealed higher levels of Iba1 in the dorsolateral striatum of rats 12 d after poly(I:C) injection (Fig. 2 J). In parallel, we found a long-lasting astrocytic reaction starting at 12 d after the injection, which persisted up to 33 d (Fig. 2 E–H).
Poly(I:C) is a synthetic double-strand RNA that selectively binds to TLR-3 (Alexopoulou et al., 2001), an innate immune receptor expressed in neurons, microglia, and astrocytes (Bsibsi et al., 2002). Our analysis revealed that TLR-3 was highly expressed in DA neurons within the SNc and VTA in control animals (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). In contrast, microglia and astrocytes expressed lower levels of TLR-3 (supplemental Fig. 2C,E, available at www.jneurosci.org as supplemental material). Poly(I:C) injection resulted in an upregulation of TLR-3 in neurons at 12 d after the injection (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material), whereas microglia and astrocytes showed a moderate upregulation (supplemental Fig. 2D–F, available at www.jneurosci.org as supplemental material). Western blot analysis confirmed increased levels of TLR-3 in the SN of poly(I:C)-injected animals 12 d after the injection, whereas striatal levels remained unchanged (Fig. 2 I,J).
The intranigral injection of poly(I:C) increases the level of proinflammatory cytokines and chemokines in the SN and striatum
To profile the neuroinflammatory response to poly(I:C), we measured the protein level of proinflammatory cytokines and chemokines in the SN and in the dorsolateral striatum of rats at 12 d after the injection using multiplex ELISA. We chose this time point because, as revealed by our immunohistochemical analysis (Figs. 1, 2), both the poly(I:C)-injected SN and the dorsolateral striatum showed robust astrocytic and microglia activation at 12 d after poly (I:C) injection. In addition, mouse models of viral infections and human pathology show that active viral infections lasts ∼10 d (de Jong et al., 2005; Jang et al., 2009). Therefore, this time point most closely mimics post-acute viral infections when immune reactions may persist within the brain parenchyma in the absence of viral particles. Our analysis revealed that, in the SN, the levels of IL-1α and the chemokines monocyte chemoattractant protein MCP-1 and RANTES (regulated upon activation, normal T-cell expressed and secreted) were significantly elevated (Table 1) (t = 2.194, p < 0.05; t = 2.179, p < 0.05; t = 3.846, p < 0.01, respectively), whereas IL-1β, IL-6, tumor necrosis factor α (TNF-α), MCP-1, and TGF-β1 were elevated in the dorsolateral striatum (Table 1) (t = 2.962, p < 0.01; t = 2.164, p < 0.05; t = 2.590, p < 0.05; t = 2.494, p < 0.05; t = 3.838, p < 0.05, respectively).
Table 1.
Intranigral administration of poly(I:C) increases the level of proinflammatory cytokines and chemokines
| Cytokine chemokine | Vehicle | Poly(I:C) |
|---|---|---|
| Substantia nigra protein level 12 d after poly(I:C) (pg/mg) | ||
| IL-1α | 7.993 ± 1.558 | 12.32 ± 1.208* |
| IL-1β | 5.564 ± 0.5115 | 6.183 ± 0.3726 |
| IL-2 | 4.926 ± 0.7362 | 4.772 ± 0.3633 |
| IL-4 | 5.910 ± 1.034 | 5.279 ± 0.4931 |
| IL-6 | 55.19 ± 3.111 | 51.55 ± 2.167 |
| IL-10 | ND | ND |
| IFN-γ | 6.618 ± 0.4500 | 8.275 ± 1.364 |
| TNF-α | 23.05 ± 3.670 | 25.25 ± 6.779 |
| MCP-1 | 6.501 ± 0.9393 | 9.541 ± 0.9878* |
| MDC | 1.959 ± 0.1989 | 2.195 ± 0.1701 |
| Fractalkine | 67.93 ± 3.127 | 69.00 ± 2.411 |
| MIP-1α | 10.12 ± 1.698 | 11.04 ± 1.224 |
| MIP-2α | 4.860 ± 0.6166 | 5.827 ± 0.5258 |
| MIP-3α | 96.18 ± 7.500 | 105.4 ± 7.355 |
| RANTES | 63.96 ± 3.495 | 96.27 ± 5.275** |
| TGF-β1 | 258.1 ± 24.64 | 246.1 ± 15.16 |
| Striatum protein level 12 d after poly(I:C) (pg/mg) | ||
| IL-1α | 13.29 ± 2.285 | 11.98 ± 1.279 |
| IL-1β | 4.728 ± 0.4303 | 6.937 ± 0.5908** |
| IL-2 | 5.124 ± 0.4348 | 5.790 ± 0.4551 |
| IL-4 | 6.394 ± 0.9461 | 5.232 ± 0.4566 |
| IL-6 | 95.59 ± 6.001 | 114.5 ± 5.941* |
| IL-10 | ND | ND |
| IFN-γ | 9.206 ± 2.411 | 9.822 ± 2.148 |
| TNF-α | 24.31 ± 4.697 | 39.42 ± 3.461* |
| MCP-1 | 2.174 ± 0.3089 | 3.365 ± 0.3347* |
| MDC | 1.959 ± 0.1989 | 2.195 ± 0.1701 |
| Fractalkine | 142.8 ± 4.006 | 136.3 ± 3.928 |
| MIP-1α | 5.000 ± 0.5016 | 5.479 ± 0.5343 |
| MIP-2α | 4.228 ± 0.4588 | 4.770 ± 0.3614 |
| MIP-3α | 81.08 ± 4.540 | 86.95 ± 6.358 |
| RANTES | 73.06 ± 6.524 | 76.51 ± 7.677 |
| TGF-β1 | 161.3 ± 10.06 | 202.4 ± 5.722** |
Cytokine and chemokine levels were analyzed in the SN and striatum at 12 d after poly(I:C) injection using multiplex ELISA. Poly(I:C) injection caused increased levels of IL-1α, MCP-1, and RANTES within the SN and IL-1β, IL-6, TNF-α, MCP-1, and TGF-β in the striatum. Data are shown as mean ± SEM. Vehicle, n = 9; poly(I:C), n = 10.
*p < 0.05,
**p < 0.001, unpaired t test. IFN, Interferon; MCP, monocyte chemoattractant protein; MDC, macrophage-derived chemokine; MIP, macrophage inflammatory protein; TGF, transforming growth factor.
The intranigral injection of poly(I:C) alters the expression of axonal and synaptic proteins in the SN and striatum
To investigate whether poly(I:C) injection influenced neuronal properties and synaptic function, we measured the levels of various axonal and synaptic proteins in the SN and in the dorsolateral striatum at 12 d after injection. Among axonal transport proteins, levels of the anterograde transport motor proteins KIF1A and KIF1B were increased in the SN, whereas KIF3A and KIF17 were reduced (Fig. 3 A,B). Levels of dynein, a retrograde transport motor protein, were also decreased (Fig. 3 A,B). In the dorsolateral striatum, we found decreased levels of KIF17 (Fig. 4 A,B). Cytoskeletal protein (actin, α-Tubulin, β-Tubulin, and γ-Tubulin) levels were not altered by the intranigral injection of poly(I:C) (Figs. 3, 4). Among synaptic proteins, RAB3B and α-Syn levels were decreased in the SN (Fig. 5 A,B). In the dorsolateral striatum, levels of the postsynaptic scaffold PSD-95 and NMDA receptor subunit 1 (NR1) were increased, whereas levels of RAB3B, MUNC18, Drebrin, and SAP102 were reduced (Fig. 6 A,B). Overexpression of PSD-95 and NR1 was confirmed by immunohistochemistry, showing that synaptic PSD-95 and NR1 staining was more intense in the striatum of poly(I:C)-injected animals (supplemental Fig. 2G–J, available at www.jneurosci.org as supplemental material).
Figure 3.

Nigral changes in the levels of axonal proteins at 12 d after poly(I:C) injection. A, Anterograde transport motor proteins (KIF1A and KIF1B) were increased, whereas KIF3A, KIF17, and dynein were decreased. Levels of TDP-43 were increased. B, Optical densities of the individual bands were quantified using NIH ImageJ. Optical densities were normalized by the averaged value of GAPDH and expressed as percentage of vehicle-injected conditions. Data are shown as mean ± SEM. Vehicle, n = 9; poly(I:C), n = 10; *p < 0.05, **p < 0.01, unpaired t test.
Figure 4.

Striatal changes in the levels of axonal proteins at 12 d after poly(I:C) injection. A, Poly(I:C) injection induced decreased levels of KIF17 and TDP-43. B, Optical densities of the individual bands. Data are shown as mean ± SEM. Vehicle, n = 9; poly(I:C), n = 10; **p < 0.01, unpaired t test.
Figure 5.

Nigral changes in the levels of synaptic proteins at 12 d after poly(I:C) injection. A, Poly(I:C) injection induced decreased levels of RAB3B and α-Syn. B, Optical densities of the individual bands. Data are shown as mean ± SEM. Vehicle, n = 9; poly(I:C), n = 10; *p < 0.05, **p < 0.01, unpaired t test.
Figure 6.

Striatal changes in the levels of synaptic proteins at 12 d after poly(I:C) injection. A, Poly(I:C) injection induced decreased levels of RAB3B, MUNC18, Drebrin, and SAP102. Levels of the postsynaptic proteins NMDAR1 and PSD-95 were significantly increased. B, Optical densities of the individual bands. Data are shown as mean ± SEM. Vehicle, n = 9; poly(I:C), n = 10; *p < 0.05, ***p < 0.001, unpaired t test.
Poly(I:C) injection induces cytosolic mislocalization of neuronal TDP-43
Levels of TDP-43 were increased in the SN and decreased in the dorsolateral striatum 12 d after the injection (Figs. 3, 4). We therefore analyzed the effect of poly(I:C) injection on the subcellular localization of TDP-43. In vehicle-injected animals, TDP-43 was primarily restricted to the nucleus, whereas diffuse cytoplasmic TDP-43 neuronal staining was observed in the SN after poly(I:C) injection (supplemental Fig. 2K–N, available at www.jneurosci.org as supplemental material). Neither intranuclear nor cytoplasmic TDP-43-immunoreactive protein aggregates were detected.
The TLR-3 agonist poly(I:C) increases vulnerability to nigral DA neuron loss induced by low-dose 6-OHDA
To test the hypothesis that viral-like neuroinflammation induced by the TLR-3 agonist poly(I:C) increases the vulnerability of the DA nigrostriatal system to degeneration, we injected rats with 20 μg of poly(I:C), followed after 12 d by an intrastriatal injection of a low dose of 6-OHDA (5 μg). Neuronal cell loss was analyzed 21 d after the injection of 6-OHDA (Fig. 7 A). We previously reported the use of low-dose 6-OHDA in combination with the bacterial mimic lipopolysaccharide in a double-hit model of DA neuron degeneration (Koprich et al., 2008). This allows the investigation of the combined role of neuroinflammation and neurotoxic triggers in the onset of DA neuron degeneration. Unbiased stereological analyses of TH-IR neurons within the SN revealed that intranigral poly(I:C) followed by intrastriatal injection of low-dose 6-OHDA produced a greater TH-IR cell loss compared with the injection of low-dose 6-OHDA alone (Fig. 7 B,F,G) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material) (3011 ± 332 and 5020 ± 492, respectively; p < 0.05). Stereological counts of NeuN-immunopositive neurons further confirmed this neuronal loss (Fig. 7 B,J,K) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material) (9238 ± 320 and 15893 ± 1297, respectively; p < 0.05). Either vehicle or poly(I:C) alone did not induced significant cell loss (Fig. 7 B,D,E,H,I) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material) (TH+ cells, 6952.3 ± 345 and 6783.3 ± 515, respectively; NeuN+ cells, 22684.9 ± 1171 and 25042.9 ± 1923, respectively). Unbiased stereological analysis indicated no loss of TH-positive or NeuN-positive neurons in the VTA in any of the groups (Fig. 7 C) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material) [TH+ cells: vehicle alone (9523 ± 1228), poly(I:C)/vehicle (9138.8 ± 659), vehicle/6-OHDA (8984.7 ± 543), poly(I:C)/6-OHDA (9638.5 ± 1165); NeuN+ cells: vehicle alone (12500 ± 1234), poly(I:C)/vehicle (12477.3 ± 2000), vehicle/6-OHDA (13368.9 ± 1799), poly(I:C)/6-OHDA (11414.1 ± 1254)]. Moreover, the different treatments [poly(I:C), low-dose 6-OHDA, poly(I:C) followed by low-dose 6-OHDA] did not alter the qualitative expression pattern of GAD67 staining in either cell bodies or presynaptic terminal density (supplemental Fig. 4G–I, available at www.jneurosci.org as supplemental material). It is therefore likely that DA neurons of the SNc are selectively vulnerable to the actions of inflammatory stimuli. We then compared the neuroinflammatory and astrocytic reaction in animals injected with low-dose 6-OHDA alone and in the combined insult model. Neuronal cell loss was accompanied by microgliosis and astrocytic activation within the SN in both groups (supplemental Fig. 4A–F, available at www.jneurosci.org as supplemental material). However, microglial activation as assessed by Iba1 immunohistochemistry was prominent in animals injected with poly(I:C) followed by low-dose 6-OHDA (supplemental Fig. 4A–C, available at www.jneurosci.org as supplemental material). In parallel, animals injected with poly(I:C) and 6-OHDA displayed a more prominent astrocytic activation when compared with animals injected with 6-OHDA alone (supplemental Fig. 4D–F, available at www.jneurosci.org as supplemental material).
Figure 7.

Poly(I:C) injection increases vulnerability to nigral DA neuron loss induced by low-dose 6-OHDA. A, Experimental timeline: rats received a single intranigral injection of 20 μg of poly(I:C) and 12 d later an intrastriatal injection of 5 μg of 6-OHDA. The lesion was allowed to progress for 3 weeks, after which animals were killed for postmortem analyses. B, Unbiased stereological analysis indicates that rats exposed to poly(I:C) injection, followed by a low-dose injection of 6-OHDA, had significant reduction of TH+ neurons in the SNc when compared with animals injected with low-dose 6-OHDA. The loss of TH+ neurons was reflected in the loss of NeuN+ neurons, confirming true neuronal loss. Either vehicle or poly(I:C) injection alone did not induce neuronal cell loss. C, Unbiased stereological analysis indicates no loss of TH+ or NeuN+ neurons in the VTA in any of the groups. D–G, Images of TH immunohistochemistry from single coronal sections of rats injected with vehicle, poly(I:C) alone, 6-OHDA alone, or combined insult. H–K, Images of TH (brown) and NeuN (red) immunohistochemistry from single coronal sections of rats injected with vehicle, poly(I:C) alone, 6-OHDA alone, or combined insult. Scale bar: D–G, 1.0 mm; H–K, 0.25 mm. SNr, Substantia nigra pars reticulata; CP, cerebral peduncle. Animals/group: vehicle/vehicle, n = 8; poly(I:C)/vehicle, n = 8; vehicle/6-OHDA, n = 8; poly(I:C)/6-OHDA, n = 8. *p < 0.05, ***p < 0.001, one-way ANOVA.
Intranigral injection of poly(I:C) increases the susceptibility of dopaminergic nerve terminals in the striatum to 6-OHDA toxicity
To investigate whether poly(I:C) injection produced changes in the striatal DA terminals in response to 6-OHDA neurotoxicity, we stained striatal sections for TH and performed densitometric analysis. TH immunostaining through the striatum revealed a marked reduction in DA innervation on the side ipsilateral to the combined 6-OHDA and poly(I:C)/6-OHDA lesion compared with the intact hemisphere (Fig. 8 A–D). Densitometric quantification of TH-positive fibers showed a significant reduction in animals injected with 6-OHDA alone or poly(I:C) followed by a subtoxic dose of 6-OHDA compared with control animals (79.1 ± 4.9, 64.5 ± 3.7, 97.9 ± 1.8% of the contralateral side, p < 0.01 and p < 0.001, respectively) (Fig. 8 E). There was a significant reduction of TH density in animals injected with poly(I:C) followed by 6-OHDA compared with animals injected with 6-OHDA alone (p < 0.05) (Fig. 8 E). TH fibers were preserved in the striatum of rats injected with poly(I:C) alone (95.4 ± 7.3% of the contralateral side) (Fig. 8 B,E). These data indicate that poly(I:C) injection increases the vulnerability of TH terminals in the striatum to 6-OHDA toxicity. Iba1 immunohistochemistry revealed activated microglial cells within the dorsolateral striatum of animals injected with 6-OHDA alone or poly(I:C)/6-OHDA (Fig. 8 F,G). However, the microglial reaction was more prominent in animals injected with poly(I:C) combined with a low dose of 6-OHDA (Fig. 8 G). Hypertrophied, activated GFAP+ astrocytes were detected in the dorsolateral striatum of both groups (Fig. 8 H,I).
Figure 8.

Intranigral injection of poly(I:C) increases the susceptibility of striatal DA nerve terminals to 6-OHDA toxicity. A–D, Representative striatal sections stained for TH from rats treated with vehicle (A), 20 μg of poly(I:C) (B), 5 μg of 6-OHDA (C), and 20 μg of poly(I:C) followed by 5 μg of 6-OHDA injection (D). E, Optical densitometric quantification of the TH-immunoreactive fibers in the striatum. The injection of 6-OHDA induced a significant loss of TH fibers in the striatum (p ≤ 0.01). The volume of striatal TH+ fibers in the 6-OHDA compared with poly(I:C)/6-OHDA treated rats, expressed relative to the unlesioned side, was significantly higher (p ≤ 0.05). There was no difference between vehicle-treated rats and poly(I:C)-injected rats. F, G, Immunohistochemistry against Iba1 in the striatum of 6-OHDA-lesioned and poly(I:C)/6-OHDA-lesioned rats. Microglia in the striatum of poly(I:C)/6-OHDA-lesioned rats showed activated morphology. H, I, Immunohistochemical staining for GFAP (green) demonstrated similar astrocytic reaction in 6-OHDA-lesioned and poly(I:C)/6-OHDA-lesioned rats. Scale bars: A–D, 2.0 mm; F–I, 50 μm. Animals/group: vehicle/vehicle, n = 8; poly(I:C)/vehicle, n = 8; vehicle/6-OHDA, n = 8; poly(I:C)/6-OHDA, n = 8. *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA.
Neutralization of IL-1 reduces cell loss produced by poly(I:C) and 6-OHDA
The intranigral injection of poly(I:C) induced an upregulation of IL-1 in the SN and in the dorsolateral striatum (Table 1). The proinflammatory cytokine IL-1 has been implicated as a central mediator of acute neuronal loss in experimental studies and in chronic neurodegenerative disorders (Allan et al., 2005). To investigate the potential role of IL-1 in the poly(I:C)-induced sensitization of the nigrostriatal DA system, we analyzed whether IL-1ra treatment prevented the degeneration produced by the combination of poly(I:C) and 6-OHDA. Importantly, IL-1ra has already been used in clinical settings with minor side effects, and it can be systemically delivered with favorable kinetics in crossing the BBB (Fisher et al., 1994; Nuki et al., 2002; Clark et al., 2008). All animals received an intranigral injection of poly(I:C), followed by the subcutaneous infusion of either IL-1ra (n = 12) or vehicle (n = 11) from day 9 until the animals were killed, and, finally, all animals received an intrastriatal injection of 5 μg of 6-OHDA on day 12 (Fig. 9 A). Postmortem analysis revealed that IL-1ra significantly prevented TH-IR cell death in the SN when compared with vehicle-treated controls (2916.111 ± 972.034 and 5734.3 ± 601.275; p < 0.01) (Fig. 9 B,C,F) and TH-fiber loss (91.34 ± 4.8 and 68.95 ± 4.5% of the contralateral side, respectively; p < 0.01) in the dorsolateral striatum (Fig. 9 D,E,G). These data indicate that blocking IL-1 signaling significantly reduces the degree of degeneration produced by the combination of poly(I:C) and 6-OHDA.
Figure 9.

Neutralization of IL-1 reduces cell loss induced by poly(I:C) and 6-OHDA. A, Experimental timeline: rats received a single intranigral injection of 20 μg of poly(I:C) and 9 d later started on either IL-1ra or vehicle, which continued through the experiment. All animals received an intrastriatal injection of 5.0 μg of 6-OHDA 12 d after vehicle/poly(I:C) injection and were allowed 21 d until postmortem analyses. B–E, Representative nigral and striatal sections stained for TH from rats treated with vehicle (B, D) or IL-1ra (C, E). F, Stereological quantification TH+ cells in the SNc revealed that IL-1ra prevented dopaminergic cell loss associated with poly(I:C) and 6-OHDA. G, Optical densitometric quantification of the TH-immunoreactive fibers in the striatum revealed that IL-1ra prevented TH fiber loss in the striatum. Data are shown as mean ± SEM. Vehicle, n = 11; IL-1ra, n = 12. **p < 0.01, unpaired t test. Scale bars: B, C, 0.25 mm; D, E, 1.0 mm. SNr, Substantia nigra pars reticulata.
Discussion
In this study, we developed a new model of neuroinflammation by the intraparenchymal injection of the TLR-3 agonist poly(I:C). This model reproduces the activation of the brain innate immune system occurring during noncytopathic CNS viral infections. We demonstrated that such neuroinflammatory responses activated a cascade of events (changes in the levels of proteins relevant to axonal transport and synaptic transmission, neuronal TDP-43 cytoplasmic mislocalization) that reflect the changes observed in early neurodegenerative stages. The activation of the brain innate immunity primed the nigrostriatal DA system to oxidative stress-induced degeneration. Importantly, IL-1ra attenuated degeneration in this model, confirming the neurotoxic role of cytokines.
The relationship between neurodegeneration and inflammation in PD remains essentially unknown. One intriguing hypothesis is that environmental stressors activate brain immune cells, which then cause or contribute to the degeneration of vulnerable neuronal populations, such as the DA neurons of the nigrostriatal system (Perry, 2004). Inflammation in the CNS can be initiated by immunological stimuli, such as bacterial or viral infections (Kreutzberg, 1996). Genetic susceptibility can also accelerate nigral DA neuron loss in response to inflammatory triggers (Frank-Cannon et al., 2008; Gao et al., 2008). In a theoretical framework, the fact that all humans experience bacterial and viral infections suggest that such inflammatory processes can contribute to the well known age-associated aggregate risk of PD in susceptible individuals (de Lau and Breteler, 2006). Peripheral immune responses generate inflammatory mediators that alter BBB permeability, allowing viruses to enter the CNS (Wang et al., 2004). Viruses can induce CNS dysfunction by direct cytolytic effect or bystander inflammatory reactions (van den Pol, 2006).
Poly(I:C)-induced long-lasting neuroinflammatory response in the SN and dorsolateral striatum
The intranigral injection of poly(I:C) induced a sustained neuroinflammatory reaction in the SN and in the dorsolateral striatum. Such a propagation of inflammatory responses could be important in neurodegenerative conditions as a contributor to the progressive damage (Cagnin et al., 2001; Cicchetti et al., 2002). This propagation could be explained by the result of glial–glial as well as neuron–glia communication mechanisms (Giaume et al., 2010). Focal neuronal demise can result in widespread astrocytic glial reactions, which follow the anatomical connectivity of the lesioned area (Isacson et al., 1987; de Jong et al., 2005). This hypothesis could include an inflammatory response propagated by DA neurons in the SN, which were triggered by poly(I:C) and released proinflammatory molecules at the axon terminals in the striatum. In line with this evidence, poly(I:C) injection induced an upregulation of TLR-3 that was more prominent in neurons.
Poly(I:C)-induced changes in levels of proteins involved in synaptic transmission and axonal transport
Our study shows that a viral challenge results in important changes in the levels of synaptic and axonal transport proteins in the absence of overt neurodegeneration. Notably, similar defects precede DA neuron degeneration and are coupled with neuroinflammation in a rat model of α-synucleinopathy (Chung et al., 2009). The most significant change was the increase of the postsynaptic proteins PSD-95 and NMDAR1 in the dorsolateral striatum. Increase in the expression of PSD-95 has been reported in the entorhinal and frontal cortex of Alzheimer's disease (AD) patients (Leuba et al., 2008). These changes could be attributable to compensatory mechanisms related to the reduced levels of presynaptic proteins implicated in synaptic transmission and exocytosis (RAB3B and MUNC18) or to an altered subcellular distribution. In this study, we show that axonal transport proteins are also altered after poly(I:C) injection. Therefore, PSD-95 and other spine proteins could accumulate in the dendritic shafts, showing a pattern of abnormal distribution and increased density. Importantly, a decrease of Drebrin was found in the dorsolateral striatum. Drebrin A is an actin-binding protein specifically localized at dendritic spines of mature neurons and plays a role in synaptic function and spinous clustering of PSD-95 (Takahashi et al., 2003; Aoki et al., 2005). A major loss of Drebrin A in dendritic spines has been reported in patients with AD, Down's syndrome, and aging (Harigaya et al., 1996; Hatanpää et al., 1999; Shim and Lubec, 2002).
Defects in axonal transport can be early pathogenic events in many human neurodegenerative diseases (Morfini et al., 2009). Our study revealed that anterograde transport motor proteins that carry synaptic vesicles (KIF1A and KIF1B) were markedly elevated in the SN. Other cargo containing vesicles (KIF3A and KIF17) to synaptic terminals were markedly reduced. In parallel, we found a significant decrease of KIF17 in the dorsolateral striatum. Levels of the retrograde transport microtubule motor protein dynein were decreased in the SN. Interestingly, loss of dynein function results in the misaccumulation of synaptic proteins and has been linked to neurodegeneration (Koushika et al., 2004; Banks and Fisher, 2008). This could explain the reduction of presynaptic proteins described in our model.
In summary, our findings provide evidence that a viral challenge induces changes of proteins involved in synaptic transmission and axonal transport, which reflect the early stages of human neurodegenerative diseases. This suggests that viral-mediated neuronal dysfunction may be causally linked to the onset of neurodegeneration.
Given the expression of TLR-3 on microglia, astrocytes, and neurons (Bsibsi et al., 2002), future studies will investigate whether such changes directly correlate with neuronal TLR-3 activation rather than with bystander inflammatory reactions.
Poly(I:C)-induced cytosolic mislocalization of neuronal TDP-43
The axonal transport changes observed after poly(I:C) injection were coupled with an accumulation of TDP-43 in the dorsolateral striatum and a decrease of this protein in the SN. TDP-43 pathology has been identified in 19% of PD dementia and 7% of PD cases (Nakashima-Yasuda et al., 2007). However, it is not clear whether TDP-43 has a physiological role in the response to neuronal injury and whether inflammation may alter its function. Our studies demonstrate that poly(I:C) injection induces TDP-43 redistribution to the neuronal cytosol in the absence of pathological aggregates. The increase of TDP-43 levels in the dorsolateral striatum after poly(I:C) injection could occur as a consequence of the impairment of the axonal transport machinery. Alternatively, one cannot exclude that a defective autophagic clearance could contribute to TDP-43 redistribution and accumulation.
Poly(I:C)-induced sensitization lowers threshold to neurotoxic triggers
Poly(I:C) delivered to the SN before the intrastriatal injection of a low dose of 6-OHDA produced an increase of DA cell death. These data support the hypotheses that neuroinflammation exerts a sensitizing action on the brain, inducing an aberrant response to subsequent neurotoxic challenges. Brain microglia, which were persistently activated after poly(I:C) injection, could react to 6-OHDA injection with an abnormal release of neurotoxic mediators. We also hypothesize that the initial neuroinflammatory reaction and neuronal TLR-3 activation elicited by the viral challenge may exert a priming action directly on neurons, rendering them more vulnerable to external stressors. This reasoning is suggested by the prominent upregulation of neuronal TLR-3 after poly(I:C) injection. Interestingly, TLR-3 is a negative regulator of embryonic neural progenitor cell proliferation and neurite extension (Cameron et al., 2007; Lathia et al., 2008).
IL-1ra neuroprotective effect against combined inflammatory and neurotoxic oxidative stress
Inhibition of IL-1 signaling by systemic administration of IL-1ra prevented the increased vulnerability produced by poly(I:C) and eliminated its contribution to 6-OHDA-induced DA cell death. IL-1β is one of the key proinflammatory cytokines in neurodegeneration (Allan et al., 2005). Importantly, IL-1ra can cross the BBB with favorable kinetics, and it has been proven safe in patients with sepsis, rheumatoid arthritis, and subarachnoid hemorrhage (Fisher et al., 1994; Nuki et al., 2002; Clark et al., 2008). Notably, IL-1ra determines a significant decrease of TNF-α levels, a proinflammatory cytokine that plays a key role in DA neuron degeneration (Koprich et al., 2008; McCoy et al., 2008). We therefore believe that IL-1ra may exert its neuroprotective action through multiple mechanisms, including blockade of IL-1 signaling, but also regulating other cytokines such as TNF-α.
Summary and conclusion
We have developed a new model of neuroinflammation by the intraparenchymal injection of the TLR-3 agonist poly(I:C) and performed a systematic analysis of the pathophysiological changes occurring in the DA nigrostriatal system. Our data show that the brain innate immune responses triggered by TLR-3 activation can initiate a cascade of neurodegenerative events, which reflect the changes described in the earliest stages in animal models of DA neuron degeneration (Chung et al., 2009). Overall, these data support the hypothesis that neuroinflammation plays a critical role in priming vulnerable neuronal populations to degeneration. DA midbrain neurons are highly sensitive to inflammatory “stressors,” and, over a lifetime, such neuroinflammatory events could substantially increase the number of potential degenerative changes. Based on these data, new neuroprotective therapies can be examined by targeting critical inflammatory mechanisms and molecules such as cytokines. In view of the beneficial and detrimental effects of neuroinflammation (Town et al., 2005; Colton and Wilcock, 2010), the challenging goal will be to develop immunomodulatory interventions that can restore the homeostatic level of the immune system harnessing its neuroprotective arm.
Footnotes
This study was conducted at McLean Hospital and was supported by funds from the National Institutes of Health/National Institute of Neurological Diseases and Stroke P50 Parkinson's Disease Udall Research Centers of Excellence to McLean/Harvard Medical School Grant NS39793, the Michael Stern Foundation for Parkinson's Disease Research, the Consolidated Anti-Aging Foundation, and the Harold and Ronna Cooper Family (O.I.). We thank Alyssa Yow, Kristen Lee, and Adam Levy for their excellent technical assistance.
The authors have no financial conflicts of interest.
References
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Aoki C, Sekino Y, Hanamura K, Fujisawa S, Mahadomrongkul V, Ren Y, Shirao T. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. J Comp Neurol. 2005;483:383–402. doi: 10.1002/cne.20449. [DOI] [PubMed] [Google Scholar]
- Banks GT, Fisher EM. Cytoplasmic dynein could be key to understanding neurodegeneration. Genome Biol. 2008;9:214. doi: 10.1186/gb-2008-9-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berth SH, Leopold PL, Morfini GN. Virus-induced neuronal dysfunction and degeneration. Front Biosci. 2009;14:5239–5259. doi: 10.2741/3595. [DOI] [PubMed] [Google Scholar]
- Bjorklund LM, Sánchez-Pernaute R, Chung S, Andersson T, Chen IY, McNaught KS, Brownell AL, Jenkins BG, Wahlestedt C, Kim KS, Isacson O. Embryonic stem cells develop into functional dopaminergic neurons after transplantation in a Parkinson rat model. Proc Natl Acad Sci U S A. 2002;99:2344–2349. doi: 10.1073/pnas.022438099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Alexopoulou L, Sloane JA, DiBernardo AB, Ma Y, Kosaras B, Flavell R, Strittmatter SM, Volpe J, Sidman R, Vartanian T. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007;27:13033–13041. doi: 10.1523/JNEUROSCI.4290-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Endo S, Isacson O. An endogenous serine/threonine protein phosphatase inhibitor, G-substrate, reduces vulnerability in models of Parkinson's disease. J Neurosci. 2007;27:8314–8323. doi: 10.1523/JNEUROSCI.1972-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci. 2002;15:991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- Clark SR, McMahon CJ, Gueorguieva I, Rowland M, Scarth S, Georgiou R, Tyrrell PJ, Hopkins SJ, Rothwell NJ. Interleukin-1 receptor antagonist penetrates human brain at experimentally therapeutic concentrations. J Cereb Blood Flow Metab. 2008;28:387–394. doi: 10.1038/sj.jcbfm.9600537. [DOI] [PubMed] [Google Scholar]
- Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- Dale RC, Church AJ, Surtees RA, Lees AJ, Adcock JE, Harding B, Neville BG, Giovannoni G. Encephalitis lethargica syndrome: 20 new cases and evidence of basal ganglia autoimmunity. Brain. 2004;127:21–33. doi: 10.1093/brain/awh008. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- de Jong EK, Dijkstra IM, Hensens M, Brouwer N, van Amerongen M, Liem RS, Boddeke HW, Biber K. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. J Neurosci. 2005;25:7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- Economo Von C. Encephalytis lethargica: its sequelae and treatment. London: Oxford UP; 1931. [Google Scholar]
- Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–318. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- Fisher CJ, Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, Reines HD, Shelly MP, Thompson BW, LaBreque JF, Catalano MA, Knaus WA, Sadoff JC, Astiz M, Rackow EC, Carpati C, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271:1836–1843. [PubMed] [Google Scholar]
- Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Treviño I, O'Brien DE, Casey B, Goldberg MS, Tansey MG. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28:10825–10834. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM. Neuroinflammation and oxidation/nitration of α-synuclein linked to dopaminergic neurodegeneration. J Neurosci. 2008;28:7687–7698. doi: 10.1523/JNEUROSCI.0143-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci. 2010;11:87–99. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- Harigaya Y, Shoji M, Shirao T, Hirai S. Disappearance of actin-binding protein, drebrin, from hippocampal synapses in Alzheimer's disease. J Neurosci Res. 1996;43:87–92. doi: 10.1002/jnr.490430111. [DOI] [PubMed] [Google Scholar]
- Hatanpää K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:637–643. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Isacson O, Fischer W, Wictorin K, Dawbarn D, Björklund A. Astroglial response in the excitotoxically lesioned neostriatum and its projection areas in the rat. Neuroscience. 1987;20:1043–1056. doi: 10.1016/0306-4522(87)90262-4. [DOI] [PubMed] [Google Scholar]
- Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, Smeyne RJ. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A. 2009;106:14063–14068. doi: 10.1073/pnas.0900096106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten M, Chade A, Tanner CM. Epidemiology of Parkinson's disease. Handb Clin Neurol. 2007;83:129–151. doi: 10.1016/S0072-9752(07)83006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson's disease. J Neuroinflammation. 2008;5:8. doi: 10.1186/1742-2094-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika SP, Schaefer AM, Vincent R, Willis JH, Bowerman B, Nonet ML. Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J Neurosci. 2004;24:3907–3916. doi: 10.1523/JNEUROSCI.5039-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Okun E, Tang SC, Griffioen K, Cheng A, Mughal MR, Laryea G, Selvaraj PK, ffrench-Constant C, Magnus T, Arumugam TV, Mattson MP. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. J Neurosci. 2008;28:13978–13984. doi: 10.1523/JNEUROSCI.2140-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba G, Savioz A, Vernay A, Carnal B, Kraftsik R, Tardif E, Riederer I, Riederer BM. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis. 2008;15:139–151. doi: 10.3233/jad-2008-15112. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev. 2004;3:105–120. doi: 10.1016/j.arr.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Ruhn KA, Martinez TN, McAlpine FE, Blesch A, Tansey MG. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiparkinsonian rats. Mol Ther. 2008;16:1572–1579. doi: 10.1038/mt.2008.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114:221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- Nuki G, Bresnihan B, Bear MB, McCabe D European Group Of Clinical Investigators. Long-term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:2838–2846. doi: 10.1002/art.10578. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- Perry VH. The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun. 2004;18:407–413. doi: 10.1016/j.bbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Kim JH, Greenamyre JT. Selective microglial activation in the rat rotenone model of Parkinson's disease. Neurosci Lett. 2003;341:87–90. doi: 10.1016/s0304-3940(03)00172-1. [DOI] [PubMed] [Google Scholar]
- Shim KS, Lubec G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer's disease and Down syndrome. Neurosci Lett. 2002;324:209–212. doi: 10.1016/s0304-3940(02)00210-0. [DOI] [PubMed] [Google Scholar]
- Slomianka L, West MJ. Estimators of the precision of stereological estimates: an example based on the CA1 pyramidal cell layer of rats. Neuroscience. 2005;136:757–767. doi: 10.1016/j.neuroscience.2005.06.086. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23:6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA. Microglia recognize double-stranded RNA via TLR3. J Immunol. 2006;176:3804–3812. doi: 10.4049/jimmunol.176.6.3804. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Viral infections in the developing and mature brain. Trends Neurosci. 2006;29:398–406. doi: 10.1016/j.tins.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Weller M, Stevens A, Sommer N, Melms A, Dichgans J, Wiethölter H. Comparative analysis of cytokine patterns in immunological, infectious, and oncological neurological disorders. J Neurol Sci. 1991;104:215–221. doi: 10.1016/0022-510x(91)90313-v. [DOI] [PubMed] [Google Scholar]