

Abstract
New advances in polarized light microscopy were used to image Congo red-stained cerebral amyloidosis in sharp relief. The rotating-polarizer method was used to separate the optical effects of transmission, linear birefringence, extinction, linear dichroism, and orientation of the electric dipole transition moments and to display them as false-color maps. These effects are typically convolved in an ordinary polarized light microscope. In this way, we show that the amyloid deposits in Alzheimer's disease plaques contain structurally disordered centers, providing clues to mechanisms of crystallization of amyloid in vivo. Comparisons are made with plaques from tissues of subjects having Down's syndrome and a prion disease. In plaques characteristic of each disease, the Congo red molecules are oriented radially. The optical orientation in amyloid deposited in blood vessels from subjects having cerebral amyloid angiopathy was 90° out of phase from that in the plaques, suggesting that the fibrils run tangentially with respect to the circumference of the blood vessels. Our result supports an early model in which Congo red molecules are aligned along the long fiber axis and is in contrast to the most recent binding models that are based on computation. This investigation illustrates that the latest methods for the optical analysis of heterogeneous substances are useful for in situ study of amyloid.
The abnormal transformation of proteins to amyloid fibrils is closely related to the so-called conformational diseases that include the common neurodegenerative disorders such as Alzheimer's disease (AD) and prion diseases. The kinetic and structural bases of fibrillogenesis in these diseases are as yet undetermined. Nevertheless, the presence of amyloid in diseased tissues has been used for the purpose of pathological diagnosis and construction of theories of pathogenesis. A structural characterization and categorization of various forms of amyloid aid accurate diagnosis of amyloid disorders and further our mechanistic understanding of an increasing list of conformational diseases.
The principal diagnostic criterion of amyloidosis, established by Divry and Florkin (1), is the detection with a polarizing optical microscope of so-called apple-green birefringence (2–6) from Congo red (CR)-stained tissue sections (7). Despite the durability of this assay, the optical characterization of amyloid has not progressed and is ambiguous (8, 9). The birefringence is rarely quantified, a problem further confounded by the fact that CR does not stain amyloid consistently, and diagnosis by staining depends on the skill of the investigator. Clearly, new optical-contrast mechanisms are required for simple, reliable amyloid diagnosis (10, 11).
Here, we show that recent advances in polarized light microscopy can be used to quickly quantify and refine our description of CR-stained amyloid. In particular, we applied a newly developed imaging system to separate the optical transmission, refractive index anisotropy [linear birefringence (LB)], and optical extinction, which are otherwise convolved to produce the ill-defined apple-green birefringence by conventional techniques. Moreover, we show that the system can be used to determine the absorption anisotropy [linear dichroism (LD)] and the average orientation of the electric dipole transition moment. The resulting micrographs that rely on sensitive charge-coupled device intensity measurements provide polarized light images of congophilic amyloid with greater structural resolution than reported previously. In this way, we have studied two forms of amyloid seen in the brains of AD subjects: amyloid in neuritic plaques, one of the hallmarks of Alzheimer pathology, and in the blood vessels characteristic of cerebral amyloid angiopathy (CAA). These two forms of amyloid, albeit in different anatomic structures, are both largely composed of amyloid-β protein species. For comparison, we also studied amyloid seen in Gerstmann–Sträussler–Scheinker disease (GSS), composed principally of the transformed prion protein, and plaques seen in Down's syndrome (DS), also composed of amyloid-β protein.
Materials and Methods
Tissue Preparation. All brain tissues used were obtained from the brain bank of the Alzheimer's Disease Research Center at the University of Washington. The protocols used in this study were approved by the University of Washington Institutional Review Board. The average postmortem interval for the tissues was 10 h. Five to 10 plaques from temporal cortex sections of the following subjects were analyzed for LB and LD anisotropy: 16 cases of sporadic AD (age 71–92 years), all with CAA; 5 cases of DS (age 45–68 years); and 3 cases of the telencephalic form of GSS (age 32–38 years) with the Ala-117 → Val mutation of prion protein (12). Sections were typically obtained from the midtemporal gyrus. CR staining was performed by the method of Puchtler et al. (13). Briefly, 10-μm formalin-fixed, paraffin-embedded sections were deparaffinized and hydrated. Sections then were sequentially dipped in Meyer's hematoxylin for 3 min, alkaline sodium chloride for 20 min, and CR for 50 min before dehydration with three changes of absolute alcohol and mounted.
Optical Imaging.
Microscopy. The polarizing microscope, a prototype of the MetriPol System (www.metripol.com) now available from Oxford Cryosystems (Oxford), is adapted with a stepper motor, rotating-polarizer, circular analyzer consisting of a linear analyzer and quarter waveplate aligned at 45°, an 8-bit monochrome charge-coupled device digital camera, and a personal computer with custom software able to deduce the absolute phase. The modified polarizing microscope is operated in two modes, in which the full optical path (rotating-polarizer/sample circular analyzer) and reduced path (rotating-polarizer/sample) are used to measure LB and LD, respectively. All measurements were made at ×40 magnification. Three wavelengths, 547, 589, and 610 nm, were accessed with interference filters. The measurements were calibrated for a linear camera response, quarter waveplate alignment, and polarization bias of the light source, camera, and objective. Triplets of optical images associated with transmission, birefringence or dichroism, and optical orientation appear on the computer monitor within 15 sec of centering the structure beneath the microscope objective. Image sensitivity is ≈0.5% of the transmission, 0.2 nm of the retardance LΔn, 0.2 mm of the absorbance difference LΔk, and ≈0.1° of the orientation (14).
Theory. By modulating the intensity signal as a function of the polarizer angle α, the intensity ratio I/Io (α) for each pixel is subject to a Fourier sum of the disparate optical contributions that then are displayed in false-color images representing the overall transmission, the anisotropy [refraction (LB) and absorption (LD)], and the orientation [optical extinction (LB) and transition dipole moment (LD)]. The expressions for transmitted intensity for the full and reduced optical paths are given in Eqs. 1 and 2 and are derived with Jones matrices (15).
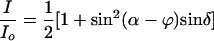 |
[1] |
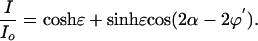 |
[2] |
Here, δ = 2πLΔn/λ (L is the thickness of the sample, Δn is the difference between the principle refractive indices or LB, and λ is the wavelength of light) is the phase shift of the extraordinary and ordinary rays at the interface of the sample, and ϕ is the orientation of the slow vibration direction as measured counterclockwise from the horizontal axis. When using the reduced light path, φ′ is the orientation of the most absorbing direction as measured counterclockwise from the horizontal direction.
The LD is measured in terms of the scaled differential transmission Δk along the eigenmodes of the sample, where ε = 2πLΔk/λ and Δk = 2(T0°–T90°)/(T0° + T90°) (T0°,90° are the transmissions along the primary polarization directions). In this way, we precisely quantified LB and LD, creating an optical matrix of the diseased tissue.
Fourier analysis of the intensity data gives images of LB that are expressed as the absolute value of the sine of the phase difference (|sin δ|). Because |sin δ| is a periodic function, the absolute value, or order, of δ is not determined by this method. This “order dilemma” was resolved by measuring the phase δ at three close wavelengths and computing the derivatives of the resulting relative phases (16). The set of signs of the three derivatives unambiguously defines the absolute phase δ and therefore the absolute birefringence.
Results
CR stains the core structure of two kinds of plaques distinguishable by Bielschowsky's silver stain: classical plaques consisting of an amyloid core and a corona and compact plaques that are cores without coronas. Representative micrographs of tissue sections containing CR-stained amyloid cores are shown in Fig. 1. Each plaque is red (λmax = 515–538 nm) (17) in linearly polarized white light (Fig. 1I). Between crossed polarizers the apple-green birefringence is apparent (Fig. 1II). Fig. 2 shows images of each pathologic structure in Fig. 1 and an additional DS plaque indicated in D. The Fourier separation of the intensity signal as a function of polarizer rotation angle yields the anisotropies of refraction, |sinδ| (I), and absorption, tanhε (III), with the related orientations of the optical extinction position, φ (II), and transition dipole moment, φ′ (IV).
Fig. 1.

CR-stained amyloid plaques characteristic of three diseases: AD (A), GSS (B), and DS (C). I, amyloid in linearly polarized white light; II, amyloid between crossed polarizers showing apple-green birefringence.
Fig. 2.

Optical images of CR-stained plaques characteristic of AD (A), GSS (B), and DS (C and D). Scales for |sin δ| and tanhε: AI, 0.07–0.23; BI, 0.07–0.28; CI, 0.05–0.24; DI, 0.05–0.18; AIII, 0.05–0.25; BIII, 0.06–0.46; CIII, 0.04–0.29; DIII, 0.04–0.16. I, |sin δ| = the absolute value of the sine of the phase difference where δ = 2πΔnL/λ; II, φ = the optical extinction angle in degrees as measured counterclockwise from the horizontal axis (I and II are measured off-resonance at 610 nm); III, hyperbolic tangent of the extinction where ε = 2πLΔk/λ and Δk = 2(T0°–T90°)/(T0° + T90°); IV, φ′ = angle in degrees from the most absorptive direction as measured counterclockwise from the horizontal axis (III and IV are measured on-resonance at 5547 nm). Pixels with values below the minimum specified here were set to black. In the centers of the plaques, sinδ and tanhε were ≈0.02.
AD plaque amyloid is characterized unambiguously by its affinity for CR (Fig. 1 AI) and apple-green birefringence (Fig. 1 AII), punctuated by a dark cross of extinction, indicating a radially ordered spherical body (1, 18). These features are reiterated in the orientation images of LB (Fig. 2 AII) and LD (Fig. 2 AIV). The colors that represent the angular orientation of the most refractive and absorbing direction, respectively, change continuously around the centers, regions below which the birefringence and dichroism are vanishingly small. Perhaps the most striking feature of the MetriPol images (see Fig. 2 AI–CIV) is the disordered center within the amyloid core, which is disguised by the extinction condition in Fig. 1II. These centers have an average diameter of 5 μm (range, 2–8 μm). In maps of transmission (not shown) these black holes clearly contain CR and presumably protein, but nevertheless anisotropy cannot be detected.
The AD plaque in Fig. 1 is compact and round, the GSS plaque is stellated and larger, and the DS plaque is somewhere in between. These morphological observations are consistent with those revealed by other microscopic methods (19, 20). Although these general characteristics were most frequently representative of each disease, all three morphologies were observed sometimes in tissue samples from subjects having suffered from each of the three diseases. A subset of DS plaques in each subject shows an alternative amyloid morphology, broken rather than radial (Fig. 2D). Morphologically they appear similar to the “fibrous plaques” described in DS (20).
The π/2 ambiguity inherent in the sinusoidal Eq. 1 governing LB was resolved by using the order method as described (16). All samples showed first-order birefringence (δ < π/2).
Fig. 3 shows CR-stained amyloid from the blood vessels of a subject burdened with CAA. Again the CR molecules are oriented radially, but there is a 90° phase shift as compared with the plaques. The phase shift is apparent when comparing the red colors representing the angle of the optical orientation in Fig. 2 AII–CII and AIV–CIV (horizontal), with Fig. 3 Lower (vertical).
Fig. 3.

Optical images of CR-stained amyloid associated with a blood vessel from a subject burdened with CAA. (A) |sin δ| (Upper) and φ (Lower). (B) tanh ε (Upper) and φ′ (Lower).
Discussion
Our in situ investigation of CR-stained amyloid illustrates a way to visualize and quantify birefringence. The images presented in Results reveal previously obscured structural features including disordered centers and well defined contours. Using the MetriPol method, we demonstrated an absolute first-order LB as well as the LD in amyloid sections. Our results define the orientation of the CR molecules with respect to the fibrils, a long-debated issue. Our results also provide clues to the in vivo mechanism of amyloid formation. Significantly, in contrast to methods used in many previous biophysical studies of amyloid, our method does not require isolation of fibrils from their tissue environment, the procedures of which potentially alter the structure of amyloid.
As well stated by Steensma (21): “At the dawn of the 21st century... the diagnostic test of choice for amyloidosis has not changed in decades... CR is still the `king of dyes'.” Therefore, it may well be of value to bring the latest methods for the optical analysis of heterogeneous substances to bear on the characterization of CR-stained amyloid. A standard procedure for staining amyloid with CR was established by Puchtler et al. (13), who surmised that the dye was attached via unspecified hydrogen bonds and “ionic linkages.” Lillie (22) invoked hydrogen bonding as the major mechanism of association, as did Glenner et al. (23), Mera and Davies (24), and Turnell and Finch (25); but Glenner et al. assigned equal importance to hydrophobic interactions, the principal noncovalent interaction according to Pigorsch et al. (26), whereas Mera and Davies invoked van der Waals forces, the principle noncovalent interaction according to Horobin (27). Turnell and Finch, leaving no stone unturned, cited the importance of hydrophobic and van der Waals interactions in addition to hydrogen bonding. Ionic forces were specified by Klunk et al. (28) as well as Puchtler (29), who later favored “nonionic” interactions, similarly preferred by Katenkamp and Stiller (30). Recent experiments were aimed at specifying more precisely the CR point(s) of attachment. Kirschner and coworkers (31, 32) identified histidine residues as the most “congophilic.” Cavillon et al. (33) preferred arginine, whereas Li et al. (34) liked lysines (as well as hydrophobic interactions). It is safe to conclude that the process of amyloid staining by CR is not fully understood.
Irrespective of the staining mechanism, simple inspection with a polarizing microscope can nevertheless provide information about the orientation of the dyes within amyloid plaques. However, there does not seem to be a consensus as to whether the transition dipole moments of the CR molecules are parallel (10, 30, 35) or perpendicular (34, 36, 37) to the fiber axes. The φ and φ′ images in Figs. 2 and 3 clearly show the radial orientation of anisotropic structures. Furthermore, their relationship suggests a solution to this fundamental question regarding the mechanism of staining of amyloid.
It is known that the amyloid fibrils in AD plaques form radial aggregates and that the transition moment in CR runs along the long molecular axis (38). Therefore, the CR molecules are aligned along the fiber as required by the fact that the fibrils lying on the horizontal axis have φ and φ′ values of ≈ 0° in Fig. 2 II and IV. In CAA there is a 90° phase shift. Assuming that the molecular mechanism of CR staining is the same in both CAA and amyloid plaques, this indicates that the fibrils run in the blood vessels. A summary of our view of CR orientation in plaques and blood vessels is shown in Fig. 4. Our judgments in this regard merely confirm what the earliest polarized light microscopists proposed; however, this judgment has curiously been disregarded in recent years.
Fig. 4.

Cartoon showing fibril and CR orientations in plaques (a) and blood vessels (b). The state of amyloid in the disorder center in a is not represented because it is unresolved.
A recent contribution to the debate on CR orientation comes from Carter and Chou (39), who built an atomic model of CR bound to amyloid by analogy with a published crystal structure of CR bound to insulin (25). In this model, CR is interleaved between the two strands of the amyloid β-sheet. Other model builders have similarly supported orthogonal CR and amyloid fibril long axes (40). This picture no longer can be supported in light of the MetriPol images that make the dichroism and the orientation of the transition moment strikingly apparent.
The anisotropy maps (Fig. 2 I and III) demonstrate disordered centers of amyloid cores despite the fact that these centers incorporated CR. This finding is reminiscent of the thioflavine S-positive and nonamyloid-β protein-immunoreactive centers of AD amyloid cores described by Schmidt et al. (20). However, the structurally defined centers observed here may not exactly correspond to the reported cytochemically defined centers. First, the former, on average, are smaller than the latter. Second, the former are equally observed in DS amyloid, whereas the latter are not. In any event, our findings of the sharp change in the order of the medium undoubtedly holds clues to the biopathological crystallization mechanism of amyloid. Are the fibrils organizing around a catalytic, disordered core, or is the mass crystallizing from the outside in, similar to a geode? An answer to this question has significant implications. Amyloid formation is considered to be a nucleation-dependent process analogous to the seeding of a crystal (41). However, the molecular composition of the “nucleus” and its seeding mechanism in amyloid formation is not understood. In vitro and in vivo evidence has suggested that amyloid formation can be seeded by a preformed fibril (11, 41). Assuming the fibrils grow around the disordered center, then the center may be composed of preformed fibrils (with affinity to CR, but not yet organized to give detectable anisotropy) as seeds for amyloid crystallization and may be composed of additional molecules that have been shown to promote amyloid formation, such as cholesterol, fatty acids, or proteoglycan. Alternatively, if the fibrils grow from the outside in, the disordered center may represent the partially structured intermediates undergoing active organization, catalyzed by surrounding structured fibrils. Our results point out the necessity of analyzing the molecular composition of the disordered centers to understand the nature of either the catalytic center or the transforming intermediates that are instrumental in the development of structured fibrils.
Comparable methods of orientation-independent imaging have been developed. These include the PolScope (42) as well as differential polarized light microscopies developed by Bustamante et al. (43), Tower and Tranquillo (44), Poenie and coworkers (45), and Ho and coworkers (46). These techniques, in principle, should provide a similar view of CR-stained amyloid.
In a very forward-thinking article, Benditt and coworkers (17) realized that a complete characterization of CR-stained amyloid ultimately would include optical rotary dispersion and circular dichroism. Such research is possible with the recently constructed circular extinction imaging microscope (47).
Acknowledgments
This work was supported by National Science Foundation Grant CHE-0092617 and National Institutes of Health/National Institute on Aging Grant 2P50 AG 05136.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AD, Alzheimer's disease; CR, Congo red; LB, linear birefringence; LD, linear dichroism; CAA, cerebral amyloid angiopathy; GSS, Gerstmann–Sträussler–Scheinker disease; DS, Down's syndrome.
References
- 1.Divry, P. & Florkin, M. (1927) C. R. Soc. Biol. 97, 1808–1810. [Google Scholar]
- 2.Missmahl, H. P. & Hartwig, M. (1952) Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 324, 489–508. [DOI] [PubMed] [Google Scholar]
- 3.Cohen, A. S. (1967) N. Engl. J. Med. 277, 522–530. [DOI] [PubMed] [Google Scholar]
- 4.Elghetany, M. T. & Saleem, A. (1988) Stain Technol. 63, 201–212. [DOI] [PubMed] [Google Scholar]
- 5.Sipe, J. D. & Cohen, A. S. (2000) J. Struct. Biol. 130, 88–98. [DOI] [PubMed] [Google Scholar]
- 6.Rosenblum, W. I. (2002) Neurobiol. Aging 23, 225–230. [DOI] [PubMed] [Google Scholar]
- 7.Bennhold, H. (1922) Muench. Med. Wochenschr. 69, 1537–1538. [Google Scholar]
- 8.Hawkins, P. N., Lavender, J. P. & Pepys, B. (1990) N. Engl. J. Med. 324, 508–513. [DOI] [PubMed] [Google Scholar]
- 9.Linke, R. P., Gärtner, H. V. & Michels, H. (1995) J. Histochem. Cytochem. 9, 863–869. [DOI] [PubMed] [Google Scholar]
- 10.Wolman, M. & Bubis, J. J. (1965) Histochemie 4, 351–356. [DOI] [PubMed] [Google Scholar]
- 11.Kisilevsky, R. (2000) J. Struct. Biol. 130, 99–108. [DOI] [PubMed] [Google Scholar]
- 12.Nochlin, D., Sumi, S. M., Bird, T. D., Snow, A. D., Leventhal, C. M., Beyreuther, K. & Masters, C. L. (1989) Neurology 39, 910–918. [DOI] [PubMed] [Google Scholar]
- 13.Puchtler, H., Sweat, F. & Levine, M. (1962) J. Histochem. Cytochem. 10, 355–364. [Google Scholar]
- 14.Glazer, A. M., Lewis, J. G. & Kaminsky, W. (1996) Proc. R. Soc. London Ser. A 425, 2751–2765. [Google Scholar]
- 15.Jones, R. C. (1941) J. Opt. Soc. Am. 31, 488–503. [Google Scholar]
- 16.Geday, M. A., Kaminsky, W., Lewis, J. G. & Glazer, A. M. (2000) J. Microsc. (Oxford) 198, 1–9. [DOI] [PubMed] [Google Scholar]
- 17.Taylor, D. L., Allen, R. D. & Benditt, E. P. (1974) J. Histochem. Cytochem. 22, 1105–1112. [DOI] [PubMed] [Google Scholar]
- 18.Kelényi, G. (1967) Acta Neuropathol. 7, 336–348. [DOI] [PubMed] [Google Scholar]
- 19.Allsop, D., Kidd, M., Landon, M. & Tomlinson, A. (1986) J. Neurol. Neurosurg. Psychiatry 49, 886–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt, M. L., Robinson, K. A., Lee, V. M. & Trojanowski, J. Q. (1995) Am. J. Pathol. 147, 503–515. [PMC free article] [PubMed] [Google Scholar]
- 21.Steensma, D. P. (2001) Arch. Pathol. Lab. Med. 125, 250–252. [DOI] [PubMed] [Google Scholar]
- 22.Lillie, R. D. (1977) in H. J. Conn's Biological Stains (Williams & Wilkins, Baltimore), 9th Ed., pp. 147–148.
- 23.Glenner, G. G., Eanes, E. D., Bladen, H. A., Linke, R. P. & Termine, J. D. (1974) J. Histochem. Cytochem. 22, 1141–1158. [DOI] [PubMed] [Google Scholar]
- 24.Mera, S. L. & Davies, J. D. (1983) J. Pathol. 141, 547–547. [Google Scholar]
- 25.Turnell, W. G. & Finch, J. T. (1992) J. Mol. Biol. 227, 1205–1223. [DOI] [PubMed] [Google Scholar]
- 26.Pigorsch, E., Elhaddaoui, A. & Turrell, S. (1994) Spectrochim. Acta, Part A 50, 2145–2152. [Google Scholar]
- 27.Horobin, R. W. (1980) J. Microsc. (Oxford) 119, 345–355. [DOI] [PubMed] [Google Scholar]
- 28.Klunk, W. E., Pettegrew, J. W. & Abraham, D. J. (1989) J. Histochem. Cytochem. 37, 1273–1281. [DOI] [PubMed] [Google Scholar]
- 29.Puchtler, H., Waldrop, F. S. & Meloan, S. N. (1985) Appl. Pathol. 3, 5–17. [PubMed] [Google Scholar]
- 30.Katenkamp, D. & Stiller, D. (1972) Histochemie 29, 37–43. [DOI] [PubMed] [Google Scholar]
- 31.Kirschner, D. A., Inouye, H., Duffy, L. K., Sinclair, A., Lind, M. & Selkoe, D. J. (1987) Proc. Natl. Acad. Sci. USA 84, 6953–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inouye, H., Nguyen, J. T., Fraser, P. E., Shinchuk, L. M., Packard, A. B. & Kirschner, D. A. (2000) Amyloid 7, 179–188. [DOI] [PubMed] [Google Scholar]
- 33.Cavillon, F., Elhaddaoui, A., Alix, A. J. P., Turrell, S. & Dauchez, M. (1997) J. Mol. Struct. 408–409, 185–189. [Google Scholar]
- 34.Li, L., Darden, T. A., Bartolotti, L., Kominos, D. & Pedersen, L. G. (1999) Biophys. J. 76, 2871–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glenner, G. G. & Page, D. L. (1976) Int. Rev. Exp. Pathol. 15, 1–92. [PubMed] [Google Scholar]
- 36.Cooper, J. H. (1974) Lab. Invest. 31, 232–238. [PubMed] [Google Scholar]
- 37.Elhaddaoui, A., Pigorsch, E., Delacourte, A. & Turrell, S. (1995) J. Mol. Struct. 347, 363–369. [Google Scholar]
- 38.Gueft, B. & Ghidoni, J. J. (1963) Am. J. Pathol. 43, 837–854. [PMC free article] [PubMed] [Google Scholar]
- 39.Carter, D. B. & Chou, K.-C. (1998) Neurobiol. Aging 19, 37–40. [DOI] [PubMed] [Google Scholar]
- 40.George, A. R. & Howlett, D. R. (1999) Biopolymers 50, 733–741. [DOI] [PubMed] [Google Scholar]
- 41.Harper, J. D. & Lansbury, P. T., Jr. (1997) Annu. Rev. Biochem. 66, 385–407. [DOI] [PubMed] [Google Scholar]
- 42.Oldenbourg, R. & Mei, G. (1995) J. Microsc. (Oxford) 180, 140–147. [DOI] [PubMed] [Google Scholar]
- 43.Bustamante, C., Kim, M. & Beach, D. A. (1988) Polarization Spectrosc. Ordered Syst. 242, 313–356. [Google Scholar]
- 44.Tower, T. T. & Tranquillo, R. T. (2001) Biophys. J. 81, 2954–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhn, J. R., Wu, Z. & Poenie, M. (2001) Biophys. J. 80, 972–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newton, R. H., Haffegee, J. P. & Ho, M. W. (1995) J. Microsc. (Oxford) 180, 127–130. [Google Scholar]
- 47.Claborn, K., Puklin-Faucher, E., Kurimoto, M., Kaminsky, W. & Kahr, B. (2003) J. Am. Chem. Soc., in press. [DOI] [PubMed]