

Abstract
Bone is one of Nature’s most remarkable materials, not only for its mechanical properties but also for its ability to repair fractures and remodel its microstructure in response to stress. At the nanoscale bone is a supramolecular matrix of collagen fibers reinforced by hydroxyapatite crystals with a high degree of order. Emulating elements of the biological synthesis of this composite could help develop strategies for advanced materials. Previous work has demonstrated the use of functionalized peptide amphiphile nanofibers in a two-dimensional system to emulate hydroxyapatite mineralization in natural bone. We describe here an artificial, in vitro biomineralization process that allows a similar process to occur in three dimensions. The system employs the natural enzyme alkaline phosphatase and a phosphorylated, anionic nanofiber gel matrix to template hydroxyapatite nanocrystals with size, shape, and crystallographic orientation resembling natural bone mineral. The formation of this biomimetic mineral in three dimensions results from the synergy of fiber-induced nucleation and the temporal control of phosphate ion harvesting by the enzyme. Gradual enzymatic harvesting of ions for crystal growth and the strong nucleating ability of the phosphorylated fibers suppresses uncontrolled precipitation of mineral. The strategy could lead to biomimetic materials to promote bone regeneration or the synthesis of hybrid materials with crystallographically defined structures.
Keywords: Biomineralization, Hydroxyapatite Nanocrystals, Supramolecular Materials, Synthetic Biology, Gels
1. Introduction
Bone is one of the most remarkable materials in Nature, a hard composite material with a hierarchical structure that hosts cells with ability to dynamically remodel in response to stress. In addition to mechano-responsive properties, bone has some capacity to repair but novel bioactive materials are still needed to promote its regeneration in large defects and extensive fractures. The matrix structure that supports bone functions consists of hydroxyapatite nanocrystals embedded within a nanofibrous framework of collagen triple helices. Intimately distributed into this composite matrix is a collection of proteins that regulate crystal growth rates, nanoscale organization, and the crystallographic orientation of the hydroxyapatite (HA) nanocrystals. [1, 2] Alkaline phosphatase (ALP) is one particularly important enzyme involved in these processes. Secreted by osteoblasts, ALP liberates phosphates necessary for HA mineralization from organic phosphates. In addition, other proteins such as osteopontin, osteocalcin, osteonectin, and bone sialoprotein, are believed to play more functional roles in regulating mineral growth.[3–7] The important functional and enzymatic roles of these non-collagenous proteins indicate that mimicking the natural “templating” of bone mineral systems may therefore require more than just providing a physical template for calcium phosphate nucleation. Mimicking some aspects of these biological processes in synthetic systems may offer interesting strategies in the design of advanced materials.
Artificial materials containing bone mineral with biomimetic features are of interest as matrices for bone regeneration.[8–11] If these matrices were to grow their mineral components in vivo, they would integrate more seamlessly with surrounding pre-existing or regenerated bone. A biomimetic mineral component in these matrices, for example nanoscale plate-like crystals, may be more readily remodeled in the biological environment. Finally, biomimetic systems could also incorporate, by design, bioactivity to promote bone regeneration. Efforts to artificially produce biomimetic mineral on organic matrices so far have focused mostly on design and function of the structural template using modified collagen, peptides, and polymers.[12–16]. In previous work we demonstrated that peptide amphiphile nanofibers could nucleate hydroxyapatite nanocrystals with biomimetic features but only in two-dimensional systems [13]. The substrate for mineralization consisted of nanofibers coated on a two-dimensional porous surface. When these nanofibers were exposed to calcium and phosphate ion solutions on top and bottom surfaces of the construct mineralization occurred producing HA nanocrystals with biomimetic features including size, shape, and crystallographic orientation of the HA with respect to organic nanofibers. The present study attempts to extend this biomimetic process to a three-dimensional gel matrix that could be used for bone regeneration.
The artificial nanofiber matrix described in the present work is a three-dimensional network of peptide amphiphile nanofibers.[13, 17] These artificial supramolecular assemblies are not intended to mimic, functionally or structurally, any single bone protein. The PA nanofibers were designed to assemble into a scaffolding framework such as that established naturally by collagen. In addition the nanofibers’ surfaces are functionally enriched in concentrated arrays of acidic and phosphorylated residues believed to be important in bone mineralization.[3, 18–21] This scaffold would provide, by design, the nucleation sites for enzymatically-mediated hydroxyapatite formation. Employing this designer matrix to direct the spatial component of hydroxyapatite templating, we investigate here the role of the natural enzyme alkaline phosphatase to control the temporal variables involved in three-dimensional mineral templating. Our motivation is to understand the complex mineralization processes involved in bone matrix mineralization and adapt the strategies to the design of materials.
2. Results and Discussion
The PA used to form our nanofiber gel scaffolds is shown in Figure 1. When calcium chloride was added to 1 % by weight aqueous solution of these molecules, charge neutralization and ionic bridging of negatively charged scaffold molecules led to the formation of a self-supporting, three-dimensional gel. This gel is composed of cylindrical nanofibers, approximately 5–7 nm in diameter, that form as the amphiphilic molecules self-assemble to emulate the basic nanofibrous architecture of a collagen matrix. Calcium ions bind to phosphorylated serine and aspartic acid residues decorating the nanofiber exterior. The ability of the fibers to bind calcium can mimic proteins that naturally template mineral. The gel could therefore be viewed as an extremely primitive form of unmineralized osteoid in bone. The in vitro enzyme-mediated mineralization of these nanofiber networks was investigated here using PA gels formed in calcium-supplemented culture medium. These gels were covered with standard osteoblastic culture medium that contained both the natural enzyme alkaline phosphatase (291 mUnits/mL in fetal bovine serum additive), and β-glycerolphosphate as a supplementary organic phosphate source. Samples were thus incubated at 37°C, refreshing 50% of the culture medium every 2 days. The macroscopic three-dimensional mineralization of the PA nanofiber gels is visually illustrated by the photographs in Figure 2. Shortly after gel formation, the gel was visibly clear. By the 4th day of incubation, the gel became slightly cloudy but the culture medium above it remained transparent. By day 8, mineralization became visibly apparent, with coarse aggregates of mineral forming throughout the volume of the gel.
Figure 1.

Schematic illustration of peptide amphiphile self-assembly into a nanofiber. The scanning electron micrograph (top right) and transmission electron micrograph (bottom right) show fibrous bundles, made up of peptide amphiphile nanofibers approximately 5–7 nm in diameter. The scanning electron micrograph was obtained from a critical-point dried gel, while the transmission electron micrograph shows nanofibers dried on a TEM grid and stained with phosphotungstic acid.
Figure 2.

Time sequence photographs of a mineralizing, three-dimensional peptide amphiphile matrix. The self-supporting gel was initially clear. After 4 days, the gel became noticeably cloudy. By 8 days, a coarse mineralized texture indicates large scale hydroxyapatite mineralization.
Fourier transform infrared (FTIR) studies of a lyophilized, mineralized PA gel revealed apatitic phosphate bands between 563 and 1215 cm−1 and the emergence of a broad hydroxyl band around 3400 cm−1 suggesting the formation of hydroxyapatite. Furthermore, amide I, amide II, and amide III bands from the peptide portion of the PA were observed in both mineralized and unmineralized gels between 1350 and 1900 cm−1, and bands from the aliphatic tail of the PA were visible between 2800 and 3000 cm−1. Deconvolution of the amide I region showed a band at 1633 cm−1 in both the original and mineralized gels, suggesting the persistence of nanofibers with β-sheet internal structure during mineralization. As expected, there were no apatitic phosphate bands present in the spectra of unmineralized gels, but there were relatively broad phosphate bands, produced by the phosphorylated serine present in PA nanofibers in the range 563 to 1215 cm−1.
Transmission electron microscopy (TEM) revealed mineralized structures approximately 200–250 nm long and 75–100 nm wide. These structures were made up of stacks of irregularly shaped, plate-like crystals approximately 50–75 nm wide, 100–200 nm long, and only 4–6 nm thick. (Figure 3a, c). Electron diffraction (Figure 3b, d) and energy-dispersive x-ray spectroscopy in the TEM, collectively confirmed that the emerging mineral phase was hydroxyapatite (Ca5(PO4)3OH) with a calcium to phosphorus ratio of 1.675 ± 0.015. Furthermore, arcing of the (002) diffraction ring (Figure 3d) observed by electron diffraction further revealed that the crystallographic c-axes of the crystals forming these aggregates were aligned along the long axis of each individual ellipsoidal aggregate. The morphology of these ellipsoidal nanocrystal clusters closely resembles the crystallographically-aligned HA nanoclusters observed in biologically mineralized tissues such as bone and dentin.[1, 2]
Figure 3.

Transmission electron micrographs of elongated hydroxyapatite nanocrystal aggregates grown on peptide amphiphile nanofibers (a) and (c). Nanofibers are visible as dim lines connecting the HA aggregates in (c). (b) Selected area electron diffraction pattern from (a) with diffraction rings matching diffraction spacings for HA. (d) Selected area diffraction pattern from the circular region marked in (c). Arcing of the (002) reflection (arrows) reveals the crystallographic alignment of the nanocrystals with the long axis of the elliptical aggregate.
The earliest stages of mineralization can be observed in the transmission electron micrograph of Figure 4a. These nanofibers are contained within a slice of fixed and epoxy-embedded three-dimensional gel obtained after 2 days of incubation. The dark contrast highlighting the nanofibers indicates they are decorated with relatively electron-dense inorganic calcium phosphate. Neither visually nor by electron diffraction is there evidence of crystal growth observed in these samples.
Figure 4.
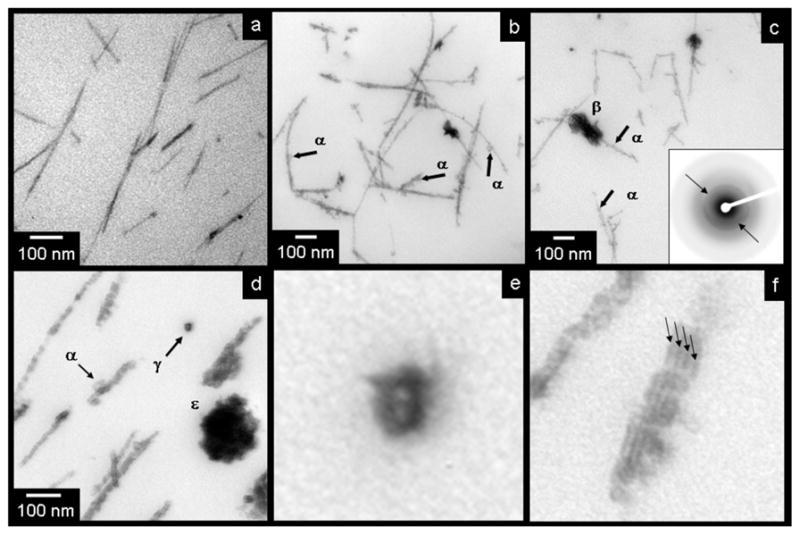
Transmission electron micrographs taken from slides of epoxy-embedded three-dimensional nanofiber gels showing stages of mineral evolution. (a) Peptide amphiphile nanofibers stained with calcium phosphate “premineral” after 2 days of mineralization. (b–c) Nanofibers with nucleated hydroxyapatite crystals after 5 days of mineralization. Crystal nuclei indicated by “α” while aggregates of stacked crystal plates are visible as well (β). (d) Advanced crystal growth (11 days), visible on peptide amphiphile nanofibers. (α) shows another growing crystal seed. (ε) shows an ellipsoidal aggregate viewed in cross-section, and (γ) shows two bundled, mineralizing nanofibers in cross section. (e) and (f) are high magnifications of features in (d) illustrating the formation of mineral around the nanofiber exteriors and between bundled nanofibers. Arrows in (e) illustrate dark bands associated with mineral growing at the junctions between bundled fibers.
TEM images in Figures 4b–d reveal later stages of mineralization. The images in Figures 4b from gel samples that had mineralized significantly after 5 days (Figure 4b, 4c), and samples extensively mineralized after 11 days (4d). The initial mineralization seen on the nanofiber surfaces after 2 days evolves into a nanocrystalline seed phase (examples indicated by arrows labeled (α) in Figures 4b–d in these later stages. These individual “seeds” then eventually grew into the stacked nanocrystal aggregates seen in Figure 3. Appearing as elongated ellipses from the side (β) and as circles in cross-section (ε), these nanocrystalline aggregates are believed to be three-dimensional ellipsoids, forming stacks of hydroxyapatite plates, much like those found in natural bone tissue.[2] These crystallographically-aligned, oblong aggregates were oriented along the long axis of the templating nanofibers, as evidenced by the arcing seen in the (002) diffraction pattern inset in Figure 4c. This is a crystallographic feature observed in natural bone with respect to collagen morphology.[2, 13]
The label γ in Figure 4d identifies 2 bundled nanofibers viewed in cross-section and coated with mineral. At low magnification, this appears as a dark ring around a lighter center. In the higher magnification image of Figure 4e, one can discern two bright spots, separated by a thin dark band of higher contrast material. This contrast pattern suggests that the inorganic coating formed around the negatively-charged nanofiber surfaces. The image further suggests that mineral was capable of forming between bundled nanofibers. This effect can also be seen in the high magnification image of Figure 4f, showing the dark bands of mineral formed between the periodic junctions among bundled nanofibers. These observations support strongly the role of nanofiber surfaces in nucleating the mineral phase.
We probed directly the role of a matrix in the templated three-dimensional mineralization observed. When a matrix was not present at all, mineral formation was not observed even in the presence of β-glycerolphosphate and ALP. Furthermore, we replaced the PA by collagen or alginate gels and carried out identical mineralization experiments. The mineralization behavior of the PA nanofiber gel was compared to that of control gels of collagen and alginate, commonly used for tissue engineering applications. Solubilized collagen was gelled by warming to 37°C in standard culture medium, while alginate gels were prepared by mixing soluble alginate with heavily calcium-enriched (170 mM Ca2+) culture medium. As indicated by the scanning electron micrographs of Figure 5, two of the matrices used, PA and collagen, were composed of nanofibers where the acid-rich PA nanofibers were relatively enriched with calcium ions bound during PA gelation. The alginate gel, on the other hand, was a non-fibrous matrix, strongly enriched with calcium. The photographs below each scanning electron micrograph in Figure 5 show that after 14 days of incubation, the PA gels formed coarse, white mineral, as expected. In contrast, however, neither the collagen nor the alginate gels formed mineral detectable either visibly or by TEM. These data suggest that neither a nanofiber matrix alone, nor calcium enrichment alone will promote the ALP-mediated, templated mineralization observed in the PA gels. These results are consistent with previously published reports using ALP to promote mineralization of collagenous substrates.[21–23] Successful mineralization in these cases was demonstrated using collagen nanofibers matrices covalently functionalized with the heavily phosphorylated glycoprotein phosphovitin. The engineered PA matrix studied here integrates by design negatively charged phosphorylated residues on its constituent nanofiber surfaces.
Figure 5.

Top, scanning electron micrographs of unmineralized gels of (a) reconstituted collagen, (b) alginate, and (c) peptide amphiphile nanofibers, alginate. All gels were critical point dried to preserve their three-dimensional structure. Bottom, photographs comparing mineral formation after 14 days in a collagen gel, an alginate gel, and a peptide amphiphile gel. The coarse white mineral visible in the PA gel was not observed in collagen or alginate gels.
To better understand the specific, functional role the PA may be playing in this process, we considered biological examples such as osteocalcin’s interaction with hydroxyapatite. Based on the recently obtained crystal structure of osteocalcin, one might hypothesize its mineral nucleation role during bone formation. This is suggested on the basis that the distance between calcium ions bound by acidic residues in this protein appear to coincide with the distance between calcium ions in the hydroxyapatite crystal lattice.[24] Others have suggested that in heavily phosphorylated proteins, such as phosphophoryn in dentin, anionic residues may have an “epitaxial match” to the surface of hydroxyapatite that may influence hydroxyapatite growth.[18, 19, 25] In the present work, x-ray diffraction analysis of PA nanofibers gelled in aqueous calcium chloride show no explicit peaks associated with hydroxyapatite crystallization, but do show a diffraction peak corresponding to 5.46 Ǻ. (Figure 6a) This spacing matches remarkably well the interatomic spacing (5.45 Ǻ) between calcium ions in the (002) planes of the HA crystal (Figure 6b). Interestingly, these calcium ions sit on a plane rich in phosphate ions with each calcium ion bound and supported by multiple phosphates in the inorganic lattice. (Figure 6c)
Figure 6.

(a) A powder XRD scan of calcium gelled peptide amphiphile showing a significant peak at 5.46 Ǻ. Peaks corresponding to hydroxyapatite were not observed, and this peak was not present in peptide amphiphile not exposed to calcium ions. (b) Schematic representation of the hydroxyapatite crystal structure as viewed down the c-axis. Atoms are shown as Ca = ( ), P =
), P =  ), O =
), O =  ), H = (○). The white lines depict the borders of the unit cell, while the yellow lines trace interatomic spacings corresponding to 5.45 Ǻ. These calcium ions are arranged hexagonally throughout the 002 planes of the hydroxyapatite crystal. (c) A model of the hydroxyapatite crystal structure as viewed along the [110] direction, showing the planes containing the 5.45 Ǻ-spaced calcium ions (yellow arrows) sitting immediately above planes rich in phosphates (blue arrows). (d) A visualization of a hydroxyapatite crystal nucleating off calcium ions spaced 5.46 Ǻ apart on the PA nanofiber. The hydroxyapatite crystal is shown with the c-axis parallel to the long axis of the PA nanofiber.
), H = (○). The white lines depict the borders of the unit cell, while the yellow lines trace interatomic spacings corresponding to 5.45 Ǻ. These calcium ions are arranged hexagonally throughout the 002 planes of the hydroxyapatite crystal. (c) A model of the hydroxyapatite crystal structure as viewed along the [110] direction, showing the planes containing the 5.45 Ǻ-spaced calcium ions (yellow arrows) sitting immediately above planes rich in phosphates (blue arrows). (d) A visualization of a hydroxyapatite crystal nucleating off calcium ions spaced 5.46 Ǻ apart on the PA nanofiber. The hydroxyapatite crystal is shown with the c-axis parallel to the long axis of the PA nanofiber.
In analogy to osteocalcin, we suggest that the negatively-charged, phosphorylated nanofiber exterior may have served to similarly coordinate and support calcium ions bound to the PA nanofibers during their assembly, creating the periodicity observed by x-ray diffraction. This calcium-enriched, nanofibrous matrix would then have presented ideal nucleation sites for subsequent hydroxyapatite mineralization. In figure 6d, we illustrate one possibility of how these calcium ions could have led to a primitive epitaxial nucleation such that the c-axis of newly growing HA crystals would be normal to the arrayed calcium ions on the PA, parallel to the long axis of the nanofiber, a result observed experimentally. Though complete confirmation of this idea warrants a comprehensive investigation beyond the scope of the present work, our hypothesis fits well with the observations presented here and offers a valuable insight on bone matrix mineralization.
This strong association between the nanofibers and the mineral can also be inferred from the selectivity of the mineral formed. Although mineral was found throughout the volume of the gel, it was grown exclusively on the nanofibers. This point is consistent with the observation that during mineralization, the culture medium above the mineralizing gels remained clear and mineral free. We deduce that this selectivity for the nanofiber surface is an important consequence not only of the PA’s functional affinity for HA, but also of the gradual, enzymatically-templated mineral formation. This idea is suggested on the basis that a three dimensional mineralization nucleated by organic fibers is very much dependent on the source of phosphate ions for hydroxyapatite formation. Phosphates gradually introduced through ALP hydrolysis of β-glycerolphosphate produced the mineralization described above. When β-glycerolphosphate was not added to the incubating solution, mineralization did not occur. When the organic phosphate was replaced with molar equivalents of soluble inorganic sodium phosphate (Na2HPO4), visible amorphous calcium phosphate quickly precipitated non-selectively out of solution (< 1 hour) (Figure 7). Clearly, the presence of the organic phosphates as the anion source for mineralization was a key requirement for nanofiber-nucleated mineralization. Just as important, however, was the presence of the enzyme needed to harvest phosphate ions to incorporate into the growing crystals. Experiments in which FBS was not added to the incubating solutions, mineralization was not observed over the course of several weeks. If, at this advanced time point however, 0.5 units of purified alkaline phosphatase were added to the incubation medium, the progression of templated gel mineralization described above began with visible mineral forming within 2–4 days.
Figure 7.

Amorphous calcium phosphate formed when β-glycerophosphate was replaced by sodium phosphate (Na2HPO4) as the phosphate source in the mineralization solution. X-ray diffraction and energy dispersive x-ray spectroscopy confirm that the aggregated spherical materials visible in the scanning electron micrograph are amorphous calcium phosphate.
These data clearly point to the enzyme-mediated nature of this mineralization process, and the ALP may play multiple roles in this complex process. Since ALP-mediated mineralization does not occur in the absence of β-glycerolphosphate it is clear that one of the key roles of the ALP is to harvest phosphates from the organophosphates for subsequent mineralization. The gradual nature of this enzymatic process provides a critical regulation of free phosphate concentration, preventing rapid, uncontrolled nonspecific mineralization in the three dimensional incubating medium. The use of phosphates bound as β-glycerophosphate keeps the phosphate concentration undersaturated with respect to homogeneous precipitation of amorphous calcium phosphate. This process allows all the reagents, calcium, phosphates, and alkaline phosphatase, to uniformly disperse among the three-dimensional nanofiber matrix, prior to mineralization. As the ALP begins to hydrolyze the β-glycerophosphate, free phosphate concentration first becomes saturated with respect to heterogeneous nucleation of calcium phosphate on nanofiber surfaces. Nanofiber surfaces are strongly enriched with negatively-charged and phosphorylated amino acid residues and we expect they are densely decorated with calcium ions bound during gelation. As discussed above these “premineralization” surfaces should be extremely favorable sites for heterogeneous nucleation of apatite crystals. As the nanocrystals are nucleated and grow, they consume newly enzyme-liberated ions, thus establishing an equilibrated phosphate concentration below the saturation level required for non-specific, homogeneous mineralization.
The phosphorylated nanofiber matrix might have additionally played a more direct role in mineralization in the presence of ALP. Using a Malachite Green assay that indicates the presence of free phosphates, we observed spectroscopically that free phosphates are enzymatically liberated from PA nanofibers incubated with ALP. Average absorbance measured at 650 nm for the PA gel in the presence of ALP was 1.57+/− 0.016, significantly higher than the average value of 0.289 +/− 0.052 measured for the PA in the absence of ALP. Hydrolysis of the phosphorylated residues in our synthetic peptide amphiphile assembly is consistent with reports by Schnepp, et al, [26] who described the use of ALP to hydrolyze phosphorylated residues in a synthetic peptide as a mechanism to drive the assembly of a mineralization competent matrix. While this hydrolysis in our system was not intended to drive assembly, our observation has several potentially important implications. First, it suggests that the ALP has an inherent affinity for the PA matrix that may locally concentrate ALP around the nanofibers. Second, our results suggest that the phosphorylated PA nanofibers serve as a source of phosphates available specifically through enzymatic hydrolysis. Both of these two scenarios would increase the free phosphate concentration immediately surrounding the PA nanofibers. Not only could these ions be incorporated into newly forming mineral, but the proximity of these ions may also contribute to the observed selectivity of the templating process. Finally, reports have described that although phosphorylated proteins commonly induce hydroxyapatite mineralization, phosphorylated residues, particularly at high concentration may inhibit hydroxyapatite mineralization.[20, 27, 28] Others have suggested that a possible role for ALP in biomineralization involves inducing mineral formation through the enzymatic dephosphorylation of these inhibitory agents.[29, 30] In the present system, it is possible that as ALP hydrolyzes some fraction of the heavily phosphorylated PA nanofibers, potential inhibitory influences of excess phosphorylation in the PA will be reduced. Subsequently, the strong HA nucleating mechanism described above would lead to the spatially selective three-dimensional mineralization we observed.
3. Conclusions
We have described here a gel matrix that templates biomimetic hydroxyapatite mineralization. Using a functionalized PA nanofiber scaffold, we have shown that enzymatically-mediated harvesting of phosphate ions combined with nanofiber surface nucleation can lead to a spatially selective and biomimetic mineralization in a three dimensional environment. This work suggests that both spatial and temporal elements are necessary to achieve biomimetic mineralization in synthetic materials.
4. Experimental
Solvents and reagents were purchased from Fisher Scientific and Sigma-Aldrich respectively, while amino acids and resins used in peptide synthesis were provided by NovaBiochem (San Diego, CA) and Applied Biosystems (Foster City, CA). Culture medium was purchased from Invitrogen, Inc (Carlsbad, CA). Fetal bovine serum was obtained from Hyclone, Inc (Logan, UT). All materials and reagents for electron microscopy were purchased from Electron Microscopy Sciences (Fort Washington, PA).
The synthesis of the peptide portion of the molecule was performed using standard solid phase synthesis on an Applied Biosystems 433A automated peptide synthesizer. The peptide was grown on an aspartic acid-functionalized Wang polystyrene resin, using 0.95 molar equivalents of HBTU and 6 equivalents of diisopropylethylamine (DIEA) for each new amino acid coupled to the resin. A C16 alkyl tail was subsequently added to the N-terminus of the peptide manually, by adding 3 molar equivalents of palmitic acid to the peptide, in the presence of 0.95 palmitic acid molar equivalents HBTU and 12 peptide molar equivalents of DIEA. The peptide amphiphile was then cleaved from the polystyrene resin and amino acid side groups were deprotected in 95% trifluoroacetic acid (TFA), 2.5% triisopropylsilane (TIS), 2.5% deionized water. TFA was removed in a rotary evaporator and the peptide was collected by precipitation in cold diethyl ether. Filtered product was dried and then redissolved at 10 mg/mL in slightly basic water (pH = 7.5–8) and distributed in 1 mL aliquots. At this stage the replacement of trifluoroacetate bound to positively charged residues was achieved by treating these aliquots with 100 mL of 0.1 M hydrochloric acid (HCl). These aliquots were lyophilized and stored at −30°C. Electrospray Ionization Mass Spectrometry performed on solutions diluted to 1 mg/mL was used to confirm the peptide sequence.
Lyophilized PA was reconstituted at 10 mg/mL in deionized water and 50 mL of this clear solution were delivered to individual wells of 8-well chamber slides. 50 mL of calcium-supplemented culture medium were mixed with the solutions taking care not to disrupt newly forming gel. Collagen control samples were prepared using 3 mg/mL soluble type I bovine collagen (Vitrogen: Cohesion Technologies, Inc., Palo Alto, CA). The collagen solution was mixed in equal volumes with the calcium-supplemented cell suspension and incubated for 5 minutes at 37 °C. 100mL of this mixture were then placed in individual wells of 8- well chamber slides where they were gelled by incubation at 37°C. Alginate gels were prepared using 2% sodium alginate from brown algae (Sigma-Aldrich), dissolved in hank’s balanced salt solution. 50 mL of this solution were delivered to individual wells of 8-well chamber slides. Gelation was achieved by addition of 50 mL of culture medium supplemented with calcium to a concentration of 170 mM CaCl2. All gels were then incubated at 37°C and 100% humidity in 5% CO2 for 2 hours to insure complete gelation. Subsequently, 500 mL of culture medium were added to chamber wells to cover the bubble of each gel. Samples were incubated as above, changing 250 mL of culture medium every 2 days.
Unmineralized gels were examined by wicking dilute suspensions of PA nanofibers from TEM grids. Grids were then negative stained by wicking an aqueous solution of 2.5% phosphotungstic acid from the grids and allowing to air dry. Gelled samples to be evaluated by electron microscopy were removed from their culture wells, fixed for 1 hour in 2.5% glutaraldehyde in 0.1M sodium cacodylate buffer. Fixed samples were then dehydrated in graded ethanol solutions (50%, 70%, 80%, 90%, 95%, 100%). Ethanol was exchanged in two changes of propylene oxide, followed by a 50% propylene oxide/EPON epoxy resin mix. Samples were infiltrated with resin by successive exchanges with pure EPON before final epoxy curing at 50°C for 24 hours, 60°C for 24 hours, 70°C for 24 hours. Sections were microtomed to 100 nm sections and placed onto copper TEM grids. Sections were viewed on a JEOL 100CX transmission electron microscope at 80kV. Gels to be examined by scanning electron microscopy were fixed and dehydrated as above, but were then critical point dried by CO2 exchange. Dried gels were sputter-coated with 6 nm Au-Pd and examined in a Hitachi S4500 Scanning Electron Microscope at 5–10 kV.
Both Transmission Fourier transform infrared spectroscopy (FTIR) and powder X-ray Diffraction (XRD) were performed on lyophilized powders of mineralized and non-mineralized gels. For FTIR the powders were mixed with potassium bromide and pressed into pellets for analysis using a Bio-Rad FTS-40 FTIR spectrophotometer. XRD studies were performed by dispersing the lyophilized PA on a “zero-background” quartz holder for examination with a Siemens D500 theta/2-theta diffractometer using CuKa radiation at 40kV and 30mA.
To determine if ALP could enzymatically liberate phosphates from the PA, samples of both assembled and non-assembled PA were probed using a commercial Malachite Green Assay (BioAssay Systems, Hayward, CA.) Free phosphates produced were determined by the absorbance measured at 650 nm on a plate-reader. The PA was tested at a concentration of 10 mg/mL in pure water (or the aqueous calcium chloride solution used to gel the PA), and was treated with 0.5 units of alkaline phosphatase for 10 minutes before measurement.
Supplementary Material
Footnotes
This work was supported by Department of Energy grant DEFG02-00ER45810 and NIH grant 5R01DE015920. The authors are grateful to Krista L. Niece from the authors’ laboratory for assistance with peptide amphiphile synthesis as well as Bonnie McKenzie and Ralph Tissot from SNL for assistance with scanning electron microscopy of amorphous calcium phosphates, and powder x-ray diffraction respectively. EDS acknowledges support by the Department of Energy Basic Energy Sciences program and use of x-ray and electron microscopy facilities at Sandia National Laboratories. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy’s National Nuclear Security Administration under Contract DE-AC04-94AL85000.
References
- 1.Olszta M, Cheng X, Jee S, Kumar R, Kim Y, Kaufman M, Douglas E, Gower L. Mater Sci and Eng R-Reports. 2007;58:77. [Google Scholar]
- 2.Weiner S, Wagner H. Annu Rev Mater Sci. 1998;28:271. [Google Scholar]
- 3.Gorski J. Calcif Tiss Int. 1992;50:391. doi: 10.1007/BF00296767. [DOI] [PubMed] [Google Scholar]
- 4.Hunter G, Goldberg H. Proc Natl Acad Sci U S A. 1993;90:8562. doi: 10.1073/pnas.90.18.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunter G, Goldberg H. Biochem J. 1994;302:175. doi: 10.1042/bj3020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salgado A, Coutinho O, Reis R. Macromol Biosci. 2004;4:743. doi: 10.1002/mabi.200400026. [DOI] [PubMed] [Google Scholar]
- 7.Termine J, Kleinman H, Whitson S, Conn K, McGarvey M, Martin G. Cell. 1981;1:99. doi: 10.1016/0092-8674(81)90037-4. [DOI] [PubMed] [Google Scholar]
- 8.Mullermai C, Stupp S, Voigt C, Gross U. J Biomed Mater Res. 1995;29:9. doi: 10.1002/jbm.820290103. [DOI] [PubMed] [Google Scholar]
- 9.Stupp S, Ciegler G. J Biomed Mater Res. 1992;26:169. doi: 10.1002/jbm.820260204. [DOI] [PubMed] [Google Scholar]
- 10.Stupp S, Braun P. Science. 1997;277:1242. doi: 10.1126/science.277.5330.1242. [DOI] [PubMed] [Google Scholar]
- 11.Stupp S, Hanson J, Eurell J, Ciegler G, Johnson A. J Biomed Mater Res. 1993;27:301. doi: 10.1002/jbm.820270304. [DOI] [PubMed] [Google Scholar]
- 12.Cao B, Mao C. Langmuir. 2007;23:10701. doi: 10.1021/la7014435. [DOI] [PubMed] [Google Scholar]
- 13.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 14.Kim H, Knowles J, Kim H. J Biomed Mater Res Part B - Appl Biomater. 2005;74B:686. doi: 10.1002/jbm.b.30236. [DOI] [PubMed] [Google Scholar]
- 15.Song J, Saiz E, Bertozzi C. JACS. 2003;125:1236. doi: 10.1021/ja028559h. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Yang C, Chen X, Zhao N. Adv Eng Mater. 2006;8:97. [Google Scholar]
- 17.Hartgerink JD, Beniash E, Stupp SI. Proc Natl Acad Sci U S A. 2002;99:5133. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.George A, Bannon L, Sabsay B, Dillon J, Malone J, Veis A, Jenkins N, Gilbert C, NG J Biol Chem. 1996;271:32869. doi: 10.1074/jbc.271.51.32869. [DOI] [PubMed] [Google Scholar]
- 19.He G, Ramachandran A, Dahl T, George S, Schultz D, Cookson D, Veis A, George A. J Biol Chem. 2005;280:33109. doi: 10.1074/jbc.M500159200. [DOI] [PubMed] [Google Scholar]
- 20.Pampena D, Robertson K, Litvinova O, Lajoie G, Goldberg H, Hunter G. Biochem J. 2004;378:1083. doi: 10.1042/BJ20031150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandenbos T, Beersten W. J Bone Mineral Res. 1994;9:1205. [Google Scholar]
- 22.Doi Y, Horiguchi T, Moriwaki Y, Kitago H, Kajimoto T, Iwayama Y. J Biomed Mater Res. 1996;31:43. doi: 10.1002/(SICI)1097-4636(199605)31:1<43::AID-JBM6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 23.Tomomatsu O, Tachibana A, Yamauchi K, Tanabe T. J Ceram Soc Japan. 2008;116:10. [Google Scholar]
- 24.Hoang Q, Sicheri F, Howard A, Yang D. Nature. 2003;45:977. doi: 10.1038/nature02079. [DOI] [PubMed] [Google Scholar]
- 25.Dahlin S, Angstrom J, Linde A. Eur J Oral Sci. 1998;106:239. doi: 10.1111/j.1600-0722.1998.tb02182.x. [DOI] [PubMed] [Google Scholar]
- 26.Schnepp Z, Gonazales-McQuire R, Mann S. Adv Mater. 2006;18:1869. [Google Scholar]
- 27.Boskey A, Maresca M, Doty S, Sabsay B, Veis A. Bone Miner. 1990;11:55. doi: 10.1016/0169-6009(90)90015-8. [DOI] [PubMed] [Google Scholar]
- 28.Hunter G, Hauschka P, Poole A, Rosenberg L, Goldberg H. Biochem J. 1996;317:59. doi: 10.1042/bj3170059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eanes E. In: Calcification in Biological Systems. Bonucci E, editor. CRC Press; Boca Raton, FL: 1992. [Google Scholar]
- 30.Morris D, Anderson H, Yoshikawa H, Nakahara H, Takaoka K, Ono K. Bone. 1990;11:281. doi: 10.1016/8756-3282(90)90081-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.