

Abstract
MDM2 is an E3 ubiquitin ligase that binds and ubiquitinates the tumor suppressor protein p53, leading to its proteasomal degradation. Nutlin-3a (Nutlin) is a preclinical drug that binds MDM2 and prevents the interaction between MDM2 and p53, leading to p53 stabilization and activation of p53 signaling events. Previous studies have reported that Nutlin promotes growth arrest and/or apoptosis in cancer cells that express wild-type p53. In the current study, Nutlin treatment caused a cytoskeletal rearrangement in p53 wild-type human cancer cells from multiple etiologies. Specifically, Nutlin decreased actin stress fibers and reduced the size and number of focal adhesions in treated cells. This process was dependent on p53 expression, but was independent of p21 expression and growth arrest. Consistent with this, Nutlin treated cells failed to form F-actin based motility structures (lamellipodia) and displayed significantly decreased directional persistence in response to migratory cues. Finally, chemotactic assays demonstrated a p53-dependent/p21-independent decrease in migratory and invasive capacity of Nutlin treated cells. Taken together these findings reveal that Nutlin treatment can inhibit the migration and invasion capacity of p53 wild type cells, adding to the potential therapeutic benefit of Nutlin and other small molecule MDM2 inhibitors.
Keywords: p53, Nutlin, Cytoskeleton, Motility, Invasion
Introduction
The tumor suppressor, P53, is one of the most commonly mutated proteins in human cancers. Wild-type p53 is a transcription factor that induces cell cycle arrest, apoptosis, or senescence in response to internal or external stresses that might otherwise predispose normal cells to cancer (1, 2). When p53 is mutated it is unable to inhibit growth in response to these stresses, leading to tumor development. Approximately fifty percent of human tumors express wild type p53, however p53 function is generally compromised in these cancers due to abnormalities in the way in which p53 is regulated or defects in the p53 signaling pathway (3). MDM2 is the major negative regulator of p53, and mdm2 gene amplification is a common abnormality observed in cancers that retain wild-type p53 (4). MDM2 is an E3 ligase that binds to and ubiquitinates p53, leading to its proteasomal degradation (5, 6). P53 and MDM2 form an autoregulatory feedback loop in which p53 transcriptionally activates the expression of MDM2 and MDM2 stimulates the degradation of p53, thereby controlling the levels of both proteins. Nutlin-3a (Nutlin) is a cis-imidazoline compound that specifically binds MDM2 and prevents the interaction between MDM2 and p53 (7). Therefore, in the presence of Nutlin, p53 does not undergo proteasomal degradation and p53 protein accumulates in the cell. P53 can then inhibit proliferation and/or cause cell death (7, 8). Nutlin treatment has been shown to inhibit the growth of human tumors that express wild-type p53 in nude mice xenograft models (7).
Metastasis from a tumor tissue of origin to distant sites constitutes the most life-threatening aspect of cancer (9). In order to metastasize, cells must become motile and invasive to surrounding tissues. The actin cytoskeleton is remodeled during cell movement, primarily through activity of the Rho-family of small GTPases (RhoA, Rac1, and Cdc42) (10). In response to migratory cues, cells initially become polarized forming a leading edge in the direction of movement. Active Cdc42 localizes at the leading edge of the cell and induces localized de novo polymerization of actin to form filipodia (spike-like structures of actin) through regulation of the activity of the WASP (Wiskott-Aldrich syndrome protein) protein complex (11, 12). Active RAC1 at the leading edge of cells stimulates the formation of lamellipodia (flat sheet-like structures of actin) through the regulation of WAVE (WASP family verprolin-homolous protein) complexes. These multiprotein complexes are recruited to the membrane at the cell leading edge and once active associate with and stimulate the activity of the actin nucleating proteins ARP 2/3 (Actin related proteins 2 and 3), leading to actin polymerization. Integrin dependent focal adhesions form between these newly formed actin-rich protrusions (lamellipodia) and the extracellular matrix (ECM) (11, 12). The cell body follows the leading edge while retracting the lagging tail. Rho A regulates cell contractility through formation of acto-myosin stress fibers and focal adhesions. RhoA has been shown to be required for retraction of the lagging tail of the cell during cell movement (11). An amoeboid form of cancer cell movement has also been described (13). Amoeboid movement leads to faster migration rates and is controlled by the action of RhoA signaling through its effector ROCK. In this form of movement RhoA-ROCK promotes remodeling of cortical actin that generates propulsion through the ECM.
Recent reports support a relationship between p53 and the cytoskeleton. P53 over-expression decreased actin stress fiber levels and focal adhesions in p53-null mouse fibroblasts (14). Other studies demonstrated reduced Cdc42 dependent filipodia formation in MEFs over-expressing p53, which prevented cell polarization and inhibited cell spreading (15). Similarly, deletion of p53 in MEFs (p53 null) led to cytoskeletal rearrangement and increased cell motility and invasion associated with increased Rac1 activity (16, 17). More recently, deletion of p53 in MEFs was reported to promote RhoA-ROCK-dependent amoeboid cell movement in 3D matrices (18). Taken together these data suggest a role for p53 in the control of cell migration/invasion through its effects on the actin cytoskeleton. Results from the current study demonstrate that Nutlin treated cancer cells display p53 dependent rearrangement of the actin cytoskeleton, failure to form actin based motility structures in response to migratory cues, and have diminished chemotactic migration and invasion capacity. These results indicate that clinical use of Nutlin may inhibit directional migration and have anti-metastatic potential against p53 wild-type cancers, in addition to its already established anti-proliferative effects.
Materials and methods
Cells
HT1080, U2OS, SAOS and A549 cells were obtained from ATCC and had been passaged for less than 6 months. Cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 100 units per ml penicillin, 100 μg/ml streptomycin and 10% fetal bovine serum (FBS). Cells were plated 24h before treatment with Nutlin. Nutlin was purchased as a racemic mix of Nutlin-3a (active enantiomer) and Nutlin-3b (inactive enantiomer) from Sigma Aldrich (St Louis, MO, USA). All stated doses of Nutlin refer to the active Nutlin-3A enantiomer. Where indicated plates/coverslips were coated with rat tail collagen I, mouse collagen IV or human fibronectin (5μg/cm2, BD Biosciences, San Jose, CA, USA) as the manufacturer describes prior to plating cells.
Immunoblots
Cells were lysed in 50mM Tris, pH 7.5, 150mM NaCl, 0.5% Nonidet P-40, phenylmethylsulfonyl fluoride and leupeptin. Lysates resolved by SDS–polyacrylamide gel electrophoresis were transferred to nitrocellulose, blocked with 5% milk, incubated in primary antibody overnight and secondary antibody for 1h, and visualized by chemiluminescence. Primary antibodies were: p53 (ab-2) (Calbiochem); Actin, Tubulin, MDM2 (SMP14), p21 (H164) (Santa Cruz, Santa Cruz, CA, USA). Densitometry was performed using NIH image J software and values corrected for Actin/Tubulin loading control. Significance was determined using Student’s t-test (P<0.05).
Immunofluorescence/Cytoskeletal Staining
Immunofluorescent cytoskeletal staining was performed using an Actin Cytoskeleton/Focal Adhesion Staining Kit (Millipore, Billerica, MA, USA). Briefly, cells were fixed on cover glass with 4% paraformaldehyde, blocked in 1% bovine serum albumin (BSA) and 0.1% Triton X-100 and incubated with primary antibodies (1:100 dilution) for 1.5 h. Cells were next incubated with TRITC-conjugated phalloidin (Millipore, 1:100 dilution) and Alexa 488-conjugated secondary antibody (1:2000 dilution; Invitrogen, Carlsbad, CA, USA) and/or Cy5 conjugated secondary antibody (1:2000, Jackson Immunoresearch) for 1h. Primary antibodies were anti vinculin (Millipore); anti p21 (H-164) and anti p53 (FL-393) (Santa Cruz, Santa Cruz, CA, USA). To visualize lamellipodia formation, cells were grown to confluency on collagen coated coverslips and cell monolayer was wounded 2h prior to fixation. Samples were visualized and imaged using a Zeiss Axiovert 200M fluorescence microscope. Cy5 staining was pseudocolored orange in all images. Image analysis was performed using NIH Image J analysis software.
Cell Cycle Analysis
Cells were trypsinized and resuspended in PBS (0.1% BSA). A total of 1.5×106 cells per ml were fixed in ethanol (4 °C), resuspended in RNase A (0.7 mg/ml)/propidium iodide (50 mg/ml) and incubated at 37 °C. Analysis was done using FACScanto flow cytometer (BD Biosciences, San Jose, CA, USA) and data were analyzed using Flowjo software.
siRNA-mediated transient knock-down
p53 RNAi, p21 RNAi, and control RNAi (On-target plus siControl nontargeting pool) were purchased from Dharmacon and transfected according to the manufacturer’s guidelines using DharmaFECT I reagent. Treatments were applied 24 h after transfection.
Time Lapse Microscopy
HT1080 cells were grown to confluency in collagen coated 6 well dishes. Cell monolayer was wounded using a sterile micropipette tip and cells at multiple points along the wound edge were imaged every 5 minutes over a 4 hour period using a Zeiss Axiovert 100 microscope. Cells were maintained at 5% CO2 and 37°C during the course of these experiments. Cell track analysis of individual cells, determination of velocity and directional persistence were performed using Metamorph version 7.6 image analysis software (50 cells per wound edge).
Chemotactic Migration Assay
Chemotactic migration was studied using BD Biosciences 24 well uncoated migration chambers (8.0-μm pore size; BD Biosciences, San Jose, CA, USA) as follows. Approximately 2.5 × 104 (1.25×104 for HT1080) untreated or Nutlin pretreated (8μM; 24h) cells were seeded in the upper chambers of plate in triplicate in 500 μL serum free DMEM (+/− Nutlin). DMEM with 10% FBS (+/− Nutlin) was added to the lower chamber as a chemoattractant. After 24h of incubation at 37°C with 5% CO2, cells remaining on the upper membrane surface were removed with a cotton swab. Cells on the lower surface of the membrane were stained with crystal violet and fixed in methanol. The invasive cells on the lower surface of membrane were counted using a microscope. Each experiment was repeated three times. In order to control for effects on cell proliferation, cells were dislodged from additional intact membranes using trypsin (15min at 37°C) and vortexing. Total cell number was counted using a hemocytometer.
Matrigel Invasion Assay
Invasion of cells through matrigel coated membranes was determined using 24-well BD invasion chambers (8.0-μm pore size; BD Biosciences, San Jose, CA, USA) in accordance with the manufacturer’s instructions with the following modifications. Briefly, cells were trypsinized and 2.5 × 104 (1.25×104 for HT1080) untreated or Nutlin pretreated (8μM; 24h) cells were placed into the upper compartment of the invasion plates in triplicate in 500 μL serum free DMEM (+/−Nutlin). The lower compartment was filled with 750 μl of 10% FBS DMEM (+/− Nutlin). After 40 h of incubation at 37°C with 5% CO2, cells remaining on the upper membrane surface were removed with a cotton swab. Cells on the lower surface of the membrane were stained with crystal violet and fixed in methanol. The invasive cells on the lower surface of membrane were counted using a microscope. Each experiment was repeated three times. In order to control for effects on cell proliferation, cells were dislodged from additional intact matrigel membranes using trypsin (2h @ 37°C) and vortexing. Total cell number was counted using a hemocytometer.
Results
Previous studies describe increased p53 levels and activity in p53 wild type cancer cells (p53wt) upon treatment with the MDM2 inhibitor, Nutlin-3A (Nutlin) (7, 8). We treated human cancer cell lines, U2OS (osteosarcoma; p53wt), A549 (lung adenocarcinoma; p53wt), HT1080 (fibrosarcoma; p53wt) and SAOS (osteosarcoma; p53 null) with increasing doses of Nutlin (0–16μM; 24h) and monitored the expression of p53, p21 and MDM2 by immunoblotting (data not shown). Nutlin treatment caused a dose dependent increase in p53 and its downstream targets, p21 and MDM2, in p53wt cells. A robust increase in p53 was observed in p53wt cell lines in our study at an 8μM Nutlin dose (Figure 1A). As expected, Nutlin did not induce p53, p21 or MDM2 expression in SAOS cells. Next, we analyzed cell cycle profiles in Nutlin treated cells. DNA content analysis demonstrated that Nutlin treatment of p53wt cell lines (U2OS, A549, HT1080) caused these cells to arrest with 2N and 4N DNA content, suggestive of G1 and G2-phase cell cycle arrest. In contrast, the same Nutlin treatment had no obvious effect on the cell cycle profile of p53-null SAOS cells (Figure 1B). Analysis of sub G1 populations in these cells showed no detectable increase in cell death at 24h or 48h after Nutlin treatment.
Figure 1. Nutlin increases p53 expression/activity and induces growth arrest in p53wt human cancer cells.
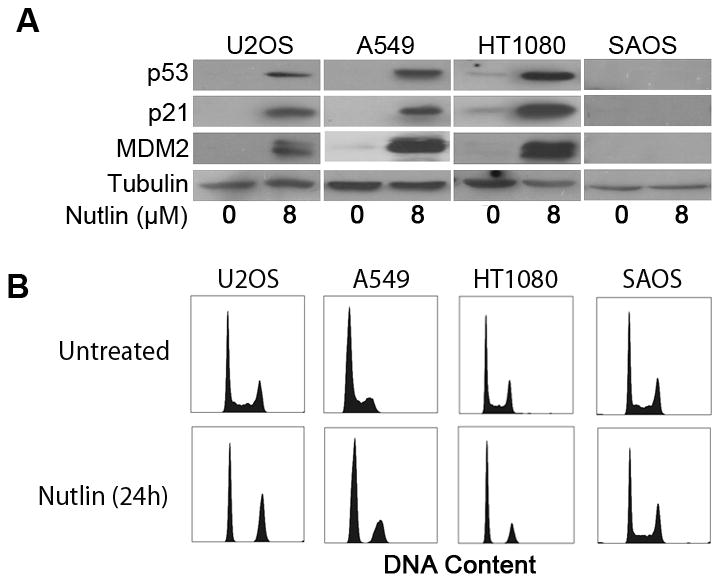
(A) Representative immunoblots from U2OS, A549, HT1080 and SAOS cell lysates collected 24h after Nutlin treatment (8μM) probed using antibodies specific against p53, p21 and MDM2. Actin expression was detected as a loading control. (B) Representative DNA content profiles of untreated and Nutlin treated (8μM) U2OS, A549, HT1080 and SAOS cells. Cells were fixed, stained with propidium iodide and analyzed by flow cytometry for DNA content. DNA profiles are representative of three independent experiments.
During the course of our studies, we observed a dose dependent alteration in cellular morphology in Nutlin treated p53wt cells. Typically, cell structure changed from a well spread polygonal structure to a more elongated morphology (Supplementary File 1) during Nutlin treatment, becoming apparent at 24h and occurring in approximately 70% of cells at 40h after treatment (16μM Nutlin). Morphological changes were most apparent at higher Nutlin doses (8–16μM), coincident with increased levels of p53. This alteration in morphology was reversible and cells returned to a spread/polygonal morphology within 8h after Nutlin removal (data not shown). These morphologic changes were also observed when Nutlin-treated cells were plated on individual ECM surfaces including fibronectin, collagen IV and collagen I (data not shown). Because cellular morphology is controlled by the actin cytoskeleton, we examined cytoskeletal organization of Nutlin treated p53wt and p53 null cells 40h after treatment. Cells were plated on collagen coated coverslips, treated with Nutlin (8μM) and stained with rhodamine conjugated phalloidin to detect F-actin and anti-vinculin antibody to detect focal adhesions. Untreated A549 and U2OS cells display numerous large actin stress fibers and large focal adhesions throughout the cell body. Treatment with Nutlin decreased F-actin stress fibers, focal adhesion size and number in A549 and U2OS cells (Figure 2A). Image analysis showed significantly decreased phalloidin staining intensity in treated versus untreated cells confirming the decrease in F-Actin expression after treatment (Figure 2B). In addition, image analysis indicated a decrease in the size and number of focal adhesions with Nutlin treatment (Figure 2C & 2D). Nutlin also induced cytoskeletal rearrangement in HT1080 cells (Supplementary File 2). These effects appeared to depend on p53-status since F-actin expression/cytoskeletal arrangement were not diminished/altered by Nutlin treatment in p53-null SAOS cells, and there was also little to no effect of Nutlin on focal adhesion number or size in these cells (Figure 2A-2D). It is worth noting that Nutlin treatment was also associated with an increase in cytoplasmic (non adhesion associated) vinculin staining in all cell types. This, however, does not appear to relate to the decrease in focal adhesion size and number as it was also apparent in SAOS cells where adhesions remain unaffected by Nutlin treatment.
Figure 2. Nutlin decreases F-Actin stress fibers, focal adhesion size and number in p53wt human cancer cells.

(A) Representative immunofluorescent images of Nutlin and untreated U2OS, A549 and SAOS cells. Cells seeded on collagen coated coverslips were treated with Nutlin (8μM; 40h) or left untreated and stained for focal adhesions (green) and F-Actin stress fibers (red). Nuclei were visualized by DAPI staining (blue). Images are representative of three independent experiments each with 200 cells analyzed per experiment. (B) Mean fluorescent intensity of rhodamine conjugated phalloidin staining per cell was determined using image J analysis software in U2OS, A549 and SAOS cells seeded on collagen coated coverslips with or without Nutlin treatment (8μM; 40h). (C,D) Mean focal adhesion size and number stained with anti-vinculin antibody was determined using image J analysis software in U2OS, A549 and SAOS cells seeded on collagen coated coverslips with or without Nutlin treatment (8μM; 40h). Data is representative of three independent experiments each with 200 cells analyzed per experiment. (*p<0.05 Nutlin treated versus untreated)
Next we wished to test the dependency of these effects on p53 and the p53 downstream effector, p21. U2OS cells were transfected with siRNA targeting p53 (sip53), p21 (sip21) or a non-targeting siRNA sequence (siCTRL) prior to treatment with Nutlin (8μM; 24h later). U2OS cells were used for these studies as they showed the most efficient knockdown of p53 and p21 expression compared to other cells. Lysates from Nutlin treated and untreated cells were collected 40h after Nutlin treatment and immunoblotted for p53 and p21 expression. Cells transfected with siCTRL showed increased expression of p53 and p21 with Nutlin treatment (Figure 3A, 4A). In contrast, sip53 transfected cells did not show a detectable increase in p53 or p21 expression after Nutlin treatment (Figure 3A), and cells transfected with sip21 showed increased p53 but no increase in p21 (Figure 4A, 4C). Cell cycle analysis demonstrated that sip53 (Figure 3B) and sip21 (Figure 4B) ablated Nutlin induced cell cycle arrest in U2OS cells.
Figure 3. Nutlin induced cytoskeletal rearrangement is dependent on p53 expression.

U2OS cells were transfected with siRNA targeting p53 (sip53) or a non targeting control sequence (siCTRL). Cells were treated with Nutlin 24h later or left untreated. (A) Representative immunoblots from siCTRL and sip53 transfected U2OS cell lysates collected 24h after Nutlin treatment (8μM) probed using antibodies specific against p53 and p21. Actin was detected as a loading control. (B) Representative DNA content profiles of untreated and Nutlin treated (8μM) sip53 and siCTRL transfected U2OS. Cells were fixed, stained with propidium iodide and analyzed by flow cytometry for DNA content. DNA profiles are representative of three independent experiments. (C) Representative fluorescent images of Nutlin and untreated sip53 and siCTRL U2OS cells. Cells seeded on collagen coated coverslips were treated with Nutlin (8μM; 40h) or left untreated, and stained for focal adhesions (green), p53 (orange) and F-Actin stress fibers (red). Nuclei were visualized by DAPI staining (blue). Images are representative of three independent experiments each with 200 cells analyzed per experiment.
Figure 4. Nutlin induced cytoskeletal rearrangement is not dependent on p21 expression/growth arrest.

U2OS cells were transfected with siRNA targeting p21 (sip21) or a non targeting control sequence (siCTRL). Cells were treated with Nutlin 24h later or left untreated. (A) Representative immunoblots from siCTRL and sip21 transfected U2OS cell lysates collected 24h after Nutlin treatment (8μM) probed using antibodies specific against p53 and p21. Actin expression was detected as a loading control. (B) Representative DNA content profiles of untreated and Nutlin treated (8μM) sip21 and siCTRL transfected U2OS. Cells were fixed, stained with propidium iodide and analyzed by flow cytometry for DNA content. DNA profiles are representative of three independent experiments. (C) Representative fluorescent image of Nutlin treated (8μM) sip21 transfected U2OS cells. Cells seeded on collagen coated coverslips were treated with Nutlin (8μM; 40h) and stained with an antibody specific against p53 (orange).(D) Representative fluorescent images of Nutlin and untreated sip21 and siCTRL U2OS cells. Cells seeded on collagen coated coverslips were treated with Nutlin (8μM; 40h), and stained for focal adhesions (green), p21 (orange) and F-Actin stress fibers (red). Nuclei were visualized by DAPI staining (blue). Images are representative of three independent experiments each with 200 cells analyzed per experiment..
Untreated and Nutlin treated sip53, sip21 and siCTRL transfected U2OS cells were next stained for stress fibers/focal adhesions. As expected, cells transfected with control siRNA showed diminished F-actin stress fiber levels, as well as decreased focal adhesion size and number with Nutlin treatment (Figure 3C). In contrast, sip53 transfected cells retained normal actin stress fiber levels with Nutlin treatment, and the number and size of focal adhesions were unaffected (Figure 3C). These results confirm that cytoskeletal changes induced by Nutlin treatment are p53 dependent. Interestingly, cells in which p21 was knocked down also displayed decreased F-actin stress fiber levels and a decrease in the number and size of focal adhesions after Nutlin treatment, similar to control cells (Figure 4D). This indicates that p21-dependent cell cycle arrest is not associated with or required for these cytoskeletal changes.
Reorganization of the actin cytoskeleton is essential for cell migration and invasion (11, 12). We therefore hypothesized that Nutlin might also affect the formation of actin based motility structures in cancer cells. In a wound assay, cells grown in a confluent monolayer form large sheets of actin (lamellipodia) and strong focal adhesions at the leading edge of the cell as they migrate into the wound (19). To investigate the effect of Nutlin on this process, we wounded a confluent monolayer of Nutlin treated or untreated U2OS (mildly invasive), SAOS (mildly invasive) and HT1080 (highly invasive) cells on collagen coated coverslips using a sterile micropipette tip. Two hours after wounding, cells were stained for stress fibers/focal adhesions and the percentage of wound-edge cells displaying lamellipodia were evaluated. Untreated U2OS, SAOS and HT1080 cells formed large F-actin rich lamellipodia at the leading edge of wounded cells (Figure 5A, 5B). Nutlin treatment (4μM & 8μM) significantly decreased the percentage of cells displaying lamellipodia in HT1080 and U2OS cells but had no effect on lamellipodia formation in SAOS cells (Figure 5B). To investigate the dependency of this effect on p53 or p21 expression, Nutlin treated or untreated U2OS cells transfected with sip53 or sip21 were wounded in a similar assay. Lamellipodia formation was lost in Nutlin treated siCTRL and sip21 cells; however, lamellipodia formation in sip53 transfected cells remained unaffected by Nutlin treatment (Figure 5C, 5D).
Figure 5. Nutlin decreases lamellipodia formation in wound edge cells in a p53 dependent/p21 independent manner in human cancer cells.

(A) Confluent U2OS, HT1080 and SAOS cells seeded on collagen coated coverslips were treated with Nutlin (4μM/8μM; 40h) or left untreated. Cells were scratched using a sterile micropipette tip and fixed 2h later. (A) Representative fluorescent images of Nutlin treated and untreated U2OS wound edge cells. Cells were stained for focal adhesions (green) and F-Actin stress fibers (red). Nuclei were visualized by DAPI staining (blue). Lamellipodia were identified as flat sheets of F-actin (white arrows) at cell leading edges. Images are representative of three independent experiments each with 200 cells analyzed per experiment.. (B) Percentage of wound edge cells displaying lamellipodia were evaluated and expressed as a percentage of untreated control cells. (* p<0.05 Nutlin treated versus untreated) (C) U2OS cells were transfected with siRNA targeting p21 (sip21), p53 (sip53) or a non targeting control sequence (siCTRL). Cells were treated with Nutlin (8μM) 24h later or left untreated. After 40h, cells were scratched using a sterile micropipette tip and fixed 2h later. Cells were stained for F-Actin stress fibers (red). Nuclei were visualized by DAPI staining (blue). Lamellipodia were identified as flat sheets of F-actin (white arrows) at cell leading edges. Images are representative of three independent experiments each with 200 cells analyzed per experiment.. (D) Percentage of siRNA transfected wound edge cells displaying lamellipodia were evaluated and expressed as a percentage of untreated control cells. (* p<0.05 Nutlin treated versus untreated)
Next we analyzed the effects of Nutlin on cancer cell motility. Highly motile HT1080 cells were grown to confluence on collagen coated plates and wounded using a sterile micropipette tip. Untreated or Nutlin treated (8μM; 40h) cells were monitored using time lapse microscopy for 4 hours after wounding. Untreated cells formed large lamellipodia at the leading edge within 5 minutes after wounding and continued to move unidirectionally into the wound throughout the 4h assay (Figure 6A). In contrast, Nutlin treated cells formed narrow protrusions and appeared to move randomly rather than in a directional manner. Some Nutlin treated cells were observed to lose and regain adhesion to the matrix within the 4h visualization period (Figure 6). Migration track analysis of cells that remained attached to the plate (Figure 6B) demonstrated that Nutlin treated and untreated cells could move with similar velocities (Figure 6C); however, Nutlin significantly decreased directional persistence in treated cells (Figure 6D). In summary, Nutlin treatment did not inhibit the overall motility of HT1080 cells, but did inhibit their ability to move in a persistent, directional manner. This lack of directional movement was correlated with altered cytoskeleton in Nutlin treated cells and decreased formation of actin-rich motility structures (lamellipodia). HT1080, cells are a rapidly migrating cell type and are particularly suitable for this form of directional motility assay, having the ability to close the wound approximately 5–6h after wounding. The slower moving U2OS cell is less suitable for this type of assay (wound closure in 24–28h), however we did perform live cell image analysis on untreated and Nutlin treated U20S cells seeded sparsely (10–20% confluency) in collagen coated plates (data not shown). Similar to HT1080 cells, Nutlin treated U2OS cells displayed multiple narrow protrusions and more frequent oscillations when compared to untreated cells. The slow moving nature of this cell made it unsuitable for image track analysis.
Figure 6. Nutlin decreases directional persistence in HT1080 cells.

Confluent HT1080 cells seeded on collagen coated plates were treated with Nutlin (8μM; 40h) or left untreated. Cells were scratched using a sterile micropipette tip and imaged on a time lapse microscope at 5 minute intervals over a period of 4 hours. (A) Representative images of Nutlin treated and untreated HT1080 cells at a wound edge 0h and 4h after scratching. Dotted line represents wound edge immediately after wounding (0h). Images are representative of three independent experiments (B) Representative plots of individual Nutlin treated and untreated HT1080 cell tracks prepared from time lapse microsopy images using Metamorph analysis software. Data is representative of 50 cells per wound edge in three independent experiments. (C, D) Comparison of average velocity and directional persistence in Nutlin treated versus untreated HT1080 cells calculated from time lapse microscopy images using Metamorph analysis software. Data is representative of 50 cells per wound edge in three independent experiments. (* p<0.05 Nutlin treated versus untreated)
Finally, we examined the effect of Nutlin on chemotactic cancer cell migration and invasion. Nutlin treated and untreated U2OS, HT1080 and SAOS cells were initially analyzed for migration and invasion in uncoated boyden chamber or matrigel coated invasion chamber assays, respectively. Total cell numbers were also evaluated to control for Nutlin’s effect on cell proliferation. Nutlin treatment significantly decreased both migration and invasion of U2OS and HT1080 cells (Figure 7A and 7B). Surprisingly, SAOS cell migration and invasion was consistently enhanced by Nutlin treatment in these assays. Similar migration and invasion assays were performed in U2OS cells transfected with sip53, sip21, or control siRNA. Nutlin treatment significantly inhibited migration and invasion of U2OS cells tranfected with either sip21 or control siRNA (Figure 7C and 7D). However, Nutlin did not inhibit migration or invasion of cells transfected with sip53. Thus, Nutlin treatment inhibited the migration and invasion capability of cancer cells in a p53-dependent but p21 independent manner.
Figure 7. Nutlin decreases cancer cell migration and invasion in a p53 dependent/p21 independent manner.

(A,B) U2OS, HT1080 and SAOS were treated with Nutlin (8μM) or left untreated in (A) uncoated or (B) matrigel coated boyden chambers. 10% FBS was added to the lower chamber to act as chemoattractant. After 24h (migration) or 48h (invasion) cells were scrubbed from upper surface and migrated/invaded cells were crystal violet stained and counted. Total cell number was calculated by releasing cells on separate membranes and direct counting on a hemocytometer. Data is normalized to total cell number at end of assay. Data is representative of three independent experiments. (C,D) U2OS cells were transfected with siRNA targeting p21 (sip21), p53 (sip53) or a non targeting control sequence (siCTRL) and were left untreated or treated with Nutlin (8μM) 24h later. Migration and invasion assays were performed as described above. Data is representative of three independent experiments. (* p<0.05 Nutlin treated versus untreated)
Discussion
Nutlin is a small molecule MDM2 antagonist and activator of p53. Results from this study demonstrate that Nutlin causes a p53-dependent, but p21-independent, decrease in the level of F-actin stress fibers as well as the number and size of focal adhesions. Consistent with this, Nutlin treated cells failed to form F-actin based motility structures (lamellipodia) and displayed significantly decreased directional persistence in response to migratory cues. Finally, chemotactic assays demonstrated a p53 dependent decrease in the migratory and invasive capacity of Nutlin treated cells. Taken together this data reveals a previously undescribed cellular response to Nutlin treatment that will likely contribute to its therapeutic and anti-tumor potential.
The non-genotoxic activation of p53 in p53wt cancer cells represents a promising therapeutic strategy. Previous studies have shown that Nutlin effectively growth arrests p53wt cancer cells in G1 and G2 phases, induces senescence and/or induces apoptosis at low micromolar concentrations in multiple cancer types (7, 8, 20). Moreover, Nutlin administration in nude mice bearing established human cancer xenografts showed effective tumor growth inhibition and tumor shrinkage at non toxic doses (7). More recently, Sechierro et al described an inhibitory effect of Nutlin on angiogenesis which was related at least partially to decreased migratory capacity of treated endothelial cells (21). It has also been shown that Nutlin can inhibit cancer cell migration and invasion indirectly through repression of SDF-1/CXCL12 expression in stromal fibroblasts (22). In the current study, we show for the first time that Nutlin can directly inhibit the migration and invasion capacity of cancer cells that express wild-type p53. Collectively, these results highlight the possibility that Nutlin and similar compounds may act as combination anti-proliferative, anti-angiogenic, and anti-metastatic agents, thereby targeting three important events in cancer progression.
In our study, Nutlin altered cell morphology, and decreased both actin stress fibers and focal adhesions specifically in p53wt cancer cells. We confirmed the dependency of this phenotype on p53 expression using siRNA targeting p53. Previous studies reported altered morphology and increased focal adhesions in p53-null MEFs (16, 17). Similarly, Alexandrova et al reported that overexpression of p53 in mouse fibroblasts results in altered cell morphology and reduction in the number and size of actin stress fibers and focal adhesions (14). We considered that Nutlin’s effect on the actin cytoskeleton could be an indirect consequence of either cell cycle arrest or apoptosis mediated by p53. However, Nutlin-treated cells in our study exhibit little or no apoptosis as determined by DNA content (sub-G1) analysis. This allows us to differentiate the cytoskeletal effects of p53 from an apoptotic response. In addition, siRNA knockdown of p21 ablated growth arrest in Nutlin treated cells but did not prevent the cytoskeletal effects. Based on these findings, we conclude the cytoskeletal rearrangements induced by Nutlin are independent of either apoptosis or cell cycle arrest mediated by p53.
We identify p53 dependent inhibition of chemotactic/directional migration and invasion in Nutlin treated cancer cells. We have also discovered abnormalities in the response of Nutlin treated cells to migratory cues. Specifically, Nutlin treated cells fail to form lamellipodia in the direction of a wound and display decreased directional persistence when compared to untreated cells. It is clear that such abnormalities in cell motility may account at least in part for the effect of Nutlin on chemotactic migration and invasion. It is perhaps surprising that Nutlin treated cells move with similar velocities to untreated cells. However it is worth noting that similar findings have been reported in studies examining the effects of overexpression of tumor suppressors, DAPK and Scribble (23, 24). In each case overexpressing cells migrate with decreased directional persistence and display impaired lamellipodia formation but retain migrational velocity. Taken together these findings and our study suggest that lamellipodia formation alone is not predictive of migration rate (velocity) in cancer cells. Previous studies have suggested an inhibitory effect of p53 on cell migration and/or invasion (14, 15, 17). In each case the inhibitory effects of p53 were linked closely with cytoskeletal rearrangement and/or abnormal cell polarization. The mechanism through which p53 mediates its effects on lamellipodia formation and directional persistence in Nutlin treated cells remains unclear. During cell movement, Rac 1 is responsible for the formation of large actin-rich lamellipodial protrusions at the leading edge. In contrast, Cdc42 is responsible for the formation of small, spike-like protrusions (filipodia), and Rho1 functions in the formation of actin stress fibers throughout the cell body, as well as in retraction of the lagging tail. The predominant effect of Nutlin treatment was to diminish F-actin stress fiber levels and inhibit lamellipodia formation at the leading edge, and we therefore monitored Rac1 and RhoA activity in Nutlin treated HT1080 cells (data not shown). In some but not all experiments both Rac1 and RhoA activity were inhibited in Nutlin treated HT1080 cells, suggesting decreased invasion and migration in Nutlin treated cells might result in part from an inhibition of Rho A and/or Rac1. This would be consistent with previous studies that have reported that high levels of p53 can inhibit RhoA and Rac1 (14, 15, 17).
Taken together, our data uncovers novel effects of Nutlin treatment on cancer cell invasion. The most deadly aspect of cancer is its ability to spread or metastasize. During metastasis, primary tumor cells invade the surrounding stroma and migrate towards blood vessels or lymphatics (25, 26). Tumor cells enter blood vessels (intravasation) and are carried by the circulatory system to distant sites where they typically arrest in the capillary bed of a distant organ. The arrested cells extravasate from the circulation system to the organ parenchyma and proliferate to form a secondary tumor. Accumulating evidence suggests that this is not a random process but rather one that is directed by host/tumor signaling. Tumor cells detect and directionally move towards chemoattractant gradients. For example, the chemokine Stromal Derived Factor-1 (SDF-1) can stimulate tumor cell migration away from the primary tumor mass and toward stromal sites in distal organs (27–29). This process is believed to contribute to the metastasis of cancer cells to preferred sites in the body such as bone, lung and liver where SDF-1 expression is high (30). Similarly, Epidermal Growth Factor (EGF) and Hepatocyte Growth Factor (HGF) which are secreted by cancer associated fibroblasts and tumor associated macrophages have been linked to the guidance of motile/invasive cancer cells (31–33). Directional movement of cancer cells is involved in stromal invasion, intravasation and extravasation during the metastatic cascade. Interference with directional movement of cancer cells has been shown to decrease metastasis in animal models of cancer (27, 34, 35). Based on our results, Nutlin’s inhibitory effects on directional movement and invasion have the possibility to translate into anti-metastatic activity. In vivo studies to examine the potential anti-metastatic effects of Nutlin will be beneficial in order to establish the relevance of our findings in suitable animal models of cancer.
Supplementary Material
Acknowledgments
Financial Support: This work was funded by NIH grant numbers 1RO1CA137598–01A1 and RO1CA108843.
References
- 1.Giono L, Manfredi J. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J Cell Physiol. 2006;209:13–20. doi: 10.1002/jcp.20689. [DOI] [PubMed] [Google Scholar]
- 2.Harris S, Levine A. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 3.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 4.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453–9. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levav-Cohen Y, Haupt S, Haupt Y. Mdm2 in growth signaling and cancer. Growth Factors. 2005;23:183–92. doi: 10.1080/08977190500196218. [DOI] [PubMed] [Google Scholar]
- 6.Iwakuma T, Lozano G. MDM2, an introduction. Mol Cancer Res. 2003;1:993–1000. [PubMed] [Google Scholar]
- 7.Vassilev L, Vu B, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 8.Tovar C, Rosinski J, Filipovic Z, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103:1888–93. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 10.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 12.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 13.Sahai E, Marshall C. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–9. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 14.Alexandrova A, Ivanov A, Chumakov P, Kopnin B, Vasiliev J. Changes in p53 expression in mouse fibroblasts can modify motility and extracellular matrix organization. Oncogene. 2000;19:5826–30. doi: 10.1038/sj.onc.1203944. [DOI] [PubMed] [Google Scholar]
- 15.Gadéa G, Lapasset L, Gauthier-Rouvière C, Roux P. Regulation of Cdc42-mediated morphological effects: a novel function for p53. EMBO J. 2002;21:2373–82. doi: 10.1093/emboj/21.10.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo F, Gao Y, Wang L, Zheng Y. p19Arf-p53 tumor suppressor pathway regulates cell motility by suppression of phosphoinositide 3-kinase and Rac1 GTPase activities. J Biol Chem. 2003;278:14414–9. doi: 10.1074/jbc.M300341200. [DOI] [PubMed] [Google Scholar]
- 17.Guo F, Zheng Y. Rho family GTPases cooperate with p53 deletion to promote primary mouse embryonic fibroblast cell invasion. Oncogene. 2004;23:5577–85. doi: 10.1038/sj.onc.1207752. [DOI] [PubMed] [Google Scholar]
- 18.Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol. 2007;178:23–30. doi: 10.1083/jcb.200701120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev. 2005;15:87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Van Maerken T, Speleman F, Vermeulen J, et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006;66:9646–55. doi: 10.1158/0008-5472.CAN-06-0792. [DOI] [PubMed] [Google Scholar]
- 21.Secchiero P, Corallini F, Gonelli A, et al. Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ Res. 2007;100:61–9. doi: 10.1161/01.RES.0000253975.76198.ff. [DOI] [PubMed] [Google Scholar]
- 22.Moskovits N, Kalinkovich A, Bar J, Lapidot T, Oren M. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006;66:10671–6. doi: 10.1158/0008-5472.CAN-06-2323. [DOI] [PubMed] [Google Scholar]
- 23.Dow L, Kauffman J, Caddy J, et al. The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene. 2007;26:2272–82. doi: 10.1038/sj.onc.1210016. [DOI] [PubMed] [Google Scholar]
- 24.Kuo J, Wang W, Yao C, Wu P, Chen R. The tumor suppressor DAPK inhibits cell motility by blocking the integrin-mediated polarity pathway. J Cell Biol. 2006;172:619–31. doi: 10.1083/jcb.200505138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–56. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 27.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 28.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–7. [PubMed] [Google Scholar]
- 29.Koizumi K, Hojo S, Akashi T, Yasumoto K, Saiki I. Chemokine receptors in cancer metastasis and cancer cell-derived chemokines in host immune response. Cancer Sci. 2007;98:1652–8. doi: 10.1111/j.1349-7006.2007.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kedrin D, van Rheenen J, Hernandez L, Condeelis J, Segall J. Cell motility and cytoskeletal regulation in invasion and metastasis. J Mammary Gland Biol Neoplasia. 2007;12:143–52. doi: 10.1007/s10911-007-9046-4. [DOI] [PubMed] [Google Scholar]
- 31.Wyckoff J, Wang W, Lin EY, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 32.Goswami S, Sahai E, Wyckoff JB, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65:5278–83. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 33.Qian LW, Mizumoto K, Maehara N, et al. Co-cultivation of pancreatic cancer cells with orthotopic tumor-derived fibroblasts: fibroblasts stimulate tumor cell invasion via HGF secretion whereas cancer cells exert a minor regulative effect on fibroblasts HGF production. Cancer Lett. 2003;190:105–12. doi: 10.1016/s0304-3835(02)00517-7. [DOI] [PubMed] [Google Scholar]
- 34.Murakami T, Maki W, Cardones AR, et al. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328–34. [PubMed] [Google Scholar]
- 35.Sun YX, Schneider A, Jung Y, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res. 2005;20:318–29. doi: 10.1359/JBMR.041109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.