

Abstract
ABL2 (also known as ARG (ABL related gene)) is closely related to the well-studied Abelson kinase cABL. ABL2 is involved in human neoplastic diseases and is deregulated in solid tumors. Oncogenic gene translocations occur in acute leukemia. So far no structural information for ABL2 has been reported. To elucidate structural determinants for inhibitor interaction, we determined the cocrystal structure of ABL2 with the oncology drug imatinib. Interestingly, imatinib not only interacted with the ATP binding site of the inactive kinase but was also bound to the regulatory myristate binding site. This structure may therefore serve as a tool for the development of allosteric ABL inhibitors. In addition, we determined the structures of ABL2 in complex with VX-680 and with an ATP-mimetic type I inhibitor, which revealed an interesting position of the DFG motif intermediate between active and inactive conformations, that may also serve as a template for future inhibitor design.
Introduction
ABL2 (v-ABL Abelson murine leukemia viral oncogene homologue 2), also known as ARG (ABL related gene), is a member of the Abelson family of nonreceptor tyrosine kinases.1−3 ABL2 and ABL1 (c-Abl) share a high degree of sequence conservation and have a similar domain organization comprising an N-terminal “cap” that is important for autoinhibition, followed by an SH3−SH2 kinase domain and a large C-terminal domain containing docking sites for SH3 domains, F-actin, and microtubules (Figure 1A).(3) The ABL kinases are ubiquitously expressed and regulate many cellular functions including reorganization of the cytoskeleton, cell proliferation, adhesion, and migration in response to stimulation of cell surface receptors. Knockout mouse studies have revealed that ABL1 and ABL2 play overlapping roles and are required in development and T cell function.4,5 Despite their homology, ABL2 has a number of distinct cellular functions including an important role in neurulation, and it is required for adhesion-dependent neurite branching, synapse/dendrite stability, as well as fibroblastic and epithelial cell adhesion and migration.
Figure 1.

Structural comparison of ABL2 and ABL1. (A) Domain organization of ABL2, showing residue numbering and the percentage sequence identity to ABL1 for each domain. (B) Ribbon diagram of the structure of ABL2 in complex with the type I inhibitor 2 to show the orientation of the molecule underneath the surface depicted in (C). (Figure 5 shows this interaction in more detail.) (C) Two views of the surface representation of the ABL2 structure are shown, separated by a rotation of 180°. Residues that are conserved with ABL1 are shown as gray surface, semiconserved residues as yellow surface, and residue differences as red surface. The residue differences are numbered in black for ABL2, and the residue letters in red correspond to their counterparts in ABL1. The representation on the left is the same orientation as in (B).
Oncogenic forms of ABL are produced by retroviral transduction (v-Abl) or chromosomal translocation events. Fusion of the ABL1 gene with the breakpoint cluster (BCR) gene gives rise to an active, oncogenic tyrosine kinase BCR-ABL and is associated with chronic myeloid leukemia (CML).6,7 Other chimeric ABL fusion gene products between ABL1 or ABL2 and ETV6 (ETS translocation variant 6), also known as TEL (translocation ETS leukemia), are associated with rare cases of CML and acute myelogenous leukemia (AML).(8)
Activity of ABL is regulated by an autoinhibitory mechanism, and cellular activity is normally low. The inhibitory mechanism in ABL differs from that found in the closely related Src kinase where interactions between a phosphorylated tyrosine residue in the C-terminal tail and the SH2 domain maintain the repressed state of Src.(9) ABL kinases lack this critical tyrosine residue, and the inactive conformation is maintained by an N-terminal myristoyl group binding to a hydrophobic pocket in the kinase domain which places the SH2−SH3 domains on the large and small lobes of the kinase, respectively, preventing its orientation to an active kinase. Two splice variants of ABL1 exist (1a and 1b) that differ only in their N-terminal region. ABL 1b is myristoylated, whereas ABL 1a is not, and it has been suggested that hydrophobic residues in the cap domain of ABL 1a may functionally substitute for the myristate. The fusion of BCR, or TEL, with ABL disrupts the autoinhibitory mechanism, and the enhanced tyrosine kinase activity leads to leukemia.
Several inhibitors have been developed for the ABL kinases, and these are currently used for the treatment of leukemia such as CML. The most commonly used inhibitor imatinib mesylate (STI571, CGP 57148B) (Chart 1) selectively inhibits BCR-ABL, ABL1, and ABL2, as well as certain other kinases including KIT, a receptor tyrosine kinase that is a target of imatinib for treatment of gastrointestinal stromal tumor.10−12 Clinical trials with imatinib showed that more than 90% of CML patients responded to treatment;(13) however, a high percentage of patients in the advanced phases of the disease developed resistance to the treatment, mainly because of BCR-ABL mutants with no, or reduced, sensitivity to imatinib inhibition.14−16 This has led to the development of alternative inhibitors such as dasatinib, which is effective against imatinib-resistant BCR-ABL mutants.(17) Unfortunately, these inhibitors are not effective against the common BCR-ABL T315I mutation often found in relapsed CML patients. The Aurora kinase inhibitors such as 1 (VX-680)(18) (Chart 1) and 3 (PHA-739358)(19) have considerable antitumor activity, and 3 inhibits imatinib-resistant BCR-ABL mutations including T315I.(20) A new approach involving allosteric inhibitors that bind to the myristate binding pocket has potential: Gray and colleagues recently reported the inhibitors GNF-2 and GNF-5 that bind in the myristate binding pocket and, when used together with ATP-competitive inhibitors, can be used to overcome resistance to ATP-site inhibitors.(21)
Chart 1. Chemical Structures of Imatinib, 1, and 2.

Crystal structures have been solved for ABL1 in complex with several inhibitors including imatinib,(22) dasatinib,(23)1,(24) and 3(25) that explain the observed potency profiles of these inhibitors based on the structural conformations of the protein−inhibitor complexes. Recently, the structure of ABL1 with GNF-2 bound was reported.(21)
Here we present three structures of ABL2 kinase domain, in complex with the inhibitors imatinib, 1, and 5-amino-3-((4-(aminosulfonyl)phenyl)amino)-N-(2,6-difluorophenyl)-1H-1,2,4-triazole-1-carbothioamide 2 (JNJ-7706621) (Chart 1), a 1-acyl-1H-[1,2,4]triazole-3,5-diamine analogue that has been reported as a potent antitumor cyclin-dependent kinase inhibitor.(26) The cocrystallized compound 2 bound to an inactive ABL2 conformation that can be considered as an intermediate between the canonical active DFG Asp in and inactive DFG Asp out conformations. This conformation places the DFG phenylanaline into the ATP site, providing the potential of aromatic stacking interactions with inhibitors.
Results
Structural Comparison of ABL2 and ABL1
We initiated structural studies on multiple constructs of ABL2 using baculovirus-mediated expression in insect cells. A construct spanning residues 279−546 (Figure 1A), encompassing the kinase domain, gave high levels of soluble protein expression in Hi5 cells, and purification yielded protein of suitable purity for crystallization. Three crystal structures of the kinase domain of ABL2 in complex with 1, imatinib, and 2 (Figure 1B) were determined at 2.05, 1.65, and 2.81 Å resolution, respectively. All structures are of the unphosphorylated form of ABL2. The overall structure of the kinase domain in each of the structures is similar, with the typical structural features of a protein kinase: the kinase domain is made up of two lobes. The N-terminal lobe is the smaller subdomain, composed of a five-stranded β-sheet and a prominent α-helix. The larger C-terminal lobe consisting of residues 367−546 is mostly helical. Connecting the two lobes is a linker that acts as a hinge about which the two lobes can rotate with respect to each other upon ATP and substrate binding. The ATP-binding site is located in a pocket between the two lobes. ABL2 is 93% identical to ABL1 over the kinase domain (Figure 1A), and the residue differences are all surface-exposed (Figure 1C). Interestingly, the hinge residue T319 in ABL1 is a proline residue (P365) in ABL2 which may influence the interaction of ATP mimetic inhibitors with the hinge backbone.
Structures of ABL2 in Complex with Imatinib and 1
The structures of the ABL2 kinase domain in complex with imatinib and 1 were determined to confirm the anticipated binding arrangement based on the ABL1:imatinib or ABL1:1 structures. As expected given the sequence similarity between ABL2 and ABL1, the binding of both molecules in the ATP-binding site was essentially identical to the equivalent ABL1 structure.
Compound 1 is bound with the N-methylpiperazine group extending out of the ATP pocket, and the cyclopropane group bound just past the gatekeeper residue Thr361 to form hydrophobic packing interactions with the methyl of Thr361 and the side chain of Met336 (Figure 2A). As with ABL1,(24)1 binds to ABL2 such that the activation segment is in an almost active conformation, with the Asp of the DFG motif pointing “in” to the ATP binding site. Also as in ABL1:1 (and ABL1:imatinib), the P-loop has a distorted conformation to allow the side chain of Tyr299 to form an aromatic π-stacking interaction with the inhibitor, although the interaction involves less of Tyr299 than is the case in ABL1:imatinib where the aromatic ring of Tyr299 packs perpendicularly to the pyridine−aminopyrimidine rings of imatinib. The ABL1:1 complex was of the imatinib-resistant mutant of ABL1, H396P, which is a mutation in the activation segment; however, the conformation of the wild-type ABL2:1 (also His at this position) was identical in this region. This observation provides additional conformation that the H396P mutation hinders formation of the inactive (DFG Asp out) conformation recognized by imatinib, without altering the active activation segment conformation recognized by 1.
Figure 2.

Comparison of ABL2:imatinib and ABL2:1 with ABL1:imatinib and ABL1:1. (A) Imatinib bound in the active sites of ABL1 and ABL2. The structure of the active site of ABL1 with imatinib bound (blue) (PDB code 1IEP)(22) is shown superimposed on ABL2:imatinib (orange), showing that the inhibitor molecules bind in the same conformation in both structures. Imatinib is shown in yellow for ABL2 and green for ABL1. (B) 1 bound in the active sites of ABL1 and ABL2. The structure of the active site of ABL1 with 1 bound (PDB code 2F4J)(24) is shown superimposed on ABL2:1. Coloring is as for (A) except that 1 is in yellow for ABL2 and purple for ABL1.
While ABL2:1 was as anticipated, the structure of ABL2:imatinib showed an unexpected result: two imatinib molecules were found bound to ABL2, one in the active site (ATP binding site) of the kinase (Figure 2A) and the other at a remote site equivalent to the myristate binding site (Figure 4). Isothermal titration calorimetry (ITC) with ABL2 and imatinib was carried out to determine whether both binding sites are occupied in solution. The ITC data support a model with two molecules of imatinib bound to the ABL2 construct, with dissociation constants (KD) of 6 nM and 0.5 μM (Figure 3).
Figure 4.

Myristate binding pocket of ABL2. (A) Surface of the myristate binding pocket of ABL2, with imatinib shown as a yellow ball-and-stick representation. (B) Overlay of the ABL1:myristic acid complex (PDB code 1OPL) (orange and black) and ABL2:imatinib (blue and yellow) structures at the myristate binding site. Phe516 (ABL1) and Phe543 (ABL2) are shown in orange and blue, respectively. Residues 516, 521, and 525 of ABL1, from the bent αI′ helix, occupy part of the space occupied by imatinib in the ABL2 structure. These residues were not present in the ABL2 construct used to determine the structure.
Figure 3.

Isothermal titration calorimetry (ITC) of ABL2 and imatinib. The data show that ABL2 binds imatinib at two sites.
The molecule of imatinib bound to the active site is in the same conformation as reported for the imatinib:ABL1 complex (PDB code 1IEP) (Figure 2A), and the structures were superimposed with an rmsd of 0.61 Å over 258 Cα atoms. Hydrogen-bonding interactions with the active site residues are conserved (Table 1). As in the case of ABL1,(27) imatinib recognized the inactive, “DFG out”, conformation of ABL2 with the activation segment in a similar inhibitory conformation, although partly disordered. Asp381 of the DFG motif (equivalent to Asp427 in ABL1) is pointing away from the ATP binding site, whereas Phe382 (ABL1:Phe428) is pointing toward the ATP binding site and forming an aromatic stacking interaction with the inhibitor. The gatekeeper residue Thr361 (ABL1:Thr315) and Glu332 (ABL1:Glu286) from αC form hydrogen bonds to imatinib (Figure 2A, Table 1).
Table 1. Comparison of Hydrogen Bond Distances to Imatinib from ABL and ABL2.
| ABL | ABL2a |
|---|---|
| N51−Ile360 O = 2.7 Å | N51−Ile406 O = 2.8 Å |
| N51−His361 O = 3.1 Å | N51−His407 O = 3.2 Å |
| O29−Asp381 N = 3.0 Å | O29−Asp427 N = 2.8 Å |
| N21−Glu286 OE2 = 3.1 Å | N21−Glu332 OE2 = 3.0 Å |
| N13−Thr315 OG1 = 3.0 Å | N13−Thr361 OG1 = 3.1 Å |
| N3−Met318 N = 2.9 Å | N3−Met364 N = 2.9 Å |
The ABL2 numbering refers to ABL2 isoform B (gi∣6382062).
The second imatinib molecule in ABL2 bound in an extended conformation in the myristate binding pocket (Figure 4A) and forms a hydrogen bond with the carbonyl group of Ala383 on helix αE. Superimposition of the ABL1/myristic acid complex (PDB code 1OPL) with ABL2:imatinib (Figure 4B) shows how imatinib mimics myristic acid binding. The superimposition with the C-terminally extended ABL1 structure shows how our shorter ABL2 construct (residues 279−546, numbering from isoform B) used for crystallization permitted the binding of imatinib at this site; however, the full length protein (with the ABL1 or ABL2 C-terminus) would be unlikely to bind imatinib at this position because of a steric clash with the αI′ helix, which was omitted from our construct. With the shorter construct, Phe543 points away from the myristic acid binding pocket, also avoiding a steric clash that would occur in the full-length protein, as can be seen from the conformation of ABL1 Phe516 (Figure 4B). If binding did occur between imatinib and full-length ABL1 or ABL2 at the myristate binding pocket, it would not be accompanied by the bending of helix αI′ that is necessary for inhibition.(28) Apart from the shorter construct used for ABL2, there are no sequence differences between ABL1 and ABL2 in the region of the myristic acid binding pocket, and the results here for ABL2 presumably would apply also to ABL1 given a similar construct.
Structure of ABL2 in Complex with 2
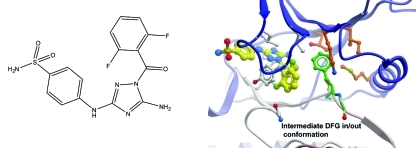
The structure of ABL2 kinase domain in complex with 2(26) was determined to investigate the binding mode of this inhibitor. Compound 2 is a potent, ATP-competitive type I inhibitor, targeting CDK1/CDK2, which shows significant antitumor activity.(26) The structure of ABL2:2 superimposes with ABL2:imatinib with an rmsd of 1.05 Å over 238 Cα atoms. Compound 2 binds to the ATP binding site, with the aminothiazole moiety occupying the adenine binding site (Figure 5A). The ligand is bound to the protein by hydrogen bonds to the backbone carbonyls of residues Glu362 and Met364 in the hinge region, in common with many ATP-mimetic inhibitors, as well as an aromatic−aromatic ring interaction with Tyr299 from the P-loop, which is oriented toward the active site (Figure 5A). The conformation of 2 is almost identical to that observed in the various other structures of kinases in complex with this ligand in the PDB (discussed below).
Figure 5.

ABL2 bound to a type I inhibitor 2. (A) ABL2:2, showing the compound bound to the ATP binding site, and the ordered activation loop. Compound 2 is shown in yellow. The DFG motif is shown in green, and the hydrophobic pocket in which the DFG Phe binds is shown in brown. (B) ABL1:PD166326 (PDB code 1OPK),(30) showing PD166326 in purple. Other coloring is as in (A). This shows the dichlorophenyl group of PD166325 occupying the same volume as the DFG Phe side chain in (A). (C) ABL2:imatinib. (D) ABL2:2 from a different angle to show clearly the rotation of the DFG motif and the intermediate position of the DFG Phe residue relative to (C) and (E). (E) ABL1:ADP (PDB code 2G2I).(29)
In ABL2:2 the activation loop is fully ordered in an “activated” conformation, which can be seen from a comparison with the structure of ABL1:ADP (PDB code 2G2I)(29) (Figure 5D,E); the activation segment is well ordered and is stabilized by a salt bridge between Arg432 and the αC glutamate (Glu338) as well as a stacking interaction between the activation segment tyrosine (Tyr439) and the catalytic HRD motif arginine (Arg408).
Compared to the structure of ABL2:imatinib, the loop region N-terminal to αC of ABL2:2 is in a significantly different conformation (Figure 5C,D), although this may be influenced by crystal packing interactions. Helix αC itself is correctly oriented as indicated by the presence of the salt bridge between the active site lysine residue Lys317 and the αC glutamate residue Glu332. However, the helix does not bend inward as in the ABL1:ADP structure (Figure 5E), where the lack of a bound inhibitor allows the hydrophobic pocket formed by Ile313, Thr315, and Met290 (ABL1 numbering) to shrink, pulling the middle of helix αC toward the ATP binding site.
The most unusual feature of the ABL2:2 structure is the arrangement of the DFG motif. Typically in an inactive (DFG Asp out) conformation such as that in ABL2:imatinib (Figure 5C), the Asp from the DFG motif is “out”, pointing away from the ATP binding site, while the Phe points into the binding site. In many complexes of kinases with inhibitors the Phe interacts with a hydrophobic part of the inhibitor molecule. In an active conformation the backbone rotates to allow the Asp to point “in” and coordinate the Mg2+. However, in ABL2:2, while the backbone conformation of the DFG motif is in the Asp “out” conformation, the Phe side chain is rotated 120° up into the hydrophobic pocket adjacent to the ATP binding site. A comparison of ABL2:2 with the structure of ABL1 in complex with PD166326 (PDB code 1OPK),(30) an inhibitor of similar chemical structure, shows that in the rotated position Phe428 occupies the position of the dichlorophenyl ring of the larger inhibitor in ABL1:PD166326 (Figure 5A,B). In this position it binds in the hydrophobic pocket formed by Ile359, Thr361, Met336, and Lys317. It is presumably for this reason that ABL2:2 does not have the bent αC conformation observed in ABL1:ADP (Figure 5E), despite also having a smaller molecule bound to the ATP binding site.
Discussion
The overall structure and binding mode of the inhibitors imatinib and 1 is conserved between ABL1 and ABL2; however, the discovery that in a truncated construct of ABL2, imatinib also binds in the myristate binding pocket represents an important and novel finding in light of the recent interest in developing allosteric modulators of this site such as GNF-2 (3-[6-[[4-trifluoromethoxy)phenyl]amino]-4-pyrimidinyl]benzamide)(31) which binds in the myristate binding pocket, as recently confirmed by a cocrystal structure.(21) It is clear from the ABL2:imatinib structure that in the full-length protein imatinib would not bind in the myristate binding pocket in such a way that would cause inhibition by promoting the bending of helix αI′ and the docking of the SH2 and SH3 domains,(28) due to a steric clash with a bent helix αI′. However, a truncated imatinib molecule presumably would bind in this context and may offer a starting point for the design of alternative allosteric ABL inhibitors. The linear region of imatinib (excluding the pyridine−aminopyrimidine rings) is colinear with the binding position of myristate (Figure 4B) and therefore represents a useful template fragment. Such allosteric inhibitors are potentially valuable for overcoming the resistance of various mutant ABL proteins to ATP-mimetic inhibitors, and it has recently been shown that it is also possible to target this pocket for activation of ABL if the molecule binds but does not induce a bending of helix αI.(28) Therefore, molecules that are developed to bind in this pocket would need characterization of antagonist versus agonist activity.
We have previously crystallized the type I inhibitor 2 with a number of other human protein kinases. In all of these other structures a rotated DFG motif Phe was not observed, and all showed a typical DFG Asp “out” conformation. The distinguishing feature in ABL2 that makes the unusual Phe conformation possible is the larger volume between the Thr361 gatekeeper residue Lys317 (from the salt bridge to αC) and the other residues of the hydrophobic pocket (Figure 5). All of the other kinases we have crystallized with 2 have a larger, hydrophobic gatekeeper residue, for example, CSNK1G3 (casein kinase 1 γ3, PDB code 2CHL, leucine gatekeeper), SLK (Ste20-like kinase, PDB code 2J51, Ile gatekeeper),(32) CAMK4 (PDB code 2W4O, leucine gatekeeper), and CLK3 (CDC-like kinase 3, PDB code 2WU6, Phe gatekeeper),(33) and also a generally smaller hydrophobic pocket. For example, ABL2 has a distance of 9.2 Å between its gatekeeper Cα backbone atom and Lys317 Cα backbone atom, while CSNK1G3, SLK, CAMK4, and CLK3 have distances of 7.8, 8.1, 7.5, and 8.9 Å, respectively. The combination of the larger side chains and the smaller Cα−Cα distances means that in these other structures there is not enough space for the DFG Phe side chain to bind in the same hydrophobic pocket. Compared to these other structures, ABL2 and ABL1 have more space that allows the rotation of Phe428. All of the structures presented here have a hydrophobic entity bound in this position: in the imatinib complex it is an aromatic ring; in the compound 1 complex it is the cyclopropane ring; and in the compound 2 complex, it is the side chain of Phe428.
The ABL2:2 structure suggests it is possible that Phe428 could exist in this rotated “intermediate in/out” position even in the presence of the imatinib-resistant gatekeeper mutation T315I (ABL1 numbering) (Figure 5A and Figure 5D), and it may be that this conformation of the kinase could be targeted for second-line treatment following imatinib resistance, especially since it is also compatible with the ordered activation loop seen with the alternative imatinib-resistant activation loop mutation H396P. In the same way that 1 successfully targets gatekeeper mutations by allowing sufficient space around the gatekeeper residue by its “Y”-shape (compare Figure 2A and Figure 2B) and its small cyclopropane ring bound in the hydrophobic pocket, this Phe428 conformation might also be compatible with a mutated gatekeeper residue. Indeed a superimposition of ABL2:1 and ABL2:2 shows that the Phe428-bound conformation actually allows more space around the gatekeeper than 1 (data not shown). Thus, targeting this conformation may also provide additional opportunities for achieving inhibitor specificity.
Materials and Methods
Cloning of ABL2 Kinase Domain
Residues 279−546 of the kinase domain of human ABL2 (gi|6382062, Arg tyrosine kinase isoform B) were cloned into a pFastBac-derived transfer vector in-frame with an N-terminal hexahistidine tag and TEV protease cleavage site. This plasmid was transformed into Escherichia coli strain DH10Bac which contains bacmid DNA. The recombinant bacmid DNA was isolated and verified by PCR. In order to generate the baculoviruses, the recombinant bacmid DNA was used for transfection of Sf9 insect cells followed by two rounds of amplification in Sf9 cells.
Protein Expression and Purification of ABL2 Kinase Domain
For expression of the protein, baculovirus obtained from the Sf9 cell culture was used to infect Trichoplusia ni (Hi5) cells grown in suspension to a density of 2 × 106 cells/mL. At 48 h postinfection the cells were harvested by centrifugation and cell pellets were stored at −80 °C. Cells were resuspended in a buffer consisting of 5 mM imidazole, 500 mM NaCl, 50 mM Hepes, pH 7.4, 5% glycerol, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), supplemented with complete protease inhibitor mixture (Roche Applied Sciences), and lysed by sonication. The lysate was centrifuged at 45000g for 1 h at 4 °C. The supernatant was filtered and loaded onto nickel-chelating resin. After being washed, the protein was eluted with the above buffer plus 50−300 mM imidazole and the eluates were combined. The hexahistidine tag was removed by overnight treatment with TEV protease at 4 °C. The digested ABL2 was concentrated to 2.5 mL volume and loaded onto a Superdex75 gel filtration column (HiLoad 16/60, GE Healthcare) equilibrated in 10 mM Hepes, pH 8.0, 300 mM NaCl, and 0.5 mM TCEP.
The protein identity was verified by electrospray ionization time-of-flight mass spectrometry (Agilent LC/MSD). Prior to removal of the hexahistidine tag, the observed mass was 33 414 Da compared to a calculated mass of 33 502; it is likely that the difference in mass was due to removal of the N-terminal methionine followed by acetylation. After removal of the hexahistidine tag the observed mass was 30 980 Da, matching exactly the calculated mass.
Crystallization and Data Collection
The ABL2:imatinib complex (PDB code 3GVU, Table 2) was crystallized at 4 °C in 200 nL drops from a 1:1 ratio of ABL2:imatinib (4 mg/mL protein containing 1 mM imatinib) and reservoir solution (20% PEG3350, 0.1 M citrate, pH 5.5). The crystals were then cryoprotected in reservoir solution with 20% (v/v) PEG300 and flash-frozen in liquid nitrogen. X-ray diffraction data was collected at 100 K on beamline X10SA at the Swiss Light Source (SLS).
Table 2. Data Collection and Refinement Statistics.
| ABL2:imatinib | ABL2:2 | ABL2:1 | |
|---|---|---|---|
| Crystal Form | |||
| unit cell | |||
| a (Å) | 88.5 | 165.9 | 170.5 |
| b (Å) | 99.4 | 39.8 | 170.5 |
| c (Å) | 87.8 | 43.1 | 100.7 |
| α (deg) | 90.0 | 90.0 | 90.0 |
| β (deg) | 90.0 | 95.69 | 90.0 |
| γ (deg) | 90.0 | 90.0 | 120.0 |
| space group | C2221 | C21 | P3221 |
| no. of molecules/asu | 1 | 1 | 3 |
| Data Collection | |||
| resolution rangea (Å) | 43.90−2.05 (2.16−2.05) | 39.72−1.65 (1.74−1.65) | 73.83−2.81 (2.96−2.81) |
| completenessa (%) | 99.3 (95.5) | 85.1 (85.4) | 99.9 (100.0) |
| multiplicitya | 4.8 (3.6) | 3.0 (3.1) | 7.3 (7.5) |
| Rmerge a (%) | 0.086 (0.445) | 0.066 (0.802) | 0.095 (1.025) |
| ⟨I/σ(I)⟩ a | 12.8 (3.2) | 11.8 (2.2) | 10.8 (1.9) |
| Refinement | |||
| R factor (%) | 18.9 | 18.8 | 24.6 |
| Rfree (%) | 22.6 | 23.4 | 28.4 |
| rmsd bond length (Å) | 0.013 | 0.014 | 0.011 |
| rmsd bond angle (deg) | 1.474 | 1.465 | 1.35 |
| PDB code | 3GVU | 3HMI | 2XYN |
Values in parentheses are for the highest resolution shell.
The ABL2:2 complex (PDB code 3HMI) was crystallized at 4 °C in 200 nL drops from a 3:1 ratio of ABL2:2 (8 mg/mL protein containing 1 mM compound 2 (5-amino-3-((4-(aminosulfonyl)phenyl)amino)-N-(2,6-difluorophenyl)-1H-1,2,4-triazole-1-carbothioamide, Calbiochem product no. 217714) and reservoir solution (0.1 M lithium sulfate, 0.05 M disodium hydrogen phosphate, 0.05 M citric acid, 19% (v/v) PEG1000). The crystals were cryoprotected in reservoir solution with 25% (v/v) ethylene glycol and flash-frozen in liquid nitrogen. X-ray diffraction data was collected at 100 K on beamline I03 at DIAMOND.
The ABL2:1 complex (PDB code 2XYN) was crystallized at 4 °C in 200 nL drops from a 2:1 ratio of ABL2:2 (10 mg/mL protein containing 1 mM 1 (GSK487830B)) and reservoir solution (0.8 M sodium succinate). The crystals were cryoprotected in reservoir solution with 25% (v/v) glycerol and flash-frozen in liquid nitrogen. X-ray diffraction data was collected at 100 K on beamline I03 at DIAMOND.
Structure Determination and Refinement
The diffraction images were processed using MOSFLM(34) and SCALA in the CCP4 suite of programs.(35) Structures were solved by molecular replacement using Phaser(36) with ABL1 as a search model and were refined against maximum likelihood targets using restrained refinement and TLS parameters (where appropriate), as implemented in the program REFMAC.(37) Iterative rounds of refinement were interspersed with manual rebuilding of the models using COOT.(38) Progress of the refinement for each structure was judged throughout by following a reduction in Rfree (calculated from a random 5% of the data that was excluded from the refinement).
Acknowledgments
The Structural Genomics Consortium is a registered charity (No. 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research, and the Wellcome Trust. We thank the staff of the Swiss Light Source and Diamond synchrotrons for assistance with data collection.
Glossary
Abbreviations Used
- ABL2
v-abl Abelson murine leukemia viral oncogene homologue 2
- ARG
Abl related gene
- ETV6
ETS variant gene 6
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- SH3
Src homology 3
- SH2
Src homology 2
- BCR
breakpoint cluster region
- CML
chronic myeloid leukemia
- TEL
translocation ETS leukemia
- AML
acute myelogenous leukemia
- PEG
polyethylene glycol
PDB codes: 3GVU, 3HMI, and 2XYN.
References
- Goff S. P.; Gilboa E.; Witte O. N.; Baltimore D. Structure of the Abelson murine leukemia virus genome and the homologous cellular gene: studies with cloned viral DNA. Cell 1980, 22, 777–785. [DOI] [PubMed] [Google Scholar]
- Kruh G. D.; King C. R.; Kraus M. H.; Popescu N. C.; Amsbaugh S. C.; McBride W. O.; Aaronson S. A. A novel human gene closely related to the abl proto-oncogene. Science 1986, 234, 1545–1548. [DOI] [PubMed] [Google Scholar]
- Kruh G. D.; Perego R.; Miki T.; Aaronson S. A. The complete coding sequence of arg defines the Abelson subfamily of cytoplasmic tyrosine kinases. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 5802–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske A. J.; Gifford A. M.; Scott M. L.; Nee M.; Bronson R. T.; Miczek K. A.; Baltimore D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron 1998, 21, 1259–1272. [DOI] [PubMed] [Google Scholar]
- Gu J. J.; Zhang N.; He Y. W.; Koleske A. J.; Pendergast A. M. Defective T cell development and function in the absence of Abelson kinases. J. Immunol. 2007, 179, 7334–7343. [DOI] [PubMed] [Google Scholar]
- Sawyers C. L. Chronic myeloid leukemia. N. Engl. J. Med. 1999, 340, 1330–1340. [DOI] [PubMed] [Google Scholar]
- Daley G. Q.; Van Etten R. A.; Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990, 247, 824–830. [DOI] [PubMed] [Google Scholar]
- Iijima Y.; Ito T.; Oikawa T.; Eguchi M.; Eguchi-Ishimae M.; Kamada N.; Kishi K.; Asano S.; Sakaki Y.; Sato Y. A new ETV6/TEL partner gene, ARG (ABL-related gene or ABL2), identified in an AML-M3 cell line with a t(1;12)(q25;p13) translocation. Blood 2000, 95, 2126–2131. [PubMed] [Google Scholar]
- Xu W.; Doshi A.; Lei M.; Eck M. J.; Harrison S. C. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 1999, 3, 629–638. [DOI] [PubMed] [Google Scholar]
- Buchdunger E.; Zimmermann J.; Mett H.; Meyer T.; Muller M.; Druker B. J.; Lydon N. B. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996, 56, 100–104. [PubMed] [Google Scholar]
- Okuda K.; Weisberg E.; Gilliland D. G.; Griffin J. D. ARG tyrosine kinase activity is inhibited by STI571. Blood 2001, 97, 2440–2448. [DOI] [PubMed] [Google Scholar]
- Antonescu C. R. The GIST paradigm: lessons for other kinase-driven cancers. J. Pathol. 2011, 223, 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H.; Sawyers C.; Hochhaus A.; Guilhot F.; Schiffer C.; Gambacorti-Passerini C.; Niederwieser D.; Resta D.; Capdeville R.; Zoellner U.; Talpaz M.; Druker B.; Goldman J.; O’Brien S. G.; Russell N.; Fischer T.; Ottmann O.; Cony-Makhoul P.; Facon T.; Stone R.; Miller C.; Tallman M.; Brown R.; Schuster M.; Loughran T.; Gratwohl A.; Mandelli F.; Saglio G.; Lazzarino M.; Russo D.; Baccarani M.; Morra E. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 2002, 346, 645–652. [DOI] [PubMed] [Google Scholar]
- Druker B. J.; Sawyers C. L.; Kantarjian H.; Resta D. J.; Reese S. F.; Ford J. M.; Capdeville R.; Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [DOI] [PubMed] [Google Scholar]
- Gorre M. E.; Mohammed M.; Ellwood K.; Hsu N.; Paquette R.; Rao P. N.; Sawyers C. L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [DOI] [PubMed] [Google Scholar]
- Shah N. P.; Nicoll J. M.; Nagar B.; Gorre M. E.; Paquette R. L.; Kuriyan J.; Sawyers C. L. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002, 2, 117–125. [DOI] [PubMed] [Google Scholar]
- Quintas-Cardama A.; Kantarjian H.; Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat. Rev. Drug Discovery 2007, 6, 834–848. [DOI] [PubMed] [Google Scholar]
- Harrington E. A.; Bebbington D.; Moore J.; Rasmussen R. K.; Ajose-Adeogun A. O.; Nakayama T.; Graham J. A.; Demur C.; Hercend T.; Diu-Hercend A.; Su M.; Golec J. M.; Miller K. M. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004, 10, 262–267. [DOI] [PubMed] [Google Scholar]
- Carpinelli P.; Ceruti R.; Giorgini M. L.; Cappella P.; Gianellini L.; Croci V.; Degrassi A.; Texido G.; Rocchetti M.; Vianello P.; Rusconi L.; Storici P.; Zugnoni P.; Arrigoni C.; Soncini C.; Alli C.; Patton V.; Marsiglio A.; Ballinari D.; Pesenti E.; Fancelli D.; Moll J. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol. Cancer Ther. 2007, 6, 3158–3168. [DOI] [PubMed] [Google Scholar]
- Gontarewicz A.; Balabanov S.; Keller G.; Colombo R.; Graziano A.; Pesenti E.; Benten D.; Bokemeyer C.; Fiedler W.; Moll J.; Brümmendorf T. H. Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant BCR-ABL mutations including T315I. Blood 2008, 111, 4355–4364. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Adrián F. J.; Jahnke W.; Cowan-Jacob S. W.; Li A. G.; Iacob R. E.; Sim T.; Powers J.; Dierks C.; Sun F.; Guo G.-R.; Ding Q.; Okram B.; Choi Y.; Wojciechowski A.; Deng X.; Liu G.; Fendrich G.; Strauss A.; Vajpai N.; Grzesiek S.; Tuntland T.; Liu Y.; Bursulaya B.; Azam M.; Manley P. W.; Engen J. R.; Daley G. Q.; Warmuth M.; Gray N. S. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010, 463, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler T.; Bornmann W.; Pellicena P.; Miller W. T.; Clarkson B.; Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 2000, 289, 1938–1942. [DOI] [PubMed] [Google Scholar]
- Tokarski J. S.; Newitt J. A.; Chang C. Y.; Cheng J. D.; Wittekind M.; Kiefer S. E.; Kish K.; Lee F. Y.; Borzillerri R.; Lombardo L. J.; Xie D.; Zhang Y.; Klei H. E. The structure of dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006, 66, 5790–5797. [DOI] [PubMed] [Google Scholar]
- Young M. A.; Shah N. P.; Chao L. H.; Seeliger M.; Milanov Z. V.; Biggs W. H. 3rd; Treiber D. K.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J.; Sawyers C. L.; Kuriyan J. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the aurora kinase inhibitor VX-680. Cancer Res. 2006, 66, 1007–1014. [DOI] [PubMed] [Google Scholar]
- Modugno M.; Casale E.; Soncini C.; Rosettani P.; Colombo R.; Lupi R.; Rusconi L.; Fancelli D.; Carpinelli P.; Cameron A. D.; Isacchi A.; Moll J. Crystal structure of the T315I Abl mutant in complex with the aurora kinases inhibitor PHA-739358. Cancer Res. 2007, 67, 7987–7990. [DOI] [PubMed] [Google Scholar]
- Lin R.; Connolly P. J.; Huang S.; Wetter S. K.; Lu Y.; Murray W. V.; Emanuel S. L.; Gruninger R. H.; Fuentes-Pesquera A. R.; Rugg C. A.; Middleton S. A.; Jolliffe L. K. 1-Acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: synthesis and evaluation of biological activities. J. Med. Chem. 2005, 48, 4208–4211. [DOI] [PubMed] [Google Scholar]
- Nagar B.; Bornmann W. G.; Pellicena P.; Schindler T.; Veach D. R.; Miller W. T.; Clarkson B.; Kuriyan J. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [PubMed] [Google Scholar]
- Jahnke W.; Grotzfeld R. M.; Pellé X.; Strauss A.; Fendrich G.; Cowan-Jacob S. W.; Cotesta S.; Fabbro D.; Furet P.; Mestan J.; Marzinzik A. L. Binding or bending: bistinction of allosteric abl kinase agonists from antagonists by an NMR-based conformational assay. J. Am. Chem. Soc. 2010, 132, 7043–7048. [DOI] [PubMed] [Google Scholar]
- Levinson N. M.; Kuchment O.; Shen K.; Young M. A.; Koldobskiy M.; Karplus M.; Cole P. A.; Kuriyan J. A Src-like inactive conformation in the Abl tyrosine kinase domain. PLoS Biol. 2006, 4, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B.; Hantschel O.; Young M. A.; Scheffzek K.; Veach D.; Bornmann W.; Clarkson B.; Superti-Furga G.; Kuriyan J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 2003, 112, 859–871. [DOI] [PubMed] [Google Scholar]
- Adrián F. J.; Ding Q.; Sim T.; Velentza A.; Sloan C.; Liu Y.; Zhang G.; Hur W.; Ding S.; Manley P.; Mestan J.; Fabbro D.; Gray N. S. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat. Chem. Biol. 2006, 2, 95–102. [DOI] [PubMed] [Google Scholar]
- Pike A. C.; Rellos P.; Niesen F. H.; Turnbull A.; Oliver A. W.; Parker S. A.; Turk B. E.; Pearl L. H.; Knapp S. Activation segment dimerization: a mechanism of kinase autophosphorylation of non-consensus sites. EMBO J. 2008, 27, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock A. N.; Das S.; Debreczeni J. E.; Rellos P.; Fedorov O.; Niesen F. H.; Guo K.; Papagrigoriou E.; Amos A. L.; Cho S.; Turk B. E.; Ghosh G.; Knapp S. Kinase domain insertions define distinct roles of CLK kinases in SR protein phosphorylation. Structure 2009, 17, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. G. Integration of macromolecular diffraction data. Acta Crystallogr. 1999, D55, 1696–1702. [DOI] [PubMed] [Google Scholar]
- Collaborative computational project number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. 1994, D50, 760−763. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997, D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004, D60, 2126–2132. [DOI] [PubMed] [Google Scholar]