

Abstract
Biotin protein ligases catalyze specific covalent linkage of the coenzyme biotin to biotin-dependent carboxylases. The reaction proceeds in two steps, including synthesis of an adenylated intermediate followed by biotin transfer to the carboxylase substrate. In this work specificity in the transfer reaction was investigated using single turnover stopped-flow and quench-flow assays. Cognate and noncognate reactions were measured using the enzymes and minimal biotin acceptor substrates from Escherichia coli, Pyrococcus horikoshii, and Homo sapiens. The kinetic analysis demonstrates that for all enzyme-substrate pairs the bimolecular rate of association of enzyme with substrate limits post-translational biotinylation. In addition, in noncognate reactions the three enzymes displayed a range of selectivities. These results highlight the importance of protein-protein binding kinetics for specific biotin addition to carboxylases and provide one mechanism for determining biotin distribution in metabolism.
Keywords: Biotin, Enzyme Turnover, Metabolism, Post-translational Modification, Pre-steady-state Kinetics, Protein-Protein Interactions
Introduction
Post-translational protein modification is ubiquitous in biology and influences critical processes including metabolism, protein degradation, and gene expression. The water-soluble vitamin biotin, a required cofactor for biotin-dependent carboxylases, functions as a transient carrier of carboxyl groups in their transfer from bicarbonate to small molecule metabolites, such as acetyl-CoA. The five mammalian biotin-dependent carboxylases function in gluconeogenesis, amino acid catabolism, and fatty acid synthesis and degradation (1). In its coenzyme function biotin must be covalently linked to a carboxylase in a highly specific post-translational modification reaction catalyzed by biotin protein ligases (BPLs).2 In this two-step reaction (Fig. 1A) an intermediate, bio-5′-AMP, is first formed from biotin and ATP substrates, and the biotin is then linked via its carboxyl group to the ϵ amine of a specific lysine residue on the carboxylase (2). The target lysine residue is on the biotin carboxylase carrier protein (BCCP) domain of the carboxylase, an enzyme complex composed of multiple copies of BCCP, biotin carboxylase, and carboxyl transferase moieties (1).
FIGURE 1.

A, ligase-catalyzed biotin transfer occurs in two steps in which an activated biotin intermediate is first synthesized from biotin and ATP with the release of pyrophosphate, and the biotin moiety of bio-5′-AMP is then covalently attached to a specific lysine residue on BCCP. B, alignment (33) of the E. coli, H. sapiens, and P. horikoshii BPL catalytic domain sequences. The identities (black) and similarities (gray) are highlighted. The N-terminal domains of the E. coli and H. sapiens sequences are not shown.
Previous studies of specificity in post-translational biotin addition are inconclusive. In any single organism, with few exceptions, only the specific target lysine on carboxylase substrates is biotinylated. Consistent with this specificity, the Pyrococcus horikoshii ligase-BCCP structure, in which BCCP refers to a minimal biotin accepting domain, is characterized by an extensive protein-protein interface (3). However, a lack of specificity is evident from numerous examples of interspecies biotinylation. For example, in vivo the Escherichia coli ligase biotinylates BCCP domains from yeast (4), human (5), plant (6), and other bacteria (7), and the human p67, a commonly used substrate that includes propionyl-CoA carboxylase BCCP, can be biotinylated by partially purified BPLs from at least nine other organisms (8, 9). This interspecies biotinylation may be attributable to the high degree of sequence conservation among ligases and BCCP domains. Alternatively, the qualitative nature of the methods used to measure interspecies reactivity may have precluded detection of selectivity in the reaction.
Comparison of BPL-BCCP pairs from E. coli (Ec), P. horikoshii (Ph) and Homo sapiens (Hs), which represent three phylogenetic domains, illustrates the sequence and structural conservation among the enzymes and substrates. For example, the most disparate ligase catalytic domain sequences of E. coli and human (Fig. 1B) maintain 26% sequence identity and 43% similarity (10). Moreover, the structures of the catalytic domains of the E. coli and P. horikoshi enzymes are nearly superimposable (11, 12). Overlay of BCCP structures from the three organisms reveals that, with the exception of the “thumb” on the E. coli domain, they are identical (Fig. 2A). At the primary sequence level (Fig. 2B), the most disparate BCCP sequences from human and E. coli display 35% identity and 54% similarity (10).
FIGURE 2.

Alignment of structures and sequences of BCCP fragments. A, superimposition of PhBCCP (gray, 2EJG), HsBCCP (dark gray, 2JKU), and EcBCCP (light gray, 1A6X) structures generated with PyMOL software. B, sequence alignment of the three BCCPs, generated using Jalview (33, 34). The working HsBCCP sequence has an exogenous tyrosine residue at the N terminus, which was added in subcloning to allow for spectrophotometric protein concentration determination.
As indicated above, ligase-catalyzed biotinylation occurs in two steps, and steady-state measurements with the human ligase revealed that bio-5′-AMP synthesis is the rate-limiting step in the overall reaction. However, a single-turnover stopped-flow fluorescence assay in which the preformed enzyme-intermediate complex is mixed with the BCCP substrate provides specific information about the ligase-acceptor protein interaction independent of the bio-5′-AMP synthesis step. Stopped-flow measurements of the HsBPL-catalyzed biotin transfer from the intermediate to a human BCCP substrate revealed a linear dependence of the apparent rate on substrate protein concentration with no evidence of leveling off (13, 14). The simplest interpretation of this kinetic behavior is that the assay reports on the bimolecular collision of the enzyme with substrate protein. However, it is not known whether this is a general feature of the BPL-catalyzed reaction. Additionally, the stopped-flow measurements, which monitor the disappearance of the enzyme-intermediate complex, provide no information about product accumulation in the reaction and, consequently, about which step limits turnover in the second half-reaction.
In this work, the single turnover stopped-flow and a quench-flow assay that allows monitoring of product formation are applied to studies of the second step in BPL-catalyzed biotinylation. Measurements performed on three cognate enzyme-substrate pairs from E. coli, P. horikoshii, and H. sapiens yield linear rate versus substrate concentration profiles in both assays. Furthermore, for each cognate enzyme-acceptor protein pair the bimolecular rates obtained using the stopped-flow and quench-flow assays are identical. Thus, bimolecular association of the enzyme-intermediate complex with BCCP substrate limits post-translational biotin transfer from bio-5′-AMP to the acceptor protein. In addition, measurements performed on noncognate enzyme-substrate pairs reveal distinct levels of substrate selectivity by the three enzymes. The control of the biotin transfer rate by enzyme-substrate collision coupled with the discrimination in noncognate reactions stresses the importance of protein-protein recognition in post-translational biotin addition and has implications for control of biotin distribution in metabolism.
EXPERIMENTAL PROCEDURES
Chemicals and Buffers
All chemicals used in buffer preparation were at least reagent grade. Bio-5′-AMP was synthesized and purified as previously described (2, 15). Biotin d-[2,3,4,6-3H] was purchased from American Radiolabeled Chemicals and stored under argon at −70 °C. Unlabeled d-biotin and ATP were obtained from Sigma-Aldrich. The ATP concentration in stock solutions, which were prepared by dissolving adenosine 5′-triphosphate disodium salt into water and adjusting the pH to 7.5, were determined by UV absorbance using an extinction coefficient at 259 nm of 15,400 m cm−1. Biotin solutions were prepared by dissolving the desired amount of powder in reaction buffer that had not been pH-adjusted, adjusting the pH to 7.5, and bringing the solution to its final volume in a volumetric flask. The biotin stock was filtered, divided into 1-ml aliquots, and stored at −70 °C. The reaction buffer was composed of 10 mm Tris-HCl, pH 7.50 ± 0.02, at either 20 °C (Ec and Hs) or 40 °C (Ph), 2.5 mm MgCl2, and 500 mm KCl (Ph) or 200 mm KCl (Ec and Hs).
Protein Expression and Purification
PhBPL, EcBPL, and HsBPL were recombinantly expressed and purified from E. coli as previously described (13, 16, 17). The E. coli biotin acceptor substrate fragment EcBCCP, which comprises the C-terminal 87 amino acids of the acetyl-CoA carboxylase BCCP subunit, was purified as described by Nenortas et al. (14). All chromatographic steps were carried out on an AKTA prime FPLC platform at 4 °C (GE Healthcare).
PhBCCP corresponds to the 73 C-terminal amino acids of P. horikoshii BCCP domain of acetyl-CoA carboxylase and was expressed from a pet11-a plasmid derivative that was a generous gift from RIKEN (18). The plasmid was transformed into E. coli BL21(λDE3)-RIL strain by electroporation, and selection was carried out on Luria-Bertani (LB) agar plates containing 50 μg/ml kanamycin. A 1-ml volume from a 5-ml overnight culture was diluted into 50 ml of LB medium containing antibiotic and grown for 8 h at 37 °C with shaking at 250 rpm. A second 500-ml culture was started by addition of 10 ml of the previous growth and grown overnight. The final four 1-liter cultures were inoculated with the previous culture at a dilution of 1:20. Protein expression was induced at an A600 of 0.7 by the addition of isopropyl β-d-thiogalactopyranoside to 350 μm, and allowed to proceed for 2.5 h. Cells were harvested by centrifugation at 7000 rpm at 4 °C for 40 min, the pellet was washed with 40 ml of lysis buffer/liter of culture, and the sample was centrifuged again for 20 min at 9000 rpm. The cell paste was resuspended in twice its weight of lysis buffer (50 mm Tris, pH 7.5, at 4 °C, 500 mm NaCl, 5% (v/v) glycerol, 0.1 mm DTT, and 0.1 mm PMSF), and cells were disrupted by sonication using 1-min bursts until the A600 decreased to <10% of the original value. The lysate was diluted to 200 ml with lysis buffer and cleared of cell debris by centrifugation at 9000 rpm for 40 min. After adding CaCl2, MgCl2, DNase I, and RNase, to 1 mm, 2.5 mm, and 0.03 mg/ml (both enzymes) final concentrations, respectively, the sample was stirred at room temperature for 2 h. The solution was then heated to 90 °C for 15 min with stirring, cooled to room temperature, and the precipitated contaminating proteins were cleared by centrifuging at 9000 rpm for 30 min at 4 °C. To eliminate residual nucleic acid contamination PEI was added to the supernatant at a final concentration of 0.2% (w/v), the sample was stirred at 4 °C for 15 min, and the precipitate was removed by centrifugation at 12,000 rpm for 20 min. Solid ammonium sulfate was stirred slowly into the supernatant to 90% (w/v) saturation, and precipitation was allowed to proceed overnight. The precipitate was collected by centrifugation at 12,000 rpm for 1.5 h and resuspended in 30 ml of 50 mm Tris, pH 7.5, at 4 °C, 5% (v/v) glycerol. This sample was dialyzed against 2 mm potassium phosphate buffer, pH 7.0, 5% (v/v) glycerol and loaded on a hydroxyapatite column (Pall Lifesciences). The flow-through was collected, dialyzed against storage buffer (10 mm Tris, pH 7.5, at 4 °C, 200 mm KCl, 5% (v/v) glycerol), and stored in aliquots at −70 °C. Because the PhBCCP does not possess tryptophan or tyrosine residues, the stock concentration was determined by comparing the density of the bands from dilutions electrophoresed on a 20% acrylamide-SDS-Tricine gel against bands with known amounts of PhBCCP(19). Lanes with known amounts of PhBCCP were obtained by loading dilutions of a PhBCCP desalted “standard” solution, the concentration of which had been determined in triplicate by quantitative amino acid analysis at the protein facility of the Iowa State University. Band density quantitation was performed using a Molecular Dynamics Laser Scanning Personal Densitometer (GE Healthcare) and ImageQuant software.
The HsBCCP fragment corresponds to the C-terminal 67 amino acids of the BCCP domain of propionyl-CoA carboxylase. To facilitate removal of the histidine tag and eliminate any exogenous residues originating from the vector, the p67 sequence from a pDEST17 derivative (a generous gift from Dr. Roy Gravel) was subcloned into pSUMO-pro (LifeSensors) (20). A tyrosine residue was added at the N terminus to allow for concentration determination using UV absorption spectroscopy. The plasmid encoding the SUMO-HsBCCP fusion was introduced into E. coli strain Rosetta (DE3) by electroporation, and transformants were selected by growth on LB agar containing 34 μg/ml chloramphenicol and 100 μg/ml ampicillin. A single colony was transferred to 5 ml of LB medium supplemented with antibiotics and grown for 8 h with shaking at 37 °C. A 1:100 dilution was performed to initiate a 50-ml overnight culture, and 1-liter cultures were inoculated with 20 ml of the overnight culture and grown at 37 °C, and protein expression was induced at an A600 of 0.8 by addition of lactose to 0.5% (w/v). Following overnight growth at 30 °C, cells were harvested by centrifugation at 4500 rpm at 4 °C, resuspended in a volume of lysis buffer corresponding to five times the cell pellet weight, and lysed by sonication in HsBCCP lysis buffer (50 mm sodium phosphate buffer, pH 8, 300 mm sodium chloride, 10 mm imidazole, 5% (v/v) glycerol, 3 mm 2-mercaptoethanol, 1 mm PMSF). The crude lysate was cleared by centrifugation at 9000 rpm for 30 min at 4 °C, and the supernatant was subjected to chromatography on 8 ml of Ni-nitrilotriacetic acid resin (Qiagen) packed in a 1.5 × 10-cm Econo-Column (Bio-Rad) according to the manufacturer's instructions. The eluted protein was dialyzed against 10 mm sodium phosphate, pH 8.0, 60 mm NaCl, 5% (v/v) glycerol, 5 mm 2-mercaptoethanol and digested overnight at room temperature with SUMO-protease-1. To remove the protease, the His6-SUMO tag, and undigested protein, the sample was passed through Ni-nitrilotriacetic acid resin. The flow-through was collected and dialyzed against 50 mm Tris, pH 7.5, at 4 °C, 20 mm KCl, 1 mm 2-mercaptoethanol, 5% (v/v) glycerol. The resulting sample was loaded onto a 8-ml DEAE anion exchange (GE Healthcare) column and eluted with a linear gradient of 20–400 mm KCl (VT = 200 ml) at a flow rate of 2 ml/min. The pooled fractions (2 ml each) containing HsBCCP were concentrated and loaded at 0.1 ml/min onto a 150-ml S-100 Sephacryl resin (GE Healthcare) packed in a 1.5 × 100-cm Econo-Column and equilibrated with 10 mm Tris, pH 7.5, at 4 °C, 0.8 m KCl, 5 mm 2-mercaptoethanol, 5% (v/v) glycerol. The eluted sample was exchanged into 10 mm Tris-HCl, pH 8, at 4 °C, 200 mm KCl, and 5% (v/v) glycerol by dialysis and stored at −70 °C. The yield was 10 mg of protein per liter of bacterial culture, and the concentration was determined spectrophotometrically using an extinction coefficient of 1450 m cm−1 at 276 nm (21).
Stopped-flow Measurements of Biotin Transfer
Biotin transfer was measured by monitoring the intrinsic fluorescence increase that occurs upon rapid mixing the enzyme-intermediate complex with the substrate biotin acceptor protein (13, 14). A solution of 1–2 μm ligase was incubated with half its concentration of biotin and 500 μm ATP for 20 min to allow for bio-5′-AMP synthesis. To avoid the complication from the large fluorescence change that accompanies ATP binding to the enzyme, PhBPL-catalyzed reactions were carried out using chemically synthesized bio-5′-AMP (17). Reactions catalyzed by the human and E. coli enzyme were carried out in standard reaction buffer, which contains 10 mm Tris, pH 7.5, at 20 °C, 200 mm KCl, 2.5 mm MgCl2. In contrast, those reactions catalyzed by PhBPL were performed at pH 7.5, 40 °C, and a KCl concentration of 500 mm. The higher salt concentration and temperature are necessary to obtain complete activity of the thermophilic enzyme (17), a reflection of the fact that the P. horikoshii intracellular salt concentration is about 500 mm, and the organism grows optimally at 98 °C (22). Each ligase-intermediate complex solution was rapidly mixed with varying concentrations of BCCP using a Kintek SF-2001 stopped-flow instrument equipped with fluorescence detection. The excitation wavelength was 295 nm, and fluorescence emission was monitored above 340 nm using a cutoff filter (Corion Corp.). At least six traces spanning 10 half-lives were collected at each BCCP concentration. The resulting transients were fit to an appropriate model (single or double exponential), and the apparent rates were plotted as a function of BCCP concentration. Further analysis and interpretation were performed as described under “Results.”
Quench-flow Measurements of Biotin Incorporation
Single turnover measurements of enzyme-catalyzed biotin transfer to the BCCP fragments were monitored as a function of time using a Kintek QF-3 quench-flow apparatus. One syringe contained a solution of 1 μm ligase, 0.463 μm biotin, 37 nm [3H]biotin, and 500 μm ATP that had been incubated for 30 min at the working temperature to allow for bio-5′-AMP synthesis. The second syringe contained variable concentrations of each BCCP fragment, ranging from 10 to 200 μm. All samples were prepared in the appropriate working buffer. Equal volumes of the two syringe solutions were mixed, and the resulting reactions were allowed to age for a specific amount of time, after which they were quenched with a 2 m HCl solution. Free and BCCP-incorporated biotin were separated using TCA precipitation (23). In reactions containing PhBCCP, 0.02% (w/v) sodium deoxycholate was included, in addition to BSA (0.1 mg/ml), to aid in precipitation. The samples were spotted on dried Whatman 3MM squares to which 200 μl of 800 μm unlabeled biotin had been added after soaking in 10% TCA. After drying, the papers were washed twice in batch mode in 250 ml of ice-cold 10% TCA and once in 100 ml of cold ethanol. The radioactivity retained on the filters was quantified in a LS6500 Beckman counter using ReadyProtein+ (Beckman) scintillation fluid. Background corrections were performed with values obtained from control reactions that contained no BCCP. Correction for precipitation efficiency (typically above 80%) and conversion from dpm to “μm biotin” were carried out using the relation between dpm and μm obtained from the precipitation of a reaction incubated for 30 min to allow for quantitative 3H incorporation. Each discontinuous transient was fit to a single exponential equation, and the resulting apparent rates as a function of BCCP concentration were subjected to linear least squares analysis. The slope of the line yielded the rate of biotin incorporation.
RESULTS
Oligomeric States of the Reacting Species in Biotin Transfer
Interpretation of kinetic and binding data requires knowledge of the assembly states of all species participating in a reaction. The oligomerization properties of the three biotin protein ligases have previously been characterized using sedimentation equilibrium. The human ligase is monomeric in both its unliganded and liganded forms (13). By contrast, the P. horikoshii ligase is a constitutive dimer (17). However, in the dimer the active sites for biotin transfer are available for interaction with the acceptor substrate (3). The E. coli enzyme can also form a homodimer, albeit in a reaction that depends on the intermediate in biotin transfer, bio-5′-AMP (24). Furthermore, the surface utilized for the homodimerization is identical to that used for heterodimerization with the biotin acceptor protein (25). The equilibrium dissociation constant governing the homodimerization reaction in buffer conditions identical to those used in the present studies is ∼10 μm (24). Consequently, to avoid any complication from this competing homodimerization reaction, all kinetic measurements in these studies were performed at a total enzyme concentration of 0.5 μm.
The assembly states of the three acceptor protein substrates employed in these studies were also characterized by sedimentation equilibrium. The EcBCCP has previously been shown to be monomeric over a broad concentration range (14). For this work, sedimentation equilibrium measurements were performed on HsBCCP and PhBCCP (data not shown). Global analysis of the data obtained at three loading concentrations and two speeds using a single species model yielded molecular masses for the proteins consistent with monomers.
Single Turnover Assays of the Second Half-reaction in Biotin Transfer
This work is focused on the interaction of the ligase with the biotin acceptor protein in post-translational biotinylation. Because the rate-determining step in the overall reaction is intermediate synthesis, steady-state measurements provide limited information about this protein-protein interaction. However, the reaction proceeds through a double displacement mechanism that can be halted after formation of the kinetically stable ligase-bio-5′-AMP complex (Fig. 1). Mixing of the preformed complex with BCCP allows monitoring of the second half-reaction independent of the first.
Biotin Transfer Rates to Cognate Substrates Are Similar for the Three Biotin Protein Ligases
In these experiments, BPL is preincubated with biotin at half of the enzyme concentration and excess ATP to allow for bio-5′-AMP synthesis. The BPL excess over biotin ensures that only one turnover of biotin transfer occurs in the stopped-flow measurement. Mixing of the preformed complex with BCCP yields a time-dependent increase in the intrinsic fluorescence (Fig. 3A) that has been shown through measurements of the spectra of the two species to be consistent with conversion of the bio-5′-AMP-bound enzyme to the free enzyme (13, 14). Transients acquired upon mixing the ligase-bio-5′-AMP complex at each BCCP concentration were fit to either a single or double exponential model to obtain apparent rates. For example, the transient shown in Fig. 3A, which was obtained by mixing HsBCCP-bio-5′-AMP with 200 μm HsBCCP, contains two well separated exponential phases. Although both the human- and E. coli-catalyzed reactions displayed this biphasic behavior at higher BCCP concentrations, all PhBCCP traces were monophasic.
FIGURE 3.

Stopped-flow measurements of biotin transfer. A, fluorescence (arbitrary units (a.u.)) versus time trace obtained upon 1:1 (v/v) mixing of 0.5 μm HsBPL-bio-5′-AMP with 200 μm HsBCCP. To illustrate the double exponential behavior, data were collected in two time windows (0–2.5 and 0–30 s). The solid line represents the best fit of the data to a double exponential model. B, plots of the dependence of the apparent rate of biotin transfer on BCCP concentration: HsBPL-HsBCCP (■), PhBPL-PhBCCP (♦), and EcBPL-EcBCCP (●). The data points represent the average of the apparent rate measured at each BCCP concentration in three independent experiments with S.D. shown as error bars. The solid lines represent the best fits of the rate versus concentration profiles to a linear equation.
Plots of the faster apparent rate versus biotin-accepting substrate concentration for all three cognate ligase-substrate pairs were linear and showed no evidence of saturation (Fig. 3B). Consequently, the slope of each line is interpreted as reporting on k1, or BCCP association with the ligase-bio-5′-AMP complex, in the following mechanism.
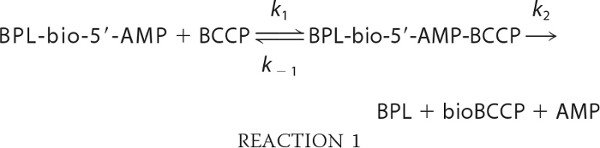 |
The bimolecular association rates for the three BPLs with their cognate substrates (Table 1), obtained from linear regression of the apparent rate versus BCCP concentration profiles (Fig. 3B), reveal that all three reactions are characterized by bimolecular association rates on the order of 104 m−1s−1. However, the human system displays the fastest rate.
TABLE 1.
Biotin transfer rates for cognate ligase-BCCP pairs
| Pair | Stopped-flowa | Quench-flowb |
|---|---|---|
| m−1s−1 | m−1s−1 | |
| HsBPL | 35,000 ± 3,000 | 31,000 ± 3,000 |
| EcBPL | 11,500 ± 700 | 10,500 ± 800 |
| PhBPL | 15,000 ± 600c | 15,000 ± 1000c |
a The errors represent the S.D. of three independent stopped-flow experiments.
b The errors correspond to the S.D. of results obtained in two independent quench-flow experiments.
c Measurements with PhBPL were carried out at 40 °C, in 10 mm Tris, pH 7.5, 500 mm KCl, 2.5 mm MgCl2. All other experiments were performed at 20 °C, in 10 mm Tris, pH 7.5, 200 mm KCl, 2.5 mm MgCl2.
For reactions that exhibited two exponentials, the rate of the slower phase exhibited no dependence on BCCP concentration, which supports its assignment to holo-BCCP dissociation from the ligase (14). The rates of HsBPL and EcBPL dissociation from their cognate holo-BCCPs are both ∼0.2 s−1 (Table 1).
Product Formation Is Limited by Bimolecular Association of Enzyme with Substrate
The stopped-flow assay reports on the disappearance of the ligase-bio-5′-AMP complex but provides no information on product accumulation in the second half-reaction. Consequently, a quench-flow assay was developed to quantify the production of biotinylated BCCP.
In the assay the reactants are prepared similarly to those used in the stopped-flow experiments, with the exception that the cold biotin is spiked with tritium-labeled biotin. Once sufficient time has elapsed for bio-5′-AMP synthesis, an aliquot of the ligase-intermediate complex solution is rapidly mixed with BCCP. After aging for the appropriate time interval, the reaction is quenched by decreasing the pH. This low pH also destroys the noncovalent ligase-bio-5′-AMP complex while leaving the covalent amide linkage between biotin and BCCP intact. In contrast to the stopped-flow assay, the quench-flow assay of biotin transfer is discontinuous. Therefore, the transient acquired at each BCCP concentration (Fig. 4A) results from multiple measurements in which reactions prepared at a given acceptor protein concentration are aged for variable amounts of time prior to quenching. Separation of free biotin from biotinylated BCCP is achieved using a modification of the TCA precipitation method previously used in steady-state kinetic measurements performed on EcBPL (23). The TCA precipitates BCCP and holo-BCCP, but not free biotin or bio-5′-AMP. Quantitation of the radioactivity in the acid-insoluble material by scintillation counting yields information on the amount of biotin incorporated at a specific time and ultimately a transient (Fig. 4A). Transients for all cognate enzyme-substrate pairs are well described by a single exponential model, further supporting the assignment of the slow rate observed in the stopped-flow traces with HsBPL and EcBPL to the enzyme-product dissociation rate. Plots of the apparent rates obtained from the transients versus BCCP concentration are linear and reveal no evidence of saturation (Fig. 4B). Furthermore, for all three enzymes the rates obtained from the slopes of the lines are identical to the bimolecular association rates obtained from the stopped-flow measurements (Table 1). Thus, for all cognate pairs, the rate of bimolecular association of enzyme with substrate limits accumulation of the biotinylated acceptor protein.
FIGURE 4.

Quench-flow measurements of biotin transfer for cognate ligase-BCCP pairs. A, transient obtained upon mixing equal volumes 0.5 μm HsBPL-bio-5′-AMP and 200 μm HsBCCP for different times before quenching. The solid line represents the best fit of the data to a single exponential model. B, plots of apparent rate versus substrate concentration for: HsBPL-HsBCCP (□), PhBPL-PhBCCP (♢), and EcBPL-EcBCCP (○). The data points represent the average of the apparent rate measured at each BCCP concentration in two independent experiments with S.D. shown as error bars. The solid lines represent the best fits of the rate versus concentration profiles to a linear equation.
Measurements of Biotin Transfer to Noncognate Acceptor Proteins Indicate Substrate Specificity
The specificity of biotin transfer was investigated by performing stopped-flow experiments with each BPL and noncognate substrates (Table 2). Like the cognate reactions, noncognate biotin transfer yielded linear apparent rate versus substrate concentration profiles. The bimolecular association rates obtained from linear regression of the data span a large range, from undetectable to 43,000 m−1s−1 with the fastest rate observed for PhBPL-catalyzed biotin transfer to HsBCCP, a noncognate substrate. The only enzyme capable of biotinylating all three BCCP substrates is PhBPL. No time-dependent change in the fluorescence signal is observed upon mixing either HsBPL or EcBPL with PhBCCP, consistent with the absence of biotin transfer. This result was confirmed by MALDI-TOF MS analysis of reactions containing either the human or E. coli ligase and PhBCCP (data not shown).
TABLE 2.
Stopped-flow measurements of biotin transfer rates for cognate and noncognate ligase-BCCP pairs
| Pair | HsBCCPa | EcBCCPa | PhBCCPa |
|---|---|---|---|
| m−1s−1 | m−1s−1 | m−1s−1 | |
| HsBPL | 35,000 ± 3000 | 270 ± 40 | NDb |
| (0.23 ± 0.02)c | |||
| EcBPL | 32,000 ± 2000 | 11,500 ± 700 | NDb |
| (0.33 ± 0.03)c | (0.21 ± 0.02)c | ||
| PhBPL | 43,000 ± 5000d | 4,200 ± 300d | 15,000 ± 600d |
a The errors correspond to the S.D. of three independent stopped-flow experiments.
b ND, not detectable.
c BCCP dissociation rate, in s−1.
d Measurements with PhBPL were carried out at 40 °C, in 10 mm Tris, pH 7.5, 500 mm KCl, 2.5 mm MgCl2. All other experiments were performed at 20 °C, in 10 mm Tris, pH 7.5, 200 mm KCl, 2.5 mm MgCl2.
For all cognate BPL-BCCP pairs the stopped-flow and quench-flow assays of biotin transfer from bio-5′-AMP yield identical results. To determine whether this is true for noncognate reactions, quench-flow measurements of PhBPL-catalyzed transfer to noncognate substrates were performed. The PhBPL was chosen for this comparison because of its ability to catalyze biotin transfer to all three BCCP substrates. Results of quench-flow measurements indicate holo-BCCP accumulation rates identical to the rates of enzyme-intermediate depletion measured by stopped-flow (Fig. 5 and Table 3). Thus, even with noncognate substrates, bimolecular enzyme-substrate association limits the rate of biotin transfer from the intermediate to the BCCP substrate.
FIGURE 5.
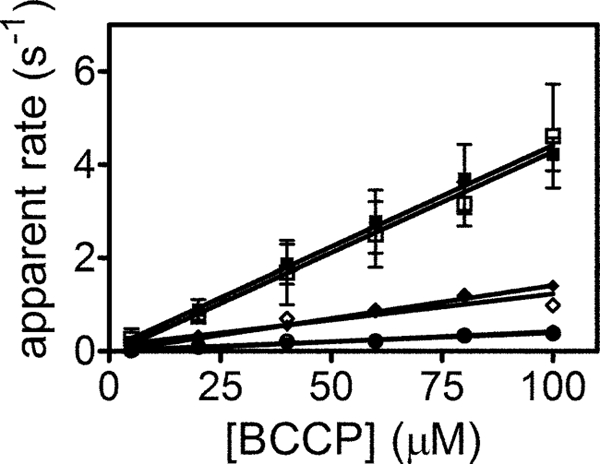
Stopped-flow and quench-flow measurements of PhBPL-catalyzed biotin transfer to cognate and noncognate substrates. Rate versus concentration profiles obtained by stopped-flow for HsBCCP (■), PhBCCP (♦), and EcBCCP (●) and quench-flow for HsBCCP (□), PhBCCP (♢), and EcBCCP (○). The solid lines were obtained from linear regression of the data.
TABLE 3.
Rates of PhBPL-catalyzed biotin transfer to cognate and noncognate substrates
| Pair | Stopped-flowa | Quench-flowb |
|---|---|---|
| m−1s−1 | m−1s−1 | |
| HsBCCP | 43,000 ± 5,000 | 42,000 ± 7,000 |
| EcBCCP | 4,200 ± 300 | 4,000 ± 1,000 |
| PhBCCP | 15,000 ± 600c | 15,000 ± 1,000c |
a The errors represent the S.D. of three independent stopped-flow experiments.
b The errors correspond to the S.D. of results obtained in two independent quench-flow experiments.
c Measurements with PhBPL were carried out at 40 °C, in 10 mm Tris, pH 7.5, 500 mm KCl, 2.5 mm MgCl2. All other measurements were performed at 20 °C, in 10 mm Tris, pH 7.5, 200 mm KCl, 2.5 mm MgCl2.
DISCUSSION
The structural and energetic basis of the specific enzyme-catalyzed post-translational biotin addition is not known. Previous steady-state measurements with HsBPL indicated that the first step, bio-5′-AMP synthesis, is the rate-limiting step in the two-step reaction (13). In this work the second half-reaction in biotin transfer was measured independent of the first using single turnover stopped-flow and quench-flow assays. The measurements performed on ligases and substrates from three different organisms demonstrate that rate of bimolecular association of enzyme with substrate limits post-translational biotin addition. Furthermore, measurements on noncognate enzyme-substrate pairs reveal specificity in post-translational biotin addition.
The stopped-flow measurements of biotin transfer from the bio-5′-AMP to BCCP, which monitors disappearance of the enzyme-intermediate complex, provides information about bimolecular association of enzyme with substrate for all ligase-BCCP pairs. This conclusion is based on the observed linear dependences of the apparent rates on BCCP concentration. To obtain values for the rate of the chemistry of biotin transfer, a quench-flow assay for the second half-reaction, in which product holo-BCCP formation is monitored, was applied to the three ligase-BCCP systems. The linear dependences and resulting bimolecular rate constants obtained with this assay are identical those obtained using the stopped-flow assay. Therefore, for all three cognate BPL-BCCP pairs the chemistry of biotin transfer from the intermediate to acceptor protein is limited by the enzyme-substrate association rate. As indicated by measurements performed using PhBPL and the noncognate BCCP substrates, this is also true for reactions involving noncognate pairs.
The three enzymes display association rates with their cognate substrates of 1–5 × 104 m−1s−1, values that are well within range of the commonly observed association rates for protein-protein interactions (26). The rates also agree with previous measurements of the EcBPL-EcBCCP interaction performed in the same substrate concentration range (14). In addition, the results obtained with the human system are comparable with those previously obtained with p67, a biotin acceptor substrate that contains in addition to the HsBCCP sequence some “extra” residues originating from the vector sequence (13). However, the measured association rate with p67 was only 2-fold slower than that measured for HsBCCP.
Measurements performed with noncognate substrates indicate that PhBPL, which biotinylates the two noncognate substrates at rates comparable with the cognate reaction, is the least discriminating of the three enzymes. By contrast, neither the HsBPL nor EcBPL can transfer biotin to PhBCCP. The human enzyme, which fails to biotinylate PhBCCP and reacts with the E. coli substrate with a slow rate of 3 × 102 m−1s−1, is the most discriminating of the three ligases. Precedent for such slow bimolecular protein-protein association rates exist, including a value of ∼2 m−1s−1 measured for the binding of the p66 and p51 subunits of HIV reverse transcriptase (27).
Previous studies provide limited information on the structural features that are important for substrate recognition by biotin protein ligases. In the PhBPL-PhBCCP complex structure the interface is characterized by multiple hydrogen bonds mediated by backbone residues as well as numerous van der Waals contacts. Functional studies of both E. coli BCCP and ligase variants have also been performed. Mutational studies on E. coli BCCP revealed that, in addition to the target lysine, a glutamic acid at position 119 is important for recognition (23). In the E. coli ligase several surface loops are proposed to function in BCCP recognition. Functional studies of EcBPL variants at amino acids Arg-116, Arg-119, and Ala-147 confirm the importance of three of these loop residues for the process (28). In addition to the catalytic domain, the human ligase is characterized by a ∼450-amino acid N-terminal extension, which has been shown both to influence the association rate with p67 (13) and to interact directly the BCCP domain of acetyl-CoA carboxylase II (29). Additional studies to elucidate the structural origins of the range of bimolecular association rates exhibited by the BPL-BCCP pairs are in progress.
The control of biotin transfer by enzyme-BCCP association has potential biological consequences. First, consider any single organisms such as H. sapiens in which there are multiple biotin-dependent carboxylases. All organisms have a limited biotin supply (30) and, therefore, a limited amount of BPL-bio-5′-AMP. Therefore, the carboxylase that associates most rapidly with the enzyme-intermediate complex should acquire the greatest portion of the available biotin. The association rate should depend on both the intrinsic bimolecular rate constant for a ligase-carboxylase pair and on the carboxylase substrate concentration. Thus, the relative intracellular apocarboxylase concentrations should be an important factor in determining biotin distribution in metabolism. The observed collision-controlled reaction is also important for bifunctional ligases such as EcBPL, which, in addition to forming the heterodimer with BCCP, homodimerizes to regulate transcription. Because a single surface on the ligase-intermediate complex is utilized for the both dimerization reactions (25), the two are mutually exclusive. However, homodimerization is governed by an association rate that is much slower than heterodimerization (31, 32). This gives rise to a competition between hetero- and homodimerization in which kinetic preference is given to biotin transfer to BCCP. Only after the unbiotinylated BCCP pool has been depleted and metabolic demand for biotin is satisfied does the enzyme-intermediate complex linger sufficiently long to allow homodimerization and transcription repression.
This work quantitatively demonstrates specificity of post-translational biotin addition to BCCP substrates. The specificity exists despite the high degree of evolutionary conservation of sequence and structures in both ligases and BCCP substrates. In addition, the measurements indicate that the association of ligases with biotin acceptor substrate limits the biotinylation reaction rate. Together, the results highlight the importance of protein-protein interactions in post-translational biotinylation and provide a mechanism for determining the distribution of biotin in carboxylases and, therefore, in metabolism.
Acknowledgments
We thank the RIKEN group for supplying the expression plasmids for PhBPL and PhBCCP, Dr. Chrisopher Lima for the plasmid used for purification of SUMO protease, Yishan Zhou for the purification of PhBCCP, Kyle Daniels for the purification of PhBPL, and Emily Streaker for the purification of EcBPL.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-GM46511 and S10-RR15899.
- BPL
- biotin protein ligase
- BCCP
- biotin carboxyl carrier protein
- bio-5′-AMP
- biotinyl-5′-adenylate
- Ec
- Escherichia coli
- Hs
- Homo sapiens
- p67
- carboxyl-terminal fragment of the propionyl-CoA carboxylase α subunit
- Ph
- Pyrococcus horikoshii
- SUMO protein
- small ubiquitin-like modifier protein
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Jitrapakdee S., Wallace J. C. (2003) Curr. Protein Pept. Sci. 4, 217–229 [DOI] [PubMed] [Google Scholar]
- 2. Lane M. D., Rominger K. L., Young D. L., Lynen F. (1964) J. Biol. Chem. 239, 2865–2871 [PubMed] [Google Scholar]
- 3. Bagautdinov B., Matsuura Y., Bagautdinova S., Kunishima N. (2008) J. Biol. Chem. 283, 14739–14750 [DOI] [PubMed] [Google Scholar]
- 4. Polyak S. W., Chapman-Smith A., Brautigan P. J., Wallace J. C. (1999) J. Biol. Chem. 274, 32847–32854 [DOI] [PubMed] [Google Scholar]
- 5. Leon-Del-Rio A., Gravel R. A. (1994) J. Biol. Chem. 269, 22964–22968 [PubMed] [Google Scholar]
- 6. Cronan J. E., Jr. (1990) J. Biol. Chem. 265, 10327–10333 [PubMed] [Google Scholar]
- 7. Marini P., Li S. J., Gardiol D., Cronan J. E., Jr., de Mendoza D. (1995) J. Bacteriol. 177, 7003–7006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Slavoff S. A., Chen I., Choi Y. A., Ting A. Y. (2008) J. Am. Chem. Soc. 130, 1160–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. León-Del-Rio A., Leclerc D., Akerman B., Wakamatsu N., Gravel R. A. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 4626–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rice P., Longden I., Bleasby A. (2000) Trends Genet. 16, 276–277 [DOI] [PubMed] [Google Scholar]
- 11. Wilson K. P., Shewchuk L. M., Brennan R. G., Otsuka A. J., Matthews B. W. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 9257–9261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bagautdinov B., Kuroishi C., Sugahara M., Kunishima N. (2005) J. Mol. Biol. 353, 322–333 [DOI] [PubMed] [Google Scholar]
- 13. Ingaramo M., Beckett D. (2009) J. Biol. Chem. 284, 30862–30870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nenortas E., Beckett D. (1996) J. Biol. Chem. 271, 7559–7567 [DOI] [PubMed] [Google Scholar]
- 15. Abbott J., Beckett D. (1993) Biochemistry 32, 9649–9656 [DOI] [PubMed] [Google Scholar]
- 16. Brown P. H., Cronan J. E., Grøtli M., Beckett D. (2004) J. Mol. Biol. 337, 857–869 [DOI] [PubMed] [Google Scholar]
- 17. Daniels K. G., Beckett D. (2010) Biochemistry 49, 5358–5365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bagautdinov B., Matsuura Y., Bagautdinova S., Kunishima N. (2007) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 63, 334–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 20. Healy S., Heightman T. D., Hohmann L., Schriemer D., Gravel R. A. (2009) Protein Sci. 18, 314–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gill S. C., von Hippel P. H. (1989) Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 22. González J. M., Masuchi Y., Robb F. T., Ammerman J. W., Maeder D. L., Yanagibayashi M., Tamaoka J., Kato C. (1998) Extremophiles 2, 123–130 [DOI] [PubMed] [Google Scholar]
- 23. Chapman-Smith A., Morris T. W., Wallace J. C., Cronan J. E., Jr. (1999) J. Biol. Chem. 274, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 24. Eisenstein E., Beckett D. (1999) Biochemistry 38, 13077–13084 [DOI] [PubMed] [Google Scholar]
- 25. Weaver L. H., Kwon K., Beckett D., Matthews B. W. (2001) Protein Sci. 10, 2618–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koren R., Hammes G. G. (1976) Biochemistry 15, 1165–1171 [DOI] [PubMed] [Google Scholar]
- 27. Venezia C. F., Meany B. J., Braz V. A., Barkley M. D. (2009) Biochemistry 48, 9084–9093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao H., Naganathan S., Beckett D. (2009) J. Mol. Biol. 389, 336–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee C. K., Cheong C., Jeon Y. H. (2010) FEBS Lett. 584, 675–680 [DOI] [PubMed] [Google Scholar]
- 30. Baker H., Frank O., Matovitch V. B., Pasher I., Aaronson S., Hutner S. H., Sobotka H. (1962) Anal. Biochem. 3, 31–39 [DOI] [PubMed] [Google Scholar]
- 31. Streaker E. D., Beckett D. (2006) Biochemistry 45, 6417–6425 [DOI] [PubMed] [Google Scholar]
- 32. Zhao H., Beckett D. (2008) J. Mol. Biol. 380, 223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 34. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]