

Abstract
The prodrug nifurtimox has been used for more than 40 years to treat Chagas disease and forms part of a recently approved combinational therapy that targets West African trypanosomiasis. Despite this, its mode of action is poorly understood. Detection of reactive oxygen and nitrogen intermediates in nifurtimox-treated extracts led to the proposal that this drug induces oxidative stress in the target cell. Here, we outline an alternative mechanism involving reductive activation by a eukaryotic type I nitroreductase. Several enzymes proposed to metabolize nifurtimox, including prostaglandin F2α synthase and cytochrome P450 reductase, were overexpressed in bloodstream-form Trypanosoma brucei. Only cells with elevated levels of the nitroreductase displayed altered susceptibility to this nitrofuran, implying a key role in drug action. Reduction of nifurtimox by this enzyme was shown to be insensitive to oxygen and yields a product characterized by LC/MS as an unsaturated open-chain nitrile. This metabolite was shown to inhibit both parasite and mammalian cell growth at equivalent concentrations, in marked contrast to the parental prodrug. These experiments indicate that the basis for the selectivity of nifurtimox against T. brucei lies in the expression of a parasite-encoded type I nitroreductase.
Keywords: Drug Action, Drug Metabolism, Oxidation Reduction, Parasite Metabolism, Trypanosome, Prodrug, Nitrile, Nitrofuran, Type I Nitroreductase
Introduction
Across the tropics, 10 million people are infected by the parasites Trypanosoma cruzi and Trypanosoma brucei, the causative agents of Chagas disease and human African trypanosomiasis, respectively (1, 2). They are responsible for 60,000 deaths per year and represent a major public health problem in regions of the world least able to deal with the associated economic burden. Their primary route of transmission is by blood-sucking insect vectors. However, other pathways, notably blood transfusion, organ transplantation, and illicit drug usage coupled with population migration, have resulted in both infections becoming problematic in non-endemic areas (3–5). Chagas disease is an emerging problem with as many as 300,000 people in the United States infected by T. cruzi, and blood supplies are now routinely screened for the parasite. Current treatment of both infections is restricted to a series of drugs whose mode(s) of action are poorly understood (6). Establishing how existing therapies work could contribute to more effective treatment and aid drug development.
The 5-nitrofuran drug nifurtimox has been used for more than 40 years to treat Chagas disease. Its use is controversial as it is toxic, reportedly carcinogenic, and of limited efficacy. Additionally, T. cruzi strains refractory to treatment have been isolated, a situation compounded by failure to complete the recommended drug schedules. Nifurtimox drug regimes can take up to 4 months and are frequently associated with unpleasant side effects. Despite these problems, nifurtimox, as part of the nifurtimox-eflornithine combination therapy, is now recommended for late-stage West African sleeping sickness (7, 8) and is also undergoing assessment as a treatment for pediatric neuroblastoma (9, 10).
As with most nitroheterocyclic agents, nifurtimox functions as a prodrug and must undergo activation by nitroreduction. Two classes of enzyme can catalyze this process, the type I and type II nitroreductases (11). Type I nitroreductases are NAD(P)H-dependent, FMN binding proteins commonly found in bacteria but rare in eukaryotes. They mediate a two-electron reduction of the nitro group to generate a nitroso intermediate (Reaction 1) that rapidly undergoes reduction to a hydroxylamine derivative (Reaction 2):
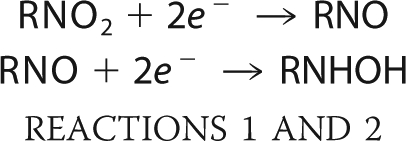 |
For nitrofuran compounds, the hydroxylamine can then be processed further to generate either the amine, which is believed to be inert, or nitrenium cations that promote DNA breakage (12–14). Alternatively, fragmentation of the furan ring may occur, yielding open-chain nitriles (11, 15, 16). Because reduction by type I nitroreductase does not involve oxygen and does not result in the production of reactive oxygen species, this activity is said to be “oxygen-insensitive.”
In contrast, the ubiquitous type II nitroreductases contain FMN or FAD as a co-factor, and their activity is “oxygen-sensitive.” They catalyze the one-electron reduction of a substrate, forming a nitro anion radical (Reaction 3) (17). In the presence of oxygen, this radical undergoes futile cycling, resulting in the production of superoxide anions and regeneration of the parent nitro-compound (Reaction 4) (18).
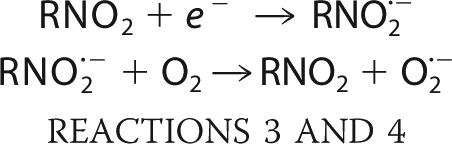 |
Following observations that nifurtimox-treated trypanosomal extracts generated superoxide anions and nitro anion radicals, it was proposed that this compound mediated its activity through induction of oxidative stress in reactions catalyzed by type II nitroreductases (19–21). Several flavoproteins, including dihydrolipoamide dehydrogenase, cytochrome P450 reductase, and trypanothione reductase, were subsequently shown to mediate the one-electron reduction of nifurtimox in vitro (21–23). To date there is insufficient functional evidence to suggest that this occurs in vivo and that trypanosomes overexpressing trypanothione reductase display the same susceptibility to nifurtimox as control cells (24). The only experimental evidence for superoxide anion involvement in nifurtimox toxicity is indirect and comes from studies on the trypanosomal superoxide dismutase repertoire. T. brucei TbSODB1 null mutants displayed enhanced sensitivity to nifurtimox (25). The same pattern of redox cycling seen in T. cruzi is observed in mammalian cells, and the selectivity of this prodrug toward the parasite is not explained by this model. Despite these issues, oxidative stress resulting from type II nitroreductase activity has been generally accepted as the main trypanocidal mechanism of nifurtimox, although this has been questioned (26–28).
Recently, an alternative activation mechanism has been proposed following the identification of two trypanosomal enzymes that can catalyze the two-electron reduction of nifurtimox (27, 29). One, a prostaglandin F2α synthase, also known as the Old Yellow Enzyme, expressed by T. cruzi, can reduce the nitrofuran in vitro, but oxidoreductase activity only occurs under anaerobic conditions. The contribution, if any, that this enzyme makes to drug metabolism within the parasite has not been established. The second pathway involves a trypanosomal type I nitroreductase (NTR)2 that displays characteristics shown by many of its bacterial homologues; the activity is oxygen-insensitive, the enzyme contains FMN as co-factor, and it can metabolize a wide range of nitro- and quinone-based compounds. Using T. cruzi and T. brucei lines with altered levels of this enzyme, a clear link between this nitroreductase activity and nifurtimox activation has been demonstrated (27). Null mutant/heterozygous cells display resistance to various nitroheterocyclic agents, including nifurtimox, whereas overexpression confers hypersensitivity. Additionally, T. cruzi selected for resistance to nifurtimox were found to have lost one copy of the chromosome containing the TcNTR gene.
The aim of this work was to determine the role of NTRs in nifurtimox action. We show that although low levels of oxygen consumption can be detected during nifurtimox reduction, NTRs are predominantly oxygen-insensitive enzymes that catalyze the four-electron reduction of the nitrofuran under both aerobic and anaerobic conditions. This produces an unsaturated open-chain nitrile that displays equivalent cytotoxicity toward mammalian and parasite cells, unlike the parental prodrug.
EXPERIMENTAL PROCEDURES
Cell Culturing
T. brucei (Lister 427, clone 221a and a derivative (2T1) engineered to constitutively express the tetracycline repressor protein) bloodstream-form parasites were grown in HMI-11 medium (30, 31). The 2T1 line was grown in the presence of 1 μg ml−1 of phleomycin. Transformed 2T1 parasite lines overexpressing TbNTR, TbPGS, TbCPR2, or TbCPR3 were maintained in this medium supplemented with 2.5 μg ml−1 of hygromycin.
A human acute monocytic leukemia cell line (THP-1) was grown at 37 °C under a 5% CO2 atmosphere in RPMI 1640 medium supplemented with 10% fetal calf serum, 20 mm HEPES (pH 8.0), 2 mm sodium glutamate, 2 mm sodium pyruvate, 2.5 units ml−1 penicillin, and 2.5 μg ml−1 streptomycin.
Enzyme Activity
Recombinant TcNTR and TbNTR were purified as described (27, 32). Type I nitroreductase activity was measured spectrophotometrically by following nifurtimox reduction (λ = 435 nm, ϵ = 18,000 M−1 cm−1). A standard reaction mixture (1 ml) containing 50 mm Tris-Cl (pH 7.5), NADH (100 μm), and nifurtimox (100 μm) was incubated at room temperature for 5 min. The background rate of nitrofuran reduction was determined, and the reaction was initiated by addition of trypanosomal enzyme (20 μg). Type I nitroreductase activity was expressed as micromoles of nifurtimox reduced min−1 mg−1 protein. For hypoxic conditions, all reagents were preincubated in an anaerobic glove box (Belle Technology Ltd, Weymouth, UK) under a 100% nitrogen (oxygen-free) atmosphere for at least 1 h. Assays were performed in sealed cuvettes, and reactions were initiated by injection of the parasite enzyme in the reaction mix. Data were evaluated by non-linear regression analysis using GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
Oxygen Consumption Assay
Oxygen consumption was followed using the Oxygen Biosensor System (BD Biosciences) (33). A standard 200-μl assay containing 50 mm Tris-Cl (pH 7.5) and nifurtimox (100 μm) was equilibrated at 27 °C for 15 min in the presence of TcNTR, TbNTR (both 4 μg), or cytochrome P450/cytochrome P450 reductase-enriched microsomal fractions (0.1 μg) (Sigma-Aldrich). Reactions were initiated by addition of prewarmed NAD(P)H (100 μm). Glucose dehydrogenase (1 unit ml−1) and glucose (3 mm) were included in the initial mixture to maintain the levels of the reduced electron donor. The change in fluorescence (excitation λ = 486 nm, emission λ = 620 nm) was monitored.
LC/MS Analysis of Nifurtimox Metabolites
In an NTR-mediated reaction, nifurtimox (100 μm) was completely reduced, and the residual enzyme was removed by binding to nickel-nitrilotriacetic acid followed by centrifugation. Supernatants were analyzed using a Series 1100 LC with SL Ion Trap MS (Agilent, Wokingham, UK). Separation was carried out on a Hypurity Elite 5-μm C18 column (15 mm × 2.1 mm) (Thermo Scientific, Hemel Hempstead, UK), eluting with a 10–30% acetonitrile gradient at a flow rate of 0.2 ml min−1. Metabolites were detected using a diode array (λ = at 250, 300, 340, and 450 nm), and their m/z values were determined by positive electrospray ionization. MS was carried out with a drying gas temperature of 325 °C, drying gas flow of 10 liters min−1, nebulizer gas pressure of 25 p.s.i., and capillary voltage of 3500 V in full scan mode in the m/z range of 50–400. Positive and negative tandem MS was performed in automatic mode using SmartFrag for ion fragmentation. Preparative HPLC of the unsaturated open-chain nitrile was performed on a Kromasil C18 column using a 10–30% acetonitrile gradient with a flow rate of 2 ml min−1. The peak fraction, as detected by absorbance at 340 nm, was collected, and the solvent was removed under vacuum. The product was resuspended in dimethyl sulfoxide, and its purity was confirmed by mass spectroscopy and absorbance spectrum.
Antiproliferative Assays
T. brucei bloodstream-form parasites (1 × 103 ml−1) or THP-1 cells (1 × 104 ml−1) were seeded in 200 μl of growth medium containing different concentrations of nifurtimox or unsaturated open-chain nitrile. After incubation at 37 °C for 3 days (T. brucei) or 6 days (THP-1), Alamar Blue (20 μl) (Invitrogen) was added to each well, and the plates were incubated for a further 8–16 h. Cell densities were determined by monitoring the fluorescence of each culture using a Gemini fluorescent plate reader (Molecular Devices (UK) Ltd, Wokingham, UK) at an excitation wavelength of 530 nm, an emission wavelength of 585 nm, and a filter cutoff at 550 nm, and the drug concentration that inhibits cell growth by 50% (IC50) was established.
RESULTS
Nifurtimox Activation in Trypanosomes Is Dependent on Type I Nitroreductases
Several trypanosomal enzymes have been implicated in the metabolism of nifurtimox, including prostaglandin F2α synthase, cytochrome P450 reductase, and NTR (27, 29, 34). To date, only NTR has been demonstrated to activate this nitrofuran prodrug in vivo. Trypanosomes with reduced NTR levels show resistance to nifurtimox, whereas cells overexpressing the enzyme are hypersensitive (27). To assess whether the other enzymes can metabolize nifurtimox, the T. brucei genes encoding for prostaglandin F2α synthase (TbPGS, AB034727) and two cytochrome P450 reductases (TbCPR2, XM_822460 and TbCPR3, XM_823737) (numbering in accordance with Portal et al. (34)) were cloned in-frame with the 9e10 epitope from the human c-myc protein in a vector that facilitated tetracycline-inducible gene expression (31). The constructs were used to transform bloodstream-form T. brucei generating the inducible overexpression lines designated TbPGSex, TbCPR2ex, and TbCPR3ex. To demonstrate that protein expression was occurring, extracts were produced from parasites grown in the presence of tetracycline and used in Western blot analyses with a monoclonal antibody raised against the 9e10 epitope. In all cases, a band of the size corresponding to the recombinant protein was detected (Fig. 1A). T. brucei bloodstream-form lines overexpressing TbPGS, TbCPR2, or TbCPR3 were then treated with nifurtimox. Parasites with elevated levels of TbNTR were examined in parallel. Only cells with elevated levels of TbNTR displayed an enhanced susceptibility to the nitrofuran (Fig. 1B).
FIGURE 1.

Overexpression of the trypanosomal type I nitroreductase, but not other enzymes, enhances susceptibility of bloodstream-form T. brucei to nifurtimox. A, Western blot analyses containing extracts (10 μg) from uninduced (-) and tetracycline-induced (+) T. brucei bloodstream-form cultures expressing TbNTR, TbPGS, TbCPR2, or TbCPR3 were probed with an anti-9E10 antibody. Loading was judged by Coomassie staining. B, dose response of parental bloodstream-form parasites (●) and cells expressing TbPGSex (♦), TbCPR2ex (▴), TbCPR3ex (+), or TbNTRex (■) to nifurtimox (NFX). All curves are means from experiments performed in quadruplicate. The insert contains IC50 values ± S.D. The differences in susceptibility between NTR-expressing cells and all other lines was statistically significant (p < 0.01), as assessed by Student's t test.
Metabolism of Nifurtimox by Trypanosomal Type I Nitroreductases
TcNTR and TbNTR have all the characteristics of classic oxygen-insensitive type I nitroreductases and are able to reduce a wide range nitroheterocyclic compounds in the presence of oxygen (27, 32, 35). However, in contrast to other substrates, such as benznidazole and 2,6-dichlorophenolindophenol, the interaction between nifurtimox and both trypanosomal enzymes does not occur via classic ping-pong kinetics (Fig. 2, A and B). To study the interaction of TbNTR or TcNTR toward nifurtimox, assays were performed in the presence of a fixed amount of parasite enzyme (20 μg) using three concentrations of NADH (60, 80, and 100 μm) against various concentrations of the prodrug (0–100 μm). Double reciprocal plots were linear between 20 to 100 μm nifurtimox for all three NADH concentrations (data shown is for TbNTR, with similar results obtained for TcNTR) (Fig. 2B). However, the plots converged to suggest that the interaction of NADH, nifurtimox, and the trypanosomal enzymes is complex, possibly occurring through more than one mechanism. As oxygen consumption and production of reactive oxygen species is often associated with nifurtimox metabolism (19–21, 36), we analyzed whether oxygen levels had any effect on the enzyme activity. In three independent experiments performed under hypoxic conditions, the maximum nitroreductase activity was consistently higher than in aerobic assays performed in parallel, irrespective of the nitroreductase source (TcNTR or TbNTR) (Fig. 2C).
FIGURE 2.

Interaction of trypanosomal type I nitroreductases with nifurtimox. A, schematic for nifurtimox (NFX) reduction by trypanosomal type I nitroreductases (NTR), using NADH as electron donor. Reduced (red) and oxidized (oxid) forms of nitrofuran are shown. B, type I nitroreductase activity was assayed in the presence of nifurtimox (0–100 μm), NADH (60 (▴), 80 (■), and 100 (♦) μm), and TcNTR (20 μg). C, type I nitroreductase activity was monitored under aerobic (●) and hypoxic (■) conditions using NADH (100 μm), nifurtimox (0–300 μm), and TcNTR (20 μg). Type I nitroreductase activity is expressed as μmol of nifurtimox reduced min−1 mg−1 protein. The graph shown is representative of three independent experiments. In B and C, the data were evaluated by linear and non-linear regression analysis, respectively, using GraphPad Prism 5. D, oxygen consumption during nifurtimox reduction. Reactions containing nifurtimox (100 μm), NAD(P)H (100 μm), and microsomes enriched for cytochrome P450/cytochrome P450 reductase (0.1 μg, curve a) or TcNTR (4 μg, curve b) were incubated at 27 °C, and the change in fluorescence was followed. The amount of microsomal fraction and trypanosomal type I nitroreductase used were equivalent in their rates of NAD(P)H oxidation. The rate of NADPH oxidation by the microsomal fraction was 21 nmol of NADPH oxidized min−1, whereas the rate of NADH oxidation by TcNTR was 22 nmol of NADH oxidized min−1, as determined by parallel spectrophotometric assays. Controls lacking substrate were also carried out (curve c), and an identical curve was obtained when the enzyme was omitted. Additional controls to validate this system are shown in supplemental Fig. S1. Results were normalized by subtraction of the background signal in the absence of the enzyme. All curves are means from experiments performed in triplicate. When TbNTR was used in place of TcNTR, similar enzyme kinetic and oxygen consumption profiles were observed.
To determine whether oxygen was consumed during aerobic nifurtimox reduction, the Oxygen Biosensor System, a fluorescence-based assay using a ruthenium-based fluorophore was employed (33, 37). In this system, oxygen quenches fluorescence, and an increase in signal intensity denotes a decline in oxygen levels. A series of control reactions validating this system are shown in supplemental Fig. S1. In the presence of NADPH and microsomes enriched for human cytochrome P450/cytochrome P450 reductase, reduction of nifurtimox via a type II nitroreductase activity resulted in a time-dependent increase in fluorescence, indicating that oxygen was being consumed, and this activity was maintained throughout the duration (80 min) of the assay (Fig. 2D, curve a). By measuring the initial rate of change in fluorescence in the presence of various nifurtimox concentrations (supplemental Fig. S1), a specific activity value was calculated: 33.3 ± 4.5 of normalized fluorescence units min−1 μg protein−1 for the microsomal extract. When using TbNTR or TcNTR with NADH as electron donor, nifurtimox reduction generated an increase in fluorescence (Fig. 2D, curve b) but at a lower rate than that seen with microsomal preparations. Again, the rate of change in fluorescence was dependent on nifurtimox levels, yielding a specific activity value (0.7 ± 0.5 of normalized fluorescence units min−1 μg protein−1) 50-fold lower than microsomal reactions. In contrast to assays using microsomal fractions enriched for human cytochrome P450/cytochrome P450 reductase, the TcNTR- or TbNTR-mediated oxygen consumption activity stopped after ∼30 min because of removal of nifurtimox from the reaction. In the presence of excess NADH, the oxygen consumption activity resumed when more nifurtimox was added to the assay (supplemental Fig. S1). Thus, although oxygen consumption does occur during NTR-mediated nifurtimox reduction, the associated futile cycling appears to be limited, with most nifurtimox undergoing reduction to form an unknown product.
The Major Product of Nifurtimox Reduction Is an Open-chain Nitrile
The activity of type I nitroreductases can lead to the formation of various products (Fig. 3). To identify which metabolites are generated from nifurtimox, this nitrofuran was enzymatically reduced by TcNTR or TbNTR under both aerobic and anaerobic conditions. In all cases, LC/MS analysis of the resultant products yielded a single analyte (Fig. 4A) whose mass spectrum contained parent molecular ions for [M+H]+ and [M+Na]+ at 256 and 278, respectively (Fig. 4B). For comparison, in parallel experiments, nifurtimox eluted off the same column at 33.5 min, and the mass spectrum contained a parent molecular ion for [M+H]+ at 288. Examination of the absorbance spectrum of the reduction product gave a characteristic profile reminiscent of the nifurtimox-derived metabolite generated by radiolysis (Fig. 4C) (38). On the basis of its size and absorbance spectrum, the product was tentatively identified as an unsaturated open-chain nitrile that results from the dehydration of the furyl hydroxylamine and subsequent furan ring opening. Further reduction of this end product to its saturated form by TcNTR was observed, but this reaction occurred at a very slow rate, requiring prolonged incubation times (>24 h). Additionally, during NTR-mediated reduction of nifurtimox to the unsaturated open-chain nitrile, it is implicit that nitroso and hydroxylamine intermediaries must be formed (Fig. 3). However, peaks corresponding to these derivatives were never detected, even when using substoichiometric concentrations of NADH, indicating that these transient intermediates undergo facile reduction and as such contribute to the rapid conversion of nifurtimox to the unsaturated open-chain nitrile.
FIGURE 3.

Reduction of nitrofurans by type I nitroreductases. Type I nitroreductases reduce the conserved nitro group of nitrofurans to generate a hydroxylamine via a nitroso intermediate. The hydroxylamine can be metabolized further to form 1) a nitrenium ion, 2) the amine form, or 3) unsaturated and then saturated open-chain nitriles (11–16).
FIGURE 4.

Trypanosomal type I nitroreductase-mediated nifurtimox reduction generates an unsaturated open-chain nitrile. A, HPLC trace (λ = 340 nm) of the reaction product obtained after complete reduction of nifurtimox by the trypanosomal type I nitroreductase TcNTR. TbNTR generates exactly the same metabolic profile. B, mass spectrum of the single peak obtained after HPLC. The m/z of 256 and 278 correspond to the nifurtimox-derived metabolite +H or +Na, respectively. The calculated molecular weight of 255 corresponds to an unsaturated open-chain nitrile form (see Fig. 3). C, absorption spectrum of the unsaturated open-chain nitrile product.
The nifurtimox reduction product was unequivocally characterized as an unsaturated open-chain nitrile using tandem MS. Positive electrospray ionization (ESI) mass spectrometry of the purified reduction product gave rise to two ions with m/z values of 170 and 131, corresponding to molecular weights of 147 and 108 ([M+Na]+), respectively (Fig. 5A). These fragments are the result of eliminative cleavage of the weak N-N bond to give a truncated open-chain nitrile and a dehydrothiomorpholine (Fig. 5B). When the converse experiments were performed using negative ESI-MS, a series of peaks predicted for the unsaturated open-chain nitrile being the parental structure were detected (Fig. 5C). The major peak had a m/z value of 227, which corresponds to the parental backbone lacking the nitrile group, whereas a series of smaller fragments was also observed whose m/z values are all consistent with the proposed unsaturated open-chain nitrile (Fig. 5D).
FIGURE 5.

Tandem mass spectrometry of the open-chain nitrile. A, positive ESI tandem MS of the unsaturated open-chain nitrile ion described in Fig. 4 (m/z of 278 corresponding to [M+Na]+) generated two fragments with m/z values of 170 and 131. B, structure of precursor unsaturated open-chain nitrile ion (278) and fragment peaks after positive ESI. C, negative ESI tandem MS of the m/z 254 [M-H]+ precursor ion generated several peaks. D, structure of precursor and fragment peaks following negative ESI.
Therefore, the predominant reaction catalyzed by both TcNTR and TbNTR under aerobic or anaerobic conditions is a two-step, two-electron reduction of nifurtimox that yields the unsaturated open-chain nitrile derivative (Fig. 3).
Trypanocidal Activity of the Nifurtimox Reduction Products
To evaluate whether the nifurtimox-derived unsaturated open-chain nitrile displayed cytotoxicity, the reduction product was purified and its growth inhibitory activity assessed against bloodstream-form T. brucei and mammalian cells (Fig. 6). The concentration of unsaturated open-chain nitrile that inhibited parasite growth by 50% (IC50) was slightly higher than that determined for nifurtimox itself (Fig. 6) (IC50 of 5.3 ± 1.0 μm for the reduction product versus 2.9 ± 0.3 μm for nifurtimox). In contrast, mammalian cells were relatively resistant to nifurtimox but displayed increased susceptibility to the TbNTR-derived metabolite. Compare the IC50 of 30.0 ± 1.0 μm for the parent compound with 5.4 ± 1.1 μm for the unsaturated open-chain nitrile. Therefore, the reduced nifurtimox derivative no longer displays selectivity toward T. brucei, suggesting that it is the NTR-mediated reduction step in the pathogen that is responsible for the difference in toxicity between the mammalian and parasite cells. When the cytotoxicity of the saturated open-chain nitrile was assessed, no significant effect on parasite or mammalian cell growth was observed over the screened range.
FIGURE 6.

Cytotoxicity of the unsaturated open-chain nitrile. Dose-dependent response of bloodstream-form T. brucei (■) and mammalian THP-1 (♦) cells to nifurtimox (NFX) (solid line) and its unsaturated open-chain nitrile product (OCN) (dotted line). All curves are means from experiments performed in quadruplicate. The insert contains the mean IC50 values ± S.D.
The importance of NTR-mediated reduction of nifurtimox to its toxic product was clearly demonstrated using T. brucei cells expressing elevated levels of TbNTR. These parasites were ∼10-fold more susceptible to nifurtimox than controls (cells with elevated levels of TbNTR had an IC50 value of 0.34 ± 0.02 μm compared with 2.51 ± 0.09 μm observed for controls), whereas they displayed similar levels of sensitivity to the unsaturated open-chain nitrile (3.71 ± 0.51 μm for T. brucei overexpressing TbNTR compared with 3.65 ± 0.16 μm in controls). Together, these data show that the unsaturated open-chain nitrile metabolite derived following TcNTR or TbNTR-mediated nifurtimox reduction contributes to toxicity of this nitrofuran and that it is the presence of a type I nitroreductase in the parasite that is responsible for the selective toxicity of nifurtimox against these pathogens.
DISCUSSION
The mode of action and basis of selectivity of the anti-trypanosomal compound nifurtimox has been an enigma since its introduction as a therapy for Chagas disease. In this article we provide evidence showing that trypanosomal type I nitroreductases catalyze reduction of this prodrug to an unsaturated open-chain nitrile derivative (Figs. 4 and 5) and that the product, unlike the nitrofuran precursor, displays equivalent growth inhibitory properties against parasite and mammalian cells (Fig. 6). Together, these findings demonstrate that selective toxicity of nifurtimox may be attributable to type I nitroreductase expression by trypanosomes and indicate that this prodrug mediates a significant component of its anti-parasitic activity by a mechanism other than induction of oxidative stress.
Activation of nitrofurans can occur by two routes, involving the two-electron reduction of the compound, reactions mediated by type I nitroreductases, or a one-electron pathway, as catalyzed by type II enzymes. Bacteria express both activities, but it is the type I system that underpins the selectivity of nitrofuran antibiotics (39–41). Prokaryotes selected for resistance to nitrofurazone invariably acquire mutations in their type I nitroreductase genes. Following activation, the precise antimicrobial action of these prodrugs is largely unknown, but it is believed to rely on production of toxic intermediate metabolites that ultimately trigger DNA damage and cell death (12, 13). In contrast, nitrofuran reduction in eukaryotic cells is generally mediated by enzymes with a type II activity, leading to nitro anion radical formation. In an aerobic environment, the radical then undergoes futile cycling, resulting in regeneration of the parental nitro-compound and production of reactive oxygen species (11, 17, 19, 36). The latter may then induce a state of oxidative stress in the target cell. Mammalian cells do express enzymes that can mediate the two-electron reduction of nitroaromatic compounds (42–44), but these activities are generally low in normal tissues and elevated in tumor cells, a difference that has been exploited in the development of anti-cancer therapies (45). Whether such eukaryotic enzymes play a role in nitrofuran metabolism has not been addressed.
The predominance of type II nitroreductases in eukaryotic cells, coupled with observations that nifurtimox-treated trypanosome extracts generated superoxide anions and nitro anion radicals, supported the proposal that parasite killing activity was mediated by the induction of oxidative stress. As a consequence, the presence of trypanosomal type I enzymes and their role in nitrofuran activation has been largely overlooked. However, following the identification of NTR and demonstration of its key role in nitroaromatic drug toxicity, the mechanism of nifurtimox action needs to be re-evaluated (27). Against this backdrop, we overexpressed prostaglandin F2α synthase and two cytochrome P450 reductases, enzymes reported to interact with nifurtimox (29, 34) and demonstrated that elevation of their levels in bloodstream-form T. brucei had no effect on toxicity (Fig. 1). In contrast, the central role of NTR in this process was clearly demonstrated, as parasites with elevated levels were hypersensitive to nifurtimox (Fig. 1) in agreement with our previous work (27, 32, 35). Additionally, in a whole-genome “loss of function” screen using nifurtimox against an induced T. brucei RNAi library, the only hits generated targeted the TbNTR transcript (28). Therefore, NTR remains the only trypanosomal enzyme with a confirmed role in nifurtimox activation within the parasite.
The ability of NTR to reduce nitroheterocyclic compounds, including nifurtimox, under aerobic conditions indicates that this enzyme is a type I nitroreductase. Our observation that nifurtimox metabolism was slightly more efficient under anaerobic than aerobic conditions and the low level of oxygen consumption (Fig. 2) are fully consistent with the activity reported for Escherichia coli oxygen-insensitive enzymes, previously ascribed to protein impurities (11). The effect of oxygen on activity may reflect that certain intermediaries generated by type I catalysis are prone to oxidation (46). Some hydroxylamines can be oxidized when exposed to air. Alternatively, the type I/type II dichotomy may be less absolute than previously thought. In any case, it is clear that the predominant mode of NTR action is via a two-electron reduction pathway.
Several different reaction products have been identified from the type I-mediated reduction of nitrofurans, including amines and open-chain nitriles (11, 15, 16). However, in many cases these are reported to be biologically inactive. Instead, prodrug toxicity is believed to occur via formation of protein and nucleic acid adducts with reactive intermediates such as the hydroxylamine or nitrenium ions (12, 13, 47). Here, we demonstrated that the nitrile product generated from nifurtimox is toxic to bloodstream-form trypanosomes, displaying an IC50 value comparable with that of the parent compound (Fig. 6). Additionally, unlike nifurtimox itself, the metabolite also demonstrated significant growth inhibitory activity against a cultured mammalian cell line. For most nitrofurans, reduction to the open-chain nitrile is reported to go to completion, with the biologically inactive saturated form being the main end product. For nifurtimox reduction this is not the case, and the major open-chain nitrile formed is in the unsaturated state. One explanation for why other groups have not detected significant quantities of the unsaturated open-chain nitrile is that products were isolated after prolonged incubation (>48 h) or were derived from animals during pharmacological testing (15, 16), whereas in our system, LC/MS analysis of the nifurtimox-derived material was performed within 60 min of initiating the reaction. However, even after 24 h, the unsaturated open-chain nitrile product generated from nifurtimox was still the major peak in traces, with only small amounts of the saturated form detectable. This indicates that the unsaturated open-chain nitrile derived from this trypanocidal prodrug is relatively stable, and this stability may explain why nifurtimox is an effective anti-parasitic agent despite being a poor substrate for NTRs compared with other nitroheterocyclic compounds (32, 35). As unsaturated open-chain nitriles have rarely been observed following nitroreduction, their role in nitrofuran toxicity has been overlooked, and the mechanism of action is unknown. The unsaturated open-chain nitrile derived from nifurtimox has the potential to function as a Michael acceptor and could react non-specifically with a range of cellular components. This may explain the pleiotropic effects of nifurtimox on trypanosomes, where treatment has been reported to inhibit various enzyme activities, modify thiol levels, and cause DNA damage (21, 48–51).
We have now shown that nifurtimox reduction by a trypanosomal type I nitroreductase leads to formation of a toxic product. The precise mode of action of the resultant unsaturated open-chain nitrile product remains to be determined, but given that it is equally toxic to mammalian and parasite cells, it is plausible that the downstream targets are common to both host and pathogen. If this is the case, then the basis of nifurtimox selectivity is due to expression of the type I nitroreductase in the parasite. Understanding how nifurtimox exerts its trypanocidal effects may lead to the development of novel anti-parasitic drugs that utilize the bioreductive activity of this enzyme.
Acknowledgments
We thank John Kelly (London School of Hygiene and Tropical Medicine) and Chris Bray (Queen Mary University of London) for valuable discussions and comments on this manuscript, and Alan Scott and Ian Sanders (Queen Mary University of London) for their assistance with the analytical techniques.
This work was supported by the Wellcome Trust.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- NTR
- trypanosomal type I nitroreductase
- ESI
- electrospray ionization
- TbCPR2 and TbCPR3
- Trypanosoma brucei cytochrome P450 reductase 2 and 3, respectively
- TbNTR
- Trypanosoma brucei type I nitroreductase
- TbPGS
- Trypanosoma brucei prostaglandin F2α synthase
- TcNTR
- Trypanosoma cruzi type I nitroreductase.
REFERENCES
- 1. Brun R., Blum J., Chappuis F., Burri C. (2010) Lancet 375, 148–159 [DOI] [PubMed] [Google Scholar]
- 2. Rassi A., Jr., Rassi A., Marin-Neto J. A. (2010) Lancet 375, 1388–1402 [DOI] [PubMed] [Google Scholar]
- 3. Bern C., Montgomery S. P. (2009) Clin. Infect. Dis. 49, e52–54 [DOI] [PubMed] [Google Scholar]
- 4. Gascon J., Bern C., Pinazo M. J. (2010) Acta Trop. 115, 22–27 [DOI] [PubMed] [Google Scholar]
- 5. Gautret P., Clerinx J., Caumes E., Simon F., Jensenius M., Loutan L., Schlagenhauf P., Castelli F., Freedman D., Miller A., Bronner U., Parola P. (2009) Euro Surveill. 14, pii=19327 [PubMed] [Google Scholar]
- 6. Wilkinson S. R., Kelly J. M. (2009) Expert Rev. Mol. Med. 11, e31. [DOI] [PubMed] [Google Scholar]
- 7. Priotto G., Fogg C., Balasegaram M., Erphas O., Louga A., Checchi F., Ghabri S., Piola P. (2006) PLoS Clin. Trials 1, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Priotto G., Kasparian S., Mutombo W., Ngouama D., Ghorashian S., Arnold U., Ghabri S., Baudin E., Buard V., Kazadi-Kyanza S., Ilunga M., Mutangala W., Pohlig G., Schmid C., Karunakara U., Torreele E., Kande V. (2009) Lancet 374, 56–64 [DOI] [PubMed] [Google Scholar]
- 9. Saulnier Sholler G. L., Kalkunte S., Greenlaw C., McCarten K., Forman E. (2006) J. Pediatr. Hematol. Oncol. 28, 693–695 [DOI] [PubMed] [Google Scholar]
- 10. Saulnier Sholler G. L., Brard L., Straub J. A., Dorf L., Illeyne S., Koto K., Kalkunte S., Bosenberg M., Ashikaga T., Nishi R. (2009) J. Pediatr. Hematol. Oncol. 31, 187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peterson F. J., Mason R. P., Hovsepian J., Holtzman J. L. (1979) J. Biol. Chem. 254, 4009–4014 [PubMed] [Google Scholar]
- 12. McCalla D. R., Reuvers A., Kaiser C. (1971) Cancer Res. 31, 2184–2188 [PubMed] [Google Scholar]
- 13. Streeter A. J., Hoener B. A. (1988) Pharm. Res. 5, 434–436 [DOI] [PubMed] [Google Scholar]
- 14. Beckett A. H., Robinson A. E. (1959) J. Med. Pharm. Chem. 1, 155–164 [DOI] [PubMed] [Google Scholar]
- 15. Gavin J. J., Ebetino F. F., Freedman R., Waterbury W. E. (1966) Arch. Biochem. Biophys. 113, 399–404 [DOI] [PubMed] [Google Scholar]
- 16. Swaminathan S., Bryan G. T. (1984) Cancer Res. 44, 2331–2338 [PubMed] [Google Scholar]
- 17. Mason R. P., Holtzman J. L. (1975) Biochemistry 14, 1626–1632 [DOI] [PubMed] [Google Scholar]
- 18. Mason R. P., Holtzman J. L. (1975) Biochem. Biophys. Res. Commun. 67, 1267–1274 [DOI] [PubMed] [Google Scholar]
- 19. Docampo R., Stoppani A. O. (1979) Arch. Biochem. Biophys. 197, 317–321 [DOI] [PubMed] [Google Scholar]
- 20. Docampo R., Mason R. P., Mottley C., Muniz R. P. (1981) J. Biol. Chem. 256, 10930–10933 [PubMed] [Google Scholar]
- 21. Viodé C., Bettache N., Cenas N., Krauth-Siegel R. L., Chauvière G., Bakalara N., Périé J. (1999) Biochem. Pharmacol. 57, 549–557 [DOI] [PubMed] [Google Scholar]
- 22. Schöneck R., Billaut-Mulot O., Numrich P., Ouaissi M. A., Krauth-Siegel R. L. (1997) Eur. J. Biochem. 243, 739–747 [DOI] [PubMed] [Google Scholar]
- 23. Blumenstiel K., Schöneck R., Yardley V., Croft S. L., Krauth-Siegel R. L. (1999) Biochem. Pharmacol. 58, 1791–1799 [DOI] [PubMed] [Google Scholar]
- 24. Kelly J. M., Taylor M. C., Smith K., Hunter K. J., Fairlamb A. H. (1993) Eur. J. Biochem. 218, 29–37 [DOI] [PubMed] [Google Scholar]
- 25. Prathalingham S. R., Wilkinson S. R., Horn D., Kelly J. M. (2007) Antimicrob. Agents Chemother. 51, 755–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boiani M., Piacenza L., Hernández P., Boiani L., Cerecetto H., González M., Denicola A. (2010) Biochem. Pharmacol. 79, 1736–1745 [DOI] [PubMed] [Google Scholar]
- 27. Wilkinson S. R., Taylor M. C., Horn D., Kelly J. M., Cheeseman I. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 5022–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baker N., Alsford S., Horn D. (2011) Mol. Biochem. Parasitol. 176, 55–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kubata B. K., Kabututu Z., Nozaki T., Munday C. J., Fukuzumi S., Ohkubo K., Lazarus M., Maruyama T., Martin S. K., Duszenko M., Urade Y. (2002) J. Exp. Med. 196, 1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirumi H., Hirumi K. (1989) J. Parasitol. 75, 985–989 [PubMed] [Google Scholar]
- 31. Alsford S., Kawahara T., Glover L., Horn D. (2005) Mol. Biochem. Parasitol. 144, 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hall B. S., Wu X., Hu L., Wilkinson S. R. (2010) Antimicrob. Agents Chemother. 54, 1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olry A., Schneider-Belhaddad F., Heintz D., Werck-Reichhart D. (2007) Plant J. 51, 331–340 [DOI] [PubMed] [Google Scholar]
- 34. Portal P., Villamil S. F., Alonso G. D., De Vas M. G., Flawiá M. M., Torres H. N., Paveto C. (2008) Mol. Biochem. Parasitol. 160, 42–51 [DOI] [PubMed] [Google Scholar]
- 35. Bot C., Hall B. S., Bashir N., Taylor M. C., Helsby N. A., Wilkinson S. R. (2010) Antimicrob. Agents Chemother. 54, 4246–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moreno S. N., Mason R. P., Docampo R. (1984) J. Biol. Chem. 259, 6298–6305 [PubMed] [Google Scholar]
- 37. Xu W., McDonough R. C., 3rd, Langsdorf B., Demas J. N., DeGraff B. A. (1994) Anal. Chem. 66, 4133–4141 [DOI] [PubMed] [Google Scholar]
- 38. Filali-Mouhim A., Champion B., Jore D., Hickel. B., Ferradini C. (1991) J. Chim. Phys. 88, 937–943 [Google Scholar]
- 39. McCalla D. R., Kaiser C., Green M. H. (1978) J. Bacteriol. 133, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sastry S. S., Jayaraman R. (1984) Mol. Gen. Genet. 196, 379–380 [DOI] [PubMed] [Google Scholar]
- 41. Whiteway J., Koziarz P., Veall J., Sandhu N., Kumar P., Hoecher B., Lambert I. B. (1998) J. Bacteriol. 180, 5529–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boland M. P., Knox R. J., Roberts J. J. (1991) Biochem. Pharmacol. 41, 867–875 [DOI] [PubMed] [Google Scholar]
- 43. Wu K., Knox R., Sun X. Z., Joseph P., Jaiswal A. K., Zhang D., Deng P. S., Chen S. (1997) Arch. Biochem. Biophys. 347, 221–228 [DOI] [PubMed] [Google Scholar]
- 44. Cenas N., Prast S., Nivinskas H., Sarlauskas J., Arnér E. S. (2006) J. Biol. Chem. 281, 5593–5603 [DOI] [PubMed] [Google Scholar]
- 45. Jaiswal A. K. (2000) Free Radic. Biol. Med. 29, 254–262 [DOI] [PubMed] [Google Scholar]
- 46. Nivinskas H., Koder R. L., Anusevicius Z., Sarlauskas J., Miller A. F., Cenas N. (2001) Arch. Biochem. Biophys. 385, 170–178 [DOI] [PubMed] [Google Scholar]
- 47. Swaminathan S., Lower G. M., Jr., Bryan G. T. (1982) Cancer Res. 42, 4479–4484 [PubMed] [Google Scholar]
- 48. Maya J. D., Repetto Y., Agosín M., Ojeda J. M., Tellez R., Gaule C., Morello A. (1997) Mol. Biochem. Parasitol. 86, 101–106 [PubMed] [Google Scholar]
- 49. Goijman S. G., Frasch A. C., Stoppani A. O. (1985) Biochem. Pharmacol. 34, 1457–1461 [DOI] [PubMed] [Google Scholar]
- 50. Goijman S. G., Stoppani A. O. (1985) Biochem. Pharmacol. 34, 1331–1336 [DOI] [PubMed] [Google Scholar]
- 51. Jockers-Scherübl M. C., Schirmer R. H., Krauth-Siegel R. L. (1989) Eur. J. Biochem. 180, 267–272 [DOI] [PubMed] [Google Scholar]