

Abstract
Human splicing factor SF3a is a component of the mature U2 small nuclear ribonucleoprotein particle (snRNP) and its three subunits of 60, 66, and 120 kDa are essential for splicing in vitro and in vivo. The SF3a heterotrimer forms in the cytoplasm and enters the nucleus independently of the U2 snRNP. Here, we have analyzed domains required for in vitro interactions between the SF3a subunits. Our results indicate that the SF3a66-SF3a120 interaction is mediated by a 27-amino acid region in SF3a120 C-terminal to the second suppressor-of-white-apricot and prp21/spp91 domain and amino acids 108–210 of SF3a66. Neither of these sequences contains known structural motifs, suggesting that the interaction domains are novel. Moreover, an ∼100-amino acid region, including the SURP2 domain of SF3a120 but extending into neighboring regions, is sufficient for binding to SF3a60. Analysis of determinants for nuclear import of SF3a demonstrates that SF3a120 provides the major nuclear localization signal and SF3a60 contributes to nuclear import.
Keywords: Nucleus, Protein Domains, Protein-Protein Interactions, Ribonuclear Protein (RNP), RNA Metabolism, RNA Splicing, Spliceosome, Nuclear Localization Signal, Splicing Factor SF3a
Introduction
The U1, U2, U4/U6, and U5 small nuclear ribonucleoprotein particles (snRNPs)5 play essential roles in pre-mRNA splicing by interacting with conserved sequences at exon-intron boundaries and assembling the catalytic core of the spliceosome (1). They consist of one or two uridine-rich small nuclear RNAs (snRNAs), seven common (Sm) and several particle-specific proteins (2). Whereas U6 snRNP biogenesis is entirely nuclear, newly transcribed U1, U2, U4, and U5 snRNAs are exported to the cytoplasm. Binding of the Sm proteins generates “core snRNPs,” which are imported into the nucleus. The final maturation steps (snRNA modification and binding of particle-specific proteins) occur in Cajal bodies, followed by movement of the mature snRNPs to sites of splicing and storage (3, 4).
Except for the U1 snRNP, other snRNPs exist in different forms. U4, U5, and U6 snRNPs undergo association-dissociation cycles that are important for spliceosome assembly (2). In addition, different forms of the U2 snRNP have been isolated. The human 12 S U2 snRNP consists of the U2 snRNA, the Sm proteins, and the U2-specific proteins U2-A′ and U2-B″, whereas a 17 S U2 snRNP contains additional polypeptides representing subunits of splicing factors SF3a and SF3b (2, 5). In vitro, the 17S U2 snRNP assembles in a stepwise fashion (6). SF3b associates with the 12 S U2 snRNP to yield a 15 S intermediate, which is converted into the 17 S U2 snRNP upon SF3a binding. Only the 17 S U2 snRNP, but not the 12 S or 15 S particles, is functional (6, 7). SF3a and SF3b interact with the pre-mRNA at or in the vicinity of the branch site early during spliceosome assembly, which is thought to recruit and tether the U2 snRNP to the spliceosome (8–10). At later stages of the splicing reaction, SF3a and SF3b appear to be destabilized from the U2 snRNP because they are underrepresented in the catalytically active spliceosome (11).
SF3a comprises three evolutionarily conserved subunits (SF3a60, SF3a66, and SF3a120), all of which are necessary for the assembly of the 17 S U2 snRNP and splicing in vitro and in vivo (12–15). Human SF3a60 contains an SAF-A/B, Acinus, and PIAS motif in its central portion and a C2H2-type zinc finger domain in the C-terminal third (16). SF3a66 harbors a C2H2 zinc finger domain close to its N terminus, and the C-terminal half is composed of 22 heptad repeats (17). SF3a120 is characterized by the presence of two suppressor-of-white-apricot and prp21/spp91 (SURP) domains and a stretch of 18 consecutive, charged residues in the N-terminal half. The C-terminal half comprises Pro-rich sequences and a ubiquitin-like domain (UBL) (see Fig. 1; 18).
FIGURE 1.

A short region immediately C-terminal to the charged region of SF3a120 is sufficient for SF3a66 binding. A, scheme of in vitro-translated and GST-tagged SF3a120 proteins. Boxes indicate known protein domains as follows: S1 and S2, SURP1 and SURP 2 motifs; +/−, charged region; Pro, Pro-rich regions with PXPP motifs. Numbering above full-length SF3a120 (accession no. Q15459) refers to amino acids. Numbers on the left refer to N and C termini of SF3a120 mutants. B, GST pulldown of N- and C-terminal SF3a120 deletion mutants with GST-3a66/1–216. In vitro-translated SF3a120 proteins indicated above the figure were incubated with GST (lane 1) or GST-3a66/1–216 (lanes 2–9) bound to glutathione-agarose. Only 3a120-FL was incubated with the GST control (lane 1, co.). Bound proteins (top) and 20% of the input proteins (bottom) were separated by 10% SDS-PAGE and visualized by autoradiography. The migration of protein markers is indicated in kDa to the right of each panel. C, GST pulldown with proteins corresponding to internal segments of SF3a120. GST (lane 1) or GST-tagged SF3a120 proteins (as indicated above lanes 2–5) were bound to glutathione-agarose and incubated with in vitro-translated 3a66-FL (input, first lane in top panel; 20% of input is shown). Bound proteins were separated by 10% SDS-PAGE and detected by autoradiography (top). GST-tagged proteins used were separated in a 10% SDS gel and stained with Coomassie Blue (bottom). Full-length proteins are marked with black circles.
The SF3a heterotrimer forms by binding of SF3a60 and SF3a66 to SF3a120 (14, 17–19). In vitro interaction studies showed that amino acids 35–107 of SF3a60 are sufficient for SF3a120 binding and the N-terminal 34 amino acids may stabilize the interaction (14). In turn, residues 151–242 of SF3a120 (encompassing SURP2) are essential for binding SF3a60 (18). The NMR structure of co-expressed fragments of SF3a60 (amino acids 71–107) and SF3a120 (amino acids 134–217) revealed that SURP2 adopts an α1-α2–310-α3 topology, and SF3a120 residues 162–195 directly contact residues 80–96 of SF3a60, which form an α-helix (20). Interaction sites between SF3a66 and SF3a120 have been mapped roughly to the N-terminal 216 amino acids of SF3a66 and residues 243–372 of SF3a120, comprising the charged region and Pro-rich sequences (18).
The SF3a heterotrimer forms in the cytoplasm and is imported into the nucleus independently of the core U2 snRNP and SF3b (21). None of the SF3a subunits appears to contain a classical nuclear localization signal (NLS). However, because regions in SF3a60 and SF3a66 that mediate the interaction with SF3a120 are required for nuclear localization, it was proposed that either SF3a120 contained a NLS or that a NLS was formed upon SF3a assembly.
We have extended the structure-function analysis of SF3a and defined sequences necessary for interactions between SF3a subunits in further detail. Our results indicate that a 27-amino acid sequence C-terminal to SURP2 of SF3a120 without known structural motifs is responsible for the interaction with amino acids 108–210 of SF3a66. We also show that SF3a120 sequences encompassing SURP2 are sufficient for SF3a60 binding in vitro. Finally, we have identified a NLS in SF3a120, which represents the major signal for nuclear targeting of SF3a.
EXPERIMENTAL PROCEDURES
Cloning Procedures
All sequences were amplified by PCR with high fidelity enzymes and correct cloning was verified by sequencing. The coding sequences of truncated SF3a120 proteins were cloned into the XhoI/EcoRI sites of pRSET (Invitrogen) for in vitro translation. Sequences encoding GST-tagged, internal regions of SF3a120 were cloned into the BamHI/EcoRI sites of pGEX6P-2 (Amersham Biosciences). The sequence encoding amino acids 1–216 of SF3a66 was cloned into the BamHI/EcoRI sites of pRSET-A. Internal sequences of SF3a66 were cloned into the KpnI/EcoRI sites of pRSET-A containing the open reading frame of the maltose binding protein in the BamHI/KpnI sites. Internal 10-amino acid deletions of SF3a66/1–216 in pRSET-A were generated by replacing the deleted sequences with a KpnI site, encoding Gly and Thr. The full-length SF3a60 sequence was cloned into the BamHI/EcoRI sites of pGEX6P-2 (Amersham Biosciences).
For transient expression of N-terminal GFP fusion proteins, sequences were cloned into pEGFP (Invitrogen). The full-length SF3a120 sequence was cloned into the EcoRI site. SF3a120 sequences encoding proteins with N- or C-terminal deletions were cloned into the XhoI/EcoRI sites. The internal deletions GFP-3a120ΔSURP1 (Δ52–94), ΔSURP2 (Δ167–208), and Δ66 (Δ269–295) were generated from GFP-SF3a120-FL by replacing the deleted sequences with a KpnI site encoding Gly and Thr. Sequences encoding the pattern 7 (pat7) motif (amino acids 680–686) were deleted from GFP-3a120-FL, ΔSURP2, and Δ66 by replacement with a SpeI site encoding Thr and Ser. The deleted sequence in GFP-3a120ΔRRNK (Δ699–702) was replaced with a SpeI site. To generate GFP-tagged fusions to chicken pyruvate kinase (PyrK), the sequence encoding PyrK amino acids 17–525 was first cloned into the EcoRI site of pEGFP, followed by cloning sequences encoding the SF3a120 pat7 motif into the BglII/EcoRI sites or EcoRI/BamHI sites to create GFP-pat7-PyrK or GFP-PyrK-pat7, respectively. Sequences encoding SF3a120 residues 676–713, 680–702, and 680–699 were cloned into the BglII site of GFP-PyrK to generate GFP-3a120/676–713-PyrK, GFP-3a120/680–702-PyrK, and GFP-3a120/680–698-PyrK, respectively. The full-length sequences of SF3a60 and SF3a66 were cloned into the BamHI site of pEGFP. Sequences encoding the SF3a60 pat7 motif (amino acids 174–180) were replaced with a KpnI site. Sequences encoding the SF3a120-interaction domain of SF3a66 (amino acids 108–210) were replaced with a BssHI site encoding Ala and Arg.
In Vitro Translation and Bacterial Expression of Recombinant Proteins
Proteins were in vitro-translated with the TnT coupled reticulocyte lysate system (Promega) according to the manufacturer's instructions. For bacterial expression, plasmids were transformed into the Escherichia coli strain TOP10 (Invitrogen) by heat shock. Proteins were expressed for 4 h at 37 °C after addition of isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 1 mm. Cells were harvested by centrifugation at 5000 × g for 10 min, lysed by sonication on ice, and supplemented with Triton X-100 in PBS to a final concentration of 1%. Proteins were purified with 500 μl of a 50% suspension of glutathione agarose (Sigma) equilibrated in PBS. Unbound proteins were removed by washing three times with 10 ml of PBS, and GST-tagged proteins were eluted with 5 mm glutathione and 50 mm Tris-HCl, pH 8.0, dialyzed against buffer D (22) supplemented with 3 mm MgCl2 and stored at −80 °C.
GST Pulldown Assays
Protein-protein interactions were analyzed by GST pulldown of in vitro-translated proteins. Reactions (200 μl) contained 40 μl of glutathione-agarose beads (Sigma) and 1 μg of the GST fusion protein in NETN (20 mm Tris-HCl, pH 8.0, 50–100 mm NaCl, 0.5% Nonidet P-40, 0.5 mm EDTA). The beads were incubated for 30 min at 4 °C, washed twice with 500 μl NETN and centrifuged at 1000 rpm at 4 °C. In vitro-translated, [35S]Met-labeled proteins were added and incubated for 45 min at 4 °C in a total volume of 200 μl NETN. Unbound proteins were removed by six NETN washes as above. Proteins were eluted from the beads by boiling in SDS sample buffer and separated by SDS-PAGE followed by autoradiography.
Cell Culture and Transient Transfection
HeLa cells were grown in DMEM supplemented with 2 mm l-glutamine, 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Sigma). Cells were plated in 35-mm glass-bottomed culture dishes (MatTek) at a density of 1–3 × 105 cells/cm2 in the same medium as above 24 h prior to transfection. Cells were transfected by calcium phosphate precipitation or with the Mirus TransIT Reagent (Mirus, Madison, WI) according to the manufacturer's instructions at a confluency of 60–70%. For immunoprecipitation, cells were grown in 10-cm dishes and transfected by calcium phosphate precipitation.
Co-immunoprecipitation
HeLa cells were washed with PBS and lysed in lysis buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1% Nonidet P-40, 1 mm DTT, 2 mm EDTA, protease inhibitors (Roche Applied Science)) for 30 min at 4 °C. Lysates were centrifuged at 16,000 × g for 5 min at 4 °C and incubated in the presence of 0.3 mg/ml RNase A (Sigma) for 20 min at room temperature. After centrifugation as before lysates were precleared by incubation for 1 h at 4 °C with Dynabeads Protein G (Invitrogen) in lysis buffer. Samples were then incubated for 1 h at 4 °C with Dynabeads Protein G coated with goat anti-GFP antibodies (a kind gift by K. Neugebauer) in wash buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 0.1% Nonidet P-40, 1 mm EDTA). After six washes in wash buffer, bound proteins were eluted with 1 × SDS sample solution for 5 min at 95 °C. Input and bound material were separated in 7.5% SDS-polyacrylamide gels followed by Western blotting (15) with rabbit anti-SF3a120 (18), rabbit anti-SF3a60 (16), and monoclonal mouse anti-SF3a66 (12) antibodies. Secondary horseradish peroxidase-conjugated goat anti-rabbit (Jackson ImmunoResearch Laboratories) and rabbit anti-mouse antibodies (Dako) were detected with the SuperSignal kit (Pierce).
Fluorescence Microscopy
GFP-tagged proteins were visualized in live cells 48 h after transfection with a Zeiss inverted fluorescence microscope (Axiovert TV 135) with 63× or 100× Planapo objectives. Images were acquired with a cooled charge-coupled device camera (model CH250, Photometrics). Images were recorded with Openlab software (Improvision) and processed with Adobe Photoshop 7.0 (Adobe Systems).
RESULTS
SF3a66 Interaction Domain (ID) of SF3a120
Previous results demonstrated that amino acids 243–373 of SF3a120 are sufficient for SF3a66 binding (18). To define the SF3a66 ID more precisely, we performed GST pulldown assays. In brief, the GST-tagged, N-terminal half of SF3a66 (amino acids 1–216; GST-3a66/1–2166), which interacts with SF3a120 as efficiently as full-length SF3a66 (14), was expressed in E. coli. The purified protein was bound to glutathione-agarose beads and incubated with in vitro-translated, [35S]Met-labeled, full-length SF3a120 (3a120-FL) or N- and C-terminal deletion mutants (Fig. 1A). Bound and input proteins were separated by SDS-PAGE and visualized by autoradiography (Fig. 1B). As a control for background binding, 3a120-FL was incubated with GST (Fig. 1B, lane 1). The C-terminal deletion mutants 3a120/1–295 and 3a120/1–289 bound SF3a66 comparable to 3a120-FL (Fig. 1B, lanes 2, 4, and 5). Binding was abolished when residues immediately C-terminal to the charged region were deleted (3a120/1–268; lane 3). Deletion of the N-terminal 250 or 268 amino acids did not affect the interaction (3a120/251–793 and 3a120/269–793; lanes 6 and 7), but truncation to amino acid 295 or beyond abolished binding (lanes 8 and 9).
These results place the SF3a66 ID of SF3a120 between amino acids 269 and 289. We next tested whether the short region was also sufficient for SF3a66 binding. In this experiment, GST-tagged SF3a120 fragments were incubated with in vitro-translated 3a66/1–216, because the shortest peptide (3a120/269–295) does not contain any Met necessary for labeling by in vitro translation. Fig. 1C shows that in vitro-translated 3a66/1–216 interacted equally well with GST-3a120/269–295 and GST-3a120/164–307, indicating that the region between amino acids 269 and 295 of SF3a120 is sufficient for SF3a66 binding (Fig. 1C, lanes 4 and 5). As expected from the results shown in Fig. 1B, binding to SURP1 or SURP2 was not observed (Fig. 1C, lanes 2 and 3).
SF3a120 ID of SF3a66
We have shown previously that the N-terminal 216 amino acids of SF3a66 suffice for SF3a120 binding (14). This region also mediates the incorporation of SF3a66 into the U2 snRNP and is required for nuclear import and localization to nuclear speckles (14, 21). Because the SF3a66 ID of SF3a120 is at most 27 amino acids in length, we asked whether the entire N-terminal half of SF3a66 was necessary for SF3a120 binding. To facilitate the analysis of shorter versions of SF3a66, truncated proteins were in vitro-translated as fusions to maltose binding protein and incubated with GST-3a120/269–295 bound to glutathione-agarose beads. The N-terminal maltose binding protein tag did not interfere with the interaction between GST-3a120/269–295 and 3a66/1–216 (Fig. 2B, lane 2; data not shown). N-terminal deletions to amino acids 104 or 108 had no effect on the interaction (lanes 3 and 4), but binding to SF3a120 gradually decreased upon further truncation (lanes 5 and 6). A C-terminal deletion to amino acid 210 resulted in slightly reduced binding, and the interaction was completely abolished upon deletion to amino acid 200 (lanes 9–12). Thus, the N- and C-terminal borders of the SF3a120 ID of SF3a66 are located between amino acids 108–126 and 200–216, respectively.
FIGURE 2.

Amino acids 108–216 of SF3a66 are sufficient for the interaction with SF3a120. A, schematic representation of the SF3a66 domain structure. The SF3a66 sequence was taken from accession no. Q15428. The zinc finger domain (Zn) and Pro-rich heptad repeats (GVHPPAP22) are indicated. Numbers below the scheme refer to the maximal and minimal extent of SF3a66 deletion mutants. B, GST pulldown of N- and C-terminal SF3a66 deletion mutants. In vitro-translated SF3a66 proteins indicated above the figure were incubated with GST-3a120/269–295 bound to glutathione-agarose (lanes 2–6 and 8–12). As a control, 3a66/1–216 was incubated with GST-coupled beads (lanes 1 and 7, co.). Bound proteins (top) and 20% of the input proteins (bottom) were separated by 10% SDS-PAGE and visualized by autoradiography. C, GST pulldown of SF3a66 proteins with internal 10-amino acid deletions. The experiment was performed as in B, except that a 12% SDS gel was used. Lane 1, 3a66/1–216 was incubated with GST (co.) lanes 2–13, proteins shown above the figure were incubated with GST-3a120/269–295.
To test whether the entire region between amino acids 108 and 216 was essential for SF3a120 binding, internal, 10-amino acid deletions were introduced into 3a66/1–216. 3a66Δ105–114 bound GST-3a120/269–295 as efficiently as 3a66/1–216 (Fig. 2C, lanes 3 and 2). Binding of 3a66Δ115–124 was reduced slightly (lane 4), in agreement with the residual binding of 3a66/114–216 (cf. Fig. 2B, lane 5). Internal deletions between residues 125 and 154 and residues 165 and 184 inhibited the interaction (Fig. 2C, lanes 5–7, 9, and 10), and deletion of amino acids 185–194 and 195–204 strongly reduced the interaction (lanes 11 and 12). In contrast, 3a66Δ155–164 bound GST-3a120/269–295 almost as efficiently as 3a66/1–216 (cf. lanes 2 and 8). Together, these results indicate that amino acids 108–216 of SF3a66 are sufficient for SF3a120 binding, but not all sequences in this region contribute similarly to the interaction.
SF3a60 ID of SF3a120
Previous in vitro interaction assays indicated that residues 151–242 of SF3a120, encompassing SURP2, were necessary for SF3a60 binding (18). Determination of the NMR structure of a complex of SF3a120 SURP2 and a SF3a60 peptide required co-expression of suitable peptides in E. coli as individually expressed peptides were insoluble (20). To examine the SF3a60-SF3a120 interaction in vitro with soluble proteins, we used GST-tagged, full-length SF3a60 (GST-3a60-FL) and in vitro-translated 3a120-FL or N- and C-terminal deletion mutants in GST pulldown assays (Fig. 3A). Compared with the control with GST bound to glutathione-agarose (Fig. 3C, lane 1), 3a120-FL (lane 2) and 3a120/1–224 (lane 3) efficiently interacted with GST-3a60. However, further C-terminal truncation to residues 217 or 205 abolished binding (Fig. 3C, lanes 4 and 5). The N-terminal deletion mutants 3a120/145–793, 3a120/157–793, and 3a120/164–793 bound GST-3a60, but no interaction was seen with 3a120/180–793 (Fig. 3C, lanes 6–9).
FIGURE 3.

Amino acids 145–243 of SF3a120 are sufficient for SF3a60 binding. A, scheme of in vitro-translated SF3a120 proteins. Protein domains are as defined in the legend to Fig. 1. B, SDS-PAGE and Coomassie Blue staining of recombinant GST (lane 1) and GST-3a60-FL (lane 2). C, GST pulldown of N- and C-terminal SF3a120 deletion mutants by GST-3a60-FL. The experiment was performed as described in Fig. 1B. D, GST pulldown of internal SF3a120 deletion mutants with GST-3a60-FL. The experiment was performed as in C, except that proteins were separated by 15% SDS-PAGE.
These results indicate that the N- and C-terminal borders of the in vitro SF3a60 ID in SF3a120 lie between amino acids 164–180 and 217–224, respectively. When we tested whether amino acids 164–224 of SF3a120 were also sufficient for SF3a60 binding, the interaction was reduced to almost background levels (Fig. 3D, cf. lanes 1 and 10). N-terminal extension by seven or 19 amino acids gradually increased binding (3a120/157–224 and 3a120/145–224; Fig. 3D, lanes 7 and 5). C-terminal extension to amino acid 243 further improved the interaction (3a120/145–243 and 3a120/157–243; Fig. 3D, lanes 6 and 8), with 3a120/145–243 binding GST-3a60 similar to 3a120-FL (Fig. 3D, cf. lanes 6 and 2). Thus, although N- and C-terminal deletions to amino acids 164 and 224 did not compromise SF3a120 binding to SF3a60, the region spanning amino acids 164–224 is not sufficient for SF3a60-SF3a120 contacts, but neighboring residues contribute to the interaction. Although SURP1 is very similar in sequence to SURP2, it does not interact with SF3a60 (Fig. 3D, lane 9) (18, 20).
Determinants for Nuclear Import of SF3a
Similar to the full-length protein, the N-terminal half of SF3a66 (amino acids 1–258) localized to the nucleus (21). Deletion of the zinc finger domain (amino acids 54–78) did not compromise nuclear import but abolished localization to speckles. The same study demonstrated that the region of SF3a60 responsible for SF3a120 binding was required for nuclear targeting. We therefore asked whether the newly defined SF3a120 ID of SF3a66 was also necessary for nuclear localization. To this end, full-length SF3a66 fused to GFP (GFP-3a66-FL) and a mutant lacking the SF3a120 ID (GFP-3a66Δ108–210) were transiently expressed in HeLa cells and visualized by fluorescence in live cells 48 h post-transfection. Whereas GFP-3a66-FL was exclusively nuclear and present in speckles, GFP-3a66Δ108–210 was mainly cytoplasmic (Fig. 4). Western blotting with anti-GFP antibodies of whole cell extracts prepared from transfected cells confirmed correct expression of the proteins (data not shown). These results strongly suggest that nuclear localization of SF3a66, similar to that of SF3a60 (21), depends on its interaction with SF3a120.
FIGURE 4.
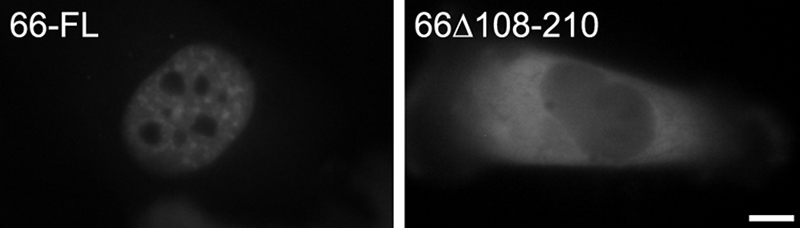
The SF3a120 ID of SF3a66 is required for nuclear localization. HeLa cells were transiently transfected with GFP-3a66-FL and GFP-66Δ108–210. Fluorescence was monitored in live cells 48 h post-transfection. Bar, 10 μm.
If SF3a120 were responsible for nuclear import of SF3a, we would expect that SF3a120 proteins deleted for the SF3a60 or SF3a66 IDs localized to the nucleus. GFP-tagged SF3a120-FL and mutant versions (Fig. 5A) were transiently expressed in HeLa cells. To confirm lack of interaction of endogenous SF3a60 and SF3a66 with GFP-3a120ΔSURP2 and -Δ66, respectively, lysates of mock-transfected cells or cells transfected with GFP-3a120-FL, -ΔSURP1, -ΔSURP2, and -Δ66 were subjected to immunoprecipitation with anti-GFP antibodies, and input and bound proteins were Western blotted with antibodies to the SF3a subunits. As expected from the data presented above (Figs. 1 and 3), SF3a60 failed to bind GFP-3a120ΔSURP2 (Fig. 5B, middle panel, lanes 7 and 8), and SF3a66 did not interact with GFP-3a120Δ66 (bottom panel, lanes 9 and 10), whereas other GFP-3a120 proteins tested were efficiently bound.7
FIGURE 5.

The C terminus of SF3a120 is essential for nuclear import. A, schematic representation of GFP-tagged SF3a120 proteins. Protein domains are as described in the legend to Fig. 1. Dashed lines indicate deleted sequences. B, HeLa lysates from mock-transfected cells or cells transiently transfected with GFP-tagged 3a120-FL, -ΔSURP1, -ΔSURP2, or -Δ66 (as indicated above the panels) were immunoprecipitated with anti-GFP antibodies. Input (IP; 10% of total) and bound (B) proteins were separated by 7.5% SDS-PAGE and detected by Western blotting with anti-SF3a120 (top), anti-SF3a60 (middle), and anti-SF3a66 antibodies (bottom). All immunoprecipitations were performed at the same time, but samples from mock-transfected cells were separated in different gels. GFP-3a120 and the endogenous SF3a subunits are indicated on the right of the panels, the migration of protein markers (in kDa) is shown on the left. C, HeLa cells were transiently transfected with plasmids encoding GFP-tagged SF3a120 proteins as indicated in the individual panels. Fluorescence was monitored in live cells 48 h post-transfection. Bar, 10 μm.
The four transiently expressed GFP-3a120 proteins were exclusively nuclear (Fig. 5C). Thus, SF3a120 can enter the nucleus independently of the smaller subunits. However, compared with GFP-3a120-FL and -ΔSURP1, GFP-3a120ΔSURP2 and -Δ66 did not localize to nuclear speckles but were distributed diffusely in the nucleus. In addition, GFP-3a120Δ66 accumulated in a few bright foci, which correspond to Cajal bodies.8 The failure of the mutants to localize to speckles was expected because only the entire SF3a heterotrimer associates with the U2 snRNP, which is a prerequisite for the movement of the 17 S U2 snRNP (and thus SF3a) away from Cajal bodies (21).
Identification of SF3a120 NLS
To determine which region in SF3a120 was responsible for nuclear import, additional mutants were tested. Proteins lacking the UBL domain (GFP-3a120ΔUBL) showed the same distribution in nuclear speckles as GFP-3a120-FL (Fig. 5C). Proteins with N-terminal deletions of 295, 369, or 555 amino acids were also exclusively nuclear but showed a diffuse localization similar to GFP-3a120ΔSURP2 and -Δ66 (Fig. 5C; data not shown). Finally, a C-terminal deletion to amino acids 268, 295, 368 or 555 resulted in different degrees of cytoplasmic localization without complete depletion of the proteins from the nucleus (Fig. 5C; data not shown). Sizes of the shorter C-terminal deletion mutants are close to the limit of ∼60 kDa for diffusion through the nuclear pore (23). However, GFP-3a120/1–555 has a molecular mass of 93 kDa and should thus be imported actively into the nucleus.
In agreement with the result that amino acids 556–713 of SF3a120 contain a sequence required for nuclear localization, the PSORTII program (24) predicted an NLS of the pattern 7 (pat7)-type between amino acids 680 and 686 (PTSKKLK; Fig. 6A). The pat7-type NLS appears to be a variation of the classical monopartite NLS and contains an N-terminal Pro followed within three residues by a basic segment containing three of four Lys or Arg residues (25). Replacement of the pat7 motif in GFP-3a120-FL by Thr-Ser impaired nuclear import (GFP-120Δpat7; Fig. 6B). However, the extent of import inhibition was not uniform. Quantification of three independent experiments indicated that in 80% of the transfected cells, GFP-120Δpat7 was exclusively cytoplasmic, in 15–20% nuclear and cytoplasmic, and in the remaining 0–5% exclusively nuclear. In contrast, GFP-3a120-FL was never observed in the cytoplasm. The residual nuclear structures detected with GFP-3a120Δpat7 represented speckles, as shown by co-localization with SC35, a marker protein for these structures (26; data not shown).
FIGURE 6.

A pat7-type motif in SF3a120 mediates its nuclear localization. A, the sequence of SF3a120 encompassing the NLS is shown below the scheme of SF3a120. The pat7 motif and the RRNK are indicated in boldface and underlined. Numbering indicates amino acids. B, HeLa cells were transiently transfected with plasmids encoding GFP-tagged SF3a120 proteins, PyrK, or PyrK fused to SF3a120 sequences as indicated. Fluorescence was monitored in live cells 48 h post-transfection. Bar, 10 μm.
To determine whether the SF3a120 pat7 motif was sufficient for nuclear targeting, it was fused to GFP-tagged chicken muscle PyrK, an exclusively cytoplasmic protein (27). GFP-PyrK did not enter the nucleus, no matter whether the pat7 motif was fused to its N or C terminus (GFP-pat7-PyrK and GFP-PyrK-pat7, respectively; Fig. 6B), indicating that the pat7 motif is necessary for nuclear localization of SF3a120 but not sufficient for nuclear import of an unrelated cytoplasmic protein.
PSORTII did not reveal any other potential NLS in SF3a120; however, a cluster of basic amino acids (RRNK) is present 12 residues C-terminal of the pat7 motif (Fig. 6A). Thus, amino acids 680–702 of SF3a120 could comprise a bipartite NLS, in which two basic residues are separated by up to 29 amino acids from a second basic region containing at least three of five basic amino acids (25). To test this possibility, the pat7 motif in GFP-pat7-PyrK was replaced by longer SF3a120 segments (Fig. 6). Whereas GFP-676–713/PyrK was entirely nuclear, GFP-680–702/PyrK, which only contained sequences from the pat7 motif to the RRNK (Fig. 6A), was mainly nuclear with faint cytoplasmic staining. In contrast, the protein lacking the RRNK (GFP-680–698/PyrK) was almost entirely excluded from the nucleus. These results demonstrate that amino acids 680–702 of SF3a120 are sufficient for nuclear uptake and may indicate that SF3a120 contains a bipartite NLS. However, although the RRNK motif was essential for import of PyrK, replacement of this motif with Thr-Ser in the context of GFP-3a120-FL only marginally affected the nuclear localization of the protein. This result suggests that, compared with the pat7 motif, the RRNK plays a minor role for nuclear targeting of SF3a120.
Contribution of SF3a60 to Nuclear Localization of SF3a
The results presented above demonstrate that SF3a120 contains a bona fide NLS. Moreover, SF3a120 mutants lacking either the SF3a60 or SF3a66 ID are nuclear, suggesting that the main signal for nuclear import of SF3a resides in SF3a120. On the other hand, C-terminal deletion mutants of SF3a120 lacking the NLS were not excluded completely from the nucleus (Fig. 5; data not shown). Thus, we could not exclude that other sequences in SF3a120 or the small subunits contributed to nuclear import. To test this possibility, the pat7 motif was deleted from GFP-3a120ΔSURP2 and -Δ66. The percentage of transfected cells with an exclusively cytoplasmic localization increased from 80% for GFP-3a120Δpat7 (see above) to >95% for GFP-3a120ΔSURP2-Δpat7 (Fig. 7), suggesting that the SF3a60-SF3a120 interaction contributes to nuclear import. In contrast, deletion of the pat7 motif from GFP-3a120Δ66 gave no additional effect (GFP-3a120Δ66-Δpat7). Inspection of the SF3a60 sequence with PSORTII predicted a pat7-type NLS between amino acids 174 and 180 (PKGRKNA). Deletion of this motif from GFP-3a60-FL caused cytoplasmic localization (Fig. 7); thus, SF3a60 also may play a role in nuclear targeting of SF3a. PSORTII did not predict any potential NLS in SF3a66. From these results, we conclude that the NLS in SF3a120 represents the major signal for SF3a nuclear import and SF3a60 contributes to efficient nuclear targeting.
FIGURE 7.
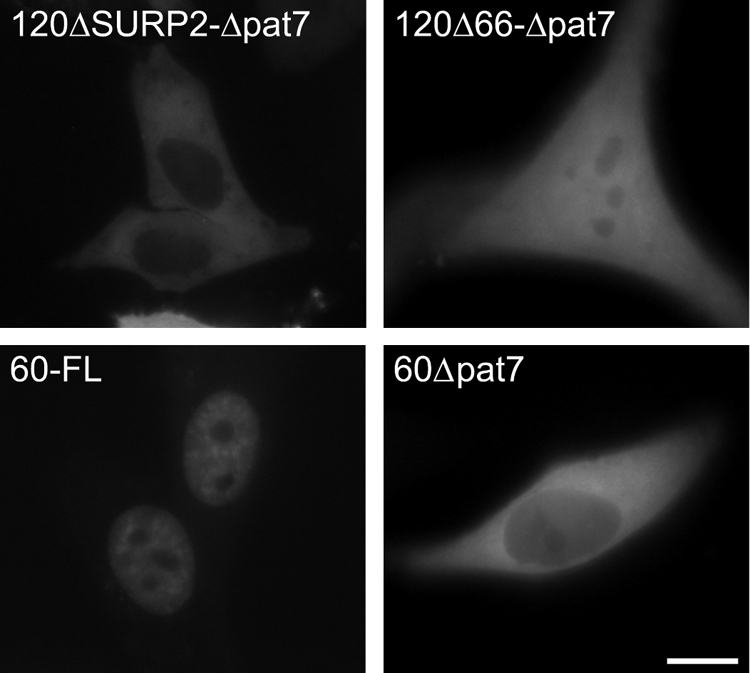
A pat7-type motif in SF3a60 contributes to nuclear localization. Live HeLa cells transiently expressing GFP-tagged SF3a120 mutants lacking the pat7 motif in addition to SURP2 or the SF3a66 ID and GFP-3a60-FL or GFP-3a60Δpat7 were analyzed by fluorescence microscopy 48 h post-transfection. Bar, 10 μm.
DISCUSSION
The SF3a heterotrimer forms by binding of SF3a60 and SF3a66 to SF3a120 (14, 18). We show here that two distinct, evolutionarily conserved regions in SF3a120 mediate binding to SF3a60 and SF3a66 in vitro. The domain in SF3a66 required for binding to SF3a120 has been defined to amino acids 108–210. We also have examined sequences that are essential for nuclear import of SF3a and show that the major NLS resides in SF3a120.
The SF3a66 ID of SF3a120 has been mapped previously to amino acids 243–372 (18). Further N- and C-terminal truncations of SF3a120 now delimit the binding domain to amino acids 269–289 (Figs. 1 and 8). This region is conserved highly between metazoan, plant, and yeast SF3a120 proteins and a mutation in the Saccharomyces cerevisiae homologue of SF3a120, Prp21p, corresponding to amino acid 282 in human SF3a120, decreases binding to Prp11p, the yeast homologue of SF3a66 (28). Our data furthermore indicate that amino acids 269–295 are sufficient for contacts with SF3a66. This region is slightly shorter than the shortest region in Prp21p tested that binds Prp11p (corresponding to residues 269–307 of human SF3a120) (28). Moreover, amino acids 253–315 of human SF3a120 supported binding of Prp11p in a two-hybrid assay (28), further emphasizing the structural and functional conservation of the SF3a66 ID of SF3a120.
FIGURE 8.

Summary of SF3a structural and functional domains. Schematic representations of SF3a120 and SF3a66 are as described in Figs. 1A and 2A, respectively. For SF3a60 (accession no. Q12874) the SAP (SAF-A/B, Acinus, and PIAS motif) and the zinc finger domain (Zn) are indicated. Thick lines represent sequences required for intersubunit interactions (Ref. 14 and this study). Black lines indicate sequences essential for interactions; gray lines mark sequences that may contribute to binding. The NLS in SF3a120 and SF3a60 as well as the zinc finger domains of SF3a60 and SF3a66 required for the assembly of the functional 17S U2 snRNP are indicated.
In contrast to the short domain in SF3a120 required for SF3a66 binding, the SF3a120 ID of SF3a66 extends over ∼100 amino acids. N-and C-terminal truncations define the borders of the maximal ID to amino acids 108 and 210 (Figs. 2 and 8). Examination of the effect of internal deletions on SF3a120 binding suggests that amino acids 108–124 and 185–204 contribute to optimal interactions, whereas residues 155–164 appear to play a minor role, if any, in binding. Thus, the core region essential for SF3a66-SF3a120 contacts spans amino acids 125–154 and 165–185. The entire ID is evolutionarily well conserved. The homologous region in S. cerevisiae Prp11p occupies almost the entire C-terminal half. (Prp11p lacks the Pro-rich repeats present in metazoans (17).) Divergent residues in Prp11p lie outside of the core ID of SF3a66, except for an insertion of nine amino acids at a position corresponding to residues 144/145 of human SF3a66. The SF3a66-SF3a120 interaction has not been studied in other organisms. However, the temperature-sensitive prp11-1 mutation was mapped to Prp11p amino acid 178, which corresponds to the conserved Pro147 in SF3a66 (29). In addition, a mutational linker-insertion screen interrogated regions in Prp11p required for growth (29). Seven of the insertions map to the region corresponding to the SF3a120 ID of SF3a66, however only two resulted in null alleles (residues 110 and 178 of human SF3a66). Of the five remaining insertions that do not have a phenotype, only one maps to the core region of the SF3a120 ID (Lys182 of SF3a66). Moreover, a deletion of the C-terminal 55 amino acids of Prp11p (starting from residue 212 of SF3a66) was lethal (29). It is likely that the null alleles result from a disruption of the Prp11p-Prp21p interaction and, as a consequence, a defect in splicing. The fact that, at least in yeast, several insertions do not result in effects on viability suggests that the structure of this region is rather flexible.
Database searches with the IDs of SF3a66 and SF3a120 did not reveal any resemblance to structural motifs in other proteins, suggesting that the domains described here are novel. Future structural determination should shed light on the molecular details of the interaction.
Distinct isoforms for human SF3a120 and SF3a60 are annotated in the Ensembl genome browser (30). Two forms containing intact N and C termini are reported for each protein. The shorter SF3a120 isoform lacks residues 106–170, encoding much of the linker between SURP1 and -2 and the first five residues of SURP2 (cf. Figs. 1A and 8). It is possible that SF3a60 binding to the shorter form is compromised, but this needs experimental verification. The short SF3a60 isoform encodes a protein deleted for residues 49–101 and thus most of the SF3a120 ID (cf. Fig. 8) (14). It is highly likely that this protein fails to bind SF3a120. Whether or not the shorter proteins play a role in processes other than splicing or negatively influence SF3a function requires further investigation.
The NMR structure of a SF3a60-SF3a120 complex was solved with protein fragments (amino acids 71–107 of SF3a60 and amino acids 134–217 of SF3a120) co-expressed in E. coli, as the individual peptides were insoluble (20). The structure revealed multiple contacts between residues 80–95 of SF3a60 and residues 162–195 of SF3a120. In contrast, earlier GST pulldown and co-immunoprecipitation assays with soluble recombinant and in vitro-translated proteins indicated that SF3a60 amino acids 35–107 were sufficient for SF3a120 binding; however, this interaction is salt-sensitive and stabilized by the N-terminal 34 amino acids (14). Moreover, the region in SF3a120 necessary for binding SF3a60 in vitro appears to differ from the domain defined by contacts in the NMR structure (Figs. 3 and 8). First, although Phe162 of SF3a120 engages in contacts with several SF3a60 residues (20), SF3a120/164–793 bound GST-SF3a60 as efficiently as SF3a120-FL. No binding was observed with SF3a120/180–793, certainly due to the deletion of the entire helix α1 of SF3a120 SURP2, mediating multiple interactions with SF3a60. Second, although the NMR structure was obtained with a SF3a120 peptide comprising residues 134–217, the N-terminal 217 amino acids are not sufficient for binding SF3a60 in vitro, whereas 224 amino acids suffice for the interaction. Moreover, when short internal, in vitro-translated fragments of SF3a120 were used in GST pulldown assays, it appeared that only 3a120/145–243 efficiently bound SF3a60. Thus, in addition to the residues defined by the NMR structure of Kuwasako et al. (20), other sequences may contribute to the interaction between SF3a120 and SF3a60, at least in vitro.
Unlike the common Sm proteins, which associate with snRNAs in the cytoplasm, proteins characteristic of individual snRNPs are thought to be imported into the nucleus independently of the core snRNPs (31). The same was proposed for SF3a, as sequences in SF3a60 and SF3a66 required for binding the partially assembled 15 S U2 snRNP are dispensable for nuclear targeting (14, 21). The proposal that SF3a enters the nucleus in form of the heterotrimer is supported by observations that the SF3a120 IDs of the small subunits are necessary for nuclear import (Figs. 3 and 4) (21). Moreover, the finding that SF3a120 mutants lacking binding sites for SF3a60 or SF3a66 are nuclear (Fig. 5) indicates that SF3a120 provides a signal for SF3a nuclear import. A pat7 motif in SF3a120 overlapping a classical monopartite NLS (25) was identified by the PSORTII program and deletion of this motif inhibited SF3a120 nuclear import (Fig. 6). However, the pat7 motif is not sufficient for import of the cytoplasmic reporter protein PyrK. Instead, an extended sequence including a cluster of basic residues (RRNK) 12 amino acids C-terminal of the pat7 motif is essential for PyrK nuclear targeting. Two basic segments separated by a linker of up to 29 residues are characteristic for bipartite NLS (25). However, as we have not tested the contribution of the potential linker sequences to nuclear import, it is unclear whether the identified NLS is bipartite or represents a more complex signal that also requires sequences between the pat7 and RRNK motifs.
Surprisingly, deletion of the RRNK motif from SF3a120 had only a minor effect on nuclear localization (Fig. 6). This result is most likely explained by the presence of a pat7-type NLS in SF3a60 (Fig. 7), suggesting that the SF3a60-SF3a120 interaction can compensate for a slight deficiency in nuclear import of SF3a120 in the absence of the RRNK motif. This notion is further supported by the observation that SF3a120/1–555 or proteins with further C-terminal deletions were not completely excluded from the nucleus (Fig. 5).
In summary, the data presented here shed further light on the interplay of the SF3a subunits to assemble the SF3a heterotrimer and target it to the nucleus (Fig. 8). Our results confirm that the SF3a subunits interact in the cytoplasm by binding of SF3a60 and SF3a66 to distinct, but neighboring domains in SF3a120. The major signal for nuclear targeting resides in SF3a120 and nuclear import is further supported by a NLS in SF3a60. Once in the nucleus, the zinc finger domains of the small subunits are required for assembly of the functional 17 S U2 snRNP in Cajal bodies (14, 15). Whether SF3a120 contributes to this process is not known at present. In addition, further experiments are required to define domains in the SF3a subunits that contact the pre-mRNA upstream of the branch site (8).
Acknowledgments
We thank past and present group members for helpful discussions and comments on the manuscript and Drs. Dobrila Nesic and Cornelia Wersig for initial experiments with SF3a120 mutants. We are grateful to Dr. Karla Neugebauer for a generous gift of anti-GFP antibodies.
This work was supported by grants from the Swiss National Science Foundation (3100-068239), the European Science Foundation EUROCORES Programme EuroDYNA (ERAS-CT-2003-980409), the European Commission (EURASNET-LSHG-CT-2005-518238), the Fondation MEDIC, and the Canton of Geneva (to A. K.).
Please note that “SF” has been omitted from the names of all plasmids.
Note that bound material was lost from the sample containing transfected GFP-3a120-FL. However, the fact that SF3a66 was efficiently co-immunoprecipitated with GFP-3a120-ΔSURP1 and GFP-3a120-ΔSURP2 demonstrates that endogenous SF3a66 can interact with the N-terminal region of GFP-3a120.
F. Ferfolgia and A. Krämer, unpublished observations.
- snRNP
- small nuclear ribonucleoprotein particle
- snRNA
- small nuclear RNA
- SURP
- suppressor-of-white-apricot and prp21/spp91
- FL
- full-length
- UBL
- ubiquitin-like domain
- NLS
- nuclear localization signal(s)
- PyrK
- pyruvate kinase
- ID
- interaction domain
- pat7
- pattern 7.
REFERENCES
- 1. Wahl M. C., Will C. L., Lührmann R. (2009) Cell 136, 701–718 [DOI] [PubMed] [Google Scholar]
- 2. Will C. L., Lührmann R. (2006) in The RNA World (Gesteland R., Cech T., Atkins J. eds.) 3rd Ed., pp. 369–400, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 3. Handwerger K. E., Gall J. G. (2006) Trends Cell Biol. 16, 19–26 [DOI] [PubMed] [Google Scholar]
- 4. Matera A. G., Shpargel K. B. (2006) Curr. Opin. Cell Biol. 18, 317–324 [DOI] [PubMed] [Google Scholar]
- 5. Krämer A., Ferfoglia F., Huang C. J., Mulhaupt F., Nesic D., Tanackovic G. (2005) Biochem. Soc. Trans. 33, 439–442 [DOI] [PubMed] [Google Scholar]
- 6. Brosi R., Gröning K., Behrens S. E., Lührmann R., Krämer A. (1993) Science 262, 102–105 [DOI] [PubMed] [Google Scholar]
- 7. Behrens S. E., Galisson F., Legrain P., Lührmann R. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8229–8233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gozani O., Feld R., Reed R. (1996) Genes Dev. 10, 233–243 [DOI] [PubMed] [Google Scholar]
- 9. Gozani O., Potashkin J., Reed R. (1998) Mol. Cell. Biol. 18, 4752–4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Will C. L., Schneider C., MacMillan A. M., Katopodis N. F., Neubauer G., Wilm M., Lührmann R., Query C. C. (2001) EMBO J. 20, 4536–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bessonov S., Anokhina M., Will C. L., Urlaub H., Lührmann R. (2008) Nature 452, 846–850 [DOI] [PubMed] [Google Scholar]
- 12. Brosi R., Hauri H. P., Krämer A. (1993) J. Biol. Chem. 268, 17640–17646 [PubMed] [Google Scholar]
- 13. Legrain P., Chapon C. (1993) Science 262, 108–110 [DOI] [PubMed] [Google Scholar]
- 14. Nesic D., Krämer A. (2001) Mol. Cell. Biol. 21, 6406–6417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanackovic G., Krämer A. (2005) Molecular biology of the cell 16, 1366–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krämer A., Legrain P., Mulhauser F., Gröning K., Brosi R., Bilbe G. (1994) Nucleic Acids Res. 22, 5223–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bennett M., Reed R. (1993) Science 262, 105–108 [DOI] [PubMed] [Google Scholar]
- 18. Krämer A., Mulhauser F., Wersig C., Gröning K., Bilbe G. (1995) RNA 1, 260–272 [PMC free article] [PubMed] [Google Scholar]
- 19. Chiara M. D., Champion-Arnaud P., Buvoli M., Nadal-Ginard B., Reed R. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 6403–6407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuwasako K., He F., Inoue M., Tanaka A., Sugano S., Güntert P., Muto Y., Yokoyama S. (2006) Structure 14, 1677–1689 [DOI] [PubMed] [Google Scholar]
- 21. Nesic D., Tanackovic G., Krämer A. (2004) J. Cell Sci. 117, 4423–4433 [DOI] [PubMed] [Google Scholar]
- 22. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Görlich D., Kutay U. (1999) Annu. Rev. Cell Dev. Biol. 15, 607–660 [DOI] [PubMed] [Google Scholar]
- 24. Nakai K., Horton P. (1999) Trends Biochem. Sci. 24, 34–36 [DOI] [PubMed] [Google Scholar]
- 25. Xu D., Farmer A., Chook Y. M. (2010) Curr. Opin. Struct. Biol. 20, 782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spector D. L., Fu X. D., Maniatis T. (1991) EMBO J. 10, 3467–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalderon D., Roberts B. L., Richardson W. D., Smith A. E. (1984) Cell 39, 499–509 [DOI] [PubMed] [Google Scholar]
- 28. Rain J. C., Tartakoff A. M., Krämer A., Legrain P. (1996) RNA 2, 535–550 [PMC free article] [PubMed] [Google Scholar]
- 29. Schappert K., Friesen J. D. (1991) Mol. Gen. Genet. 226, 277–282 [DOI] [PubMed] [Google Scholar]
- 30. Flicek P., Amode M. R., Barrell D., Beal K., Brent S., Chen Y., Clapham P., Coates G., Fairley S., Fitzgerald S., Gordon L., Hendrix M., Hourlier T., Johnson N., Kähäri A., Keefe D., Keenan S., Kinsella R., Kokocinski F., Kulesha E., Larsson P., Longden I., McLaren W., Overduin B., Pritchard B., Riat H. S., Rios D., Ritchie G. R., Ruffier M., Schuster M., Sobral D., Spudich G., Tang Y. A., Trevanion S., Vandrovcova J., Vilella A. J., White S., Wilder S. P., Zadissa A., Zamora J., Aken B. L., Birney E., Cunningham F., Dunham I., Durbin R., Fernández-Suarez X. M., Herrero J., Hubbard T. J., Parker A., Proctor G., Vogel J., Searle S. M. (2011) Nucleic Acids Res. 39, D800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Will C. L., Lührmann R. (2001) Curr. Opin. Cell Biol. 13, 290–301 [DOI] [PubMed] [Google Scholar]