

Abstract
Many stochastic simulation approaches for generating observations from a posterior distribution depend on knowing a likelihood function. However, for many complex probability models, such likelihoods are either impossible or computationally prohibitive to obtain. Here we present a Markov chain Monte Carlo method for generating observations from a posterior distribution without the use of likelihoods. It can also be used in frequentist applications, in particular for maximum-likelihood estimation. The approach is illustrated by an example of ancestral inference in population genetics. A number of open problems are highlighted in the discussion.
One of the basic problems in Bayesian statistics is the computation of posterior distributions. We imagine data 𝒟 generated from a model ℳ determined by parameters θ, the prior density of which is denoted by π(θ). We assume unless otherwise stated that the data are discrete. The posterior distribution of interest is 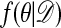 , which is given by
, which is given by
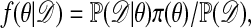 |
[1] |
where 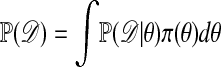 is the normalizing constant.
is the normalizing constant.
In most scientific contexts, explicit formulae for such posterior densities are few and far between, and we usually resort to stochastic simulation to generate observations from f. Perhaps the simplest approach for this is the rejection method:
A1. Generate θ from π(·).
A2. Accept θ with probability
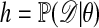 ; return to A1.
; return to A1.
Accepted observations have distribution 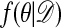 (cf. ref. 1). The computations can often be accelerated if an upper bound c for
(cf. ref. 1). The computations can often be accelerated if an upper bound c for 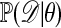 is known; h then is replaced by h/c. If
is known; h then is replaced by h/c. If 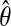 denotes the maximum-likelihood estimator of θ, we could take
denotes the maximum-likelihood estimator of θ, we could take 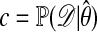 .
.
There are many variations on this theme. Of particular relevance here is the case in which the likelihood 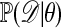 cannot be computed explicitly. One obvious approach then is:
cannot be computed explicitly. One obvious approach then is:
B1. Generate θ from π(·).
B2. Simulate
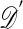 from the model ℳ with parameter θ.
from the model ℳ with parameter θ.B3. Accept θ if
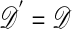 ; return to B1.
; return to B1.
The success of this approach depends on the fact that the underlying stochastic model ℳ is easy to simulate. This approach can be useful when computation of the likelihood is possible but time-consuming.
The practicality of algorithms such as these depends crucially on the size of 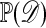 , because the probability of accepting an observation is proportional to
, because the probability of accepting an observation is proportional to 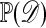 . In cases where the acceptance rate is too small, one might resort to approximate methods such as:
. In cases where the acceptance rate is too small, one might resort to approximate methods such as:
C1. Generate θ from π(·).
C2. Simulate
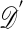 from the model ℳ with parameter θ.
from the model ℳ with parameter θ.C3. Calculate the distance
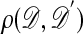 between
between 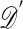 and 𝒟.
and 𝒟.C4. Accept θ if ρ ≤ ε; return to C1.
This approach requires selection of a suitable metric ρ as well as a choice of ε. As ε → ∞ it generates observations from the prior. If ε = 0, an observation 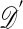 is accepted only if
is accepted only if 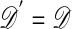 , and then accepted observations come from the density
, and then accepted observations come from the density 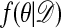 . The choice of ε therefore reflects a tension between computability and accuracy. The method is still honest in that, for a given ρ and ε, we are generating independent and identically distributed observations from
. The choice of ε therefore reflects a tension between computability and accuracy. The method is still honest in that, for a given ρ and ε, we are generating independent and identically distributed observations from  .
.
When 𝒟 is high-dimensional or continuous, this approach can be impractical as well, and then the comparison of 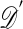 with 𝒟 can be made by using lower-dimensional summaries of the data. The motivation for this approach is that if the set of statistics S = (S1,..., Sp) is sufficient for θ, in that
with 𝒟 can be made by using lower-dimensional summaries of the data. The motivation for this approach is that if the set of statistics S = (S1,..., Sp) is sufficient for θ, in that 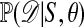 is independent of θ, then
is independent of θ, then 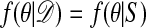 . The normalizing constant
. The normalizing constant 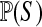 is typically larger than
is typically larger than 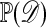 , resulting in more acceptances. In practice it will be hard, if not impossible, to identity a suitable set of sufficient statistics, and we then might resort to a more heuristic approach. Thus we seek to use knowledge of the particular problem at hand to suggest summary statistics that capture information about θ. With these statistics in hand, we have the following approximate Bayesian computation scheme for data 𝒟 summarized by S:
, resulting in more acceptances. In practice it will be hard, if not impossible, to identity a suitable set of sufficient statistics, and we then might resort to a more heuristic approach. Thus we seek to use knowledge of the particular problem at hand to suggest summary statistics that capture information about θ. With these statistics in hand, we have the following approximate Bayesian computation scheme for data 𝒟 summarized by S:
D1. Generate θ from π(·).
D2. Simulate
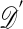 from stochastic model ℳ with parameter θ, and compute the corresponding statistics S′.
from stochastic model ℳ with parameter θ, and compute the corresponding statistics S′.D3. Calculate the distance ρ(S, S′) between S and S′.
D4. Accept θ if ρ ≤ ε, and return to D1.
There are several advantages to these rejection methods, among them the fact that they are usually easy to code, they generate independent observations (and thus can use embarrassingly parallel computation), and they readily provide estimates of Bayes factors that can be used for model comparison. On the other hand, sampling from the prior in complex probability models is unlikely to be sensible when the posterior is a long way from the prior. Later we discuss Markov chain Monte Carlo (MCMC) algorithms and provide an alternative MCMC approach that does not require the evaluation of likelihoods.
Examples from Evolutionary Biology
Examples of these algorithms have appeared in the evolutionary genetics literature. For example, inference problems in molecular population genetics can be described as follows. We sample the molecular variation present at several loci in a population, obtaining a discrete variation data set 𝒟 (DNA sequence data, for example). Inference and estimation for population parameters of interest such as mutation rates, recombination rates, migration rates, and demographic parameters are then based on a stochastic model ℳ for 𝒟.
The coalescent (2) provides a commonly used modeling framework in this setting. The coalescent is a stochastic model for the ancestral relationships between the sampled sequences. In the absence of recombination, these ancestral relationships form a binary branching tree. Because the tree is not observed, inference for parameters of interest can be thought of as a missing data problem (for reviews see, for example, refs. 3 and 4).
Examples of algorithm A are given by Tavaré et al. (5), of algorithm C by Plagnol and Tavaré (6), and of algorithm D by Fu and Li (7), Weiss and von Haeseler (8) and Pritchard et al. (9), among others. Beaumont et al. (10) describe an interesting generalization of the rejection method in which all observations (θ, S′) generated by the first two steps of algorithm D are used in a local-linear regression framework to generate observations that follow more closely the required distribution 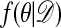 . This reference also contains a number of other examples of these approaches.
. This reference also contains a number of other examples of these approaches.
MCMC Methods
We begin by recalling the Metropolis–Hastings algorithm (11, 12) for generating observations from 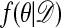 using output from a Markov chain.
using output from a Markov chain.
E1. If now at θ, propose a move to θ′ according to a transition kernel q(θ → θ′).
- E2. Calculate
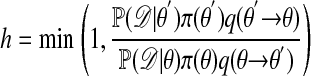
[2] E3. Move to θ′ with probability h, else remain at θ; go to E1.
Under suitable regularity conditions, f is the stationary and limiting distribution of the chain. The practical complexities of implementing MCMC are described by Gilks et al. (13) for example. In concert with dramatically increased computing power, this approach has revolutionized Bayesian statistics over the last 15 years (see, for example, refs. 14 and 15).
One comparison that can be made between algorithms A and E is the way in which they use the likelihood 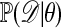 . In the rejection method, the comparison is with
. In the rejection method, the comparison is with 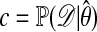 (a global comparison), whereas in the Metropolis–Hastings algorithm
(a global comparison), whereas in the Metropolis–Hastings algorithm 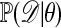 is compared to
is compared to  (a local comparison). One therefore expects that MCMC approaches accept observations more frequently, but the price paid for higher acceptance rates is dependent outcomes.
(a local comparison). One therefore expects that MCMC approaches accept observations more frequently, but the price paid for higher acceptance rates is dependent outcomes.
Approximating the Likelihood Ratio. The theme of this note is simulation of observations from a posterior when likelihoods are either hard or impossible to calculate. The first such approach is to approximate the likelihood ratio 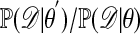 appearing in the acceptance probability in E3. This can be done by estimating each term in the ratio separately. For a given value of θ, estimate
appearing in the acceptance probability in E3. This can be done by estimating each term in the ratio separately. For a given value of θ, estimate 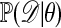 by simulation of B data sets
by simulation of B data sets 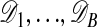 from the model ℳ with parameter θ, and form the point estimate
from the model ℳ with parameter θ, and form the point estimate
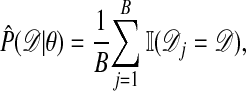 |
where 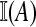 is 1 if A is true and 0 otherwise. More sophisticated estimates might also be used depending on the details of the specific application. For example, an estimate of
is 1 if A is true and 0 otherwise. More sophisticated estimates might also be used depending on the details of the specific application. For example, an estimate of 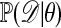 might be precomputed and stored over a grid of θ values.
might be precomputed and stored over a grid of θ values.
This method also applies when the underlying data are continuous, in which case the likelihood ratio is a ratio of densities. In this case the B simulated observations can be used in a kernel density-estimation routine, and the density at the point 𝒟 is returned. This approach can also be made dynamic, in that B need not be fixed ahead of time. See Diggle and Gratton (16) and the references contained therein for applications of this approach in frequentist settings. Of course, the same methods can be applied for the approaches described in C and D above. An example appears later.
MCMC Without Likelihoods. In this section we describe an MCMC approach that is the natural analog of algorithm B in that no likelihoods are used or estimated in its implementation. It is based on the following steps:
F1. If now at θ propose a move to θ′ according to a transition kernel q(θ → θ′).
F2. Generate
 using model ℳ with parameters θ′.
using model ℳ with parameters θ′.F3. If
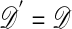 , go to F4, and otherwise stay at θ and return to F1.
, go to F4, and otherwise stay at θ and return to F1.- F4. Calculate
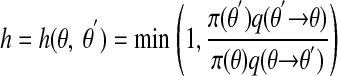
F5. Accept θ′ with probability h and otherwise stay at θ, then return to F1.
The stationary distribution of the chain is indeed 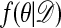 , as is demonstrated below.
, as is demonstrated below.
Theorem. 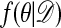 is the stationary distribution of the chain.
is the stationary distribution of the chain.
Proof: Denote the transition mechanism of the chain by r(θ → θ′), and (without loss of generality) choose θ′ ≠ θ satisfying
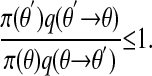 |
[3] |
Then
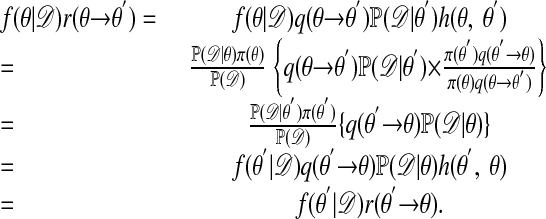 |
The argument when the ratio on the left of Eq. 3 is >1 is analogous. Thus 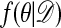 satisfies the detailed balance equations, which implies that indeed
satisfies the detailed balance equations, which implies that indeed 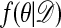 is the stationary distribution of the chain, and the proof is complete.
is the stationary distribution of the chain, and the proof is complete.
Assuming that the chain is ergodic (which occurs under the same conditions that make the chain in algorithm E ergodic), we can now simulate observations having approximately the distribution 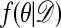 . We also mention two special cases:
. We also mention two special cases:
If q(θ′ → θ) = q(θ → θ′) then h depends only on the prior.
If q is reversible with respect to π [so that π(θ)q(θ → θ′) = π(θ′)q(θ′ → θ) for all θ ≠ θ′], then h = 1 and the algorithm reduces to a rejection method with correlated outputs.
For the reasons discussed earlier this approach also may be impractical, in which case we can resort to the equivalent of algorithms C and D by replacing step F3 above with:
F3′. If
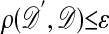 ≤ ε, go to F4, and otherwise stay at θ and return to F1, in which case the stationary distribution is
≤ ε, go to F4, and otherwise stay at θ and return to F1, in which case the stationary distribution is 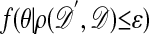 , or
, orF3″. If ρ(S′, S) ≤ ε, goto F4, and otherwise stay at θ and return to F1, in which case the stationary distribution is f(θ|ρ(S′, S) ≤ ε). These methods can also be used when 𝒟 is continuous.
An Example from Population Genetics
To illustrate these ideas, we use an example of ancestral inference from population genetics. The data are a sample of n = 63 Nuu Chah Nulth mtDNA sequences obtained by Ward et al. (17). These sequences, of 360 bp in length, come from hypervariable region I of the mitochondrial control region. The observed base frequencies in the sequences are (πA, πG, πC, πT) = (0.330, 0.112, 0.337, 0.221), there are H = 28 distinct sequences, and V = 26 base positions showed variation in the sample.
These data have been discussed in the coalescent framework by Markovtsova et al. (18) and Markovtsova et al. (19). The posterior distribution of the (rescaled) mutation parameter θ and the height ℐ of the coalescent tree of the sample [i.e., the time to the most recent common ancestor (MRCA) of the sample] were found by MCMC methods using the full sequence data; we use these results to calibrate those of the likelihood-free approach. Further details of the coalescent model and the mutation model and its parameters may be found there. In particular, we use Felsenstein's mutation model (cf. ref. 20) with a transition-transversion parameter of κ = 100.
Implementing Algorithm F
The simplest form in which we could implement our method would be to generate a new tree topology and set of mutations each time we propose a new mutation rate. However, in this example it is not effective to do so, because this rarely leads to accepted parameter values. Instead we augment the state space to include information about the tree topology and occurrence of mutations on that topology to increase the acceptance rate. See ref. 15, for example, for further information about data augmentation and auxiliary variable approaches. Intuitively speaking, the inclusion of more information within the state space makes it easier to make more local moves in that state space and therefore improve the acceptance rate. (Once the algorithm has found a state that it can accept, it is able to explore small changes to that state that will be more likely a priori to also lead to states with a high acceptance probability.) This leads to a higher acceptance rate, but the tradeoff is that the state space becomes more complex, and therefore it is slower to move within that space.
We implemented the following approach. Our state space includes both the tree topology and the times of coalescence events on that topology. Furthermore, we characterize mutations by the time at which they occur, the branch on which they happen (i.e., the individuals whose genome is modified by this mutation), and their location on the genome. We additionally include the number of mutations occurring between two coalescent events. We did not record their location on the tree, which is chosen uniformly among the branches of the tree when we simulate the data. This was the minimal set of information to include in the state space to lead to a reasonable acceptance rate.
Given that state space, we update as follows: the topology of the tree is updated by using a scheme described by Markovtsova et al. (18). We update times between coalescence events by adding a Gaussian random variable to the existing time. We update the mutation rate by adding a uniform random variable to the old rate. The new mutation rate, as well as the updated times, define a new intensity for the Poisson random variable that determines the number of mutations between each pair of coalescence events. This number was updated by using the following basic properties of a Poisson random variable:
If α < α′ and Poisson (α) and Poisson (α′ – α) are independent Poisson random variables with the indicated means, then their sum is Poisson (α′).
If α > α′ and from a Poisson (α) number of events we keep each with probability α′/α, then the number of kept events is Poisson (α′).
When a new mutation occurs we choose its location on the genome and tree uniformly at random. When the number of mutations decreases, we randomly select the necessary number of mutations and erase them. There are many variations of this scheme. For example, one could also keep track of the genotype of the MRCA or of some information about the mutations (which are transversions, for example). The underlying principle is that the more information included in the state space, the easier it is to simulate the exact data but the harder it is to move effectively around the state space.
Results
Here we compare the rejection, estimated likelihood, and likelihood-free MCMC approaches in two settings: using the summary statistic S = V and using the summary statistic S = (V, H). We also discuss the effects of varying the tolerance ε.
Using the Number of Variable Sites. We begin by summarizing the data by using the number V of variable (or segregating) sites. Data sets are accepted if |V – 26| ≤ ε. In Table 1, three methods are compared in the case ε = 2. As expected, the methods produce comparable results for the height T of the coalescent tree of the sample and the mutation parameter θ. The methods have quite different acceptance rates. In Table 2, the effects of varying the parameter ε are shown for the no-likelihood approach. Under the coalescent prior, the mean height of the coalescent tree is 1.97 units; the posterior means do not differ substantially from this. The posterior for T using the full data 𝒟 can be found by an MCMC approach (cf. table 3, column 2, in ref. 19). The posterior mean of T was estimated to be 0.68. We note the substantial difference between the results using S = V and the “true” result. This suggests that summarizing the data by using only V results in a loss of information. The effects of adding the number of haplotypes to this summary are explored in the next section.
Table 1. Comparison of the three approaches using S = V, ε = 2.
|
|
Rejection*
|
Estimated likelihood†
|
No likelihood‡
|
|---|---|---|---|
| Acceptance rate | 3.0% | 50.6% | 15.1% |
| TMRCA T | |||
| 1st quartile | 1.07 | 1.11 | 1.08 |
| Mean | 1.74 | 1.82 | 1.75 |
| Median | 1.48 | 1.55 | 1.53 |
| 3rd quartile | 2.14 | 2.23 | 2.19 |
| Mutation rate θ | |||
| 1st quartile | 0.015 | 0.014 | 0.015 |
| Mean | 0.019 | 0.019 | 0.019 |
| Median | 0.018 | 0.018 | 0.018 |
| 3rd quartile | 0.023 | 0.022 | 0.022 |
Algorithm D; based on 2,000 observations. Estimated SEM of T = 0.02.
Based on likelihoods estimated from B = 1,000 simulations; 1,000 observations after sampling every 200 steps. Estimated SEM of T = 0.03.
Algorithm F; based on 1,000 observations after sampling every 10,000 steps. Estimated SEM of T = 0.03.
Table 2. Comparison of effects of ε using algorithm F and S = V.
|
|
ε = 2*
|
ε = 1†
|
ε = 0†
|
|---|---|---|---|
| Acceptance rate | 15.1% | 11.1% | 4.8% |
| TMRCA T | |||
| 1st quartile | 1.08 | 1.12 | 1.14 |
| Mean | 1.75 | 1.77 | 1.82 |
| Median | 1.52 | 1.52 | 1.55 |
| 3rd quartile | 2.19 | 2.15 | 2.26 |
| Mutation rate θ | |||
| 1st quartile | 0.015 | 0.015 | 0.015 |
| Mean | 0.019 | 0.019 | 0.019 |
| Median | 0.018 | 0.018 | 0.018 |
| 3rd quartile | 0.022 | 0.022 | 0.022 |
Based on 1,000 observations after sampling every 10,000 steps.
Based on 1,000 observations after sampling every 50,000 steps.
Using the Number of Variable Sites and Haplotypes. We report inference about θ and T using the summary statistic S = (V, H). In this case a simulated data set was kept if
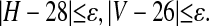 |
Results are given in Table 3 for the case ε = 2. We note that the MCMC method has a substantially higher acceptance rate than the rejection method, although it is still quite low. The estimated-likelihood approach is at the edge of feasibility in this case, but it does have a good acceptance rate. The key feature of these results is that the posterior based on these summary statistics is very close to the full posterior; addition of the summary statistic H has moved the posterior mean from ≈1.75 to 0.69, in comparison with the full posterior mean of 0.68.
Table 3. Comparison of the three approaches using S = (V, H), ε = 2.
|
|
Rejection*
|
Estimated likelihood†
|
No likelihood‡
|
|---|---|---|---|
| Acceptance rate | 0.0008% | 16.9% | 0.2% |
| TMRCA T | |||
| 1st quartile | 0.51 | 0.50 | 0.54 |
| Mean | 0.69 | 0.67 | 0.70 |
| Median | 0.64 | 0.63 | 0.66 |
| 3rd quartile | 0.81 | 0.80 | 0.81 |
| Mutation rate θ | |||
| 1st quartile | 0.024 | 0.025 | 0.024 |
| Mean | 0.029 | 0.031 | 0.029 |
| Median | 0.028 | 0.030 | 0.028 |
| 3rd quartile | 0.033 | 0.035 | 0.033 |
Algorithm D; based on 1,000 observations. Estimated SEM of T = 0.01.
Based on likelihoods estimated from B = 200 simulations; 1,000 observations after sampling every 100 steps. Estimated SEM of T = 0.01.
Algorithm F; based on 1,000 observations after sampling every 50,000 steps. Estimated SEM of T = 0.01.
In Table 4 we present results for the no-likelihood approach for various values of ε. In the cases ε = 1 and 0, the rejection method and the estimated-likelihood approach are not feasible. This example shows that the MCMC method that uses no likelihoods can provide a good approximation to the “right” answer in a case where rejection methods are too time-consuming to use. We sound a note of caution, however: The effects on the posterior of summarizing the data can be unexpected. See ref. 10 for further examples in the coalescent context.
Table 4. Comparison of effects of ε using algorithm F and S = (V, H).
|
|
ε = 2*
|
ε = 1*
|
ε = 0†
|
|---|---|---|---|
| Acceptance rate | 0.2% | 0.04% | 0.005% |
| TMRCA T | |||
| 1st quartile | 0.54 | 0.49 | 0.46 |
| Mean | 0.70 | 0.64 | 0.59 |
| Median | 0.66 | 0.60 | 0.55 |
| 3rd quartile | 0.81 | 0.74 | 0.69 |
| Mutation rate θ | |||
| 1st quartile | 0.024 | 0.025 | 0.026 |
| Mean | 0.029 | 0.030 | 0.030 |
| Median | 0.028 | 0.030 | 0.031 |
| 3rd quartile | 0.033 | 0.035 | 0.034 |
Based on 1,000 observations after sampling every 50,000 steps.
Based on 1,000 observations after sampling every 200,000 steps.
To illustrate how the likelihood-free MCMC approach works, we compared the approximate Bayesian computation results with the true result obtained for the full data. Typically this will not be possible; the point is to use this approach when there are no feasible alternatives. Further research is required to identify good methods for combining summary statistics to obtain better estimates of the posterior.
Discussion
We have described a number of approaches for simulating observations from posterior distributions when likelihoods are hard or impossible to compute. Problems such as this arise frequently in scientific applications, where it is often the case that a probability model for the data can be simulated rapidly but is sufficiently complicated that explicit formulae for the appropriate probability distributions are intractable. In particular, we provided an MCMC approach that does not require the use of likelihood ratios in its implementation. The development of more sophisticated MCMC methods that do not use likelihoods is clearly of practical importance.
In practice, these methods might not work well for complex data, and it is often useful to replace the full data by a number of judiciously chosen summary statistics. The resulting approximate Bayesian computation allows us to explore scenarios that are intractable if the full data are used. Motivated by considerations of sufficiency, the choice of summary statistics is crucial. There is scope for research on practical methods for identifying approximately sufficient statistics (cf. refs. 21 and 22), and for assessing the adequacy of the approximate posterior distributions.
Acknowledgments
We thank Duncan Thomas for helpful discussions and the referees for their comments. S.T. thanks the Statistical and Applied Mathematical Sciences Institute for its hospitality during the preparation of this article. This work was supported by National Institutes of Health Grant GM58897.
Abbreviations: MCMC, Markov chain Monte Carlo; MRCA, most recent common ancestor.
References
- 1.Ripley, B. D. (1982) Stochastic Simulation (Wiley, New York).
- 2.Kingman, J. F. C. (1982) J. Appl. Prob. 19A, 27–43. [Google Scholar]
- 3.Nordborg, M. (2001) in Handbook of Statistical Genetics, eds. Balding, D. J., Bishop, M. J. & Cannings, C. (Wiley, New York), pp. 179–208.
- 4.Stephens, M. (2001) in Handbook of Statistical Genetics, eds. Balding, D. J., Bishop, M. J. & Cannings, C. (Wiley, New York), pp. 213–238.
- 5.Tavaré, S., Balding, D. J., Griffiths, R. C. & Donnelly, P. (1997) Genetics 145, 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plagnol, V. & Tavaré, S. (2004) in Proceedings of the 5th International Conference on Monte Carlo and Quasi-Monte Carlo Methods in Scientific Computing, ed. Niederreiter, H. (Springer, Heidelberg).
- 7.Fu, Y.-X. & Li, W.-H. (1997) Mol. Biol. Evol. 14, 195–199. [DOI] [PubMed] [Google Scholar]
- 8.Weiss, G. & von Haeseler, A. (1998) Genetics 149, 1539–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pritchard, J. K., Seielstad, M. T., Perez-Lezaun, A. & Feldman, M. W. (1999) Mol. Biol. Evol. 16, 1791–1798. [DOI] [PubMed] [Google Scholar]
- 10.Beaumont, M. A., Zhang, W. & Balding, D. J. (2002) Genetics 162, 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H. & Teller, E. (1953) J. Chem. Phys. 21, 1087–1092. [Google Scholar]
- 12.Hastings, W. K. (1970) Biometrika 57, 97–109. [Google Scholar]
- 13.Gilks, W. R., Richardson, S. & Spiegelhalter, D. J. (1996) Markov Chain Monte Carlo in Practice (Chapman and Hall/CRC, Boca Raton, FL).
- 14.Gelman, A., Carlin, J. B., Stern, H. S. & Rubin, D. B. (1995) Bayesian Data Analysis (Chapman and Hall/CRC, Boca Raton, FL).
- 15.Carlin, B. P. & Louis, T. A. (2000) Bayes and Empirical Bayes Methods for Data Analysis (Chapman and Hall/CRC, Boca Raton, FL), 2nd Ed.
- 16.Diggle, P. J. & Gratton, R. J. (1984) J. R. Stat. Soc. B 46, 193–227. [Google Scholar]
- 17.Ward, R. H., Frazier, B. L., Dew, K. & Pääbo, S. (1991) Proc. Natl. Acad. Sci. USA 88, 8720–8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markovtsova, L., Marjoram, P. & Tavaré, S. (2000) Genetics 156, 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markovtsova, L., Marjoram, P. & Tavaré, S. (2000) Genetics 156, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thorne, P. H., Kishino, H. & Felsenstein, J. (1992) J. Mol. Evol. 34, 3–16. [DOI] [PubMed] [Google Scholar]
- 21.Le Cam, L. (1964) Ann. Math. Stat. 35, 1419–1455. [Google Scholar]
- 22.Cabrera, J. & Yohai, V. J. (1999) A New Computational Approach for Bayesian and Robust Bayesian Statistical Analysis, www.rci.rutgers.edu/~cabrera/pap/vic.pdf, preprint.