

Abstract
Phosphatidic acid (PA) and phytosphingosine-1-phosphate (phyto-S1P) have both been identified as lipid messengers mediating plant response to abscisic acid (ABA). To determine the relationship of these messengers, we investigated the direct interaction of PA with Arabidopsis sphingosine kinases (SPHKs) that phosphorylate phytosphingosine to generate phyto-S1P. Two unique SPHK cDNAs were cloned from the annotated At4g21540 locus of Arabidopsis, and the two transcripts are differentially expressed in Arabidopsis tissues. Both SPHKs are catalytically active, phosphorylating various long-chain sphingoid bases (LCBs) and are associated with the tonoplast. They both interact with PA as demonstrated by lipid-filter binding, liposome binding, and surface plasmon resonance (SPR). SPHK1 and SPHK2 exhibited strong binding to 18:1/18:1, 16:0/18:1, and 16:0/18:2 PA, but poor binding to 16:0/16:0, 8:0/8:0, 18:0/18:0, and 18:2/18:2 PA. Surface dilution kinetics analysis indicates that PA stimulates SPHK activity by increasing the specificity constant through decreasing KmB. The results show that the annotated At4g21540 locus is actually comprised of two separate SPHK genes. PA binds to both SPHKs, and the interaction promotes lipid substrate binding to the catalytic site of the enzyme. The PA-SPHK interaction depends on the PA molecular species. The data suggest that these two Arabidopsis SPHKs are molecular targets of PA, and the PA stimulation of SPHK is part of the signaling networks in Arabidopsis.
Keywords: Arabidopsis, Hormones, Kinetics, Phosphatidic Acid, Phospholipase D, Abscisic Acid, Lipid Signaling, Phosphatidic Acid, Phospholipase D, Phytosphingosine Kinase
Introduction
Phosphatidic acid (PA)4 has emerged as a class of pivotal lipid messengers in cell growth, development, and stress responses, and the regulatory functions of PA are being established in plants, animals, and fungi (1–3). PA is a minor membrane lipid, constituting less than 1% of total phospholipids in most plant tissues (4). However, the cellular level of PA in plants is dynamic, increasing rapidly under various conditions, including chilling, freezing, wounding, pathogen elicitation, dehydration, salt, nutrient starvation, nodule induction, and oxidative stress (1, 2, 5, 6). The functional significance of PA has been indicated by characterization of various phospholipase Ds (PLD) that produce regulatory PA and by measurements of PA changes under different stress conditions (1, 2). Characterization of genetic ablations, together with biochemical analyses, has shown that different PLDs have unique functions (1, 7). The differential activation, expression, and cellular locales, as well as substrate preferences of PLDs, indicate that the cellular location and timing of PA production are important determinants of PA function.
A series of recent results have provided mechanistic insights into how specific PLD and PA mediate the abscisic acid (ABA) promotion of stomatal closure in Arabidopsis (8, 9). Recently, PLDα1 and PA were found to regulate NADPH oxidase activity and the production of reactive oxygen species (ROS) in ABA-mediated stomatal closure (10). In addition to PA, another lipid messenger, long-chain base-1-phosphate (LCBP) including sphingosine-1-phosphate (S1P) and phyto-S1P has been found to promote the ABA effect on stomatal closure (11–13). Arabidopsis sphingosine kinase (SPHK) activity was mainly associated with the membrane fraction (13). Recent study suggests that sphingosine and S1P are not detectable in Arabidopsis leaves due to the lack of expression of sphingolipid Δ4-desaturase, indicating that sphingosine and S1P are unlikely to play a significant role in ABA-mediated stomatal closure (14). However, knock-out of Arabidopsis SPHK1 rendered the stomatal closure less sensitive to ABA, whereas overexpression of SPHK1 increased stomatal closure and ABA sensitivity (15). These results suggest that other LCBPs are involved in ABA signaling in Arabidopsis (16). Phytosphingosine is one of such LCBs in Arabidopsis leaves and its phosphorylated form, phyto-S1P, is also detectable in Arabidopsis leaves (17). Thus, ABA promotes the formation of PA and phyto-S1P, and both PLD/PA and SPHK/phyto-S1P positively regulate ABA-mediated stomatal closure (8, 13). However, the relationship between PA and phyto-S1P in plant signaling pathways is unknown.
One important mode of action by PA to regulate cell function is through its direct interaction with effector proteins (1). PA has been reported to bind to various proteins, including transcriptional factors, protein kinases, lipid kinases, protein phosphatases, and proteins involved in vesicular trafficking and cytoskeletal rearrangement (1). Several PA-interacting proteins have been identified in plants, including ABI1, PDK1, CTR1, TGD2, and NADPH oxidase (8, 10, 18, 19, 20). Additional PA-binding proteins were isolated by PA-affinity chromatography followed by mass spectrometric analysis (21). In animals, both SPHK and its product S1P are potentially important signaling molecules. Acidic phospholipids including PA have been suggested to stimulate SPHK activity (22). PLD activation up-regulated SPHK in mammalian cells (23). The PLD activator, PKC, was found to activate SPHK1 (24). PA has also been suggested to promote the intracellular translocation of cytosolic murine SPHK1 to membranes enriched in PA (25). These results suggest that SPHK is an effector protein of PA in animal cells.
To determine the relationship of the lipid messengers PA and phyto-S1P in regulating plant functions, we investigated the direct interaction of PA with Arabidopsis SPHK1 (At4g21540) that phosphorylates phytosphingosine to generate phyto-S1P. During the study, we found that the annotated At4g21540 locus of Arabidopsis actually encodes two SPHKs and both SPHKs are associated with the vacuolar membrane. PA binds to both Arabidopsis SPHKs and the interaction stimulates their activity by promoting the binding of lipid substrate to the catalytic site of the enzyme.
EXPERIMENTAL PROCEDURES
Cloning the SPHK1 and SHPK2 cDNAs
The At4g21540 locus contains a tandem repeat, and the second repeat sequence was previously cloned and named as SPHK1 (15). The coding region of SPHK1 was amplified from a stock DNA for At4g21540 obtained from ABRC (Stock U16738) using specific primers AtSPHK1-F 5′-TAGGATCCATGGATCGTCAGCCGGAGAGGGA-3′ and AtSPHK1-R 5′-TACTCGAGTTATTCAGGAGAGAAGAGAGTGGC-3′ with engineered BamHI and XhoI (underlined) sites, respectively. The cDNA of the first repeat (SPHK2) of At4g21540 was amplified from Arabidopsis leaf cDNA using the primers: 5′-ATGGAGAATGATCAATTCATGTGTC-3′ (forward) and 5′-AGCAAGATGGAGGGAGACGAGT-3′ (reverse). The cloned fragments were sequenced and a stop codon was found at the 3′-end. Then the following primers were designed to clone the coding region of SPHK2: AtSPHK2-F 5′-GCGGGATCCATGGAGAATGATCAATTCATGTGTC-3′ and AtSPHK2-R 5′-GCGCTCGAGTCAATATTCAGGAGAGAAGAGTG-3′ with engineered BamHI and XhoI (underlined) sites, respectively. Phusion High-Fidelity DNA Polymerase (New England BioLabs) was used for PCR under the condition of 98 °C 1min, 40 cycles of 98 °C 10 s, 60 °C 20 s, and 72 °C 30 s.
Expression and Purification of SPHKs
The cDNA of SPHK1 and SPHK2 were amplified using the primers described above and ligated to pET-28a-c(+) vector to produce SPHK1 and SPHK2 with 6 histidine residues at the N terminus. The recombinant plasmids were transformed into Escherichia coli BL21(DE3)pLysS. Expression of SPHKs was induced by 0.4 mm isopropyl-1- thio-β-d-galactopyranoside at room temperature for 8 h. Cells were harvested by centrifugation at 2,000 × g at 20 °C for 10 min. SPHKs were purified using Ni-NTA-agarose (Qiagen) according to the manufacturer's instructions with modifications. The cells (harvested from 200 ml cell culture) were resupended in 10 ml of lysis buffer and lysed by sonication in lysis buffer containing 1 mm phenylmethanesulfonyl fluoride (PMSF). The lysate was centrifuged at 12,000 × g at 4 °C for 20 min and the supernatant was incubated with Ni-NTA-agarose for 2 h at 4 °C with gentle rotation. The agarose beads were pelleted and washed three times with a wash buffer. Protein was eluted with an elution buffer and dialyzed with TBS buffer overnight. The dialyzed protein was centrifuged at 12,000 × g for 20 min, and protein concentration was determined using the Bradford protein assay. Purified proteins were analyzed by 10% SDS-PAGE, followed by Coomassie Blue staining. The prepared proteins for activity assay were kept in 50% glycerol at −80 °C.
RNA Extraction and Real-time PCR
Arabidopsis thaliana (ecotype Col-0) plants were grown in a growth chamber with cool white light of 200 μmol m−2·s−1 under 12-h light/12-h dark and 23 °C/19 °C cycles. Total RNA was isolated from tissues of 8-week-old Arabidopsis plants using RNeasy Plant Mini kit (Qiagen) according to the manufacturer's instruction. Total RNA was digested with RNase-free DNase I. The absence of genomic DNA contamination was confirmed by PCR using RNA as template without reverse transcription. The first-strand cDNA was synthesized from 1 μg of total RNA using an iScript cDNA synthesis kit in a total volume of 20 μl according to the manufacturer's instructions (Bio-Rad). The efficiency of the cDNA synthesis was assessed by real-time PCR amplification of a control gene encoding UBQ10 (At4g05320). cDNAs were then diluted to yield similar threshold cycle (Ct) values (20) based on the Ct of the UBQ10. The level of individual gene expression was normalized to that of UBQ10 by subtracting the Ct value of UBQ10 from the tested genes. PCR was performed with a MyiQ system (Bio-Rad) using SYBR Green. Each reaction contained 7.5 μl of 2×SYBR Green master mix reagent (Bio-Rad), 3.5 μl diluted cDNA, and 200 nm of each gene-specific primer in a final volume of 15 μl. The primers were as follows: UBQ10, 5′-CACACTCCACTTGGTCTTGCGT-3′ (forward) and 5′-TGGTCTTTCCGGTGAGAGTCTTCA-3′ (reverse); SPHK1, AGACCTTGGTTGAGAAAGGAGGAG-3′ (forward) and 5′-GATGGAACTTATCGGACCAAAGCT-3′ (reverse); SPHK2, 5′-CGGTGGACAGAGTATGGACTCC-3′ (forward) and 5′-GCAGCAGATTCCTCCTGCCT-3′ (reverse). The real-time PCR condition was: 95 °C for 3 min; and 50 cycles of 95 °C for 30 s, 57 °C for 30 s, and 72 °C for 30 s.
Subcellular Localization of SPHKs
SPHK1 and SPHK2 cDNA were cloned into p35S-FAST/eYFP, which was derived from p35S-FAST by introducing eYFP. Agro-infiltration for transient protein expression in tobacco leaves was performed as described by Voinnet et al. (26). The constructs were transformed into C58C1 Agrobacterium tumefaciens strain and grown to stationary phase. Bacterial cells were collected and resuspended in solution containing 10 mm MES (pH 5.7), 10 mm MgCl2, and 150 mg ml−1 acetosyringone. 3-week-old Nicotiana benthamiana leaves were infiltrated with the bacteria solutions through abaxial air spaces. p35S-FAST/eYFP and p35S-FAST/PLDδ:eYFP were transformed as control. The eYFP fluorescence was examined in tobacco leaves using a Zeiss LSM 510 confocal/muti-photon microscope, with a 488 nm excitation mirror and a 505–530 nm and 530–560 nm emission filter to record images.
The above SPHK:eYFP contsructs were transformed into Arabidopsis to obtain transgenic plants. To isolated subcellular fractions, total proteins from leaves of SPHK1 or 2 transgenic Arabidopsis plants were extracted with a chilled buffer containing 50 mm Tris-HCl, pH 7.5, 10 mm KCl, 1 mm EDTA, 0.5 mm PMSF, and 2 mm DTT. Total protein was centrifuged at 10,000 × g for 20 min at 4 °C to remove tissue debris, and the supernatant was centrifuged at 100,000 × g for 45 min at 4 °C. The resulting supernatant and pellet are referred to as the soluble and microsomal fractions. The plasma and intracellular membranes were prepared using an aqueous polymer two-phase system according to the method described by Fan et al. (27). To isolate tonoplasts, protoplasts were prepared from fully expanded leaves of 4–6-week-old Arabidopsis (28). Vacuoles were then purified from protoplasts following the protocol adapted from Jaquinod et al. (29). Marker enzymes for the plasma membrane, intracellular membrane, and tonoplast are ATPase, cytochrome c reductase, and α-mannosidase, respectively (27, 29). The concentration of proteins from different fractions was determined using the Bradford protein assay. Proteins from the different fractions were subjected to 10% SDS-PAGE followed by immunoblotting. SPHK1 was immunoblotted with anti-FLAG antibody and SPHK2 was detected with anti-GFP antibody.
Assaying Sphingosine Kinase Activity
Sphingosine, phytosphingosine, dihydrosphingosine (d-erythro-DHS), dl-threo-dihydrosphingosine (dl-threo-DHS), N,N-dimethylsphingosine (DMS) were purchased from Enzo Life Sciences. 4-Hydroxy-8-sphingenine (t18:1) and 4, 8-sphingadienine (d18:2) were generous gifts from Dr Daniel Lynch (Williams College). SPHK activity was measured as previously described with some modifications (30). Briefly, purified SPHK was incubated at 37 °C in 200 μl of sphingosine kinase buffer in the presence of 50 μm sphingolipid added in micellar form with 0.25% (v/v) Triton X-100, and [γ-32P]ATP (10 μCi, 1 mm) in 10 mm MgCl2. Reactions were stopped by the addition of 800 μl of chloroform/methanol/concentrated HCl (100:200:1; v/v/v). Chloroform (250 μl) and 2 m KCl (250 μl) were then added sequentially to generate a two-phase system. The labeled lipids in the organic phases were separated by TLC with chloroform/acetone/methanol/acetic acid/water (10:4:3:2:1;v/v/v/v) and visualized with a phosphorimager (Molecular Dynamics, Sunnyvale, CA). For quantification, LCBP was scraped and extracted from the TLC plate and quantified by scintillation counter. SPHK activity was expressed as nanomol of LCBP formed per minute and per milligram of protein. Michaelis-Menten plots and enzyme kinetic parameters were analyzed using SigmaPlot Enzyme Kinetics Module.
Lipid-SPHK Binding by Blotting
The filter binding was performed as described with some modifications (31). Lipids (10 μg) were spotted on a nitrocellulose filter, followed by incubation with purified His-tagged SPHK in TBST (0.1% Tween 20) overnight at 4 °C. The filter was washed three times with TBST (0.1% Tween 20). The filter was then incubated with anti-His antibody, followed by incubation with a second antibody conjugated with alkaline phosphatase. SPHK protein that bound to lipids on filters was visualized by staining alkaline phosphatase activity.
Liposome Binding Assay
Liposome binding assay was performed as described (10). PC and PA were mixed in the molar ratio of 2:1 in chloroform with the final concentration of lipids per sample being at 640 nmol. The lipids were dried under nitrogen and rehydrated for 1 h using extrusion buffer containing 250 mm raffinose, 25 mm Tris pH 7.5, and 1 mm DTT. Liposomes were produced using lipid extruder (0.2 μm filters, Avanti Polar Lipids) following the manufacturer's protocol. Liposomes were diluted in three volumes of a binding buffer containing 125 mm KCl, 25 mm Tris, pH 7.5, 1 mm DTT, and 0.5 mm EDTA and centrifuged at 50,000 × g for 15 min. The liposome pellet was resuspended in 1 ml of binding buffer, and 1.2 μg of purified SPHKs was added and incubated for 1 h at room temperature. The His-tagged SPHK protein used in the assay was preclarified by centrifugation at 16,000 × g for 30 min to remove any insoluble protein. Liposomes were harvested by centrifuging at 16,000 × g for 30 min and washed three times in the binding buffer. Liposomes were resuspended in SDS-PAGE sample buffer and were loaded on a SDS-PAGE gel. The proteins were subjected to SDS-PAGE and then transferred on a PVDF membrane, followed by immunoblotting using anti-His antibodies.
Surface Plasmon Resonance Analysis
SPR binding assays were performed using a Biacore 2000 system according to the manufacturer's instructions with some modifications. Liposomes were prepared by mixing PC and PA at a 2:1 molar ratio as described above. Liposomes were resuspended in a running buffer (0.01 m HEPES, 0.15 m NaCl, 50 μm EDTA, pH7.4). The purified His-tagged SPHK1 was dialyzed in the running buffer overnight at 4 °C, and then the protein was centrifuged at 13,000 × g to remove insoluble protein. The protein concentration was measured using the Bradford assay. Biacore Sensor Chip NTA designed to bind His-tagged proteins for interaction analysis was used to immobilize protein. For each experiment, the running buffer containing 500 μm NiCl2 was injected to saturate the NTA chip with nickel. His-tagged SPHK1 protein (2 μm) was immobilized on the sensor chip via Ni2+/NTA chelation. Lipid-SPHK interaction was monitored as di16:0 PA/di18:1 PC or di18:1 PA/di18:1 PC liposomes (100 μm) were injected in sequence over the surface of the sensor chip. The liposome made with di18:1 PC only was used as control. Sensor chip was regenerated by stripping nickel from the surface with a regeneration buffer (0.01 m HEPES, 0.15 m NaCl, 0.35 m EDTA, pH 8.3). During the evaluation, the sensorgrams from the beginning of association to the end of dissociation for each protein-liposome interaction were analyzed and plotted by SigmaPlot 10.0. Kinetic constants including Bmax, association (kon) and dissociation rate (koff) were analyzed using the BIAevaluation Software.
Preparation of Triton X-100/Phytosphingosine Mixed Micelles
Triton X-100/lipid micelles were prepared according to the method described by Qin et al. (32). Phytosphingosine dissolved in ethanol was dried under a stream of nitrogen, and de-ionized water was added to give a final concentration of 1 mm. The phytosphingosine suspension was sonicated on ice until clear. To obtain specific substrate concentrations at the desired mol fraction (MF), the phytosphingosine solution was diluted with Triton X-100 stock solution (40 mm) using the following formula: mol fractionphytosphingosine = [phytosphingosine]/([phytosphingosine]+[Triton X-100 (free)]); [Triton X-100 (free)] = [Triton X-100 (total)] − critical micelle concentration of Triton X-100 (0.24 mm). When the effect of PA mol concentration was to be tested, PA was added at this point, using the following formula: mol fractionPA = [PA]/([phytosphingosine] + [PA] + [Triton X-100 (free)]). The Triton X-100/phytosphingosine mixture was vortexed briefly and let stand at room temperature for half an hour.
RESULTS
At4g21540 Locus Encodes Two SPHK Genes
Four genes showing homology to human and mouse SPHK genes have been annotated in the Arabidopsis genome. At5g23450 encodes a long-chain base kinase, designated as AtLCBK1 (33) whereas At5g51290 was reported to be a ceramide kinase (34). At2g46090 did not have sphingosine phosphorylating activity (15). At4g21540 potentially encodes two SPHKs in tandem but was annotated as one SPHK in database (Fig. 1A). A cDNA from the second repeat was previously reported to encode an active SPHK while the cloning of the first repeat remained unsuccessful (15).
FIGURE 1.

Cloning of two sphingosine kinase genes from Arabidopsis. A, diagram showing genomic structure of At4g21540 locus containing SPHK1 and SPHK2. The black region is SPHK2 and gray region is SPHK1. B, gene structure of SPHK1 and SPHK2, gray boxes are exons, and white boxes indicate introns. The nucleotide length is shown within the box. C, comparison of amino acid sequences of Arabidopsis SPHK1 and SPHK2 with mouse SPHK1. Shaded black represents identical residues, and shaded gray represents conserved residues. The conserved domains (C1-C5) are underlined.
Utilizing primers corresponding to the first and second repeats, corresponding cDNAs were cloned and further verified by DNA sequencing (Fig. 1B). Sequencing of the first repeat revealed a stop codon at the 3′-end that is 788 bp upstream of the start codon of the second repeat SPHK1. Thus, the annotated At4g21540 is actually comprised of two separate SPHK genes (Fig. 1A). Since the second repeat was already named SPHK1(15), we thus designated the first repeat SPHK2. Both genes have 10 exons and 9 introns, and the size of exons from 2 to 9 is the same for two genes. Arabidopsis SPHK1 and SPHK2 share 72.6% identity of amino acid sequences. Like mouse SPHK1, both SPHK1 and SPHK2 have 5 conserved C domains in the deduced amino acid sequence (Fig. 1C).
SPHK1 and SPHK2 Display a Distinguishable Pattern of Expression and Are Associated with the Tonoplast
The expression of SPHK1 and SPHK2 in different Arabidopsis tissues was examined by real-time PCR (Fig. 2A). Both SPHK1 and SPHK2 were detectable in all tissues examined and they had similar levels of expression in flowers, siliques, young leaves, and roots. However, SPHK1 had a much higher expression level in inflorescence, older leaves, and stems than SPHK2 (Fig. 2A). These distinguishable patterns of expression further support the finding that SPHK1 and SPHK2 are encoded by two separate genes.
FIGURE 2.

Gene expression and subcellular localization of SPHKs. A, expression of SPHK1 and SPHK2 in Arabidopsis tissues as determined by real-time PCR normalized to UBQ10. RNA was extracted from different tissues of 8-week-old plants. Values are means ± S.E. (n = 3). B, subcellular localization of SPHK1 and SPHK2, using eYFP and PLDδ:eYFP as control. The green color represents eYFP fluorescence and red color marks chloroplasts as a reference. The constructs were transiently transformed into tobacco leaves by infiltration. C, immunoblotting of SPHK1 and SPHK2 in subcellular fractions of Arabidopsis leaves. 25 μg of protein per lane was loaded for total and soluble proteins, and 8 μg for membrane fractions. WT, wild-type total protein from leaves; Total, total protein from transgenic Arabidopsis leaves; S, soluble fraction; M, microsomal fraction; PM, plasma membrane; IM, intracellular membrane; T, tonoplast. SPHK1 was immunoblotted with anti-FLAG antibody, and SPHK2 was immunoblotted with anti-GFP antibody.
To determine the intracellular location of these enzymes, SPHK1 and SPHK2 were fused with yellow fluorescence protein (eYFP) at the C terminus and transiently expressed in tobacco leaves while eYFP and PLDδ:eYFP were used as control. eYFP alone was detected in the nucleus and cytoplasm as expected, as the eYFP fluorescence surrounded the chloroplast in the cytoplasm (Fig. 2B, panel a). PLDδ was previously documented to be associated with the plasma membrane (35), and the distribution of PLDδ:eYFP associated with the plasma membrane was consistent with the previous results (Fig. 2B, panel b). The subcellular distribution of SPHK1:eYFP and SPHK2:eYFP both were different from that of eYFP or the plasma membrane-associated PLDδ:eYFP. Using chloroplast (red color) as a reference, SPHK1:eYFP and SPHK2:eYFP fluorescence was separated from the plasma membrane by chloroplasts (Fig. 2B, panels c and d), indicating that they are not associated with the plasma membrane. Arabidopsis SPHK1 was previously reported to be localized on tonoplast (36). SPHK1:eYFP and SPHK2:eYFP exhibited the same pattern of localization, suggesting that both are localized on the tonoplast.
To further verify the subcellular association of SPHK1 and SPHK2, we generated transgenic Arabidopsis expressing SPHK1:eYFP and SPHK2:eYFP and isolated subcellular fractionations from leaves of the transformed plants. Isolation of the membranes was confirmed by assaying the marker enzymes for different membrane fractions (Table 1). The activity of vanadate-sensitive ATPase was highest in the plasma membrane fraction, but low in the other fractions. NADH-Cyt c reductase activity was highest in the intracellular membranes whereas α-mannosidase activity was mainly associated with the tonoplast-enriched fraction. The data indicate that different membrane fractions were isolated with a low level of contamination (Table 1). Proteins from different fractions were separated by SDS-PAGE followed by immunoblotting. SPHK1 and SPHK2 were present primarily in the microsomal fraction and only trace amounts of SPHKs were detected in the cytosolic fraction (Fig. 2C). When the microsomal fraction was separated into the plasma and intracellular membranes, SPHKs were associated with the intracellular membranes and not with the plasma membrane (Fig. 2C). In addition, we isolated vacuoles from leaf protoplasts. Both SPHKs were present in the tonoplast fraction (Fig. 2C). These results consistently indicate that both SPHKs were associated with the tonoplast (Fig. 2C).
TABLE 1.
Marker enzyme activity in membrane fractions of Arabidopsis leaves
| SPHK1 transgenic Arabidopsis | SPHK2 transgenic Arabidopsis | |||||||
|---|---|---|---|---|---|---|---|---|
| Marker Enzyme | M | PM | IM | T | M | PM | IM | T |
| ATPasea | 13.84b | 39.12 | 4.33 | 2.32 | 13.32 | 44.93 | 5.42 | 3.19 |
| Cyt c reductasec | 95.24 | 34.29 | 247.33 | 13.39 | 74.18 | 26.17 | 225.08 | 9.34 |
| α-mannosidased | 23.21 | 9.43 | 30.31 | 133.76 | 20.12 | 10.21 | 41.32 | 118.43 |
a nmol phosphate min−1 mg protein−1.
b Values are means of three measurements.
c μmol Cyt c min−1 mg protein−1.
d nmol p-nitrophenol min−1 mg protein−1.
SPHK1 and SPHK2 Are Both Catalytically Active
We expressed both SPHK1 and SPHK2 protein in E. coli to determine whether they were active enzymes. Proteins at about 53 kDa were produced from the cDNA of SPHK1 and SPHK2, and the size was as predicted based on the cDNA coding regions (Fig. 3, A and B). Both SPHK1 and SPHK2 phosphorylated phytosphingosine to produce phyto-S1P. The increase in phyto-S1P production was proportional to the reaction time within 15 min (Fig. 3C). It was reported previously that SPHK1 expressed in human embryonic kidney 293 (HEK 293) cells used various LCBs as substrates (15). In our study both purified SPHK1 and SPHK2 were able to utilize various LCBs including sphingosine, phytosphingosine, t18:1, d18:2 and d-erythro-DHS as substrates (Fig. 3D). However, SPHK1 and SPHK2 displayed different activities toward these substrates. SPHK1 had higher activity on sphingosine, phytosphingosine and t18:1 while SPHK2 was more active on d18:2 (Fig. 3D). In addition, SPHK2 exhibited much less activity toward dl-threo-DHS than did SPHK1 (Fig. 3D). DMS, a potent inhibitor for mammalian SPHKs, was not phosphorylated by either of SPHKs under our experiment condition (Fig. 3D).
FIGURE 3.

Expression and activity assays of SPHKs. A, immunoblotting of SPHK1 and SPHK2 expressed in E. coli. Total protein (10 μg) was loaded on a SDS-PAGE gel. SPHK1 and SPHK2 were immunoblotted with anti-polyhistidine antibody conjugated with alkaline phosphatase. B, Coomassie Blue staining of purified SPHK1 and SPHK2 from E. coli separated on a 10% SDS-PAGE gel. C, SPHK1 and SPHK2 activity as a function of reaction time. Purified SPHK1 or SPHK2 (3.2 μg) was incubated with 50 μm phytosphingosine for the indicated time. Values are means ± S.E. (n = 3). D, phosphorylation of different LCBs (50 μm) by purified SPHK1 and SPHK2. 0.25 μm enzyme was incubated with substrate for 15 min. Values are means ± S.E. (n = 3).
PA Binds to SPHK1 and SPHK2
To determine the potential interaction of PA with SPHK1 and SPHK2, we performed a filter-binding assay utilizing nitrocellulose filter spotted with different lipids. Both SPHK1 and SPHK2 exhibited binding to egg yolk PA but not other phospholipids, including PC, PE, PG, PI, PS, LPC, and LPE (Fig. 4A). Different PA molecular species showed different binding patterns as 8:0/8:0, 18:0/18:0 or 18:2/18:2 PA did not bind to either SPHK, whereas 16:0/16:0, 18:1/18:1, 16:0/18:1, and 16:0/18:2 PA exhibited binding (Fig. 4A). SPHK1 was further used to examine the PA-SPHK interaction by a liposome binding assay. The liposomes were made with 18:1/18:1 PC only as control or with a mixture of 18:1/18:1 PC and different PA species in a molar ratio of 2:1. No SPHK1 was pelleted with PC-only liposomes suggesting the binding to be specific to PA-containing liposomes. Only trace quantities of SPHK1 were pelleted with liposomes containing 18:0/18:0 or 18:2/18:2 PA, whereas substantially more SPHK1 was associated with liposomes containing 16:0/18:1, 16:0/18:2 16:0/16:0, or 18:1/18:1 PA (Fig. 4B). The result of liposome binding was consistent with that of lipid-filter binding assay.
FIGURE 4.

PA binding to SPHKs. A, lipid binding specificity of SPHK1 and SPHK2 on filters. Different lipids and PA species (10 μg) were spotted onto a nitrocellulose membrane and incubated with equal amounts of purified SPHK1, SPHK2, or total protein from E. coli transformed with empty vector. EY, egg yolk PA. B, SPHK1 binding to liposomes containing PC only or PC plus different PA species. Purified SPHK1 (20 μg) was incubated with different liposomes for 1 h at room temperature. The vesicles were pelleted by centrifugation. The protein was visualized by immunoblotting with anti-His antibody. C, SPR quantitative analysis of PA binding to SPHK1. Liposomes containing PC only or PC plus 16:0/16:0 PA or 18:1/18:1 PA were used. SPHK1 was first immobilized on the NTA chip followed by injection of liposomes.
PA-SPHK interaction was further validated with SPR, which is a highly sensitive method for quantitative detection of molecular interaction. Purified SPHK1 was first immobilized on a NTA chip followed by injection of liposomes made of PC only or PA plus PC. In the representative sensorgram, response unit (RU) increased when the liposome was composed of PA (16:0/16:0 or 18:1/18:1) plus PC. By comparison, there was almost no increase of RU when PC only liposome was injected, indicating that PA interacts with SPHK1 specifically (Fig. 4C). Compared with 16:0/16:0 PA binding to SPHK1, 18:1/18:1 PA displayed a higher association rate constant (Ka = 590.50 m−1 s−1 versus 16.93 m−1 s−1) and a lower dissociation rate constant (Kd = 2.88 × 10−4 s−1 versus 3.20 × 10−3 s−1). The maximum specific binding is estimated to be 8904 RU for 16:0/16:0 PA and 4426 RU for 18:1/18:1 PA. The equilibrium binding constant KD is calculated to be 1.89 × 10−4 m for 16:0/16:0 PA-SPHK1 interaction and 4.88 × 10−7 m for 18:1/18:1 PA-SPHK1 interaction, indicating a low affinity between 16:0/16:0 PA and SPHK1 but a high affinity between 18:1/18:1 PA and SPHK1.
PA Stimulates SPHK Activity
To determine the effect of PA binding on SPHK, we tested the activity of SPHK1 under a range of PA concentrations. Including 10 nm PA in the assay augmented SPHK1 activity by 1.5-fold (Fig. 5A). The stimulation of enzyme activity continued in a dose-dependent manner, but reached a plateau up at 50 μm PA at which a 2.5-fold increase in kinase activity was observed (Fig. 5A). When different PA species were tested for their effect on SPHK1 activity at 50 μm, egg yolk PA, 18:1/18:1 PA, 16:0/18:1 PA, and 16:0/18:2 PA significantly increased SPHK1 activity more than 2-fold whereas 16:0/16:0 PA, 18:0/18:0 PA, 18:2/18:2 PA, or egg yolk PC had no significant effect on SPHK1 activity (Fig. 5B). The pattern of stimulation of SPHK1 activity by different PA species was in agreement with that of PA binding, suggesting that PA-SPHK interaction stimulates SPHK activity.
FIGURE 5.

PA stimulates SPHK activity. A, the effect of varied PA (18:1/18:1) concentrations on SPHK1 activity. Different concentrations of PA from 10 nm to 500 μm were tested for the effect on SPHK1 activity with 0.25% Triton X-100. B, the effect of different PA species on SPHK1 activity was tested with 0.25% Triton X-100. EY PC that did not bind to SPHK1 was used as a control.
PA Stimulates SPHK Activity by Promoting Substrate Binding
To determine the kinetic behavior of SPHK and the mechanism of stimulation of SPHK activity by PA, we used a surface dilution kinetic system because SPHK catalyzes the reaction at a water-lipid interface. The surface dilution model takes into account both two-dimensional surface interaction and three-dimensional bulk interaction between an enzyme and lipid substrate (37). The principle of surface dilution kinetics is presented in Equation 1 and the rate expression for surface dilution kinetic model is given in Equation 2 (Fig. 6A). Triton X-100 is one of the most commonly used detergents for surface dilution kinetics as it forms uniformly mixed micelles with different lipids including sphingolipids (38). SPHK1 activity was measured using increasing Triton X-100 concentrations in the mixed micelles along with purified SPHK1. The result showed that Triton X-100 served as a typical neutral dilutor at a concentration range of 0.8 to 10 mm for SPHK1 (Fig. 6B).
FIGURE 6.

Surface dilution kinetic model and effect of Triton X-100 on SPHK1 activity. A, equation 1 depicts the surface dilution model, and equation 2 is the rate expression for surface dilution kinetic model. B, SPHK1 activity measured with increasing molar concentrations of Triton X-100 in the mixed micelles. The molar concentration of phytosphingosine was held at 50 μm. Values are means ± S.E. (n = 3).
When phytosphingosine concentration was kept at 50 μm, the maximum activity was achieved at 0.8 mm Triton X-100 (Fig. 6B). To determine the surface dilution kinetic parameters, SPHK1 activity was determined as a function of the sum of the molar concentration of Triton X-100 and phytosphingosine at a series of set molar fractions (MF) (Fig. 7A). As the surface concentration of phytosphingosine decreased, the apparent Vmax decreased (Fig. 7A). Double-reciprocal plot of the results in Fig. 7A indicated that SPHK1 exhibited saturation kinetics when the bulk concentration of Triton X-100 and phytosphingosine was varied at each fixed MF of phytosphingosine (Fig. 7B). According to Equation 2, the intercept of the 1/V intercept axis is equal to 1/Vmax and the intercept of the 1/B axis is equal to −1/KmB. 1/V intercepts obtained in Fig. 7B versus the reciprocal of the MF of phytosphingosine was replotted to determine the Vmax and KmB of SPHK1 (Fig. 7C). The Vmax and KmB were determined to be 12.94 nmol/min/mg and 5.49 × 10−3 MF, respectively. The slope versus 1/B from Fig. 7B was replotted to determine the dissociation constant KsA. KsA was calculated to be 18.68 nm by using the slope of the line in Fig. 7D and the Vmax and KmB determined in Fig. 7C.
FIGURE 7.

Activity of SPHK1 toward phytosphingosine in mixed micelles with Triton X-100. A, SPHK1 activity measured as a function of the sum of the molar concentrations of Triton X-100 (TX) plus phytosphingosine at a series of set mol fractions of phytosphingosine. Data represent the average of three replicates. B, reciprocal plot of the data in A. C, replot of 1/V intercepts obtained in B versus the reciprocal of the mol fraction of phytosphingosine. D, replot of slopes obtained in B versus the reciprocal of the mol fraction of phytosphingosine.
To understand the mechanism by which PA stimulates SPHK activity, we compared the two constants, KsA and KmB, in the absence or presence of PA. The effect of PA on KsA of SPHK1 activity was measured as a function of the sum of the molar concentration of Triton X-100 and phytosphingosine at three set MF of PA (0, 0.002, and 0.02) with the phytosphingosine MF fixed at 0.01 (Fig. 8A). Increasing mol fractions of PA increased the apparent Vmax but the apparent KsA was not significantly changed (Fig. 8A). The result indicates that PA does not promote the bulk binding of SPHK1 to the mixed micelles. The effect of PA on KmB for SPHK1 activity was measured as a function of the MF of phytosphingosine at the three set PA mol fractions (Fig. 8B). The apparent Vmax increased with the increase of PA mol fractions and the apparent KmB decreased by more than 50% in the presence of PA (Fig. 8B). The specificity constant (apparent Vmax/KmB) was increased by 2.44-fold in the presence of 0.005 mole fraction of PA (Fig. 8B). Overall, the surface-dilution kinetics analysis indicates that PA stimulates SPHK1 activity by promoting the binding of substrate to the catalytic site of the enzyme, but PA does not affect the binding of SPHK1 to the mixed micelle surface.
FIGURE 8.
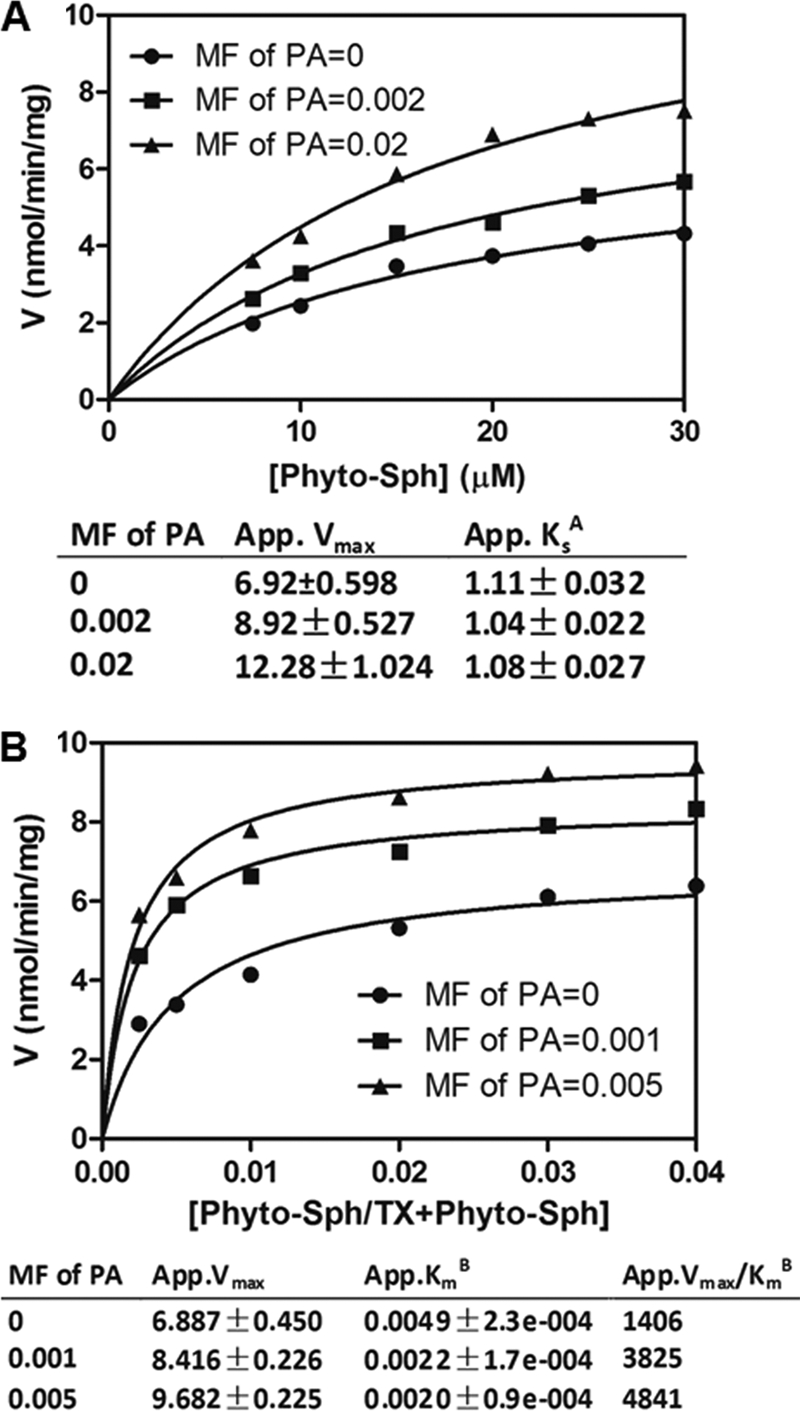
Effect of PA on the kinetic behavior of SPHK1. A, SPHK1 activity measured as a function of phytosphingosine molar concentrations at set mol fractions of PA. The molar fraction of phytosphingosine was 0.01. Data represent the average of three replicates. B, SPHK1 activity measured as a function of phytosphingosine mol fractions at set mol fractions of PA. The molar concentration of phytosphingosine was 50 μm. Data represent the average of three replicates.
DISCUSSION
Results of this study indicate that the annotated At4g21540 locus is actually comprised of two separate SPHK genes, which are both transcribed in A. thaliana. The conclusion is supported by molecular cloning, sequence analysis, and the distinguishable patterns of expression of SPHK1 and SPHK2 in Arabidopsis tissues. The stop codon of SPHK2 is 788 bp upstream of the start codon of SPHK1, and the 788 bp region may serve as the promoter of the second SPHK1. Subcellular localization indicates that both SPHK1 and SPHK2 were localized on tonoplasts, which is consistent with the finding that SPHK activity is mainly associated with membranes (15). Arabidopsis SPHK1 and SPHK2 were expressed in E. coli and both purified SPHK1 and SPHK2 were active in producing phyto-S1P. The substrate specificity of SPHK1 from the E. coli-expressed enzyme is the same as the SPHK1 expressed in human embryonic kidney 293 cells, phosphorylating sphingosine, phytosphingosine, and other plant LCBs (15). The catalytic activity of SPHK1 is slightly higher than that of SPHK2 toward several LCBs. In addition, the level of expression of SPHK1 is higher than that of SPHK2 in most tissues examined except in the silique. These results indicate that SPHK1 is more prevalent than SPHK2 in producing LCBP in vegetative tissues.
In plants, PA and phyto-S1P play important roles in transducing the ABA effect in stomatal closure. PA acts as an important regulator of various proteins by interacting with effector proteins. However, the mechanism by which PA regulates target protein function is not well understood. The present study shows that PA binds to SPHK1 and SPHK2. The binding has been demonstrated by different approaches, including lipid filter assay, liposomal binding, and SPR. PA-protein interaction can affect the protein function by changing the protein membrane association and/or directly modulating the activity of its effector enzymes. This modulation can be either activation or inhibition, depending upon the target proteins (1). The localization of signaling kinases is regarded as key to their signaling functions (39). Mouse SPHK activity was found to be simulated by acidic phospholipids including PA (22), and PA stimulated mouse SPHK1 activity by promoting the association of mouse SPHK1 to membranes which were rich in PA (25). However, unlike mammalian SPHKs, these two Arabidopsis SPHKs are associated with the tonoplast. In addition, the basal level of PA in Arabidopsis cells is estimated to be 50–100 μm, which is considerably above PA critical micelle concentration (CMC), which is in the subnanomolar range (1). Above the CMC, the concentration of PA monomer is constant, independent of the total concentration of the lipid. Thus, the accumulation of PA above the critical level during cell activation affects the concentration of membrane-associated PA, but not monomeric PA. This suggests that PA binding to target proteins occurs at the membrane, but not in solution. Our kinetic analysis data indicate that PA increases Arabidopsis SPHK1 activity by promoting the binding of lipid substrates to the catalytic site of the enzyme without altering the bulk binding of the enzyme to the micelle surface. The result is consistent with the observation that SPHKs are already associated with the tonoplast, which is rich in phospholipids and sphingolipids (40). PA stimulates SPHK activity at a 10 nm to 200 μm range, a level of PA achievable in plant cells.
In addition, the present results show that the PA binding and stimulation of SPHK depends on the PA molecular species. PA is composed of different molecular species due to variation in two fatty acyl chains. A recent study indicated that 18:1/18:1, 18:2/18:2, 16:0/18:1, 16:0/18:2, and 18:0/18:2 PAs bound to the NADPH oxidase RbohD, but 16:0/16:0 and 18:0/18:0 PAs did not (10). In the PA-SPHK interaction, the present results showed that 18:2/18:2 did not bind to SPHK, whereas 16:0/16:0 PA displayed binding although the binding is much weaker than that of 18:1/18:1 PA to SPHK. The SPHK binding to different PA species was also qualitatively different from ABI1 that displayed much stronger binding to 18:1/18:1 PA than 16:0/16:0, 18:0/18:0, or 18:2/18:2 PAs tested (8). SPHKs, ABI1, and RbohD all are involved in mediating ABA response and stomatal movement. The differential interaction with different PA species could mean that the PAs that interact with the different target proteins may result from different sources. The molecular interaction with different PAs may underlie a mechanism for the diverse function of PAs in mediating cellular response. Although more than 20 proteins have been found to interact with PA, the protein structure required for the PA-protein interaction is unknown. It has been proposed that lysine and arginine residues increase the charge of PA and induce an electrostatic/hydrogen bond switch to stabilize the protein-lipid interaction (41, 42). The requirements of different PA acyl species by different proteins suggest that not only the head group but also the acyl groups are involved in PA-protein interaction. To fully understand the function of PA in cell regulation, it is necessary to elucidate the structural requirements for such PA-protein interaction and how that interaction modulates the function in the ensuing lipid-protein complex.
Acknowledgments
We thank Dr. Daniel Lynch (Williams College) for kindly providing 4-hydroxy-8-sphingenine (t18:1) and 4,8-sphingadienine (d18:2), Dr. Howard Berg for technical help with confocal microscopy, and Dr. Sangaralingam Kumaran for suggestions on SPR analysis.
This work was supported by National Science Foundation Grant IOS-0818740 and United States Department of Agriculture Grant 2007-35318-18393.
The nucleotide sequence(s) reported in this paper has been submitted to the Gen-BankTM/EBI Data Bank with accession number(s) HQ825315.
- PA
- phosphatidic acid
- ABA
- abscisic acid
- ABI1
- ABA Insensitive 1
- PC
- phosphatidylcholine
- PG
- phosphatidylglycerol
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- PS
- phosphatidylserine
- LPC
- lyso PC
- PLD
- phospholipase D
- phyto-S1P
- phytosphingosine-1-phosphate
- SPHK
- sphingosine kinase
- LCB
- long-chain sphingoid base.
REFERENCES
- 1. Wang X., Devaiah S. P., Zhang W., Welti R. (2006) Prog. Lipid Res. 45, 250–278 [DOI] [PubMed] [Google Scholar]
- 2. Bargmann B. O., Munnik T. (2006) Curr. Opin. Plant Biol. 9, 515–522 [DOI] [PubMed] [Google Scholar]
- 3. Foster D. A. (2009) Biochim. Biophys. Acta 1791, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Welti R., Li W., Li M., Sang Y., Biesiada H., Zhou H. E., Rajashekar C. B., Williams T. D., Wang X. (2002) J. Biol. Chem. 277, 31994–32002 [DOI] [PubMed] [Google Scholar]
- 5. Testerink C., Munnik T. (2005) Trends Plant Sci. 10, 368–375 [DOI] [PubMed] [Google Scholar]
- 6. Li M., Hong Y., Wang X. (2009) Biochim. Biophys. Acta 1791, 927–935 [DOI] [PubMed] [Google Scholar]
- 7. Hong Y., Pan X., Welti R., Wang X. (2008) Plant Cell 20, 803–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang W., Qin C., Zhao J., Wang X. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9508–9513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mishra G., Zhang W., Deng F., Zhao J., Wang X. (2006) Science 312, 264–266 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y., Zhu H., Zhang Q., Li M., Yan M., Wang R., Wang L., Welti R., Zhang W., Wang X. (2009) The Plant Cell 21, 2357–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ng C. K., Carr K., McAinsh M. R., Powell B., Hetherington A. M. (2001) Nature 410, 596–599 [DOI] [PubMed] [Google Scholar]
- 12. Coursol S., Fan L. M., Le Stunff H., Spiegel S., Gilroy S., Assmann S. M. (2003) Nature 423, 651–654 [DOI] [PubMed] [Google Scholar]
- 13. Coursol S., Le Stunff H., Lynch D. V., Gilroy S., Assmann S. M., Spiegel S. (2005) Plant Physiol. 137, 724–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Michaelson L. V., Zäuner S., Markham J. E., Haslam R. P., Desikan R., Mugford S., Albrecht S., Warnecke D., Sperling P., Heinz E., Napier J. A. (2009) Plant Physiol. 149, 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Worrall D., Liang Y. K., Alvarez S., Holroyd G. H., Spiegel S., Panagopulos M., Gray J. E., Hetherington A. M. (2008) Plant J. 56, 64–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lynch D. V., Chen M., Cahoon E. B. (2009) Trends Plant Sci. 14, 463–466 [DOI] [PubMed] [Google Scholar]
- 17. Markham J. E., Jaworski J. G. (2007) Rapid Commun. Mass Spectrom. 21, 1304–1314 [DOI] [PubMed] [Google Scholar]
- 18. Anthony R. G., Henriques R., Helfer A., Mészáros T., Rios G., Testerink C., Munnik T., Deák M., Koncz C., Bögre L. (2004) EMBO J. 23, 572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Testerink C., Larsen P. B., van der Does D., van Himbergen J. A., Munnik T. (2007) J. Exp. Bot. 58, 3905–3914 [DOI] [PubMed] [Google Scholar]
- 20. Lu B., Benning C. (2009) J. Biol. Chem. 284, 17420–17427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Testerink C., Dekker H. L., Lim Z. Y., Johns M. K., Holmes A. B., Koster C. G., Ktistakis N. T., Munnik T. (2004) Plant J. 39, 527–536 [DOI] [PubMed] [Google Scholar]
- 22. Olivera A., Rosenthal J., Spiegel S. (1996) J. Cell. Biochem. 60, 529–537 [DOI] [PubMed] [Google Scholar]
- 23. Melendez A. J., Allen J. M. (2002) Semin. Immunol. 14, 49–55 [DOI] [PubMed] [Google Scholar]
- 24. Johnson K. R., Becker K. P., Facchinetti M. M., Hannun Y. A., Obeid L. M. (2002) J. Biol. Chem. 277, 35257–35262 [DOI] [PubMed] [Google Scholar]
- 25. Delon C., Manifava M., Wood E., Thompson D., Krugmann S., Pyne S., Ktistakis N. T. (2004) J. Biol. Chem. 279, 44763–44774 [DOI] [PubMed] [Google Scholar]
- 26. Voinnet O., Rivas S., Mestre P., Baulcombe D. (2003) Plant J. 33, 949–956 [DOI] [PubMed] [Google Scholar]
- 27. Fan L., Zheng S., Cui D., Wang X. (1999) Plant Physiol. 119, 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoo S. D., Cho Y. H., Sheen J. (2007) Nature Protocols 2, 1565–1572 [DOI] [PubMed] [Google Scholar]
- 29. Jaquinod M., Villiers F., Kieffer-Jaquinod S., Hugouvieux V., Bruley C., Garin J., Bourguignon J. (2007) Mol. Cell Proteomics 6, 394–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olivera A., Kohama T., Tu Z., Milstien S., Spiegel S. (1998) J. Biol. Chem. 273, 12576–12583 [DOI] [PubMed] [Google Scholar]
- 31. Stevenson J. M., Perera I. Y., Boss W. F. (1998) J. Biol. Chem. 273, 22761–22767 [DOI] [PubMed] [Google Scholar]
- 32. Qin C., Wang C., Wang X. (2002) J. Biol. Chem. 277, 49685–49690 [DOI] [PubMed] [Google Scholar]
- 33. Nishiura H., Tamura K., Morimoto Y., Imai H. (2000) Biochem. Soc. Trans. 28, 747–748 [PubMed] [Google Scholar]
- 34. Liang H., Yao N., Song J. T., Luo S., Lu H., Greenberg J. T. (2003) Genes Dev. 17, 2636–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang C., Wang X. (2001) Plant Physiol. 127, 1102–1112 [PMC free article] [PubMed] [Google Scholar]
- 36. Marion J., Bach L., Bellec Y., Meyer C., Gissot L., Faure J. D. (2008) Plant J. 56, 169–179 [DOI] [PubMed] [Google Scholar]
- 37. Dennis E. A. (1973) Arch. Biochem. Biophys. 158, 485–493 [DOI] [PubMed] [Google Scholar]
- 38. Carman G. M., Deems R. A., Dennis E. A. (1995) J. Biol. Chem. 270, 18711–18714 [DOI] [PubMed] [Google Scholar]
- 39. Wattenberg B. W., Pitson S. M., Raben D. M. (2006) J. Lipid Res. 47, 1128–1139 [DOI] [PubMed] [Google Scholar]
- 40. Yoshida S., Uemura M. (1986) Plant Physiol. 82, 807–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kooijman E. E., Tieleman D. P., Testerink C., Munnik T., Rijkers D. T., Burger K. N., de Kruijff B. (2007) J. Biol. Chem. 282, 11356–11364 [DOI] [PubMed] [Google Scholar]
- 42. Kooijman E. E., Burger K. N. (2009) Biochim. Biophys. Acta 1791, 881–888 [DOI] [PubMed] [Google Scholar]