

Abstract
The transporter associated with antigen processing (TAP) plays a key role in adaptive immunity by translocating proteasomal degradation products from the cytosol into the endoplasmic reticulum lumen for subsequent loading onto major histocompatibility (MHC) class I molecules. For functional and structural analysis of this ATP-binding cassette complex, we established the overexpression of TAP in the methylotrophic yeast Pichia pastoris. Screening of optimal solubilization and purification conditions allowed the isolation of the heterodimeric transport complex, yielding 30 mg of TAP/liter of culture. Detailed analysis of TAP function in the membrane, solubilized, purified, and reconstituted states revealed a direct influence of the native lipid environment on activity. TAP-associated phospholipids, essential for function, were profiled by liquid chromatography Fourier transform mass spectrometry. The antigen translocation activity is stimulated by phosphatidylinositol and -ethanolamine, whereas cholesterol has a negative effect on TAP activity.
Keywords: ABC Transporter, Antigen Processing, ATPases, Peptides, Reconstitution of Membrane Transporters
Introduction
The adaptive immune system of jawed vertebrates is based on recognition and elimination of cells that are either invaded by intracellular pathogens or malignantly transformed. One essential part of these processes is cell surface presentation of antigenic peptides via major histocompatibility complex (MHC) class I molecules to cytotoxic T-cells. Cells degrade defective ribosomal products and misfolded or unwanted proteins by the ubiquitin-proteasome pathway. Resulting degradation products are recognized and translocated by the transporter associated with antigen processing (TAP)2 into the ER lumen, where they are loaded onto MHC-I molecules. Peptide-MHC complexes are then shuttled by the secretory pathway to the cell surface for antigen presentation to cytotoxic T-cells, leading in the case of viral or malignant transformation to lysis and apoptosis of the presenting cell. Therefore, the TAP complex represents a key control point within the antigen presentation pathway (for review, see Refs. 1 and 2).
TAP belongs to the large family of ATP-binding cassette (ABC) proteins that couple binding and hydrolysis of ATP with the uphill transport of a broad range of substrates (3, 4). The functional TAP complex is composed of the two half-transporters, TAP1 (ABCB2) and TAP2 (ABCB3). Each subunit contains a transmembrane domain, responsible for binding and transmembrane movement of the peptide cargo (5, 6). In addition, each subunit harbors a C-terminal nucleotide-binding domain (NBD), responsible for ATP binding and hydrolysis, in cooperation with the second NBD of the adjacent subunit. Notably, peptide and ATP binding are independent of each other (7). However, peptide binding induces ATP hydrolysis (8). Recent investigations identified a sensor region at the transmission interface between transmembrane domains and NBDs involved in the allosteric coupling of peptide binding and subsequent ATP hydrolysis (9). Interestingly, cross-linking of the peptide sensor (cytosolic loop (CL) 1) or peptide-binding region (CL2) with the X-loop of the opposite NBD resulted in either arrest in a translocation-incompetent state or inhibition of substrate binding and thereby transport (10). Nevertheless, detailed mechanistic insights into the translocation cycle are incomplete. Structural information is available only for the TAP1-NBD (11, 12), whereas the organization of transmembrane helices was investigated by cysteine-scanning and cross-linking approaches (10, 13). Studies of the TAP mechanism were hampered by the difficulty in expressing sufficient amounts of the heterodimeric TAP complex.
In this study, we established the heterologous expression of TAP in Pichia pastoris. This methylotrophic yeast has been used for successful overexpression of mammalian membrane proteins, including the voltage-dependent potassium channel (Kv1.2), P-glycoprotein (ABCB1A), and the sterol transporter complex (ABCG5/G8) (14–16). Extensive solubilization and purification screens allowed purification of large amounts of the heterodimeric ABC complex TAP, thus enabling the intensive characterization of peptide and ATP binding as well as of the allosteric coupling reflected by peptide-stimulated ATPase activity. By establishing reconstitution of functional TAP into proteoliposomes, we systematically analyzed its modulation by specific lipids. We demonstrate that the membrane environment and TAP-associated lipids have a marked influence on the function of the antigen translocation machinery. TAP-associated lipids were profiled by a lipidomic approach using liquid chromatography Fourier transform mass spectrometry (LC FT-MS). Phosphatidylethanolamine (PE) and phosphatidylinositol (PI) were identified as main activators of peptide translocation, whereas cholesterol had a negative effect on peptide translocation.
EXPERIMENTAL PROCEDURES
Materials
Digitonin and Tween-20 were purchased from Carl Roth, and all other detergents were from Anatrace. 1,2-Dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phosphatidylglycerol (DOPG), phosphatidylinositol (PI), cholesterol, Escherichia coli lipids, and soybean polar extract (acetone/ether-precipitated preparations) were from Avanti Polar Lipids. Restriction enzymes were obtained from Fermentas. All standard chemicals were purchased from Sigma-Aldrich or Carl Roth.
Cloning and Expression
Full-length tap1 and tap2 were codon-optimized for expression in P. pastoris. For cloning into the pAO815 vector (Invitrogen), the tap1 gene was flanked by EcoRI restriction sites. A C-terminal glycine-serine linker (GGGS)2 followed by a tobacco etch virus (TEV) protease cleavage site (ENLYFQG) and a His10 tag were added for affinity purification. Codon-optimized tap2 was flanked by unique 5′-XhoI and 3′-NotI sites for cloning into the respective sites of pPICZ vector (Invitrogen). The 3′-extension coding for a (GGGS)2 linker and a TEV protease cleavage site were as for tap1 except that the His10 tag was replaced by the epitope PRGPDRPEGIEE recognized by the anti-C8 antibody (17). Due to very low protein expression levels of codon-optimized TAP2, the tap2 gene was replaced by wild-type tap2, which was PCR-amplified from a baculovirus expression vector (9) using Phusion high fidelity DNA polymerase (Finnzymes) and primers providing sequences 5′-ATCCCCCTTCGAAACGATGGGGCTCCCTGACCTGAGACCCTGGA-3′ and 5′-CTCCGCATGCGAGCTGGGCAAGCTTCTGCA-3′. The PCR product was cloned into the 5′-NspV and 3′-SpHI sites of pPICZ-tap2 so as to preserve the described C-terminal extensions. Integrity of the gene sequences was verified by sequencing.
Both plasmids (50 μg) were linearized with PmeI and co-transformed by electroporation into P. pastoris strain SMD1163 (his4 pep4 prb1) following standard procedures (18). Transformants were plated onto YPDS agar plates containing 0.1, 0.5, 1.0, or 2.0 mg/ml Zeocin to select for multicopy integrations. To detect high expression transformants, several clones were grown in 50-ml shaker cultures in YPD or MGY media. Expression was induced by minimal methanol medium (containing 1% methanol), and cells were harvested 24 h after induction. To monitor TAP expression, a rapid preparation of membranes similar to Ref. 19 was performed, except that the breaking buffer contained 250 mm sucrose, 50 mm potassium phosphate buffer (KH2PO4/K2HPO4) pH 7.3, 5 mm EDTA, and protease inhibitor mix (SERVA). The membrane fraction was resuspended in Standard buffer (20 mm Hepes pH 7.5, 150 mm NaCl, 8.6% glycerol) and adjusted to 5 mg of protein/ml. Membrane preparations were analyzed by SDS-PAGE (10%) and immunoblotting using monoclonal antibodies α-TAP1 (148.3) and α-TAP2 (435.3) (20).
For large-scale TAP production, P. pastoris was grown in a 7.5-liter Labfors4 reactor (Infors-HT). The fermentor, equipped with microprocessor control of dissolved oxygen, pH, temperature, agitation, nutrient feed, and methanol concentration, was loaded with basal salts (19). After sterilization, 25 ml of anti-foam 204 solution and 16 ml of PTM trace salts (Invitrogen fermentation guidelines) were added, and the system was adjusted to 30 °C. The fermentor was inoculated with 400 ml of P. pastoris grown in MGY medium to A600 = 4.0. Cells were grown at 30 °C, 1,000 rpm agitation, and a pO2 level of 30% while maintaining pH 4.5 by the addition of ammonium hydroxide (16%, w/v) until glycerol was consumed. The cell density at this stage was 90–130 g/liter. Carbon-limited growth was then continued at 30 °C and pH 5.0 by infusion of a 50% (w/v) glycerol solution containing 10 ml/liter PTM trace salts until an A600 of about 200 was reached (up to 160 g of cells/liter). Glycerol feeding was then reduced and finally stopped until cells started starving as indicated by an increase of the pO2 level. Expression was induced by a controlled methanol feed containing 10 ml/liter PTM trace salts (maximum concentration of methanol was 1%). Cells were harvested 24–36 h after induction (cells yielded up to 250 g/liter). Cells were harvested, frozen in liquid nitrogen, and stored at −80 °C.
Membrane Preparation
Frozen cells were thawed on ice and diluted with 1 volume of breaking buffer. An equal volume of glass beads was added, and cells were disrupted in an ice water-cooled bead beater (BioSpec) by five cycles of 1 min of beating, 2 min of cooling. The lysate was centrifuged at 2,000 × g for 10 min to remove cell debris. The supernatant was reserved on ice, whereas the pellet was subjected to two additional rounds of bead beating and centrifugation. Combined supernatants were centrifuged at 100,000 × g for 1 h at 4 °C. Membrane pellets were resuspended in Standard buffer using a Teflon homogenizer. Membranes were adjusted to ∼20 mg of protein/ml, frozen in liquid nitrogen, and stored at −80 °C.
Solubilization Screen
For small-scale solubilization trials, TAP-containing membrane pellets were resuspended in Standard buffer containing 2% of the respective detergent (w/v) to yield a final protein concentration of 5 mg/ml and incubated for 1 h at 4 °C. Samples were centrifuged at 100,000 × g for 45 min at 4 °C, and supernatants were collected for immunoblotting and peptide binding assays. Long-term stability of the TAP complex was determined by keeping supernatants on ice for up to 7 days. Aliquots were recentrifuged to remove precipitated protein, and the amount of remaining TAP was quantified by immunoblotting.
Detergent and salt conditions optimal for metal affinity purification were screened using a 96-well format. 10 mg of membrane protein/well were centrifuged for 30 min at 20,000 × g, 4 °C, and the pellet was resuspended to give 5 mg of protein/ml in 20 mm Hepes, pH 7.5, 15% glycerol, 150 or 500 mm NaCl, and 1 or 2% (w/v) detergent. After solubilization for 2 h at 4 °C, samples were centrifuged 30 min at 100,000 × g, 4 °C, and supernatants were loaded onto pre-equilibrated His MultiTrap FF filter plates (GE healthcare) pre-equilibrated with IMAC buffer (20 mm Hepes, pH 7.5, 15% glycerol, 150 mm NaCl) with the respective detergent reduced to two times its CMC. For affinity binding, the plates were centrifuged at 100 × g for 4 min at 4 °C before washing twice with 0.5 ml of IMAC buffer and twice with 0.5 ml of washing buffer (IMAC buffer containing 40 mm of histidine). TAP was eluted with 200 μl of elution buffer (IMAC buffer containing 200 mm of histidine). TAP recovery was analyzed by immunoblotting using α-TAP1 and α-TAP2 antibodies.
Purification of TAP
Membranes were thawed on ice and diluted with solubilization buffer (20 mm Hepes, pH 7.5, 500 mm NaCl, 8.6% glycerol, 2% digitonin (w/v)) to a final concentration of 5 mg of protein/ml. After 1 h solubilization on ice, the mixture was centrifuged for 30 min at 100,000 × g, 4 °C. The supernatant was loaded at a flow rate of 0.5 ml/min onto a Ni2+-iminodiacetic acid (IDA) column (GE Healthcare) pre-equilibrated with Standard buffer/0.05% digitonin. TAP was washed with 10 column volumes of Standard buffer/0.05% digitonin containing 0, 20, and 40 mm histidine and finally eluted with 200 mm of histidine. Eluted proteins were analyzed by SDS-PAGE (10%) and Coomassie Blue staining. To examine monodispersity by size exclusion chromatography, 50-μl aliquots of 3.5 μm purified TAP complex were separated on a Superose 6 column (PC 3.2/30, GE Healthcare) at a flow rate of 0.05 ml/min in Standard buffer containing 0.05% digitonin. Purified TAP was concentrated to 0.5 mg/ml by Amicon spin concentrators (cut-off 100 kDa, Millipore), frozen in liquid nitrogen, and stored at −80 °C.
Reconstitution of TAP
Liposomes were prepared as described previously (21). Triton X-100-destabilized large unilamellar vesicles were mixed with purified TAP at a lipid-to-protein ratio of 10:1 (w/w) and incubated for 30 min on ice. Detergent was removed by incubation with polystyrene beads (40 mg/ml wet weight, Bio-Beads SM-2; Bio-Rad) that had been prewashed with methanol, water, and Standard buffer. After incubation four times for at least 1 h at 4 °C with gentle agitation, beads were removed, and proteoliposomes were pelleted by centrifugation for 45 min at 100,000 × g at 4 °C. The pellet was resuspended in Standard buffer to a final lipid concentration of 5 mg/ml. Protein aggregates and empty vesicles were separated from proteoliposomes by centrifugation on continuous Ficoll density gradients (0–10% in Standard buffer) at 200,000 × g, 4 °C for 16 h in a SW41 Ti rotor (Beckman Coulter). Gradients were fractionated from the top using a Piston Gradient Fractionator (Biocomp). Proteoliposomes were pooled, washed twice with Standard buffer to remove residual Ficoll, and finally resuspended in Standard buffer.
After reconstitution, the orientation of the TAP complex in proteoliposomes was determined by specific cleavage of the His10 tag. Proteoliposomes containing 2.5 μg of protein were incubated in a total volume of 30 μl for 4 h at 30 °C in the presence of 2 units of TEV protease (MoBiTec). Reactions without TEV protease or with 1% Triton X-100 served as controls. After TEV cleavage, samples were analyzed by SDS-PAGE (10%) and immunoblotting using antibodies specific for TAP1, TAP2, and His6 (Novagen).
Freeze-Fracture EM Analysis
Proteoliposomes were frozen in liquid nitrogen-cooled Freon 22. Replicas were made according to standard techniques (22) on a Balzers BA 300 freeze-etching apparatus (Balzers AG) equipped with an electron gun evaporator and an oscillating quartz. Samples were fractured at −100 °C and 2 microtorrs. Replicas were obtained by shadowing the fracture surface with platinum and carbon at an angle of 38° followed by carbon at 90°. After overnight cleaning in household bleach and washing in distilled water, replicas were transferred on 200 mesh copper grids and examined in a Philips EM 410 operated at 80 kV.
Peptide Binding Assay
Specific TAP binding of 125I-labeled peptide RRYQKSTEL was determined as described (21).
ATP Hydrolysis Assay
ATPase activity of purified TAP was determined by a malachite green-based colorimetric assay (23).
Peptide Transport Assay
P. pastoris membranes (45 μg of total protein) or proteoliposomes containing TAP (1 μg) were preincubated for 15 min on ice in 100 μl of PBS reaction buffer (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 2 mm NaHPO4; pH 7.4) containing 0.5 μm fluorescein-labeled peptide (RRYC(F)KSTEL, (F) indicates fluorescein coupled to cysteine) and 10 mm MgATP. The transport reaction was performed as described previously (21). Transport specificity was verified by a 2-min preincubation with either apyrase (1 unit) or viral inhibitors. For ICP47-dependent inhibition, samples were incubated in reaction buffer containing 10 μm ICP47 (24). In the case of US6, membranes were pretreated with 1% saponin for 1 min at 37 °C, washed with ice-cold PBS buffer, and then incubated with reaction buffer containing 50 μm of either the active domain US620–146 or the inactive domain US620–125 (25). Km values were determined by varying the ATP or peptide concentration, respectively.
ATP Binding Assay
50-μl aliquots of 0.5 μm purified TAP were mixed with 100 μl of ATP-agarose pre-equilibrated in 250 μl of binding buffer (20 mm Hepes, pH 7.5, 150 mm NaCl, 5 mm MgCl2, 8.6% glycerol, 0.05% digitonin), containing various concentrations of competitor ATP. Mixtures were incubated for 30 min on ice. After washing twice with 500 μl of binding buffer lacking ATP, specifically bound protein was eluted with 50 μl of binding buffer containing 50 mm MgATP. Eluates were analyzed by SDS-PAGE (10%) and immunoblotting. Intensities were plotted against ATP concentration and fitted with a 1:1 Langmuir binding isotherm. To determine the half-maximal inhibitory concentration (IC50) of ATP, the following equation was used
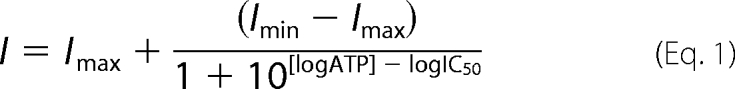 |
where I represents the protein intensity, and Imax and Imin represent the maximal and minimal intensities.
TNP-ATP Binding
Purified TAP (0.1 μm) was incubated for 15 min on ice in 1 ml of binding buffer containing various concentrations of TNP-ATP. For competitive binding assays, 6.5 μm TNP-ATP was used with various concentrations of competitor ATP. TNP-ATP fluorescence was measured at 8 °C with an excitation/emission wavelength of 410/541 nm and plotted against ATP concentration. To determine the half-maximal inhibitory ATP concentration (IC50), the following equation was used
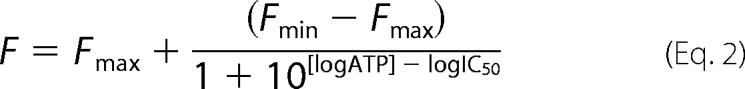 |
where F represents the fluorescence intensity, and Fmax and Fmin represent the maximal and minimal fluorescence values. Ki,ATP was determined using the Cheng-Prusoff equation (26).
 |
Phospholipid Analysis
Phospholipid concentrations of proteoliposomes were determined by a colorimetric assay based on the complex formation with ammonium ferrothiocyanate (27). 0.5-ml aliquots were mixed with 2 ml of chloroform and 1 ml of thiocyanate reagent (27 g of FeCl3·6H2O and 30 g of NH4SCN dissolved in 1 liter of H2O) and vortexed vigorously. After centrifugation for 10 min at 3,000 × g, the absorbance of the lower phase was determined at 488 nm and compared with lipid standards.
Quantification of TAP-associated Lipids
After purification, lipids bound to TAP were determined by acidic digestion and subsequent colorimetric detection of free phosphate via ammonium molybdate (28, 29). Briefly, 200 μl of digitonin or n-dodecyl-β-d-maltoside (DDM)-purified TAP (10 μm) were hydrolyzed at 120 °C for 18 h in the presence of 300 μl of 0.6 m H2SO4 and 0.21 m HClO4. Following this step, samples were clarified by centrifugation (5 min; 20,000 × g). 100 μl of supernatant were mixed with 250 μl of staining solution (0.32 mm malachite green, 45 mm (NH4)6Mo7O24, 1.5 m H2SO4). After incubation for 20 min at room temperature, absorbance was measured at 650 nm, and the amount of phosphate was determined by comparison with a NaH2PO4 standard.
Lipid Analysis by Mass Spectrometry
Lipids were extracted by chloroform/methanol (2:1, v/v) and analyzed by normal phase liquid chromatography using a PVA-SIL column (YMC Europe GmbH) interfaced with a nanoflow ion source Triversa NanoMate (Advion Biosciences, Inc.) and a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). PC, PE, and PI species were monitored in positive ion mode by recording FT-MS spectra with a target mass resolution of 100,000. Lipid species were quantified by extracting their peak intensities as previously described (30) and by normalizing to the total intensity of all monitored lipid species.
RESULTS
Expression of TAP in P. pastoris
Protein production in P. pastoris allows efficient membrane targeting and folding as well as posttranslational modifications such as glycosylation or disulfide bond formation. P. pastoris generally provides the highest cell density of any expression system and thus greater amounts of protein per culture volume. However, expression levels vary widely and are often directly correlated with the number of integrated copies of the gene of interest (31–33). In the present case, assembly of the functional antigen translocation complex required high-level co-expression of TAP1 and TAP2, so it was required to screen for transformants with multiple copies of both the tap1 and the tap2 genes. Co-transformation of codon-optimized tap1 and tap2 resulted in high-level expression of TAP1, but TAP2 was undetectable. Consequently, codon-optimized tap1 was co-transformed into P. pastoris with the wild-type tap2 gene. Highest expression of TAP1 and TAP2 was obtained at high concentration of Zeocin (2 mg/ml), suggesting the integration of multiple copies into the genome.
To scale up biomass production, we established Pichia fermentation procedures based on classical batch and feed methods (19, 34, 35), with strictly controlled methanol feed rates, growth, and feeding schedules. Samples were taken at several time points after induction, and membrane preparations were analyzed by SDS-PAGE and immunoblotting. High-level expression of TAP1 and TAP2 was observed 24 h after induction (Fig. 1A). At this time, an average of 250 g of cells (wet pellet) was harvested per liter of culture.
FIGURE 1.

TAP expression in P. pastoris. A, large-scale production of TAP by fermentation. The expression level of TAP in crude membranes (35 μg of protein) was analyzed at different time points by SDS-PAGE (10%) and immunoblotting using α-TAP1 (148.3) and α-TAP2 (435.3) antibodies. B, transport activity of TAP. Crude membranes (45 μg of protein) were incubated with fluorescein-labeled peptide (0.5 μm of RRYC(F)KSTEL) in the presence (10 mm, black bar) or absence (1 unit of apyrase, white bar) of ATP. Peptide transport was inhibited by the viral factor ICP47 (10 μm) or US620–146 (50 μm). Inactive US620–125 served as negative control. C and D, transport kinetics of TAP in crude membranes. Translocation assays were performed with increasing concentrations of either RRYC(F)KSTEL or ATP. Data were fitted to the Michaelis-Menten equation. Error bars in panels B–D show S.D.
To analyze function of the heterologously expressed TAP complex, we established a peptide transport assay in P. pastoris. As shown in Fig. 1B, transport activity was ATP-specific, yielding a 5-fold higher transport activity than controls in the absence of ATP (apyrase-treated). Importantly, translocation activity was TAP-specific because both the herpesviral inhibitor ICP47, preventing peptide binding to TAP (24), as well as the cytomegaloviral inhibitor US620–146 active domain, which inhibits ATP binding to TAP (25), completely blocked transport activity. The inactive domain US620–125 did not affect peptide transport. The Michaelis-Menten values Km,peptide of 0.34 ± 0.12 μm (Fig. 1C) and Km,ATP of 96 ± 41 μm (Fig. 1D) were similar to those observed in human and insect cell membranes (21).
Solubilization of TAP
For mechanistic analysis, purification of the TAP complex was a key requirement. As a first critical step, the TAP complex had to be extracted from its native lipid environment by detergents maintaining its stability and function. This was a challenging task due to the fact that the TAP complex is highly hydrophobic (20 transmembrane segments), prone to aggregate or to disassemble into its subunits. We used immunoblotting to analyze the solubilization efficiency of TAP1 and TAP2 in detergents commonly used for membrane protein purification and crystallization. Most detergents were able to solubilize TAP; however, some preferentially solubilized only one subunit, and others such as n-octylglucoside failed to extract either subunit from the membrane (Fig. 2A).
FIGURE 2.

Specific detergents maintain TAP function. A, solubilization efficiency in different detergents. Crude membranes (500 μg of protein) were solubilized in different detergents (2%, w/v). Detergent extracts were analyzed by SDS-PAGE (10%) and immunoblotting. The solubilization efficiency of TAP1 (black bars) and TAP2 (gray bars) was compared with the total amount of TAP in crude membranes (cm). UDM, n-undecyl-β-d-maltoside; FC-14, Fos-Choline-14; OG, n-octylglucoside; TX-100, Triton X-100; NP40, Nonidet P-40; LDAO, lauryldimethylamine-oxide. B, TAP function in different detergents. Crude membranes were solubilized (as described in A), and peptide binding of solubilized TAP was probed with 125I-labeled peptide RRYQKSTEL (1 μm) in the presence or absence of 200-fold excess of unlabeled peptide. Specific binding was normalized to that observed in digitonin, which gave the highest binding efficiency. Each data point represents the mean of triplicate measurements; error bars show S.D. Asterisk: no binding detectable. C, long-term stability of solubilized TAP. Peptide binding to TAP was analyzed after several days of incubation at 4 °C and normalized to specific binding at day 1 in the presence of digitonin. Error bars show S.D.
The function of solubilized TAP was examined in peptide binding assays. TAP solubilized in digitonin demonstrated the highest binding of peptide RRYQKSTEL, similar to TAP expressed in insect cells (21). Notably, DDM, which gave transient activity in the insect expression system, resulted in the loss of peptide binding in the Pichia system. Besides digitonin, Tween 20 also maintained binding activity, although to a lesser extent than digitonin (Fig. 2B). Importantly, TAP-dependent peptide binding demonstrated a half-life of approximately 5 days in the presence of digitonin, whereas shorter half-lives of 1–3 days were observed in Tween 20, lauryldimethylamine oxide, and DDM (Fig. 2C).
Purification of the TAP Complex
A multiscreen strategy was chosen to identify optimal conditions for purification. We used a 96-well-based metal affinity matrix to screen a broad range of detergents and buffer conditions. n-Octylglucoside and CHAPS extracted little TAP from the membrane and were therefore unsuitable for subsequent purification (supplemental Fig. S1). Fos-Choline-14 disrupted the TAP complex as demonstrated by reduced recovery of TAP2 in comparison with His-tagged TAP1 (Fig. 3A). Interestingly, the chain length of n-alkyl-β-d-maltosides strongly affected TAP solubilization and purification. Although n-undecyl-β-d-maltoside and DDM gave relatively good yields of purified TAP in the presence of high salt, little protein was recovered in n-decyl-β-d-maltoside (DM), probably due to inefficient solubilization. TAP recovery improved at high salt and detergent concentrations, especially with Tween 20, which required 500 mm NaCl and 2% detergent for solubilization (supplemental Fig. S1). Results with Triton X-100, Nonidet P-40, and DDM illustrated the importance of detergent screening. Although all three produced high yields of purified TAP, none preserved peptide binding. In summary, only digitonin allowed the purification of functional TAP complex. A complete evaluation of the screen with quantification of the relative TAP1 and TAP2 levels can be found in supplemental Fig. S1.
FIGURE 3.

Purification of the TAP complex. A, purification screen. Crude membranes (10 mg of protein) were solubilized with 1% or 2% (w/v) of detergent at either low-salt (150 mm) or high-salt (500 mm) concentrations. TAP was purified by metal affinity chromatography. Recovery was analyzed by immunoblotting using α-TAP1 and α-TAP2 antibodies. All conditions were analyzed in independent duplicates. Representative results for digitonin, Fos-Choline-14 (FC-14), DM, and DDM are shown; all other results are summarized in supplemental Fig. S1. Error bars show S.D. B, TAP purification. Crude membranes were solubilized in 2% digitonin and purified by metal affinity chromatography. Purified protein was analyzed by SDS-PAGE (10%). C, monodispersity of the purified TAP complex was verified by size exclusion chromatography (Superose 6 PC 3.2/30). V0, void volume; Vt, total volume; the elution volumes of thyroglobulin (669 kDa) and bovine serum albumin (67 kDa) are indicated. mAU, milliabsorbance units.
Based on these data, we established the following large-scale purification protocol. Crude membranes were solubilized with 2% digitonin at high salt. TAP was purified via the His10 tag on TAP1 by metal affinity chromatography. Washing and elution were accomplished in low-salt conditions, and the digitonin concentration was reduced to 0.05% (w/v), approximately two times its CMC. SDS-PAGE followed by Coomassie Blue or silver staining showed the purity of the final TAP fraction to be >95% (Fig. 3B). In size exclusion chromatography, purified TAP (theoretical mass 156 kDa) eluted as a single, symmetric peak with no evidence of aggregates or impurities. The apparent molecular mass was ∼250 kDa, consistent with a heterodimeric complex with associated detergent (Fig. 3C). The final yield after purification was 30 mg of functional TAP complexes per liter of culture, which is remarkably high for a membrane protein complex expressed in P. pastoris (19, 36) and 50-fold higher per liter than the previously described expression in cultured insect cells (21).
Activity of Purified TAP
We next compared the activity of TAP purified from P. pastoris or insect cells under optimal conditions. Binding assays using the radiolabeled peptide RRYQKSTEL revealed an affinity of ∼1 μm, independent of the expression system. However, the maximal binding capacity (Bmax) of TAP purified from P. pastoris was higher, demonstrating more functional complexes (Fig. 4A).
FIGURE 4.

Activity of purified TAP. A, peptide binding. TAP (10 μg) purified from P. pastoris (filled circles) or insect cells (open circles) was incubated with increasing amounts of 125I-labeled peptide RRYQKSTEL in the absence or presence of a 200-fold excess of unlabeled peptide. Data were fitted to a Langmuir (1:1) binding isotherm. B, ATP binding. 3.5 μg of purified TAP was bound to ATP-agarose in the presence of increasing concentrations of ATP. TAP bound to ATP-agarose was analyzed by SDS-PAGE and immunoblotting. Intensities were quantified and fitted to a dose-response curve. C, TNP-ATP binding. Purified TAP (0.1 μm) was incubated with increasing amounts of TNP-ATP. The fluorescence intensity at λex/em 410/541 nm was plotted against the respective TNP-ATP concentration, and data were fitted to a Langmuir (1:1) binding isotherm. D, competitive binding assay. Purified TAP (10 μg) was incubated with 6.5 μm TNP-ATP in the presence of increasing concentrations of ATP. Fluorescence intensity at λex/em 410/541 nm was plotted against ATP concentration. E and F, peptide stimulated ATP hydrolysis of TAP. Purified TAP (6 μg) was measured with increasing concentrations of RRYQKSTEL (E) or ATP (F) and fitted to the Michaelis-Menten equation. Each data point represents the mean of triplicate measurements. Error bars show S.D.
Based on the amount and purity of TAP, the ATP binding affinity of the purified transport complex could be determined. Because of the low affinity of ABC transporters for ATP (37), a direct binding approach with radiolabeled nucleotides is difficult. We therefore used competitive binding assays. Specific binding of TAP to ATP-agarose in the presence of increasing ATP concentrations revealed a half-maximal inhibitory concentration (IC50) of ∼20 μm (Fig. 4B). Alternatively, the ATP binding affinity of TAP was determined by competition with TNP-ATP. TNP-ATP binds to ABC proteins with higher affinity than ATP and is hydrolyzed slowly (38). TNP nucleotides are weakly fluorescent in aqueous solution, but the quantum yield is greatly enhanced in hydrophobic environments, such as in the nucleotide-binding pocket of the NBD. As for other ABC transporters (19, 37), TNP-ATP binds to TAP with high affinity, at a KD of 3.6 ± 0.5 μm (Fig. 4C). The IC50 value for competition of TNP-ATP was 150 μm ATP (Fig. 4D), from which we calculated a Ki,ATP of ∼53 μm for ATP binding to TAP.
As known for other ABC transporters, detergents can uncouple substrate binding from ATP hydrolysis (39, 40), possibly due to the lack of an optimal lipid environment. Notably, TAP purified in digitonin displayed tight coupling of ATPase activity to peptide binding, with no basal ATPase activity (Fig. 4E). ATP hydrolysis showed classical Michaelis-Menten kinetics with Km,peptide of 0.76 ± 0.18 μm (Fig. 4E) and Km,ATP 0.68 ± 0.10 mm (Fig. 4F). The peptide affinity of TAP was in excellent agreement with the value measured in ER membranes, consistent with the tight coupling of peptide binding and ATP hydrolysis. However, the Km,ATP value of solubilized TAP was significantly higher than that observed in the membrane, indicating an influence of the membrane environment.
Functional Reconstitution of TAP into Proteoliposomes
We next reconstituted TAP into Triton X-100 destabilized unilamellar liposomes (diameter ≤ 400 nm) prepared from E. coli total lipid. The lipid-to-protein ratio was 10:1 (w/w), and detergent was slowly removed via polystyrene beads. Because detergent removal can promote protein aggregation, we separated TAP-containing proteoliposomes from empty liposomes and protein aggregates by Ficoll gradient centrifugation (Fig. 5A). Gradient fractions were analyzed for lipid and protein content, revealing a reconstitution efficiency of about 90% (Fig. 5A). The orientation of protein insertion into the vesicles was determined by TEV protease cleavage of the TAP1 C-terminal His tag. Incorporation of TAP with ∼55% of the NBDs facing the outside was observed (Fig. 5B). Freeze-fracture transmission electron microscopy confirmed that TAP was incorporated into the lipid bilayer and not merely surface-bound (Fig. 5C).
FIGURE 5.

Reconstitution of TAP into proteoliposomes. A, quantification of lipid and protein after gradient centrifugation. Purified TAP was reconstituted into liposomes prepared from E. coli total lipids. Proteoliposomes were separated by Ficoll density gradient centrifugation (0–10%; left image), and gradient fractions were assayed for lipids (right panels, upper) and TAP (right panels, lower). Empty and filled liposomes are indicated as open and filled circles, respectively. Corresponding fractions are marked by triangles. B, orientation of TAP in proteoliposomes. Proteoliposomes containing TAP (2.5 μg) were incubated with TEV protease in the presence or absence of 1% TX-100. After TEV cleavage, samples were analyzed by SDS-PAGE (10%) and immunoblotting using antibodies specific for TAP1, TAP2, and His6 tag. P, purified protein; R, reconstituted protein. TX-100, Triton X-100. C, freeze-fracture EM. Particles indicate incorporated TAP complexes.
TAP Function Depends on Specific Lipids
The activity of the TAP complex reconstituted into E. coli lipids was determined by peptide binding and transport assays. Strikingly, after reconstitution, all functional parameters of TAP determined in the membrane were restored (Table 1). Km,peptide 0.32 ± 0.09 μm of TAP reconstituted in proteoliposomes was twice lower than in detergent solution (Fig. 6A, Table 1). However, Km,ATP 22 ± 5 μm was 30-fold lower than in detergent solution and much closer to values measured in cell membranes (Fig. 6B, Table 1). These findings demonstrate the influence of the lipid environment on TAP function.
TABLE 1.
Km values for ATP hydrolysis
| TAP complex | Km,peptide | Km,ATP |
|---|---|---|
| μm | μm | |
| P. pastoris membrane | 0.34 ± 0.12 | 96 ± 41 |
| Purified in 0.05% digitonin | 0.76 ± 0.18 | 680 ± 90 |
| Reconstituted into E. coli lipids | 0.32 ± 0.09 | 22 ± 5 |
FIGURE 6.

TAP transport activity depends on the lipid composition. A and B, steady-state kinetics of peptide transport by reconstituted TAP. TAP was reconstituted into liposomes prepared from E. coli total lipids, and peptide transport was measured in the presence of varying concentrations of peptide (A) or ATP (B). Data were fitted to the Michaelis-Menten equation. C, cholesterol inhibits TAP activity. TAP was reconstituted into liposomes prepared with different lipid mixtures, and transport activity was determined. D, transport activity of reconstituted TAP depends on specific lipids. Activity of TAP reconstituted into liposomes made from most abundant phospholipids of the ER was analyzed as in C. Ratios of ER lipids are DOPC/DOPE/DOPS/PI/lyso-PC/sphingomyelin (61:19:3:9:4.5:5). Error bars show S.D. Asterisk, no transport detectable. Chol, cholesterol.
To examine this effect of the lipid environment more closely, we for the first time analyzed peptide transport activity of TAP reconstituted into different lipid mixtures. Transport assays were performed with proteoliposomes normalized to peptide binding. Notably, TAP showed the highest peptide transport activity after reconstitution into E. coli or E. coli/PC lipids, whereas total lipid extracts from B lymphoblastoid Raji cells (preparation described in supplemental procedures) led to a reduction of transport activity (Fig. 6C). These differences might perhaps be explained by the presence of cholesterol. Indeed, the addition of 10% cholesterol to E. coli lipids resulted in a significant decrease (74%) in transport activity. Conversely, reconstitution of TAP into lipids mimicking the cholesterol-poor composition of the ER membrane (41) resulted in higher transport activity.
E. coli lipids, which promoted TAP activity, are composed of a relatively high ratio of PE (42), which might be required for optimal transport activity. To address the influence of the polar head group, we reconstituted TAP into mixtures of the most abundant ER lipids, namely PC, PE, and PI. The presence of PI or PE drastically increased TAP transport activity, whereas PC or PC with 10% cholesterol did not promote TAP function (Fig. 6D). Only low activity was observed in PC/PG mixtures, excluding a simple charge effect. Remarkably, PI, even at very low molar ratios, enhanced peptide transport, suggesting an important role in TAP function.
Associated Lipids Are Essential for TAP Function
To investigate the functional correlation between specific lipids and peptide binding in detail, the amount of phospholipids associated with purified TAP complex was determined. Digitonin-purified TAP binds 3.9 ± 0.6 mol of Pi per mole of TAP complex, corresponding to approximately four associated phospholipids. Interestingly, an almost complete delipidation of the translocation complex was observed in the presence of DDM (0.8 ± 0.3 mol Pi per mole TAP), suggesting that loss of TAP function is directly associated with delipidation (Fig. 7A). In line with these findings, peptide binding of DDM-purified TAP was restored by reconstitution into soybean polar extract lipids, composed of PC, PE, and PI lipid species in a ratio of about 2:1:1. Notably, reconstitution into liposomes, prepared with PC lipids only, showed no restoration (Fig. 7B). The importance of TAP-associated lipids (extraction is described in supplement) was demonstrated by extracting those bound to digitonin-purified TAP and using them for reconstitution of the TAP complex purified in DDM (Fig. 7B). In this case, a recovery of peptide binding of 69.4 ± 7.2% was achieved.
FIGURE 7.

Associated lipids modulate TAP function. A, DDM causes delipidation of the TAP complex. After purification in digitonin or DDM, TAP-associated phospholipids were determined via the amount of inorganic phosphate. Each data point represents the mean value of triplicate measurements. B, recovery of peptide binding activity after reconstitution. DDM-purified TAP was reconstituted into liposomes prepared of DOPC; PC was doped with PE and PI lipids (soybean polar extract) or into lipids extracted after purification with digitonin. Peptide binding to purified (P) and reconstituted TAP (R) was analyzed, compared with DDM-purified TAP (Ø), and normalized to protein orientation as determined by TEV cleavage. C, after purification in digitonin, TAP-associated lipids were profiled by LC FT-MS (representative spectrum, positive ion mode). Identified lipid species are annotated by m/z, and sum composition (total carbon atoms in acyl chains:double bonds) is shown. D, comparison of PE/PC and PI/PC intensity ratios from the TAP complex (black bars) and in total cell extracts (white bars). Error bars in A and B show S.D. Rel. intensity, relative intensity.
Based on these results, we focused on profiling TAP-associated lipid species by LC FT-MS analysis. Remarkably, we observed that the purified TAP complex preferentially binds PI 34:1, PI 34:2, PE 34:2, PE 36:3, PC 34:2, and PC 36:3 (Fig. 7C). Comparison with whole cell lipid extracts showed a significant increase of PI, and to a lesser extent, PE associated with TAP (supplemental Fig. S2). Comparing the PE/PC and PI/PC ratios in membranes and purified TAP confirmed these results (Fig. 7D). PE lipids were enriched 18% in the extract of the TAP complex, whereas the PI species were more than doubled (∼114%). The composition of monitored PC, PE, and PI species is given in (supplemental Fig. S3). Notably, the MS data revealed only unsaturated phospholipids associated with the TAP complex, which is consistent with the elevated concentration of lipids having unsaturated fatty acid moieties in the ER membrane (43). In conclusion, TAP function critically depends on specific lipids and the membrane environment.
DISCUSSION
Functional and structural analysis of membrane proteins is often impaired by limited yield, stability, and function of the purified protein. In the case of the ABC transporter TAP, this is even more challenging because (i) TAP functions as a heterodimeric complex, (ii) preformed TAP1 serves as a scaffold for the folding of TAP2 (44), and (iii) solubilized TAP is highly unstable and prone to aggregate or to dissociate into its subunits (21). Earlier attempts of TAP expression in human cells (45), Saccharomyces cerevisiae (46), and insect cells (20) allowed the investigation of some important aspects of the antigen translocation machinery. However, limited protein yields have so far hampered detailed mechanistic analyses of the TAP complex, in particular the influence of specific lipids on TAP function.
Recently, the production of several mammalian membrane proteins in P. pastoris has been reported, resulting in structure determination of the voltage-dependent shaker channel Kv1.2 (16) and of the multidrug transporter P-glycoprotein (14). In this study, we report the production of functional TAP complex in P. pastoris in high yields. TAP function in P. pastoris membranes was confirmed by peptide transport activity that could be blocked by TAP-specific viral inhibitors. A Km,peptide of 0.34 ± 0.12 μm and a Km,ATP of 96 ± 41 μm were determined (Table 1). After extensive screening, optimal conditions for TAP solubilization and purification were established. Digitonin fully preserved the peptide binding activity of TAP, with a half-life of approximately 5 days. All other detergents, even those with good solubilization and purification efficiencies, showed only residual or no peptide binding activity, which might be attributable to the delipidation of the translocation complex.
A procedure based on the results of these detergent screens allowed purification of functional TAP to >95% purity, with a yield of 30 mg/liter of P. pastoris culture. Size exclusion chromatography verified monodispersity of the purified TAP complex, which retained peptide and ATP binding. Importantly, the ATPase activity of detergent-solubilized, purified TAP exhibited a markedly higher Km value for ATP than did the membrane-embedded complex (Table 1). Perhaps differences in lateral membrane pressure affected the protein conformation (47, 48), thereby influencing the coupling between peptide binding and ATP hydrolysis. Indeed, reconstitution of TAP into E. coli liposomes restored the Km for ATP to that observed in cell membranes. This clearly demonstrates for the first time a strong and direct effect of the lipid bilayer on TAP function.
This result was extended further by analyzing TAP activity in different lipid mixtures. Our data demonstrate a direct influence of the phospholipid head group on peptide transport. In the ER membrane, TAP encounters mainly phospholipids such as PC, PE, and PI. Strikingly, reconstitution of purified TAP into an ER-like lipid environment restored TAP transport activity. PC as matrix lipid had no effect on peptide transport, whereas PI and PE stimulated the translocation activity of TAP. PI exerted this effect even at low concentrations. Because reconstitution into PC/PG mixtures did not increase TAP activity, we conclude that the head group of the phospholipids rather than the charge of the lipid is important for TAP function.
Based on the high yields of purified TAP, specifically associated lipids were profiled by a lipidomic approach based on LC FT-MS. Functional TAP complex is associated with PC, PI, and PE lipids. Remarkably, PI was detected at higher levels than PE, confirming reconstitution data. The functional importance of specific lipids was demonstrated by reactivation of TAP by supplementation of the identified lipids. We further propose that PC is not directly involved in TAP function but plays an important role as a matrix lipid, presumably by sustaining a required lateral membrane pressure.
The present study also showed drastic impairment of TAP function by cholesterol, which is a well known modulator of plasma membrane ABC transporters (49–51). To our knowledge, this study is the first demonstrating a negative influence of cholesterol on an intracellular ABC transporter. Although cholesterol is primarily synthesized in the ER, it distributes rapidly along the secretory pathway. As a result, the ER membrane contains only very low cholesterol concentrations (52). However, recent findings demonstrated that TAP resides not only in the ER but is also localized to the ER-Golgi intermediate compartment, Golgi compartment (53), and early endosomal compartments (54). Hence, cholesterol potentially could serve as a novel regulatory mechanism for TAP function in different subcellular compartments.
Acknowledgments
We thank Karin Schlattmann and Christina Köppler for excellent technical assistance and Ariane Zutz, Dr. Dominik Barthelme, and Dr. Rupert Abele for helpful discussions. Dr. Ellen Hildebrandt edited the manuscript.
This work was supported by the German Research Foundation (SFB 807 Transport and Communication across Biological Membranes, Grants TA157/7 and AB149/1 to R. T.), the United States Department of Defense (Grant W81XWH-05-1-0316 to I. L. U.), the Danish Council for independent Resarch (Grant DOK1155860 to C. S. E.), the Lundbeckfonden (Grant R45-A4342 to C. S. E.), and the European Drug Initiative on Channels and Transporters (EDICT to R. T.) funded by the European Commission Seventh Framework Program.

The on-line version of this article (available at http://www.jbc.org) contains supplemental experimental procedures and Figs. S1–S3.
- TAP
- transporter associated with antigen processing
- ABC
- ATP-binding cassette
- ER
- endoplasmic reticulum
- DDM
- n-dodecyl-β-d-maltopyranoside
- DM
- n-decyl-β-d-maltopyranoside
- DOPC
- 1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPE
- 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- LC FT-MS
- liquid chromatography Fourier transform-mass spectrometry
- NBD
- nucleotide-binding domain
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PG
- phosphatidylglycerol
- PI
- phosphatidylinositol
- TEV
- tobacco etch virus
- TNP-ATP
- 2′,3′-O-(2,4,6-trinitrophenyl)-ATP
- IMAC
- immobilized metal ion affinity chromatography
- CMC
- critical micelle concentration
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phosphoserine
- IDA
- iminodiacetic acid.
REFERENCES
- 1. Procko E., O'Mara M. L., Bennett W. F., Tieleman D. P., Gaudet R. (2009) FASEB. J. 23, 1287–1302 [DOI] [PubMed] [Google Scholar]
- 2. Parcej D., Tampé R. (2010) Nat. Chem. Biol. 6, 572–580 [DOI] [PubMed] [Google Scholar]
- 3. Davidson A. L., Dassa E., Orelle C., Chen J. (2008) Microbiol. Mol. Biol. Rev. 72, 317–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hollenstein K., Dawson R. J., Locher K. P. (2007) Curr. Opin. Struct. Biol. 17, 412–418 [DOI] [PubMed] [Google Scholar]
- 5. Androlewicz M. J., Ortmann B., van Endert P. M., Spies T., Cresswell P. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 12716–12720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Endert P. M., Tampé R., Meyer T. H., Tisch R., Bach J. F., McDevitt H. O. (1994) Immunity 1, 491–500 [DOI] [PubMed] [Google Scholar]
- 7. Uebel S., Meyer T. H., Kraas W., Kienle S., Jung G., Wiesmüller K. H., Tampé R. (1995) J. Biol. Chem. 270, 18512–18516 [DOI] [PubMed] [Google Scholar]
- 8. Chen M., Abele R., Tampé R. (2003) J. Biol. Chem. 278, 29686–29692 [DOI] [PubMed] [Google Scholar]
- 9. Herget M., Oancea G., Schrodt S., Karas M., Tampé R., Abele R. (2007) J. Biol. Chem. 282, 3871–3880 [DOI] [PubMed] [Google Scholar]
- 10. Oancea G., O'Mara M. L., Bennett W. F., Tieleman D. P., Abele R., Tampé R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5551–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gaudet R., Wiley D. C. (2001) EMBO. J. 20, 4964–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Procko E., Ferrin-O'Connell I., Ng S. L., Gaudet R. (2006) Mol. Cell. 24, 51–62 [DOI] [PubMed] [Google Scholar]
- 13. Schrodt S., Koch J., Tampé R. (2006) J. Biol. Chem. 281, 6455–6462 [DOI] [PubMed] [Google Scholar]
- 14. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., Chang G. (2009) Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson B. J., Lee J. Y., Pickert A., Urbatsch I. L. (2010) Biochemistry 49, 3403–3411 [DOI] [PubMed] [Google Scholar]
- 16. Long S. B., Campbell E. B., Mackinnon R. (2005) Science 309, 897–903 [DOI] [PubMed] [Google Scholar]
- 17. Abacioglu Y. H., Fouts T. R., Laman J. D., Claassen E., Pincus S. H., Moore J. P., Roby C. A., Kamin-Lewis R., Lewis G. K. (1994) AIDS Res. Hum Retroviruses 10, 371–381 [DOI] [PubMed] [Google Scholar]
- 18. Wang Z., Stalcup L. D., Harvey B. J., Weber J., Chloupkova M., Dumont M. E., Dean M., Urbatsch I. L. (2006) Biochemistry 45, 9929–9939 [DOI] [PubMed] [Google Scholar]
- 19. Lerner-Marmarosh N., Gimi K., Urbatsch I. L., Gros P., Senior A. E. (1999) J. Biol. Chem. 274, 34711–34718 [DOI] [PubMed] [Google Scholar]
- 20. Meyer T. H., van Endert P. M., Uebel S., Ehring B., Tampé R. (1994) FEBS. Lett. 351, 443–447 [DOI] [PubMed] [Google Scholar]
- 21. Herget M., Kreissig N., Kolbe C., Schölz C., Tampé R., Abele R. (2009) J. Biol. Chem. 284, 33740–33749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moor H., Mühlethaler K. (1963) J. Cell. Biol. 17, 609–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baykov A. A., Evtushenko O. A., Avaeva S. M. (1988) Anal. Biochem. 171, 266–270 [DOI] [PubMed] [Google Scholar]
- 24. Ahn K., Meyer T. H., Uebel S., Sempé P., Djaballah H., Yang Y., Peterson P. A., Früh K., Tampé R. (1996) EMBO. J. 15, 3247–3255 [PMC free article] [PubMed] [Google Scholar]
- 25. Kyritsis C., Gorbulev S., Hutschenreiter S., Pawlitschko K., Abele R., Tampé R. (2001) J. Biol. Chem. 276, 48031–48039 [DOI] [PubMed] [Google Scholar]
- 26. Cheng Y., Prusoff W. H. (1973) Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 27. Stewart J. C. (1980) Anal. Biochem. 104, 10–14 [DOI] [PubMed] [Google Scholar]
- 28. Cogan E. B., Birrell G. B., Griffith O. H. (1999) Anal. Biochem. 271, 29–35 [DOI] [PubMed] [Google Scholar]
- 29. Valiyaveetil F. I., Zhou Y., MacKinnon R. (2002) Biochemistry 41, 10771–10777 [DOI] [PubMed] [Google Scholar]
- 30. Ejsing C. S., Sampaio J. L., Surendranath V., Duchoslav E., Ekroos K., Klemm R. W., Simons K., Shevchenko A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 2136–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brierley R. A., Davis G. R., Holtz G. C., Gleeson M. A., Howard B. D. (March 18, 1997) U. S. Patent 5612198
- 32. Cregg J. M., Vedvick T. S., Raschke W. C. (1993) Biotechnology 11, 905–910 [DOI] [PubMed] [Google Scholar]
- 33. Scorer C. A., Clare J. J., McCombie W. R., Romanos M. A., Sreekrishna K. (1994) Biotechnology 12, 181–184 [DOI] [PubMed] [Google Scholar]
- 34. Zhang W., Bevins M. A., Plantz B. A., Smith L. A., Meagher M. M. (2000) Biotechnol. Bioeng. 70, 1–8 [DOI] [PubMed] [Google Scholar]
- 35. Zhang W., Inan M., Meagher M. M. (2000) Biotechnol. Bioprocess. Eng. 5, 275–287 [Google Scholar]
- 36. Chloupková M., Pickert A., Lee J. Y., Souza S., Trinh Y. T., Connelly S. M., Dumont M. E., Dean M., Urbatsch I. L. (2007) Biochemistry 46, 7992–8003 [DOI] [PubMed] [Google Scholar]
- 37. Oswald C., Jenewein S., Smits S. H., Holland I. B., Schmitt L. (2008) J. Struct Biol. 162, 85–93 [DOI] [PubMed] [Google Scholar]
- 38. Qu Q., Russell P. L., Sharom F. J. (2003) Biochemistry 42, 1170–1177 [DOI] [PubMed] [Google Scholar]
- 39. Reich-Slotky R., Panagiotidis C., Reyes M., Shuman H. A. (2000) J. Bacteriol. 182, 993–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davidson A. L., Maloney P. C. (2007) Trends Microbiol. 15, 448–455 [DOI] [PubMed] [Google Scholar]
- 41. Watanabe J., Asaka Y., Mino K., Kanamura S. (1996) J. Electron Microsc. (Tokyo) 45, 171–176 [DOI] [PubMed] [Google Scholar]
- 42. Raetz C. R., Dowhan W. (1990) J. Biol. Chem. 265, 1235–1238 [PubMed] [Google Scholar]
- 43. Shaikh S. R., Edidin M. A. (2006) Chem. Phys. Lipids. 144, 1–3 [DOI] [PubMed] [Google Scholar]
- 44. Keusekotten K., Leonhardt R. M., Ehses S., Knittler M. R. (2006) J. Biol. Chem. 281, 17545–17551 [DOI] [PubMed] [Google Scholar]
- 45. Gorbulev S., Abele R., Tampé R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3732–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Urlinger S., Kuchler K., Meyer T. H., Uebel S., Tampé R. (1997) Eur. J. Biochem. 245, 266–272 [DOI] [PubMed] [Google Scholar]
- 47. van den Brink-van der Laan E., Chupin V., Killian J. A., de Kruijff B. (2004) Biochemistry 43, 4240–4250 [DOI] [PubMed] [Google Scholar]
- 48. Marsh D. (2007) Biophys. J. 93, 3884–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang J., Sun F., Zhang D. W., Ma Y., Xu F., Belani J. D., Cohen J. C., Hobbs H. H., Xie X. S. (2006) J. Biol. Chem. 281, 27894–27904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takahashi K., Kimura Y., Kioka N., Matsuo M., Ueda K. (2006) J. Biol. Chem. 281, 10760–10768 [DOI] [PubMed] [Google Scholar]
- 51. Storch C. H., Ehehalt R., Haefeli W. E., Weiss J. (2007) J. Pharmacol Exp. Ther. 323, 257–264 [DOI] [PubMed] [Google Scholar]
- 52. van Meer G., Voelker D. R., Feigenson G. W. (2008) Nat. Rev. Mol. Cell. Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ghanem E., Fritzsche S., Al-Balushi M., Hashem J., Ghuneim L., Thomer L., Kalbacher H., van Endert P., Wiertz E., Tampé R., Springer S. (2010) J. Cell. Sci. 123, 4271–4279 [DOI] [PubMed] [Google Scholar]
- 54. Burgdorf S., Schölz C., Kautz A., Tampé R., Kurts C. (2008) Nat. Immunol. 9, 558–566 [DOI] [PubMed] [Google Scholar]