

Abstract
Prion proteins (PrP) can aggregate into toxic and possibly infectious amyloid fibrils. This particular macrostructure confers on them an extreme and still unexplained stability. To provide mechanistic insights into this self-assembly process, we used high pressure as a thermodynamic tool for perturbing the structure of mature amyloid fibrils that were prepared from recombinant full-length mouse PrP. Application of high pressure led to irreversible loss of several specific amyloid features, such as thioflavin T and 8-anilino-1-naphthalene sulfonate binding, alteration of the characteristic proteinase K digestion pattern, and a significant decrease in the β-sheet structure and cytotoxicity of amyloid fibrils. Partial disaggregation of the mature fibrils into monomeric soluble PrP was observed. The remaining amyloid fibrils underwent a change in secondary structure that led to morphologically different fibrils composed of a reduced number of proto-filaments. The kinetics of these reactions was studied by recording the pressure-induced dissociation of thioflavin T from the amyloid fibrils. Analysis of the pressure and temperature dependence of the relaxation rates revealed partly unstructured and hydrated kinetic transition states and highlighted the importance of collapsing and hydrating inter- and intramolecular cavities to overcome the high free energy barrier that stabilizes amyloid fibrils.
Keywords: Amyloid, Prions, Protein Assembly, Protein Conformation, Protein Denaturation, Protein Structure
Introduction
Amyloid fibrils are filamentous polypeptide aggregates that are associated with devastating disorders such as Alzheimer and Parkinson diseases, type II diabetes, and prion (proteinaceous infectious particle) diseases, including Creutzfeldt-Jakob and mad cow disease (1, 2). There is increasing evidence that under appropriate conditions the amyloid state is accessible also to many other proteins that are not related to diseases, suggesting that the amyloid state is a generic structural feature, which might be adopted by any polypeptide chain (3–6).
The development of in vitro model systems together with various biophysical and biochemical techniques (7–9) has improved the knowledge on the physicochemical basis of amyloid formation as well as on their structural and biochemical properties. It is now well recognized that a common property of amyloid fibrils is the extensive stacking of intermolecular β-strands that are arranged perpendicularly to the fibril axis and stabilized by a dense network of non-covalent interactions (10–13). These fibrils consist of a variable number and arrangement of thin assemblies called proto-filaments that give rise to different fibril morphologies of diameters between 5 and 30 nanometers, both in in vitro preparations or in tissues (14–22). Fibrils and their precursors are generally cytotoxic (23, 24) and are, thus, thought to be responsible for the neurodegeneration that is associated with many amyloid diseases.
Yet the fundamental parameters that govern the protein aggregation process and dictate fibril stability/clearance are not well known. As the study of protein folding has been greatly advanced by examining its reverse process (i.e. protein unfolding), the assessment of amyloid fibril disassembly could be valuable not only for determining the parameters that define their formation and stability but also for designing medical/biotechnological strategies to prevent or delay the formation of protein aggregates or to favor the clearance of amyloid deposits. Fibrils, although very stable in vitro, can undergo a continuous “molecular recycling” (25) and are susceptible to revert to monomeric forms in vivo upon a decrease in the levels of fresh amyloidogenic precursor (26–29). Dissociation of amyloid fibrils can be triggered by the addition of highly concentrated chemical denaturants such as urea or guanidinium hydrochloride or of trifluoroethanol and the use of high temperature (30–34). Although not as well studied, the application of elevated pressure has also been used to dissociate non-mature amyloid fibrils and proto-fibrils of several proteins (35–42).
We, thus, hypothesized that under high pressure mature fibrillar PrP structures should dissociate and eventually unfold. To prove this hypothesis, a high pressure and particularly the pressure-jump technique (43–46) was applied to study the activation energy parameters and structural changes involved in mature amyloid fibril disassembly by using the recombinant prion protein (PrP)3 as a model amyloidogenic protein. A rapid increase in pressure forced PrP amyloid fibrils to change irreversibly to a new, less cytotoxic state that was still partly fibrillar but lacked the typical structural features of amyloids. The analysis of the relaxation kinetics toward this new state gave information about the reaction mechanism and the transition state ensemble. The pressure-jump technique proved to be advantageous as 1) it does not require the introduction of a chemical reagent into the sample, 2) pressure propagates nearly instantaneously and homogeneously through the sample, and 3) the activation volumes of the reactions can be measured, thus providing structural (volumetric) information about the kinetic transition state, which cannot be obtained with other experimental approaches.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The gene encoding mPrP23–230 (murine full-length recombinant prion protein) was cloned into the pET22b(+) vector (Invitrogen) and expressed in Escherichia coli BL21(DE3) cells after isopropyl thio-β-d-galactoside induction. Recombinant PrP accumulated as inclusion bodies. After lysis, sonication, and solubilization of the inclusion bodies by guanidine hydrochloride, purification of PrP was performed essentially as described previously (47) using a nickel-Sepharose column. Refolding of the protein was achieved on the column by heterogeneous phase renaturation simultaneously with purification. Purified PrP was recovered in the desired buffer by elution through a G25 desalting column. The final protein concentration was measured by absorbance at 280 nm using an extinction coefficient of 63,495 m−1cm−1. Purified PrP was stored lyophilized.
Formation of Amyloid Fibrils
To form amyloid fibrils, PrP stock solutions were prepared immediately before use by resuspending lyophilized PrP in 50 mm MES, pH 6.0. The stock solution was diluted to a final protein concentration of 0.5 mg ml−1 with MES, pH 6.0 (final concentration, 50 mm) and guanidine HCl (final concentration, 2 m). Fibrillation reactions were performed in 1.5-ml conical plastic tubes in a total reaction volume of 0.6 ml at 37 °C with continuous shaking at 600 rpm using a Titramax 100 plate shaker (Heidolph). Fibril formation was monitored using a ThT binding assay (7). Fibrils were dialyzed in 10 mm sodium acetate, pH 5.0.
Fluorescence Measurements under High Pressure
Fluorescence measurements were carried out using an Aminco Bowman Series 2 fluorescence-spectrophotometer (SLM Aminco) modified to accommodate a thermostated high pressure optical cell that allowed measurements up to 700 MPa. Dialyzed fibrils were diluted in the same buffer to a final protein concentration of 0.125 mg ml−1 and placed in 5-mm diameter quartz cuvettes closed at the top with flexible polyethylene film that was kept in place by a rubber O-ring. Pressure-jumps consisted of rapid (within 30 s) changes from atmospheric pressure to a range of final pressures of 330–600 MPa. The pressure was then maintained for ∼90 min. A pressurization cycle was then completed by decompression of the sample to atmospheric pressure.
Protein disaggregation was followed by monitoring the changes in light-scattering intensity at 300 nm (4 nm slits). ThT (10 μm final concentration) was also used as a probe to measure the extrinsic fluorescence. ThT fluorescence intensity was recorded at 482 nm (16 nm slit) and excited at 385 nm using a 4-nm slit.
After each pressure-jump the relaxation profiles of the amyloid structural reaction were fitted to double exponential decays, according to Equation 1,
where I(t) and I0 are the fluorescence intensities at time t and at time 0, A and B are the phase amplitudes, and kobs is the measured apparent rate constant at the final pressure p.
The thermodynamic apparent activation parameters ΔH* and ΔS* were determined by fitting NAkBln(kobs/kobs0) =f(1/T) to Equation 2,
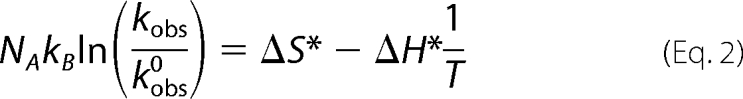 |
where kobs = 1/τ is the observed rate, kobs0 is the pre-exponential factor for the observed rate corrected for the change in medium viscosity due to change in pressure, NA is Avogadro's number, kB is the Boltzmann factor, and T is the absolute temperature in kelvin. From theory, kobs0 = 1.33kBTcNA(1/η), where c is the molar concentration of PrP protein, and η is the medium viscosity at a given pressure (48).
The change in apparent activation volume of the kinetic transition state was determined by fitting RTln(kobs/kobs0) = ΔG* to Equation 3,
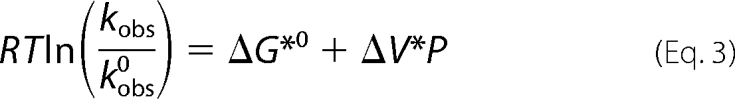 |
where ΔG*0 is the apparent activation free energy change at atmospheric pressure at a given temperature T.
Fluorescence Measurements at Atmospheric Pressure
Fluorescence measurements at atmospheric pressure were performed at 20 °C using a FluoroMax-2 fluorimeter (Jobin Yvon-Spex) and a 10 × 2-mm path length rectangular cuvette. Aliquots of soluble PrP, PrP fibrils, and PrP fibrils after pressure treatment were incubated with either 10 μm ThT for 1 min or 50 μm 8-anilino-1-naphthalene sulfonate (ANS) at room temperature for 10 min before monitoring the fluorescence. For ANS spectra, excitation was at 385 nm. ThT emission spectra were recorded after excitation at 450 nm. The excitation and emission slits widths were 4 nm. Each emission spectrum was the average of three scans.
Epifluorescence Microscopy
Sample preparation was carried out as described (7). Briefly, fibrils were diluted to 0.1 μm using the same buffer and stained with ThT (10 μm) for 3 min. Samples were analyzed with an inverted microscope (Zeiss Axiovert 200M). The emission was isolated from Rayleigh and Raman-shifted light by a GFP filter, with excitation at 455–495 nm, and emission at 505–555 nm. Digital images were acquired using an Axiocam MRm camera (Zeiss).
Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (FTIR) at Atmospheric Pressure
Infrared spectra were measured using a Bruker Tensor 27 FTIR instrument (Bruker Optics). Both untreated and pressure-treated samples were concentrated in 5 mm NaOAc, pD 5.0, to 1.5 mg ml−1 using Nanosep centrifugal devices with a 3-kDa cutoff. For each concentrated sample, 20 μl were loaded into BioATR II cell, and 512 scans were collected at 2 cm−1 resolution under constant purging with nitrogen. Average spectra were corrected for buffer and water vapor. Absorption bands were resolved by Fourier self-deconvolution using Lorentz parameters of 20 cm−1 bandwidth and a noise suppression factor of 0.3. Second derivatives of deconvoluted spectra were calculated using 13-point Savitzky-Golay smoothing.
FTIR Spectroscopy at High Pressure
PrP fibrils were concentrated by centrifugation. The fibril pellet was washed twice with 50 mm sodium acetate buffer, pH 5.0, and resuspended in the same buffer to a final protein concentration of 50 mg ml−1. The in situ pressure experiments were performed with a diamond anvil cell (DAC) using barium sulfate as an internal pressure calibrant (49). The pressure was raised to 540 MPa (within 2 min), and subsequently IR spectra were acquired every 5 min. Infrared spectra were recorded on a Bruker IFS66 FTIR spectrometer equipped with a liquid nitrogen-cooled mercury cadmium telluride detector at a nominal resolution of 2 cm−1. Each spectrum was the average of 256 interferograms. The sample compartment was continuously purged with dry air to minimize the spectral contribution of atmospheric water.
Atomic Force Microscopy
Samples of PrP fibrils before and after treatment with pressure were 100× diluted with ultrapure water to ∼5 μg ml−1. 5 μl of each sample were left on a freshly cleaved piece of mica at room temperature on the bench until dry. To analyze all the ingredients of fibril solutions, the washing step was omitted. Atomic force microscopy imaging was performed using a Pico LE system (Agilent Technologies). The atomic force microscopy scanner equipped with a silicon cantilever PPP-NCH (Nanosensors) was operated in tapping mode. Images (512 × 512 pixels) were collected at a scan rate of 1.5 lines s−1.
Transmission Electron Microscopy
Samples were deposited onto Formvar carbon-coated grids, negatively stained with freshly filtered 2% uranyl acetate, dried, and viewed in a JEOL 1200EX2 electron microscope at an accelerating voltage of 80 kV.
SDS-PAGE in Denaturing and Non-denaturing Conditions
To study whether monomeric soluble PrP was present before and after pressure treatment of PrP fibrils, samples were treated with denaturing (60 mm Tris-HCl, 2% SDS, 5% β-mercaptoethanol, 2.25 m urea, heating for 15 min at 90 °C) and non-denaturing buffer (same buffer but without SDS, β-mercaptoethanol, and urea; no heating). 12% SDS-PAGE gels (Criterion XT precast gel, Bio-Rad) were used for sample analysis. To estimate the amount of soluble protein, fibrils and insoluble aggregates were eliminated by centrifugation at 20,800 × g for 45 min.
Annealing of Fibrils and Proteinase K Digestion Assay
Tris-HCl buffer, pH 7.5, and Triton X-100 were added to untreated and pressure-treated PrP fibrils to final concentrations of 100 mm and 0.1%, respectively. Aliquots (8 μl, 0.1 mg ml−1 of PrP) were placed in 0.5-ml conical plastic tubes, incubated at 80 °C for 15 min, and cooled down. Fibrils were treated with proteinase K (proteinase K to PrP ratio of 1:100) at 37 °C for 1h. Digestion was stopped by adding PMSF. Samples were heated at 95 °C for 10 min and analyzed on 12% SDS-PAGE gels (Criterion XT precast gel, Bio-Rad) followed by silver staining.
Primary Neuronal Cells
Primary cell cultures of neurons were derived from the cerebral cortex of 17.5 E rat embryos. Cells were dissociated by enzymatic incubation in 0.05% trypsin-EDTA and by mechanical dissociation. Cells were resuspended in Neurobasal medium with 2% B27, 0.25% 200 mm glutamine, 1% Glutamax, and 1% penicillin/streptomycin and seeded to a density of 50,000 cells per dish on glass coverslips in 24-well plates previously coated with 10 μg ml−1 poly-d-lysine. Two days later, recombinant PrP isoforms (soluble, fibrils, or pressure-treated fibrils) were added at a final concentration of 1 μm. Separation of pressure-treated fibrils from soluble species was performed by centrifugation at 20,800 × g for 45 min. The pelleted fibrils, resuspended in 10 mm sodium acetate pH 5.0, and the supernatant were added to the cultured cells at a final concentration of 1 μm protein. After 72 h of treatment, the cytotoxic effect of each PrP conformer was analyzed by fixing cells in 4% paraformaldehyde solution and staining with Hoechst 33258. Apoptotic cells were identified by the characteristic nuclear bright blue fluorescence due to condensed or fragmented chromatin. Neurons were identified with primary mouse anti-β-3 tubulin antibodies at 1:400 (Sigma). Secondary antibodies were labeled with Cy3 and diluted 1:400 (Jackson ImmunoResearch). Digital images were captured from 5 random fields for each sample (∼500 cells total) using an Axiovert 200M Zeiss inverted microscope. Neuronal cell death was determined by counting the bright neurons (including those with fragmented or condensed nuclei) and expressed as a percentage of the total neuronal cell number compared with control cultures. Statistical analysis (Student's t test) was carried out on the results of three independent experiments.
RESULTS
Pressure-induced Amyloid Fibril Dissociation
By applying the method (manual set up) developed by the group of Baskakov and co-workers (7, 50), full-length mouse recombinant PrP were converted into amyloid fibrils under partially denaturing solvent conditions (2 m guanidine HCl, pH 6.0) and continuous shaking as described under “Experimental Procedures.” Mature amyloid fibrils were then dialyzed against 10 mm sodium acetate, pH 5.0.
To evaluate the effects of pressure on amyloid fibrils, 600 MPa (at 25 °C) was applied using the pressure-jump technique to PrP fibrils. The light-scattering intensity substantially decreased within ∼80 min (Fig. 1), and the initial intensity was not recovered upon depressurization, suggesting that pressurization led to irreversible, partial dissociation of PrP fibrils. The amount of resolubilized PrP rescued from the fibrillar form after the pressure treatment was about 30% that of the entire protein sample, as judged from the absorbance at 280 nm of the supernatants obtained after removing fibrils and insoluble aggregates by centrifugation. In accordance with the light-scattering results, the remaining insoluble fraction appeared to be stable, as an additional cycle of compression/decompression did not affect the balance between aggregated and soluble proteins.
FIGURE 1.

Pressure-induced amyloid fibril dissociation after a sudden increase of pressure to 600 MPa. The extent of structural changes was recorded as a decrease in light scattering intensity. The arrow denotes the point in the kinetics when a second cycle of compression (600 MPa)/decompression was carried out. The temperature was 25 °C. a.u., arbitrary units.
Analysis of ThT and ANS Binding to Pressure-treated Amyloid Fibrils
Moreover, after a cycle of compression (90 min at 600 MPa, 25 °C) and decompression to atmospheric pressure, the ThT fluorescence emission spectrum of amyloid fibrils was comparable with that of native PrP (Fig. 2A), demonstrating a complete and irreversible loss of their amyloid ThT binding capacity. In line with this observation, after decompression, amyloid fibrils could not be detected by fluorescence microscopy (Fig. 2B).
FIGURE 2.

Pressure-induced irreversible loss of ThT binding to amyloid fibrils. A, shown are ThT fluorescence emission spectra of native monomeric PrP protein (dashed and dotted line), untreated amyloid fibrils (solid line), and after a cycle of compression (600 MPa)/decompression at 25 °C (dashed line). B, shown are epifluorescence microscopy images of untreated PrP fibrils (left panel) and after a cycle of compression (600 MPa)/decompression at 25 °C (right panel). Fibrils were stained using ThT. a.u., arbitrary units.
Similar results were obtained with the fluorescent dye ANS. Indeed, ANS binding to pressure-treated fibrils resulted only in a minor increase of the fluorescence yield and did not show any evidence of a blue shift in the fluorescence spectrum. Conversely, upon binding to PrP fibrils, the ANS fluorescence spectrum was enhanced and blue-shifted in comparison to the weak fluorescence yield observed after binding to native, soluble PrP (supplemental Fig. S1).
Pressure-jump-induced Amyloid Structural Kinetics
To better understand the structural changes induced by pressure, we then measured the kinetics of amyloid fibril structural changes by monitoring ThT fluorescence after fast increases (within 30 s) of pressure to 600 MPa. Each pressure-jump produced double exponential kinetics that consisted of a relatively fast (relaxation time, τfast = 4.2 min) and a slower decay (τslow = 27.8 min), each encompassing about half of the total amplitude (Fig. 3). As shown in Fig. 3A, fitting the data to a mono-exponential decay was not satisfactory. Because both processes were irreversible and the amplitude of the fluorescence reduction did not vary significantly as a function of the pressure, we assumed that the individual rate constants for the backward reaction (k−1),
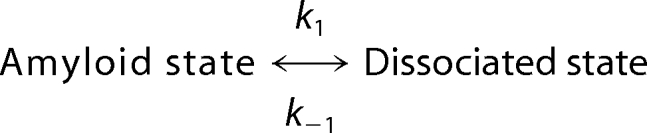 |
associated to the fast and slow phases were negligibly small, such that the relaxation time, τ = (1/kobs), predominantly reflected the inverse of the individual rate constant for the forward reaction τ = (1/k1).
FIGURE 3.

Kinetics of the pressure-induced dissociation of ThT from the amyloid fibrils. A, shown are residuals of the pressure-induced relaxation kinetics after fitting the data to a single and double exponential equation. Temperature (B) and pressure (C) dependence of the pressure-induced amyloid structural kinetics is shown. Pressure-jumps (from 0.1 to 600 MPa) were carried out at different temperatures; 15 °C (solid line), 25 °C (long dashed line), 35 °C (short dashed line), and 45 °C (dashed and dotted line). Pressure-jumps (at 25 °C) were performed to obtain different final pressures from 10 to 600 MPa (short and long dashed line), to 540 MPa (dashed and dotted line), to 470 MPa (short dashed line), to 400 MPa (long dashed line), and to 330 MPa (solid line). Insets show the pressure and temperature dependence of the observed relaxation times (τ) calculated from the fast phase (solid circles) and from the slow phase (open circles). Solid lines are linear fits to the data.
Temperature Dependence of the Relaxation Kinetics
The observed pressure-induced structural reactions accelerated when the temperature at which pressure-jumps were applied increased from 15 to 45 °C. Within our experimental temperature range, the Arrhenius plots of the relaxation times for the fast and slow phases were linear (Fig. 3B), with positive apparent activation energies of 72 ± 3 and 64 ± 8 KJ mol−1, respectively. These values suggest that the pressure-induced reaction could be ascribed to a chemical transformation process (47). Moreover, when the known contribution to binding energies of hydrogen bonds was taken into account (51), these apparent activation energy values pointed to significant conformational remodeling in the kinetic transition state.
Then, the free energy barriers associated with the pressure-induced process were estimated from the temperature dependence of the observed rate constants. To this aim, the relative contribution of apparent activation enthalpy and entropy to the rate constants was calculated using Equation 2 (see “Experimental Procedures”).
The resulting apparent activation parameters summarized in Table 1 showed that the pressure-induced reaction kinetics was controlled by competition between apparent activation enthalpy and entropy. Furthermore, the pressure-induced structural transition involved a high energy barrier, ΔG*, that accounted for the strong stability of amyloid fibrils at atmospheric pressure and physiological temperature.
TABLE 1.
Thermodynamic activation parameters
| Fast phase | Slow phase | |
|---|---|---|
| Temperature-dependent kinetics | ||
| ΔH* (kJ mol−1) | 54.4 ± 2.5 | 48.5 ± 3.3 |
| ΔS* (J mol−1 K−1) | 54.9 ± 8.1 | 21.4 ± 0.0 |
| TΔS*298K(kJ mol−1) | 16.4 ± 2.4 | 6.4 ± 0.0 |
| ΔG*298K (kJ mol−1) | 38.0 ± 3.4 | 42.1 ± 3.3 |
| Pressure-dependent kinetics | ||
| >ΔG*0298K (kJ mol−1) | 48.1 ± 0.8 | 54.6 ± 0.8 |
| ΔV* (ml mol−1) | −18.3 ± 1.7 | −23.8 ± 1.8 |
| ΔG*298K (kJ mol−1) | 37.1 ± 1.3 | 40.3 ± 1.3 |
Pressure Dependence of the Relaxation Kinetics
We performed pressure-jumps of different magnitude within a broad pressure range, with final pressures ranging from 330 to 600 MPa. The resulting relaxation kinetics of the amyloid structural changes were faster at high pressure, and the plots of relaxation times as a function of pressure were linear (Fig. 3C). The thermodynamic apparent activation parameters derived from the analysis are shown in Table 1.
The negative apparent activation volumes, ΔV* = −18 ± 2 and −24 ± 2 ml mol−1, for the fast and slow phase respectively, indicate that the fibrils in their ground state were more voluminous than in their kinetic transition state. Although the pressure-induced conformational change of mature fibrils was apparently prohibited by relatively large apparent activation free energies (ΔG*), these energetic barriers were compensated by negative contributions from ΔV*, leading to an acceleration of the reaction at increasing pressure.
Analysis of the Pressure-induced Structural Changes
To assess whether the soluble fraction produced by pressure-induced reactions contained monomeric PrP, samples (i.e. native monomeric PrP, PrP fibrils, PrP fibrils incubated at 25 °C at atmospheric pressure for 90 min, and PrP fibrils after a cycle of compression/decompression at 25 °C) were separated on SDS-PAGE gels under denaturing and non-denaturing conditions. Although, as expected, under denaturing conditions monomers were detected in all samples, under non-denaturing conditions a substantial amount of monomeric PrP was observed only after pressure treatment (Fig. 4).
FIGURE 4.

Pressure-induced partial disaggregation of amyloid fibrils into monomeric soluble PrP. Resolubilization was followed by silver staining of SDS-PAGE gels in denaturing and non-denaturing conditions. PrP samples were prepared in denaturing (60 mm Tris, 2% SDS, and 5% β-mercaptoethanol, 2.25 m urea; heating for 15 min at 90 °C) or non-denaturing sample buffer (no SDS/β-mercaptoethanol/urea/heating). Under native conditions, only non-fibrillar PrP enters the PAGE. Lane 1, PrP fibrils. Lane 2, fibrils after a cycle of compression (600 MPa)/decompression at 25 °C. SB, sample buffer.
We then evaluated whether pressure affected the generic physical properties of amyloid fibrils by using different procedures. First, changes in the protein secondary structure that accompany PrP fibrillation and fibril pressurization were assessed by FTIR spectral analysis of the amide I band (Fig. 5). The major contribution of the soluble native protein corresponded to the α-helical form (1653 cm−1), conforming to its structural prevalence (52). Amyloid fibrils showed strong peaks at 1630 and 1616 cm−1, which are characteristic of intermolecular β-sheet structures, and a peak at 1662 cm−1 that can be assigned to loop components with possible contributions from β-turns and α-helices. In contrast, pressure-treated samples showed an intermediate spectrum as the relative intensity of the band at 1630 cm−1 decreased and that of the 1653 cm−1 band increased, reflecting a partial loss of β-sheet structure with a concomitant gain in α-helical structure. As we used an attenuated total reflectance cell, which registers infrared absorption only in the thin lowest layer of the sample, these data concern predominantly structural features characteristic of PrP fibrils and much less of soluble PrP. Pressurization did not, however, lead to new spectral bands that were not present in the original native or fibrillar forms (supplemental Fig. S2). This was also confirmed by far UV CD analysis (results not shown). Hence, the observed changes in the secondary structure presumably reflect the pressure-induced partial dissociation of fibrils into soluble native PrP together with a structural reorganization of PrP subunits in fibrils.
FIGURE 5.

Pressure-induced changes in the secondary structure of amyloid fibrils. Fourier-deconvoluted IR spectra of the amide I region of native monomeric PrP (dashed and dotted line), untreated PrP fibrils (solid line), and after a cycle of compression (600 MPa)/decompression at 25 °C (dashed line).
We next measured the time course of the secondary structural changes after a pressure-jump from atmospheric pressure to 570 MPa. In agreement with the above mentioned results, an exponential decrease in band intensities assigned to β-sheet structure was observed, indicating that fibrils under pressure exhibit a lower content of β-sheet. In addition, we noticed a concomitant increase in band intensity around 1641 cm−1, indicating a conformational transition to a more disordered structure (supplemental Fig. S3).
Then the PK digestion assay was used to assess the presence and length of the PK-resistant core. Like untreated amyloid fibrils, pressure-treated fibrils showed strong resistance to PK digestion. However, their PK-digestion profile was different; the 12- and 10-kDa bands were slightly less intense and an additional 13-kDa band was detected (Fig. 6). These differences were further enhanced after annealing, a procedure specific for amyloid PrP structures that induces conformational rearrangements within PrP fibrils, accompanied by an extension of the PK-resistant core (53). Indeed, although untreated amyloid fibrils showed a substantial extension of the PK-resistant core and formation of a new 16-kDa PK-resistant band, consistent with previously reported results (53), pressure-treated fibrils were characterized by a very weak 16-kDa band, indicating a modified capacity of the resulting PrP species to undergo annealing. These results were consistent with the loss of structural and conformational integrity of PrP fibrils. Moreover, the appearance of two new bands at 13 and 24 kDa of unknown identity suggests that the PrP aggregates that persist after the pressure-induced transition are different from the original fibrils in terms of tertiary and/or quaternary structure.
FIGURE 6.

Altered digestion profile after pressure treatment of the PK-resistant core of amyloid fibrils. Silver staining of an SDS-PAGE gel of untreated and pressure-treated PrP fibrils after PK digestion is shown. The annealing procedure involves brief heating of the sample at 80 °C in the presence of detergent.
Finally, we investigated the macrostructure of this new pressure-induced conformer. An analysis of the fibril morphology in the starting PrP fibril samples by transmission electron microscopy (Fig. 7A) and atomic force microscopy (Fig. 7B) revealed polymorphic fibrils, indicating different association/twisting of individual filaments. After the pressure treatment, samples displayed remarkable differences compared with untreated amyloid fibrils; (i) the number of smaller fibrillar species and oligomers increased and (ii) the remaining fibrils appeared thinner, and in addition apparently single filaments were observed (supplemental Fig. S4).
FIGURE 7.

Macrostructure of the new pressure-induced conformer. Negative-stained transmission electron micrographs (A) and atomic force microscopy images (B) of PrP fibrils collected before (left panels) and after a cycle of compression (600 MPa)/decompression at 25 °C. Arrowheads show small oligomers and individual filaments.
Pressure Effects on Amyloid Fibril Toxicity
We then compared the neuronal cytoxicity of soluble native PrP, untreated amyloid fibrils, and pressure-treated amyloid fibrils by Hoechst nuclear staining of primary neuronal cells after incubation with the different forms (Fig. 8). Soluble native PrP caused only a minor toxic effect, whereas untreated amyloid fibrils were two times more toxic. In addition, cells clustered to form large clumps (Fig. 8A) as reported also in primary hippocampal neurons treated with fibrils (54). Pressure treatment highly reduced the toxicity of amyloid fibrils. Indeed, the toxicity of the pressurized sample was comparable with that of soluble native PrP (Fig. 8B). To evaluate whether the remaining minor toxicity of the pressure-treated sample was due to soluble or aggregated species, we separated the two fractions by centrifugation and then tested their toxicity at a final concentration of 1 μm in culture medium. Under these conditions the residual toxicity, estimated from the percentage of neuronal cell death, was due to both soluble (62.5%) and aggregate (37.5%) fractions.
FIGURE 8.

Pressure alters cytotoxicity of amyloid fibrils in primary neurons. A, photomicrographs of representative microscopic fields of cells stained with Hoechst 33258 and β3-tubulin showing the cytotoxic effect of the different PrP isoforms: soluble monomeric protein (upper panel; Soluble), fibrils (middle panel), and fibrils after a cycle of compression (600 MPa)/decompression at 25 °C (lower panel; P-treated fibrils). Scale bar, 20 μm. Neurons were cultured for 2 days and then incubated with the different PrP isoforms (final concentration, 1 μm). B, neuronal cell death quantification is shown. Each set of data is the mean value ± S.E. (in percentage) of three experiments; five independent microscopic fields were counted for each experiment. Asterisks indicate significant differences after Student's t test. ***, p < 0.001, untreated fibrils versus pressure-treated fibrils; untreated fibrils versus soluble protein. ns, non-significant difference between soluble protein and pressure-treated fibrils.
DISCUSSION
Pressure-induced Loss of Amyloid Features
Our starting hypothesis was that under high pressure mature fibrillar PrP structures should dissociate and eventually unfold. The present results show that the pressure-induced effects on mature prion amyloid fibrils are more complex than expected. About 30% of amyloid fibrils became dissociated into soluble species, whereas the remaining fibrils stayed in fibrillar form but lost some of the amyloid-specific features.
High pressure refolding of temperature-induced β-sheet-rich aggregates of a mammalian prion protein has been previously reported (55, 56). However, to our knowledge, none of the β-sheet-rich misfolded conformers was described to display amyloid fibril structure. In line with these reported effects of pressure on PrP aggregates, our in vitro results provide the first evidence that under mild physicochemical conditions (ambient temperature, absence of extraneous chemical agents) the use of high pressure can irreversible change the PrP fibril structure and unlock PrP from amyloid fibrils. It is, therefore, reasonable to think that unlocking PrP from the aggregate state could also account for the clearance process of amyloids observed in vivo (26–29). Because previous reports show that amyloid fibrils prepared from full-length recombinant mammalian prion protein are highly toxic to cultured cells and primary neurons (54) and display infectivity (57), our results may also explain the decreased resistance to proteolytic digestion and the reduction of the infectivity titer of brain homogenates, processed meat, and isolated pathogenic isoform, PrPSc (58–60) upon pressure treatment. Nevertheless, caution should be taken in extrapolating these findings, as the mechanisms linking PrP structural transformation to infectivity and neuropathological changes characteristic of prion diseases remain enigmatic and under debate (61).
After pressurization amyloid fibrils seem to form a new, not yet described PrP structural species in which the fibrillar macrostructure is similar to that of the original amyloid fibrils but the secondary, tertiary, and quaternary structures are significantly different, as judged from the lack of ANS (tertiary structure) and ThT binding (quaternary structure), the strong decrease in β-sheet content, and relative increase of α-helices (secondary structure) in comparison to untreated fibrils. The structural changes are further confirmed by the different PK digestion patterns of treated and untreated fibrils. Presumably, as a result of pressure-induced conformational rearrangements within the fibrillar substructure, the PrP central region (previously described as encompassing residues ∼90–140) (53) adopts an altered conformation with a more tightly packed structure that in turn may account for the modified annealing behavior. We, thus, suggest that pressurized fibrils should be considered as a new alternative conformation that can be reached by a protein under certain physicochemical conditions. Indeed, previous insight into unexplored conformational states of the recombinant cellular PrP isoform was obtained using high pressure (55, 56, 62–66). The results revealed that these PrP states are distinguished by their volumetric properties (i.e. hydration and packing).
How to explain these pressure-induced fibril structural changes? Due to the pressure dependence of the chemical potential under equilibrium or micro-equilibrium conditions, pressure influences chemical reactions that are characterized by a net volume change (67). For proteins, these volume changes arise from contributions of internal cavities and interactions with hydration water (68). Our findings suggest that the packing defects and hydrophobic pockets in mature PrP fibrillar structures disappear upon pressure treatment and that the sum of the dissociated prion protein conformers and the new fibrillar species occupy a smaller volume than the initial amyloid fibrils, in agreement with previous volumetric measurements of amyloid fibrils (35, 40, 69, 70). Moreover, several recent reports indicate the existence in amyloid fibrils of well defined ThT binding sites (71) that form cavities of about 8–9 Å in diameter. The absence of ThT fluorescence in pressurized fibrils can be explained by the collapse of these cavities, leading to ThT expulsion. Similarly, loss of ANS binding can be explained by the fact that ANS binds to intramolecular hydrophobic pockets, which might disappear in the pressurized fibrillar species.
Apparent Activation Energy Parameters
The kinetic data on the pressure-induced amyloid structural changes obtained with experiments performed at different temperatures and pressures permitted us to gain insight into the kinetic transition states of the reaction paths.
Particularly, the strong reduction in volume (ΔV* between −18 and −24 ml mol−1) suggests that the kinetic transition state is significantly more hydrated than the initial amyloid state. This might arise from the hydration of cavities or voids (due to their collapse) and from the polar alignment of initially bulk water around charged residues (electrostriction) at molecular surfaces that become exposed in the kinetic transition state. Water is indeed expected to occupy a 15% smaller volume when transferred from bulk to the first protein hydration shell (72).
The kinetic transition and initial amyloid states also differ significantly in both apparent activation enthalpy and entropy. Several factors may contribute to the increased apparent activation enthalpy of the kinetic transition states, such as the disruption of atomic packing interactions and the unfavorable energy of solvation of newly exposed surfaces. The significant increase in entropy suggests also a transition from a more ordered, bound structure, to a more disordered, loose structure. The indication of partial protein unfolding in the course of the activation process, which is suggested by our results, is conceptually attractive as it may help to understand why otherwise stable amyloid fibrils dissociate partly under high pressure.
Multiple Steps
The ruggedness and complexity of the amyloid energy landscape, including a wide range of different conformational stages and the multitude of available pathways, render the interpretation of the kinetic data hypothetical. Nevertheless, the existence of two kinetic rate constants for the pressure-induced structural changes of PrP fibrils generally concurs with previous kinetic data revealing that β-amyloid fibril association/dissociation occurs in multiple steps (3) and could imply two different degrees of association/dissociation between monomers and the fibril structure. However, we cannot exclude the possibility of two consecutive and dynamically well separated processes.
The heterogeneity of amyloid fibrils is further supported by the finding that their pressurization leads to partial dissociation and to a new fibrillar species that does not dissociate when subjected to a second pressurization cycle. These results can be interpreted by assuming that before pressure treatment two populations of PrP fibrils coexist in the sample; that is, a “partially locked” (reversibly bound monomers) form that can be dissociated and a “totally locked” form (irreversibly bound monomers) that cannot be dissociated. These two conformers are currently thought to be different states occurring during amyloidogenesis (73–76). Yet the structural differences between these two initial and co-existing populations of fibrils appear to be subtle. Indeed, the apparent activation parameters of both kinetic phases are rather similar. It is, therefore, likely that the kinetic transition states of both phases reflect a partially disordered or unfolded form of the amyloid fibrils. Depending on whether the transition state was attained by pressurizing partially locked or totally locked amyloid fibrils, it relaxes by dissociating or by forming a new stable fibrillar conformer, respectively.
Cytotoxicity
The issue of reversibility of amyloid formation may be of significance in assessing ways of stopping and reversing toxic and/or infectious species for therapeutic purposes in vivo, presuming that reversing amyloid deposition does not result in an increased production of toxic species. Indeed, although several reports suggest that fibrils themselves possess toxicity (54, 77–79), oligomeric species are considered to be the primary cytotoxic species. Here we show that pressure treatment of amyloid fibrils results in partial structural changes and dissociation concomitantly with a strongly reduced cytotoxicity when compared with initial PrP fibrils. These data support the finding that the aggregation state of recombinant PrP is crucial for cytotoxicity and, even under certain physicochemical conditions, for encoding prion infectivity (57, 80, 81). Intriguingly, the soluble fraction of the pressure-treated fibrils accounts for almost two-thirds of its cytotoxic properties. The structural details of this low but persisting cytotoxicity remain to be elucidated.
A Model of the Effects of Pressure on Amyloid Fibrils
On the basis of structural studies of PrP fibrils (82) and a single-fibril immunoconformational study (83), PrP protofilaments can be considered as composed of an outer and an inner layer of β-strands characterized by different degrees of compactness and packing. Based on this model, the pressure-induced structural changes of mature amyloid fibrils can be schematized as follows.
In one population of initial PrP fibrils, high pressure converts the outer layer into a random coil conformation (supplemental Figs. S2 and S3), whereas the inner layer remains intact (supplemental Fig. S2) (Fig. 9A). The higher susceptibility of the β-sheeted outer layer results from poorly optimized side-chain packing interactions that give rise to void volumes. As pressure is released, this structurally altered region tends to refold into its native secondary structure, which for PrP is likely to be α-helical, as suggested also by the changes in the second derivatives of the FTIR spectra (supplemental Fig. S2). The unfolding and the subsequent alternative refolding of the fibrillar outer layer disrupt ThT and ANS binding and lead to the altered proteolytic cleavage after PK treatment. In addition, the significant thinning of the remaining fibrils observed upon pressurization (supplemental Fig. S4) supports the hypothesis that by suppressing intermolecular interactions between the outer β-strands, pressure promotes lateral disassembly or untwisting of bundles of two or more proto-filaments. The resulting new fibrillar structures, which are insensitive to further pressure treatment, therefore consist of well packed amyloid fibrils without structured hydrophobic or β-sheeted domains at the protein surface, where compounds such as ANS or ThT can bind.
FIGURE 9.

Schematic representation of the effects of high pressure on mature PrP amyloid fibrils. We considered that each PrP proto-filament consisted of an outer and an inner layer of β-strands. This diagram illustrates the proposed model of pressure-induced structural changes, which depends on the thermodynamic state (partially locked or locked) adopted by PrP proteins in the proto-filament. A, after pressure treatment, fibrils composed of totally locked proto-filaments convert into morphologically different fibrils characterized by an irreversible loss of several amyloid specific features. B, partially locked proto-filaments, characterized by a lower degree of packing, disaggregate into native monomers after a pressure-induced unfolding-refolding process. Kinetic data reveal that both pathways proceed via a mechanistically similar unfolding process that implies a disordered and hydrated kinetic transition state. Further details are specified under “Discussion.”
In another population of initial PrP fibrils, the partial disaggregation of the sample into native PrP monomers (Fig. 9B) could be attributed to the presence of proto-filaments that are characterized by a lower degree of packing in the inner layer and by internal cavities and hydrophobic pockets. Under such conditions, the PrP assembly, being highly sensitive to pressure, could easily attain new conformational coordinates, leading to unfolded PrP that, as observed in the present work, appears to refold into the native conformation after pressure release.
Conclusion
This work highlights the value of studying pressure-induced destruction of amyloid features and dissociation of prion fibrils. Further development of this approach should allow us to gain fundamental knowledge on the solubilization and degradation mechanisms of large proteinaceous deposits and on the biological effects of prions. Moreover, because amyloid fibrils are associated with a number of diseases, such as Alzheimer disease, type 2 diabetes, senile amyloidosis, and dialysis-related amyloidosis, we envisage that the ability to study pressure-induced PrP amyloid fibril structural changes could be transferred to other specific amyloidogenic proteins. Furthermore, the finding that pressure can be used to convert an amyloid to a non-amyloid fibril conformer might be interesting for the study of proteins that are forming non-amyloid fibrils, such as lithostathine (84, 85).
Acknowledgment
We are grateful for the expert advice of Dr. R. Sabaté.
This work was supported, in whole or in part, by National Institutes of Health Grant NS045585 (to I. V. B.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- PrP
- prion protein
- ANS
- 8-anilino-1-naphthalene sulfonate
- ThT
- thioflavin T
- PK
- proteinase K
- MPa
- megapascals.
REFERENCES
- 1. Fändrich M. (2007) Cell. Mol. Life Sci. 64, 2066–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sipe J. D., Cohen A. S. (2000) J. Struct. Biol. 130, 88–98 [DOI] [PubMed] [Google Scholar]
- 3. Dobson C. M. (2003) Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 4. Fändrich M., Dobson C. M. (2002) EMBO J. 21, 5682–5690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiménez J. L., Guijarro J. I., Orlova E., Zurdo J., Dobson C. M., Sunde M., Saibil H. R. (1999) EMBO J. 18, 815–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pertinhez T. A., Bouchard M., Tomlinson E. J., Wain R., Ferguson S. J., Dobson C. M., Smith L. J. (2001) FEBS Lett. 495, 184–186 [DOI] [PubMed] [Google Scholar]
- 7. Breydo L., Makarava N., Baskakov I. V. (2008) Methods Mol. Biol. 459, 105–115 [DOI] [PubMed] [Google Scholar]
- 8. Fernàndez-Busquets X., de Groot N. S., Fernandez D., Ventura S. (2008) Curr. Med. Chem. 15, 1336–1349 [DOI] [PubMed] [Google Scholar]
- 9. Langkilde A. E., Vestergaard B. (2009) FEBS Lett. 583, 2600–2609 [DOI] [PubMed] [Google Scholar]
- 10. Chamberlain A. K., MacPhee C. E., Zurdo J., Morozova-Roche L. A., Hill H. A., Dobson C. M., Davis J. J. (2000) Biophys. J. 79, 3282–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chiti F., Dobson C. M. (2006) Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 12. Harper J. D., Lieber C. M., Lansbury P. T., Jr. (1997) Chem. Biol. 4, 951–959 [DOI] [PubMed] [Google Scholar]
- 13. Jiménez J. L., Nettleton E. J., Bouchard M., Robinson C. V., Dobson C. M., Saibil H. R. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 9196–9201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bauer H. H., Aebi U., Häner M., Hermann R., Müller M., Merkle H. P. (1995) J. Struct. Biol. 115, 1–15 [DOI] [PubMed] [Google Scholar]
- 15. Fändrich M., Meinhardt J., Grigorieff N. (2009) Prion 3, 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldsbury C. S., Cooper G. J., Goldie K. N., Müller S. A., Saafi E. L., Gruijters W. T., Misur M. P., Engel A., Aebi U., Kistler J. (1997) J. Struct. Biol. 119, 17–27 [DOI] [PubMed] [Google Scholar]
- 17. Jiménez J. L., Tennent G., Pepys M., Saibil H. R. (2001) J. Mol. Biol. 311, 241–247 [DOI] [PubMed] [Google Scholar]
- 18. Madine J., Jack E., Stockley P. G., Radford S. E., Serpell L. C., Middleton D. A. (2008) J. Am. Chem. Soc. 130, 14990–15001 [DOI] [PubMed] [Google Scholar]
- 19. Meinhardt J., Sachse C., Hortschansky P., Grigorieff N., Fändrich M. (2009) J. Mol. Biol. 386, 869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paravastu A. K., Leapman R. D., Yau W. M., Tycko R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pedersen J. S., Dikov D., Flink J. L., Hjuler H. A., Christiansen G., Otzen D. E. (2006) J. Mol. Biol. 355, 501–523 [DOI] [PubMed] [Google Scholar]
- 22. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., Tycko R. (2005) Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 23. Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., Stefani M. (2002) Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 24. Conway K. A., Lee S. J., Rochet J. C., Ding T. T., Williamson R. E., Lansbury P. T., Jr. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 571–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carulla N., Caddy G. L., Hall D. R., Zurdo J., Gairí M., Feliz M., Giralt E., Robinson C. V., Dobson C. M. (2005) Nature 436, 554–558 [DOI] [PubMed] [Google Scholar]
- 26. Hawkins P. N., Richardson S., MacSweeney J. E., King A. D., Vigushin D. M., Lavender J. P., Pepys M. B. (1993) Q. J. Med. 86, 365–374 [PubMed] [Google Scholar]
- 27. Hawkins P. N., Richardson S., Vigushin D. M., David J., Kelsey C. R., Gray R. E., Hall M. A., Woo P., Lavender J. P., Pepys M. B. (1993) Arthritis Rheum. 36, 842–851 [DOI] [PubMed] [Google Scholar]
- 28. Rydh A., Suhr O., Hietala S. O., Ahlström K. R., Pepys M. B., Hawkins P. N. (1998) Eur. J. Nucl. Med. 25, 709–713 [DOI] [PubMed] [Google Scholar]
- 29. Tan S. Y., Irish A., Winearls C. G., Brown E. A., Gower P. E., Clutterbuck E. J., Madhoo S., Lavender J. P., Pepys M. B., Hawkins P. N. (1996) Kidney Int. 50, 282–289 [DOI] [PubMed] [Google Scholar]
- 30. Calamai M., Canale C., Relini A., Stefani M., Chiti F., Dobson C. M. (2005) J. Mol. Biol. 346, 603–616 [DOI] [PubMed] [Google Scholar]
- 31. Callahan M. A., Xiong L., Caughey B. (2001) J. Biol. Chem. 276, 28022–28028 [DOI] [PubMed] [Google Scholar]
- 32. Loksztejn A., Dzwolak W. (2009) Biochemistry 48, 4846–4851 [DOI] [PubMed] [Google Scholar]
- 33. MacPhee C. E., Dobson C. M. (2000) J. Mol. Biol. 297, 1203–1215 [DOI] [PubMed] [Google Scholar]
- 34. Morel B., Varela L., Conejero-Lara F. (2010) J. Phys. Chem. B. 114, 4010–4019 [DOI] [PubMed] [Google Scholar]
- 35. Abdul Latif A. R., Kono R., Tachibana H., Akasaka K. (2007) Biophys. J. 92, 323–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chatani E., Kato M., Kawai T., Naiki H., Goto Y. (2005) J. Mol. Biol. 352, 941–951 [DOI] [PubMed] [Google Scholar]
- 37. Dirix C., Meersman F., MacPhee C. E., Dobson C. M., Heremans K. (2005) J. Mol. Biol. 347, 903–909 [DOI] [PubMed] [Google Scholar]
- 38. Foguel D., Suarez M. C., Ferrão-Gonzales A. D., Porto T. C., Palmieri L., Einsiedler C. M., Andrade L. R., Lashuel H. A., Lansbury P. T., Kelly J. W., Silva J. L. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 9831–9836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grudzielanek S., Smirnovas V., Winter R. (2006) J. Mol. Biol. 356, 497–509 [DOI] [PubMed] [Google Scholar]
- 40. Kamatari Y. O., Yokoyama S., Tachibana H., Akasaka K. (2005) J. Mol. Biol. 349, 916–921 [DOI] [PubMed] [Google Scholar]
- 41. Meersman F., Dobson C. M. (2006) Biochim. Biophys. Acta 1764, 452–460 [DOI] [PubMed] [Google Scholar]
- 42. Mishra R., Winter R. (2008) Angew. Chem. Int. Ed. Engl. 47, 6518–6521 [DOI] [PubMed] [Google Scholar]
- 43. Font J., Torrent J., Ribó M., Laurents D. V., Balny C., Vilanova M., Lange R. (2006) Biophys. J. 91, 2264–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Torrent J., Font J., Herberhold H., Marchal S., Ribó M., Ruan K., Winter R., Vilanova M., Lange R. (2006) Biochim. Biophys. Acta 1764, 489–496 [DOI] [PubMed] [Google Scholar]
- 45. Vidugiris G. J., Markley J. L., Royer C. A. (1995) Biochemistry 34, 4909–4912 [DOI] [PubMed] [Google Scholar]
- 46. Woenckhaus J., Köhling R., Thiyagarajan P., Littrell K. C., Seifert S., Royer C. A., Winter R. (2001) Biophys. J. 80, 1518–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rezaei H., Marc D., Choiset Y., Takahashi M., Hui Bon Hoa G., Haertlé T., Grosclaude J., Debey P. (2000) Eur. J. Biochem. 267, 2833–2839 [DOI] [PubMed] [Google Scholar]
- 48. Först P., Werner F., Delgado A. (2000) Rheologica Acta 39, 566–573 [Google Scholar]
- 49. Meersman F., Heremans K. (2003) Biophys. Chem. 104, 297–304 [DOI] [PubMed] [Google Scholar]
- 50. Bocharova O. V., Breydo L., Salnikov V. V., Gill A. C., Baskakov I. V. (2005) Protein Sci. 14, 1222–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pace C. N. (2009) Nat. Struct. Mol. Biol. 16, 681–682 [DOI] [PubMed] [Google Scholar]
- 52. James T. L., Liu H., Ulyanov N. B., Farr-Jones S., Zhang H., Donne D. G., Kaneko K., Groth D., Mehlhorn I., Prusiner S. B., Cohen F. E. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 10086–10091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bocharova O. V., Makarava N., Breydo L., Anderson M., Salnikov V. V., Baskakov I. V. (2006) J. Biol. Chem. 281, 2373–2379 [DOI] [PubMed] [Google Scholar]
- 54. Novitskaya V., Bocharova O. V., Bronstein I., Baskakov I. V. (2006) J. Biol. Chem. 281, 13828–13836 [DOI] [PubMed] [Google Scholar]
- 55. Cordeiro Y., Kraineva J., Ravindra R., Lima L. M., Gomes M. P., Foguel D., Winter R., Silva J. L. (2004) J. Biol. Chem. 279, 32354–32359 [DOI] [PubMed] [Google Scholar]
- 56. Torrent J., Alvarez-Martinez M. T., Heitz F., Liautard J. P., Balny C., Lange R. (2003) Biochemistry 42, 1318–1325 [DOI] [PubMed] [Google Scholar]
- 57. Makarava N., Kovacs G. G., Bocharova O., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2010) Acta Neuropathol. 119, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brown P., Meyer R., Cardone F., Pocchiari M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 6093–6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fernández García A., Heindl P., Voigt H., Büttner M., Wienhold D., Butz P., Stärke J., Tauscher B., Pfaff E. (2004) J. Gen. Virol. 85, 261–264 [DOI] [PubMed] [Google Scholar]
- 60. Heindl P., Fernández García A., Büttner M., Voigt H., Butz P., Tauscher B., Pfaff E. (2005) Braz. J. Med. Biol. Res. 38, 1223–1231 [DOI] [PubMed] [Google Scholar]
- 61. Aguzzi A., Sigurdson C., Heikenwaelder M. (2008) Annu. Rev. Pathol. 3, 11–40 [DOI] [PubMed] [Google Scholar]
- 62. Alvarez-Martinez M. T., Torrent J., Lange R., Verdier J. M., Balny C., Liautard J. P. (2003) Biochim. Biophys. Acta 1645, 228–240 [DOI] [PubMed] [Google Scholar]
- 63. El Moustaine D., Perrier V., Smeller L., Lange R., Torrent J. (2008) FEBS J. 275, 2021–2031 [DOI] [PubMed] [Google Scholar]
- 64. Silva J. L., Vieira T. C., Gomes M. P., Bom A. P., Lima L. M., Freitas M. S., Ishimaru D., Cordeiro Y., Foguel D. (2010) Acc. Chem. Res. 43, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Torrent J., Alvarez-Martinez M. T., Harricane M. C., Heitz F., Liautard J. P., Balny C., Lange R. (2004) Biochemistry 43, 7162–7170 [DOI] [PubMed] [Google Scholar]
- 66. Torrent J., Alvarez-Martinez M. T., Liautard J. P., Balny C., Lange R. (2005) Protein Sci. 14, 956–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Van Eldik R., Asano T., Le Noble W. J. (1989) Chem. Rev. 89, 549–688 [DOI] [PubMed] [Google Scholar]
- 68. Silva J. L., Foguel D., Royer C. A. (2001) Trends Biochem. Sci. 26, 612–618 [DOI] [PubMed] [Google Scholar]
- 69. Akasaka K., Latif A. R., Nakamura A., Matsuo K., Tachibana H., Gekko K. (2007) Biochemistry 46, 10444–10450 [DOI] [PubMed] [Google Scholar]
- 70. Niraula T. N., Konno T., Li H., Yamada H., Akasaka K., Tachibana H. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 4089–4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Biancalana M., Koide S. (2010) Biochim. Biophys. Acta 1804, 1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Merzel F., Smith J. C. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 5378–5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Esler W. P., Stimson E. R., Jennings J. M., Vinters H. V., Ghilardi J. R., Lee J. P., Mantyh P. W., Maggio J. E. (2000) Biochemistry 39, 6288–6295 [DOI] [PubMed] [Google Scholar]
- 74. Gobbi M., Colombo L., Morbin M., Mazzoleni G., Accardo E., Vanoni M., Del Favero E., Cantù L., Kirschner D. A., Manzoni C., Beeg M., Ceci P., Ubezio P., Forloni G., Tagliavini F., Salmona M. (2006) J. Biol. Chem. 281, 843–849 [DOI] [PubMed] [Google Scholar]
- 75. Maggio J. E., Stimson E. R., Ghilardi J. R., Allen C. J., Dahl C. E., Whitcomb D. C., Vigna S. R., Vinters H. V., Labenski M. E., Mantyh P. W. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 5462–5466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Takeda T., Klimov D. K. (2008) Biophys. J. 95, 1758–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gharibyan A. L., Zamotin V., Yanamandra K., Moskaleva O. S., Margulis B. A., Kostanyan I. A., Morozova-Roche L. A. (2007) J. Mol. Biol. 365, 1337–1349 [DOI] [PubMed] [Google Scholar]
- 78. Meyer-Luehmann M., Spires-Jones T. L., Prada C., Garcia-Alloza M., de Calignon A., Rozkalne A., Koenigsknecht-Talboo J., Holtzman D. M., Bacskai B. J., Hyman B. T. (2008) Nature 451, 720–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pieri L., Bucciantini M., Nosi D., Formigli L., Savistchenko J., Melki R., Stefani M. (2006) J. Biol. Chem. 281, 15337–15344 [DOI] [PubMed] [Google Scholar]
- 80. Legname G., Baskakov I. V., Nguyen H. O., Riesner D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2004) Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 81. Wang F., Wang X., Yuan C. G., Ma J. (2010) Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ostapchenko V. G., Sawaya M. R., Makarava N., Savtchenko R., Nilsson K. P., Eisenberg D., Baskakov I. V. (2010) J. Mol. Biol. 400, 908–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Novitskaya V., Makarava N., Bellon A., Bocharova O. V., Bronstein I. B., Williamson R. A., Baskakov I. V. (2006) J. Biol. Chem. 281, 15536–15545 [DOI] [PubMed] [Google Scholar]
- 84. Laurine E., Grégoire C., Fändrich M., Engemann S., Marchal S., Thion L., Mohr M., Monsarrat B., Michel B., Dobson C. M., Wanker E., Erard M., Verdier J. M. (2003) J. Biol. Chem. 278, 51770–51778 [DOI] [PubMed] [Google Scholar]
- 85. Milhiet P. E., Yamamoto D., Berthoumieu O., Dosset P., Le Grimellec C., Verdier J. M., Marchal S., Ando T. (2010) PLoS One 5, e13240. [DOI] [PMC free article] [PubMed] [Google Scholar]