

Abstract
Sialylated glycans serve as cell surface attachment factors for a broad range of pathogens. We report an atypical example, where desialylation increases cell surface binding and infectivity of adeno-associated virus (AAV) serotype 9, a human parvovirus isolate. Enzymatic removal of sialic acid, but not heparan sulfate or chondroitin sulfate, increased AAV9 transduction regardless of cell type. Viral binding and transduction assays on mutant Chinese hamster ovary (CHO) cell lines defective in various stages of glycan chain synthesis revealed a potential role for core glycan residues under sialic acid in AAV9 transduction. Treatment with chemical inhibitors of glycosylation and competitive inhibition studies with different lectins suggest that N-linked glycans with terminal galactosyl residues facilitate cell surface binding and transduction by AAV9. In corollary, resialylation of galactosylated glycans on the sialic acid-deficient CHO Lec2 cell line with different sialyltransferases partially blocked AAV9 transduction. Quantitative analysis of AAV9 binding to parental, sialidase-treated or sialic acid-deficient mutant CHO cells revealed a 3–15-fold increase in relative binding potential of AAV9 particles upon desialylation. Finally, pretreatment of well differentiated human airway epithelial cultures and intranasal instillation of recombinant sialidase in murine airways enhanced transduction efficiency of AAV9 by >1 order of magnitude. Taken together, the studies described herein provide a molecular basis for low infectivity of AAV9 in vitro and a biochemical strategy to enhance gene transfer by AAV9 vectors in general.
Keywords: Carbohydrate-binding Protein, Cell Surface Receptor, Glycoconjugate, Glycosaminoglycan, Virus Entry
Introduction
Cell surface glycans have been shown to play a critical role in the infectious pathways of viruses (1). Detailed studies of virus-glycan interactions have yielded significant insight into mechanisms underlying emergence and transmission of viral pathogens in different hosts. Among various glycolipids, glycoproteins, or proteoglycans anchored to the plasma membrane, sialylated glycans and heparan sulfate proteoglycans appear to serve as predominant substrates for viral attachment to the cell surface. For instance, heparan sulfate serves as a primary receptor for Herpesviridae (2) as well as certain adenoviruses (3) and parvoviruses (4, 5). Interactions between sialylated glycans and members of the Orthomyxoviridae (6), Reoviridae (7), Polyomaviridae (8) families and certain parvoviruses are also well known (9–11).
Adeno-associated viruses (AAV)2 are small, single-stranded DNA viruses that belong to the genus Dependovirus of the Parvoviridae family (12). Recombinant AAV vectors, by virtue of their lack of pathogenicity and low immunogenicity, are currently being evaluated as lead candidates in clinical gene therapy trials (13). The discovery of a large number of AAV isolates over the past decade has accelerated efforts to exploit tissue tropisms displayed by different strains for therapeutic gene transfer applications (14, 15). Successful translation from bench to bedside will require a thorough understanding of molecular mechanisms underlying AAV infection. As with other viruses, attachment to cell surface glycans constitutes the first step in the AAV infectious pathway. For instance, several AAV serotypes have been shown to bind heparan sulfate proteoglycans (AAV2 (4), AAV6 (11)), whereas others utilize sialic acid for cell surface binding and entry (AAV4 (16), AAV5 (10), AAV1/6 (11), bovine AAV (17)).
Sialylated glycans that serve as primary receptors for the latter AAV strains vary at the level of N-acetylneuraminic acid linkage to underlying sugars, i.e. α2–3 or α2–6 linked to galactose residues (11, 16). Further receptor specificity has been demonstrated at the level of N-linked or O-linked glycans displayed on the cell surface (11, 16). Selective recognition of such linkages and underlying core glycan types (18) is likely enabled by differences in the capsid surface topology of AAV serotypes (19, 20). In general, dependence of AAV infectivity on sialic acid has been demonstrated using a battery of chemical, biochemical, and genetic tools to desialylate cell surface glycans. The current study is focused on glycan interactions of a human AAV isolate, AAV serotype 9/Hu.14 (Clade F) (21). Recombinant AAV9 vectors display widespread and robust transduction following systemic administration in animal models but fail to infect cells in culture (21, 22). We demonstrate that efficient gene transfer by AAV9 vectors requires an atypical interaction with nonsialylated cell surface glycans. The results described herein provide a molecular basis for the low infectivity of AAV9 observed in cell culture.
MATERIALS AND METHODS
Plasmids and Viruses
All plasmids were obtained from the University of North Carolina (UNC) vector core. The triple plasmid transfection protocol (23) utilized for production of AAV9 vectors includes (i) the AAV helper plasmid, pXR9, containing AAV2 Rep and AAV9 Cap genes; (ii) the adenoviral helper plasmid, pXX6-80; and (iii) the vector genome cassette, pTR-CBA-Luc or pHpa-trs-SK, containing the firefly luciferase gene driven by the chicken β-actin (CBA) promoter or self-complementary GFP cassette driven by the cytomegalovirus (CMV) promoter, respectively. The vector genome cassette is flanked by inverted terminal repeats required for packaging. The inverted terminal repeats are the only elements within the vector genome cassette derived from the wild-type AAV genome, thereby eliminating 96% of viral elements. Recombinant AAV9 vectors generated thus allow quantitation of viral infectivity (or transduction efficiency) through luciferase transgene expression assays. HEK293 cells utilized for production of recombinant AAV9 vectors were obtained from the UNC vector core. Sonicated cell lysates and PEG8000 precipitates from supernatant were pooled and subjected to cesium chloride ultracentrifugation as described earlier (23). Dialyzed peak fractions were subjected to quantitative PCR using a Roche Light Cycler instrument with luc transgene-specific primers to determine viral vector titers (forward, 5′-AAA AGC ACT CTG ATT GAC AAA TAC-3′; reverse, 5′-CCT TCG CTT CAA AAA ATG GAA C-3′).
Cell Lines
All cell lines were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin, streptomycin, amphotericin B (Sigma) and maintained in 5% CO2 at 37 °C unless mentioned otherwise. COS-1 (monkey kidney), Neuro2a (mouse neuroblastoma), U87 (human glioma) and Huh7 (human hepatocarcinoma) cells were obtained from the UNC tissue culture facility and utilized in viral transduction assays. Chinese hamster ovary (CHO) Pro5 and mutant Lec1, Lec2 cell lines were a gift from Dr. Jude Samulski (UNC Chapel Hill), and the CHO Lec8 cell line was purchased from American Type Culture Collection. All CHO cells, utilized for viral binding and transduction assays, were cultured in α-MEM (GIBCO) supplemented with 10% FBS and penicillin, streptomycin, and amphotericin B as outlined above. Well differentiated human airway epithelial (HAE) cultures (4–6 weeks) grown on permeable membrane supports (Millipore, Corning, NY) at the air-liquid interface were provided by the Cell Culture Models Core and the UNC Cystic Fibrosis/Pulmonary Research Center.
Transduction Assays
Different cell lines were seeded at 105 cells/well in 24-well plates and allowed to adhere overnight at 37 °C. Plates were then prechilled at 4 °C for 30 min and incubated with AAV9 vectors at a multiplicity of infection (m.o.i.) of 1000 vector genomes (vg)/cell to allow binding to the cell surface for 1.5 h at 4 °C. Unbound virions were then removed by washing three times with ice-cold 1× phosphate-buffered saline (1× PBS), and 0.5 ml of DMEM added to each well. Luciferase transgene expression levels were quantitated after incubation for 24 h from cell lysates using a Victor 2 luminometer (PerkinElmer Life Sciences). For studies with HAE, well differentiated cultures were pretreated with 1.25 units/ml sialidase A (Prozyme GK80040) at 37 °C for 3 h followed by three washes with ice-cold 1× PBS. Cultures were then incubated with scAAV9-CMV-GFP vectors (m.o.i. = 105 vg/cell) as outlined above. Fluorescence micrographs of green fluorescent protein (GFP) expression in HAE cultures at 2 weeks after transduction were obtained using an Olympus epifluorescence microscope equipped with a 20× objective and a Hamamatsu camera.
Enzymatic Desialylation Assays
Different cell lines were treated with heparinase I and III (from Flavobacterium heparinum; Sigma H2519 and H8891), chondroitinase ABC (from Proteus vulgaris; Sigma C2905), and neuraminidase type III (from Vibrio cholerae; Sigma N7885) to determine the role of cell surface glycans in AAV9 infection. Briefly, COS-1 cells were seeded at 105 cells/well in 24-well plates and pretreated with 50 milliunits/ml neuraminidase, 3 units/ml heparinase I, 1.5 units/ml heparinase III, and 1.5 units/ml chondroitinase ABC in DMEM at 37 °C for 2 h. Neuro2a, U87, HEK293, and Huh7 cells were treated with neuraminidase alone. Cells were then washed three times with 1× PBS and subjected to AAV9 infection at an m.o.i. of 1000 vg/cell. Luciferase transgene expression assays were carried out as described earlier at 24 h after infection.
Chemical Inhibition Assays
CHO Lec2 cells were seeded at 105 cells/well in 24-well plates and pretreated for 24 h with small molecule inhibitors of glycosylation, Swainsonine (10 μm; Sigma S8195) and α-benzyl-GalNAc (1 μg/ml; Sigma B4894) to determine the role of N- and O-glycans in AAV9 infection. Cells pretreated with chemicals were subjected to AAV9 infection at an m.o.i. of 1000 vg/cell and luciferase transgene expression assays carried out as described earlier.
Enzymatic Resialylation Assays
The sialic acid-deficient cell line, CHO Lec2 was treated with 50 milliunits/ml each of α2,3-(N)-sialyltransferase (Calbiochem 566218), α2,6-(N)-sialyltransferase (Calbiochem 566222), or α2,3-(O)-sialyltransferase (Calbiochem 566227) and 1 mm CMP-sialic acid (Sigma) in medium for 3 h at 37 °C. Untreated CHO Pro5 cells with endogenous sialic acid were included as control. Cells were then rinsed three times with 1× PBS and subjected to AAV9 infection at an m.o.i. of 1000 vg/cell and luciferase transgene expression assays carried out as described earlier.
Lectin Inhibition and Staining Assays
Competitive inhibition of AAV9 infection was carried out using a panel of lectins, concanavalin A (ConA), wheat germ agglutinin (WGA), Maackia amurensis lectin (MAL I), Sambucus nigra lectin (SNA), and Erythrina cristagalli lectin (ECL) obtained from Vector Laboratories (Burlingame, CA). Briefly, prechilled CHO Pro5 or mutant Lec2 cells were incubated with 100 μg/ml FITC-conjugated lectin (fluorescent labeling assay) in α-MEM or along with AAV9 particles (transduction assay at m.o.i. = 10,000 vg/cell) for a period 1.5 h at 4 °C. After three washes with ice-cold 1× PBS to remove unbound virions and/or lectins, cells were imaged using an Olympus epifluorescence microscope equipped with a Hamamatsu camera or incubated for 24 h at 37 °C prior to luciferase transgene expression analysis.
Cell Surface Binding Assays
CHO Pro5 cells were seeded at a density of 104 cells/well in 96-well plates prior to treatment with 50 milliunits/ml neuraminidase type III from V. cholerae for 2 h at 37 °C. Untreated Pro5 and Lec2 cells were included as negative and positive controls, respectively. Cells were then prechilled at 4 °C for 30 min, followed by incubation with AAV9 particles at m.o.i. of 102, 5 × 102, 103, 5 × 103, 104, 5 × 104, 105, 5 × 105 vg/cell for 1.5 h at 4 °C. Cells were then subjected to three washes with ice-cold 1× PBS to remove unbound virions. Cell surface-bound virions were collected along with cell lysates following three freeze-thaw cycles and vg copy numbers/cell determined using quantitative PCR as outlined earlier. Binding curves were generated using GraphPad Prism 5 software by applying the single site binding model (Y = Bmax*X/(Kd′ + X)), where Y represents the number of bound virions/cell determined by quantitative PCR; X represents m.o.i.; Bmax is the maximum binding capacity, and Kd′, the observed disassociation constant.
Animal Studies
All experiments were carried out with 6–8-week old female BALB/c mice (Jackson Laboratories, Bar Harbor, ME) maintained and treated in accordance with National Institutes of Health guidelines and as approved by IACUC at UNC-Chapel Hill. Mice were administered via intranasal instillation with either 100 μl of PBS (50 μl/nostril) or 100 μl of neuraminidase type III from V. cholerae (200 milliunits; Sigma). At 2 h after treatment, a dose of 5 × 1010 AAV9 particles in 1× PBS (50 μl/nostril) was administered. Luciferase transgene expression in live animals was obtained using a Xenogen IVIS Lumina® imaging system (Caliper Lifesciences, CA) after intranasal instillation of luciferin substrate (120 mg/kg; Nanolight). Image analysis was carried out using the Living Image software® (Caliper Lifesciences) and luciferase expression reported in relative light units (photons/s per cm2 per steradian).
RESULTS
Neuraminidase Treatment Selectively Increases AAV9 Transduction in Different Cell Types
Enzymatic removal of different cell surface glycans was achieved by treating cells with different glycosidases. Removal of terminal sialic acid residues using neuraminidase (from V. cholerae) enhanced AAV9 transduction by >1 log unit compared with virus alone on COS-1 cells (Fig. 1A). In contrast, hydrolysis of cell surface heparan sulfate or chondroitin sulfate proteoglycans using heparinase I/III or chondroitinase ABC, respectively, had no effect on viral transduction compared with control. Neuraminidase treatment abrogated AAV1 transduction, whereas AAV2 remained unaffected under these conditions (Fig. 1B). In addition, treatment of cell lines derived from different tissues with neuraminidase (gray bars) prior to AAV9 infection enhanced transduction efficiency by nearly 2 log units in contrast to infection with virus alone (white bars) (Fig. 1C). These results were corroborated by increased binding and internalization of AAV9, but not AAV1 vectors upon sialidase treatment in these cell lines (supplemental Figs. S1 and S2). Thus, sialic acid appears to mask cell surface glycans that selectively facilitate AAV9 infection in vitro. Further, enzymatic desialylation might serve as a facile biochemical strategy to enhance transduction efficiency of AAV9 vectors in vitro and might enable detailed analysis of intracellular trafficking pathways.
FIGURE 1.
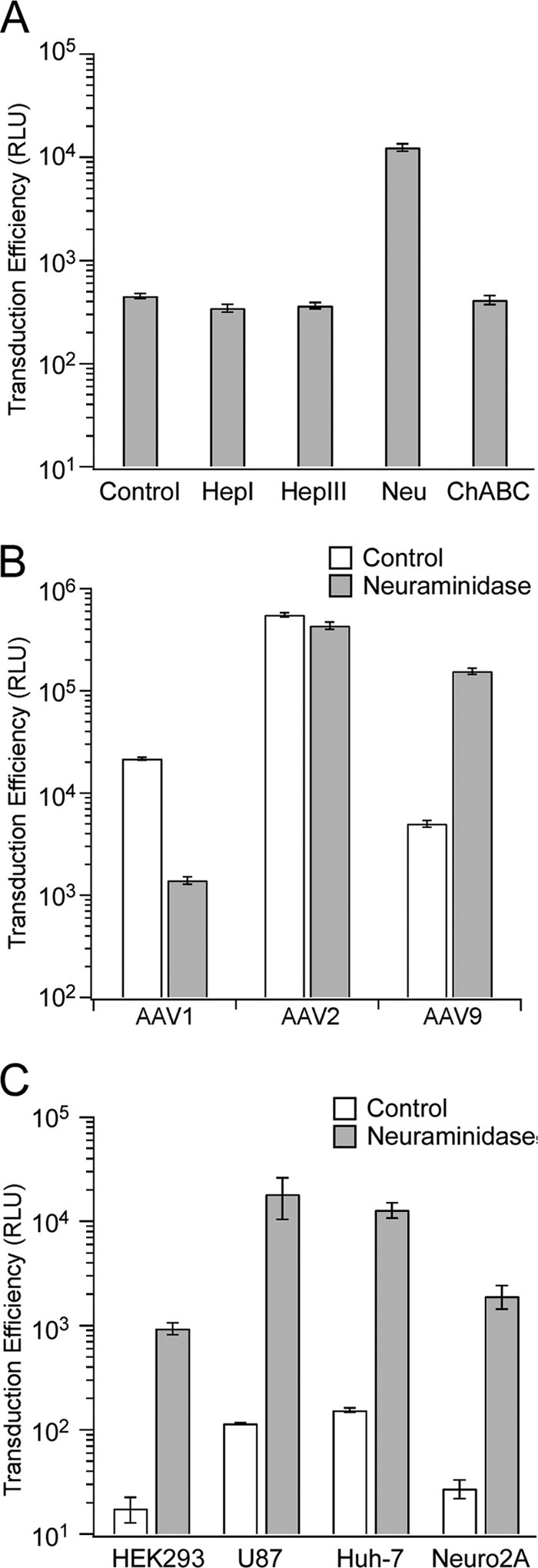
Effect of enzymatic desialylation on AAV9 transduction. A, different enzymes (heparinase I (HepI), heparinase III (HepIII), neuraminidase from V. cholerae (Neu) and chondroitinase ABC (ChABC)) were utilized to deglycosylate prominently expressed cell surface glycans on COS-1 cells. Transduction assays with AAV9 vectors (m.o.i. = 1000 vg/cell) were carried out 2 h after enzymatic treatment. B, neuraminidase treatment of COS-1 cells prior to infection with AAV1, AAV2, or AAV9 vectors (m.o.i. = 1000 vg/cell) was carried out to demonstrate serotype selective effects of enzymatic desialylation on transduction efficiency. C, different cell lines (HEK293, U87, Huh7, and Neuro2a) were untreated (white bars) or pretreated with neuraminidase from V. cholerae (gray bars) followed by subsequent infection with AAV9 vectors (m.o.i. = 1000 vg/cell). Luciferase transgene expression (relative light units, RLU) was quantified for both studies at 24 h after infection. All experiments were carried out in triplicate. Error bars represent S.E.
Mutant CHO Lec2 Cells Lacking Terminal Sialic Acid Are Highly Permissive to AAV9 Infection
Analysis of cell surface binding and infectivity of AAV1 and AAV9 on parental (Pro5) and mutant CHO cell lines was carried out to further understand the role of core glycans under sialic acid in AAV9 infection. The CHO Lec2 cell line lacks terminal sialic acid due to a defect in CMP-sialic acid transport (24), whereas Lec8 and Lec1 cell lines are defective in translocation of UDP-galactose and N-acetylglucosaminyl transferase activity (25, 26), respectively. Correspondingly, cell surface glycans on CHO Lec2 cells contain terminal galactosyl residues, whereas Lec8 and Lec1 cell lines display predominantly terminal N-acetylglucosamine and mannosylated glycans, respectively (Fig. 2A). As seen in Fig. 2, B and C, cell surface binding and transduction of AAV9 particles (gray bars) on Lec2 cells are significantly increased (>1 log unit) compared with the parental Pro5 cell line. In contrast, AAV1 (white bars), which requires sialic acid for infection, shows ∼10-fold decrease in binding and transduction in all CHO Lec mutant cell lines. In addition, no major changes in binding and infectivity are observed in case of Lec8 and Lec1 cells for AAV9 particles. These results suggest that galactosylated glycans immediately underlying sialic acid can facilitate AAV9 cell surface binding and entry. The results also support the notion that AAV9 particles might exploit an inefficient and nonspecific uptake mechanism in the parental Pro5 and mutant Lec8 and Lec1 cell lines.
FIGURE 2.

Effect of glycan chain composition on cell surface binding and transduction of AAV9. A, schematic representation of N-glycan compositions of the parental CHO Pro5 cell line and mutants Lec2, Lec8 and Lec1 (36) using nomenclature proposed by the Consortium for Functional Glycomics nomenclature committee (●, mannose; ■, GlcNAc; ○, galactose; ♦, sialic acid) is shown. B, binding of AAV1 (white bars) and AAV9 (gray bars) particles to wild-type and parental CHO cell lines was analyzed using quantitative PCR as described under “Materials and Methods.” The number of bound virions is expressed as vg/cell. C, transduction efficiency (relative light units, RLU) of AAV1 (white bars) and AAV9 (gray bars) particles (m.o.i. = 1000 vg/cell) on parental and mutant CHO cell lines was analyzed by quantifying luciferase transgene expression at 24 h after infection. All binding experiments were carried out in quadruplicate and infectivity assays in triplicate. Error bars represent S.E.
Sialylation of N-Linked Glycans Blocks AAV9 Infection
To elucidate the nature of glycans required for AAV9 infection further, we utilized small molecule inhibitors of glycosylation and sialyltransferases to modify terminal galactosyl residues on the sialic acid-deficient Lec2 cell surface. Swainsonine (27) and α-benzyl-O-GalNAc (28) are chemical inhibitors of N-linked and O-linked glycosylation, respectively. Treatment with these reagents results in a corresponding decrease in cell surface expression of N-linked glycans and O-linked glycans. As seen in Fig. 3A, AAV9 infection is significantly blocked by Swainsonine (∼ 75%), whereas α-benzyl-O-GalNAc has a modest inhibitory effect (∼25%). These results suggest that AAV9 prefers N-linked cell surface glycans for infection. In addition, resialylation of the sialic acid-deficient Lec2 cell surface was carried out to understand the nature of the sialic acid linkage that blocks AAV9 infection (Fig. 3B). The abilities of different sialyltransferases to partially block AAV9 infection was observed to follow the order α2,3-N-sialyltransferase (3′NST) > α2,6-N-sialyltransferase (6′NST) > α2,3-O-sialyltransferase (3′OST). Alteration of cell surface glycans upon resialylation was confirmed by staining with lectins recognizing different glycan residues and linkages (Fig. 3C). As expected Pro5 cells expressing sialylated glycans and Lec2 cells expressing asialoglycans demonstrate preferential staining by FITC-MAL I and FITC-ECL, respectively. Resialylation of Gal(β1,4)GlcNAc residues with 3′NST, but not 3′OST partially restores FITC-MAL-I staining concurrent with decrease in AAV9 transduction. The lack of MAL I staining in 6′NST-treated Lec2 cells is expected due to lack of recognition of α2,6-sialylated glycans by MAL I. Taken together, these results not only corroborate the important role played by core N-linked glycans in AAV9 infection, but also the potential for α2,3 and α2,6 sialic acid linkages to block infection by masking underlying glycoconjugates on the cell surface.
FIGURE 3.

A, effect of glycosylation inhibitors on AAV9 transduction. Prior to infection with AAV9 vectors (m.o.i. = 1000 vg/cell), CHO Lec2 cells were treated with α-benzyl-GalNAc (1 μg/ml) or Swainsonine (10 μm), inhibitors of O- and N-glycosylation, respectively. B, effect of enzymatic resialylation on AAV9 transduction. CHO Lec2 cells were treated with 50 milliunits/ml each of α2,3-(N)-sialyltransferase (α2,3NST), α2,6-(N)-sialyltransferase (α2,6NST), or α2,3-(O)-sialyltransferase (α2,3OST) and 1 mm CMP-sialic acid (Sigma) to resialylate cell surface asialo glycans. Luciferase transgene expression was quantified at 24 h after infection and is expressed as percent infectivity with respect to untreated or wild type (CHO Pro5) control. All experiments were carried out in triplicate. Error bars indicate S.E. C, lectin staining of CHO cell lines subjected to enzymatic resialylation. CHO Lec2 cells treated with CMP-sialic acid alone or with CMP-sialic acid and different sialyltransferases were subjected to lectin staining using FITC-labeled ECL, which exclusively recognizes Gal(β1,4)GlcNAc or FITC-labeled MAL, which recognizes α2,3-sialylated Gal(β1,4)GlcNAc. Untreated wild-type CHO Pro5 cells, which show high levels of FITC-MAL I staining and untreated Lec2 cells, which show high levels of FITC-ECL staining, were included as control.
Terminal Galactosyl Residues Are Critical for AAV9 Infection
To understand better the nature of AAV9-glycan interactions that mediate infection, we carried out competitive inhibition studies with lectins that recognize different glycan linkages on the cell surface (Fig. 4, A and B). Specifically, we utilized (i) MAL I, which recognizes native, α2,3-sialylated, or sulfated glycoconjugates having Gal(β1,4)N-GlcNAc structures (29, 30); (ii) SNA lectin, which binds preferentially to α2,6-sialylated galactose residues (31); (iii) ECL, which demonstrates specificity toward galactose residues, in particular, Gal(β1,4)N-GlcNAc (32); (iv) WGA, which binds N-GlcNAc and tolerates glycoconjugates containing sialic acid with different linkages (33); and (v) ConA, which recognizes mannose residues (34). The SNA lectin had no effect on AAV9 infection and can be explained by low levels of α2,6-sialylated glycans in both cell lines of rodent (hamster) origin (35). On the other hand, the MAL I demonstrated 5–10-fold inhibition of AAV9 infection in both Pro5 and Lec2 cell lines. More importantly, a striking difference in AAV9 transduction was observed in case of ECL-treated cells with 5-fold inhibition in parental Pro5 cells and nearly 200-fold inhibitory activity in the Lec2 cell line, demonstrating the importance of core Galβ1-linked residues. The ConA and WGA lectins demonstrated broad inhibitory activity in both parental Pro5 cells and the sialic acid-deficient Lec2 cell line suggesting that underlying N-GlcNAc and core mannose residues might contribute to AAV9-glycan interactions.
FIGURE 4.

Effect of lectin competition on AAV9 transduction. A and B, wild-type CHO Pro5 (A) and sialic acid-deficient Lec2 (B) cell lines were co-incubated with lectins recognizing different glycan linkages (100 μg/ml) and AAV9 vectors (m.o.i. = 10,000 vg/cell). Briefly, ConA lectin recognizes mannose residues, whereas WGA lectin recognizes GlcNAc residues as well as sialic acid residues. MAL and SNA lectins recognize α2,3- and α2,6-linked sialic acid residues, respectively, whereas ECL exclusively recognizes Gal(β1,4)GlcNAc residues. Luciferase transgene expression (percent transduction) was quantified at 24 h after infection. All experiments were carried out in triplicate. Error bars represent S.E. C, lectin staining of Pro5 and Lec2 cell lines was carried out using FITC-labeled lectins, and fluorescent images were obtained as described under “Materials and Methods.”
These results were corroborated by fluorescent staining of Pro5 and Lec2 cell lines with aforementioned lectins (Fig. 4C). As expected, MAL I and WGA lectin, which bind to α2,3-sialylated glycans, preferentially label Pro5 cells. In contrast, preferential ECL staining is noted in Lec2 cells. The SNA lectin, which recognizes α2,6-sialic acid, does not stain either Pro5 or Lec2 cells, confirming the lack (or modest expression) of α2,6-sialylated glycans on these hamster-derived cell lines. Further evidence supporting the important role of galactose in mediating AAV9 infection was obtained by treatment of CHO Pro5 cells with endo-β-galactosidase (supplemental Fig. S3, A and B). Removal of galactose residues, but not fucose abrogated AAV9 transduction. Taken together, these results suggest that terminal galactosyl residues serve as the primary receptor for AAV9.
Desialylation Increases the Cell Surface Binding Potential of AAV9 Particles
Cell surface binding curves were generated to establish a quantitative biochemical rationale for the observed increase in AAV9 transduction following desialylation. Binding of AAV9 particles to the surface of parental CHO Pro5 cells, sialidase-treated Pro5 cells, or sialic acid-deficient Lec2 cells was carried out over a range of multiplicities of infection (vg/cell). As seen in Fig. 5, enzymatic removal of sialic acid partially recapitulates the effect of genetic desialylation by increasing the number of cell surface-bound AAV9 particles. Nonlinear regression analyses of binding data were carried out using the single site binding model (Y = Bmax*X/(Kd′ + X)), where X and Y represent multiplicity of infection and number of bound AAV9 particles, respectively; Bmax is the maximum binding capacity, and Kd′ is the relative (observed) binding affinity. Calculation of aforementioned parameters reveals Bmax values averaging 375 vg/cell on wild-type Pro5 cells, a ∼3-fold increase to 1025 vg/cell on sialidase-treated Pro5 cells, and a ∼5-fold increase to 1971 vg/cell on the sialic acid-deficient Lec2 cell line. These results support the observation that AAV9 prefers asialo N-glycans with terminal Galβ1-linked residues. In addition, an apparent decrease in Kd′ (∼3-fold) is observed in Lec2 cells as well as a corresponding 15-fold increase in relative binding potential (Bmax/Kd′) (Table 1). These results support the notion that enhanced avidity plays a role in mediating AAV9 binding to asialo N-glycans.
FIGURE 5.
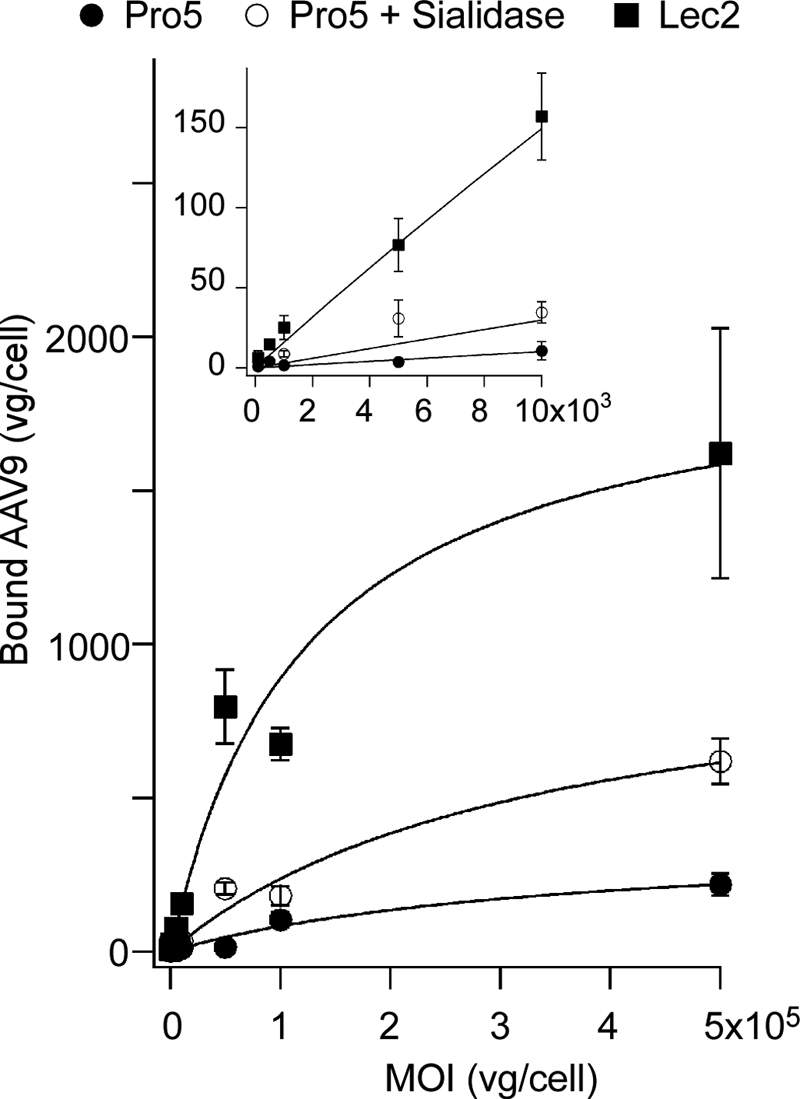
Effect of desialylation on cell surface binding of AAV9 particles. Sialic acid-deficient Lec2 cells (■), untreated wild-type Pro5 cells (●), and sialidase-treated Pro5 cells (○) were prechilled and incubated with AAV9 particles at different m.o.i. ranging from 100 to 500,000 (across 4.5 orders of magnitude) at 4 °C to allow binding, without cellular uptake. Quantitative analysis of dose-dependent AAV9 binding to cell surface asialo N-glycans was carried out by generating binding curves using a single-site binding model as described under “Materials and Methods.” Inset shows linear range of the binding curve from x axis values ranging from 100 to 10,000 vg/cell. Calculated binding parameters are listed in Table 1. All experiments were carried out in triplicate. Error bars represent S.E.
TABLE 1.
Binding parameters for AAV9 interactions with cell surface glycans
| Parameters | Pro5 | Pro5 + Sialidase | Lec2 |
|---|---|---|---|
| Bmax (vg/cell) | 3.75 × 102 ± 0.86 × 102 | 1.03 × 103 ± 0.19 × 103 | 1.97 × 103 ± 0.25 × 103 |
| Kd′ (vg/cell) | 3.58 × 105 ± 1.64 × 105 | 3.34 × 105 ± 1.26 × 105 | 1.22 × 105 ± 0.41 × 105 |
| Bmax/Kd′ | 1.05 × 10−3 | 3.07 × 10−3 | 1.62 × 10−2 |
| R2 | 0.97 | 0.96 | 0.96 |
Sialidase Pretreatment Increases AAV9 Gene Transfer Efficiency in HAE and Murine Airways
To evaluate the potential of enzymatic desialylation as a strategy to enhance gene transfer by AAV9 vectors, we evaluated the effect of sialidase pretreatment on AAV9 transduction efficiency in well differentiated HAE cultures and murine airways. A marked increase in GFP- positive HAE was observed upon treatment with neuraminidase from Arthrobacter ureafaciens (recombinant sialidase A) prior to infection with scAAV9/CMV-GFP vectors (Fig. 6, A and B). Further, quantitation of live animal bioluminescent images of BALB/c mice following intranasal instillation of sialidase showed ∼10-fold increase in luciferase transgene expression in murine airways mediated by AAV9/CBA-Luc vectors (Fig. 6, C and D and supplemental Fig. S4). These results further validate the role of asialo N-glycans as the primary receptor for AAV9 in mouse and human species as well as provide a novel adjuvant strategy to enhance AAV9 gene transfer.
FIGURE 6.

Effect of enzymatic desialylation on AAV9 transduction efficiency in HAE cultures in vitro and murine airways in vivo. A and B, well differentiated HAE cells were untreated (A) or pretreated with recombinant sialidase A (B) followed by subsequent infection with self-complementary (sc) AAV9-CMV-GFP vectors (m.o.i. = 105 vg/cell). Representative fluorescence micrographs of GFP transgene expression were obtained using an Olympus microscope equipped with a 20× objective and a Hamamatsu camera. C, representative live animal bioluminescent images of luciferase expression in BALB/c mice pretreated with intranasally administered PBS or neuraminidase (200 milliunits/50 μl per nostril) are shown. Intranasal instillation of AAV9-CBA-luciferase vectors (5 × 1010 vg/50 μl per nostril) was carried out 2 h after sialidase treatment, and bioluminescent images were obtained at 4 weeks after administration. D, bioluminescence intensity was quantified using Living Image® software and is expressed as relative light units (RLU). Error bars represent S.E.
DISCUSSION
Glycomic profiles of major N-and O-glycans expressed in the parental CHO Pro5 and the CHO Lec2 mutant (35, 36) were instrumental toward delineating the nature of glycans that play a role in cell surface binding and infection by AAV9 vectors. The N-glycan profile of CHO Pro5 cells has been shown to possess complex bi-, tri-, and tetra-antennary structures bearing multiple N-acetyllactosamine (LacNAc) extensions, capped with sialic acid (NeuAc) residues. In addition, the O-glycan profile contains Gal(β1,3)GalNAc core structures that are mono- or disialylated. In contrast, the most abundant glycans produced by the sialic acid-deficient Lec2 cell line are asialo N-glycans, possessing between 2 and 7 LacNAc units. The O-glycan profile is known to be similarly affected by altered sialylation (36). These observations suggest that LacNAc units might serve as cell surface attachment factors for AAV9. Treatment of the Lec2 cell line with chemical inhibitors and sialyltransferase enzymes confirmed that Galβ1-linked N-glycans rather than O-glycans were the preferred substrate for attachment. These results were further corroborated by competitive inhibition studies with the LacNAc-specific ECL. In addition, ConA and WGA lectins revealed a potential contributing role for mannose and GlcNAc residues underlying terminal galactose in mediating AAV9 infection. Further evidence supporting the aforementioned results was also obtained from competitive inhibition studies of AAV9 particles co-incubated with different glycan residues. Although only a modest inhibitory effect was observed potentially due to low affinity interactions, monomeric LacNAc (LN) units, but not α2,3-sialylated LN (3′-SLN) or α2,3-sialylated di-LN (3′-S-Di-LN) glycans appear to block AAV9 infection selectively (supplemental Fig. S5).
Cell surface binding studies constitute a direct approach to determine the relative binding potential (Bmax/Kd′) of AAV9 particles under physiological conditions. Upon desialylation, the observed increase in number of binding sites (Bmax) is likely due to increased availability of asialo N-glycans on the cell surface. The increase in relative binding affinity (Kd′) is potentially a consequence of the increased number of binding sites and can be explained by enhanced avidity arising from multivalent interactions. The aforementioned results suggest that the relatively low abundance of asialo N-glycans provides a molecular basis for the low transduction displayed by AAV9 in cell culture. In contrast to in vitro assays, studies in rodent, canine, and primate models have demonstrated robust and widespread systemic gene transfer using AAV9 vectors following intravenous administration (22, 37, 38). As such, the nature of AAV9-glycan interactions does not provide sufficient rationale for the observed lack of in vitro-in vivo correlation outlined above. As seen in case of other viruses, potential interactions with blood components (39) or co-receptors such as integrins (40) might play an important role in dictating the transduction profile observed in animal models. Nevertheless, it is tempting to speculate that the relative abundance of asialo N-glycans in various animal tissues likely contributes to the broad biodistribution pattern and tissue tropism displayed by AAV9 vectors in vivo. Studies with animal models lacking sialyltransferases (41, 42) might provide further insight into the mechanism(s) underlying tissue tropism displayed by AAV9.
Carbohydrate receptors utilized by members of different AAV clades appear to fall under two classes, namely, heparan sulfate proteoglycans and sialylated glycans (43). For instance, AAV2, a Clade B member, utilizes heparan sulfate proteoglycan as a primary receptor (4). The closely related strains, AAV1 and AAV6 of Clade A, appear to equally prefer α2,3- and α2,6-N-linked sialic acid for infection (11). Further, AAV6 has been shown to bind heparin, implying a potential dual mechanism of interaction with cell surface glycans (44). Serotypes AAV4 and AAV5, which have not been assigned to any clade thus far, require α2,3-O- and α2,3-N-linked sialic acid for cell surface binding, respectively (16). The current study identifies a third class of glycan receptors with terminal galactose utilized by the AAV strain Hu.14/AAV9 for infection. The latter serotype has been classified under Clade F within the AAV phylogenetic tree (21). The major capsid protein (VP3, viral protein subunit 3) of other AAV isolates within Clade F is largely similar to Hu.14/AAV9 (GenBank accession number AY530579.1). Specifically, isolate Hu.31 (GenBank accession number AY530596.1) differs from Hu.14/AAV9 by 2 amino acid residues (S386G, N716S), whereas the VP3 subunit of isolate Hu.32 (GenBank accession number AY530597.1) is identical to Hu.14/AAV9. These observations suggest that at least one other member of Clade F (Hu.32) might utilize nonsialylated glycans for cell surface binding and entry. Finally, Akache et al. (45) have previously reported the role of laminin receptor (LamR) in mediating transduction by AAV9. Whether the 2.5-fold increase in AAV9 transduction mediated by LamR in the previous report within transfected NIH 3T3 cells is due to enhanced binding or internalization is unknown. Nevertheless, low levels of LamR expressed in CHO cells (46) might account for residual transduction of AAV9 seen in the current study.
Recombinant sialidase has been proposed as a therapeutic agent in several clinical modalities. For instance, a sialidase fusion protein has been proposed as an antiviral agent for protection from influenza viruses (47, 48). The latter approach hinges on the inhibition of cell surface binding by influenza viruses due to desialylation of cell surface glycans. In another recent study, intrathecal infusion of recombinant sialidase, which reverses inhibition of axon outgrowth in neurons, has been proposed as a candidate therapy for spinal cord injury (49). In the current study, enzymatic desialylation was found to markedly enhance binding and transduction of AAV9 across different cell types regardless of host or tissue type. In addition, intranasal instillation of sialidase markedly enhanced gene transfer efficiency of AAV9 in murine airways supporting the potential application of recombinant sialidase as an adjuvant in therapeutic gene transfer applications in the lung/airways. Specifically, co-administration or pretreatment of different tissue types such as the lung, central nervous system, or eye with recombinant sialidase might serve as (i) a strategy to expose high avidity glycan receptors and consequently restrict AAV9 transduction to these specific tissue types; (ii) a facile biochemical strategy to increase gene transfer efficiency of AAV9 vectors; and (iii) a method to evaluate AAV9 vectors in desialylated preclinical models eliminating cross-species variation in sialylation patterns.
Acknowledgments
We thank the glycan synthesis core (D) and the Consortium for Functional Glycomics GM62116 for providing glycan reagents used in this study. We also thank Eric Horowitz and Nagesh Pulicherla for helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL089221-A1 and -A1S2 (to A. A.). This work was also supported by a grant from the American Heart Association (to A. A.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- AAV
- adeno-associated virus
- CBA
- chicken β-actin
- ConA
- concanavalin A
- ECL
- Erythrina cristagalli lectin
- HAE
- human airway epithelia
- MAL
- Maackia amurensis lectin
- m.o.i.
- multiplicity of infection
- SNA lectin
- Sambucus nigra lectin
- UNC
- University of North Carolina
- vg
- vector genomes
- WGA
- wheat germ agglutinin.
REFERENCES
- 1. Olofsson S., Bergström T. (2005) Ann. Med. 37, 154–172 [DOI] [PubMed] [Google Scholar]
- 2. Akhtar J., Shukla D. (2009) FEBS J. 276, 7228–7236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dechecchi M. C., Melotti P., Bonizzato A., Santacatterina M., Chilosi M., Cabrini G. (2001) J. Virol. 75, 8772–8780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Summerford C., Samulski R. J. (1998) J. Virol. 72, 1438–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmidt M., Govindasamy L., Afione S., Kaludov N., Agbandje-McKenna M., Chiorini J. A. (2008) J. Virol. 82, 8911–8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Viswanathan K., Chandrasekaran A., Srinivasan A., Raman R., Sasisekharan V., Sasisekharan R. (2010) Glycoconj. J. 27, 561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guglielmi K. M., Johnson E. M., Stehle T., Dermody T. S. (2006) Curr. Top. Microbiol. Immunol. 309, 1–38 [DOI] [PubMed] [Google Scholar]
- 8. Neu U., Stehle T., Atwood W. J. (2009) Virology 384, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nam H. J., Gurda-Whitaker B., Gan W. Y., Ilaria S., McKenna R., Mehta P., Alvarez R. A., Agbandje-McKenna M. (2006) J. Biol. Chem. 281, 25670–25677 [DOI] [PubMed] [Google Scholar]
- 10. Walters R. W., Yi S. M., Keshavjee S., Brown K. E., Welsh M. J., Chiorini J. A., Zabner J. (2001) J. Biol. Chem. 276, 20610–20616 [DOI] [PubMed] [Google Scholar]
- 11. Wu Z., Miller E., Agbandje-McKenna M., Samulski R. J. (2006) J. Virol. 80, 9093–9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bowles D. E., Rabinowitz J. E., Samulski R. J. (2006) in Parvoviruses (Kerr J. R., Cotmore S. F., Bloom M. E. eds) pp. 15–24, Edward Arnold Ltd., New York [Google Scholar]
- 13. Mueller C., Flotte T. R. (2008) Gene Ther. 15, 858–863 [DOI] [PubMed] [Google Scholar]
- 14. Gao G., Vandenberghe L. H., Wilson J. M. (2005) Curr. Gene Ther. 5, 285–297 [DOI] [PubMed] [Google Scholar]
- 15. Mitchell A. M., Nicolson S. C., Warischalk J. K., Samulski R. J. (2010) Curr. Gene Ther. 10, 319–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kaludov N., Brown K. E., Walters R. W., Zabner J., Chiorini J. A. (2001) J. Virol. 75, 6884–6893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schmidt M., Chiorini J. A. (2006) J. Virol. 80, 5516–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen M., Varki A. (2010) OMICS 14, 455–464 [DOI] [PubMed] [Google Scholar]
- 19. Govindasamy L., Padron E., McKenna R., Muzyczka N., Kaludov N., Chiorini J. A., Agbandje-McKenna M. (2006) J. Virol. 80, 11556–11570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ng R., Govindasamy L., Gurda B. L., McKenna R., Kozyreva O. G., Samulski R. J., Parent K. N., Baker T. S., Agbandje-McKenna M. (2010) J. Virol. 84, 12945–12957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao G., Vandenberghe L. H., Alvira M. R., Lu Y., Calcedo R., Zhou X., Wilson J. M. (2004) J. Virol. 78, 6381–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zincarelli C., Soltys S., Rengo G., Rabinowitz J. E. (2008) Mol. Ther. 16, 1073–1080 [DOI] [PubMed] [Google Scholar]
- 23. Grieger J. C., Choi V. W., Samulski R. J. (2006) Nat. Protoc. 1, 1412–1428 [DOI] [PubMed] [Google Scholar]
- 24. Deutscher S. L., Nuwayhid N., Stanley P., Briles E. I., Hirschberg C. B. (1984) Cell 39, 295–299 [DOI] [PubMed] [Google Scholar]
- 25. Deutscher S. L., Hirschberg C. B. (1986) J. Biol. Chem. 261, 96–100 [PubMed] [Google Scholar]
- 26. Stanley P., Chaney W. (1985) Mol. Cell. Biol. 5, 1204–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elbein A. D., Solf R., Dorling P. R., Vosbeck K. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 7393–7397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuan S. F., Byrd J. C., Basbaum C., Kim Y. S. (1989) J. Biol. Chem. 264, 19271–19277 [PubMed] [Google Scholar]
- 29. Wang W. C., Cummings R. D. (1988) J. Biol. Chem. 263, 4576–4585 [PubMed] [Google Scholar]
- 30. Bai X., Brown J. R., Varki A., Esko J. D. (2001) Glycobiology 11, 621–632 [DOI] [PubMed] [Google Scholar]
- 31. Shibuya N., Goldstein I. J., Broekaert W. F., Nsimba-Lubaki M., Peeters B., Peumans W. J. (1987) J. Biol. Chem. 262, 1596–1601 [PubMed] [Google Scholar]
- 32. Wu A. M., Wu J. H., Tsai M. S., Yang Z., Sharon N., Herp A. (2007) Glycoconj. J. 24, 591–604 [DOI] [PubMed] [Google Scholar]
- 33. Yamamoto K., Tsuji T., Matsumoto I., Osawa T. (1981) Biochemistry 20, 5894–5899 [DOI] [PubMed] [Google Scholar]
- 34. Ohyama Y., Kasai K., Nomoto H., Inoue Y. (1985) J. Biol. Chem. 260, 6882–6887 [PubMed] [Google Scholar]
- 35. Stults N. L., Fechheimer M., Cummings R. D. (1989) J. Biol. Chem. 264, 19956–19966 [PubMed] [Google Scholar]
- 36. North S. J., Huang H. H., Sundaram S., Jang-Lee J., Etienne A. T., Trollope A., Chalabi S., Dell A., Stanley P., Haslam S. M. (2010) J. Biol. Chem. 285, 5759–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kornegay J. N., Li J., Bogan J. R., Bogan D. J., Chen C., Zheng H., Wang B., Qiao C., Howard J. F., Jr., Xiao X. (2010) Mol. Ther. 18, 1501–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pacak C. A., Mah C. S., Thattaliyath B. D., Conlon T. J., Lewis M. A., Cloutier D. E., Zolotukhin I., Tarantal A. F., Byrne B. J. (2006) Circ. Res. 99, e3–9 [DOI] [PubMed] [Google Scholar]
- 39. Baker A. H., Mcvey J. H., Waddington S. N., Di Paolo N. C., Shayakhmetov D. M. (2007) Mol. Ther. 15, 1410–1416 [DOI] [PubMed] [Google Scholar]
- 40. Nemerow G. R., Pache L., Reddy V., Stewart P. L. (2009) Virology 384, 380–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hennet T., Chui D., Paulson J. C., Marth J. D. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 4504–4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin L. T., Marth J. D., Varki A., Varki N. M. (2002) J. Biol. Chem. 277, 32930–32938 [DOI] [PubMed] [Google Scholar]
- 43. Wu Z., Asokan A., Samulski R. J. (2006) Mol. Ther. 14, 316–327 [DOI] [PubMed] [Google Scholar]
- 44. Wu Z., Asokan A., Grieger J. C., Govindasamy L., Agbandje-McKenna M., Samulski R. J. (2006) J. Virol. 80, 11393–11397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Akache B., Grimm D., Shen X., Fuess S., Yant S. R., Glazer D. S., Park J., Kay M. A. (2007) Mol. Ther. 15, 330–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hundt C., Peyrin J. M., Haïk S., Gauczynski S., Leucht C., Rieger R., Riley M. L., Deslys J. P., Dormont D., Lasmézas C. I., Weiss S. M. (2001) EMBO J. 20, 5876–5886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Malakhov M. P., Aschenbrenner L. M., Smee D. F., Wandersee M. K., Sidwell R. W., Gubareva L. V., Mishin V. P., Hayden F. G., Kim D. H., Ing A., Campbell E. R., Yu M., Fang F. (2006) Antimicrob. Agents Chemother. 50, 1470–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Triana-Baltzer G. B., Gubareva L. V., Nicholls J. M., Pearce M. B., Mishin V. P., Belser J. A., Chen L. M., Chan R. W., Chan M. C., Hedlund M., Larson J. L., Moss R. B., Katz J. M., Tumpey T. M., Fang F. (2009) PLoS One 4, e7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mountney A., Zahner M. R., Lorenzini I., Oudega M., Schramm L. P., Schnaar R. L. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 11561–11566 [DOI] [PMC free article] [PubMed] [Google Scholar]