

Abstract
The Gi-coupled somatostatin 2A receptor (sst2A) mediates many of the neuromodulatory and neuroendocrine actions of somatostatin (SS) and is targeted by the SS analogs used to treat neuroendocrine tumors. As for other G protein-coupled receptors, agonists stimulate sst2A receptor phosphorylation on multiple residues, and phosphorylation at different sites has distinct effects on receptor internalization and uncoupling. To elucidate the spatial and temporal regulation of sst2A receptor phosphorylation, we examined agonist-stimulated phosphorylation of multiple receptor GPCR kinase sites using phospho-site-specific antibodies. SS increased receptor phosphorylation sequentially, first on Ser-341/343 and then on Thr-353/354, followed by receptor internalization. Reversal of receptor phosphorylation was determined by the duration of prior agonist exposure. In acutely stimulated cells, in which most receptors remained on the cell surface, dephosphorylation occurred only on Thr-353/354. In contrast, both Ser-341/343 and Thr-353/354 were rapidly dephosphorylated when cells were stimulated long enough to allow receptor internalization before agonist removal. Consistent with these observations, dephosphorylation of Thr-353/354 was not affected by either hypertonic sucrose or dynasore, which prevent receptor internalization, whereas dephosphorylation of Ser-341/343 was completely blocked. An okadaic acid- and fostriecin-sensitive phosphatase catalyzed the dephosphorylation of Thr-353/354 both intracellularly and at the cell surface. In contrast, dephosphorylation of Ser-341/343 was insensitive to these inhibitors. Our results show that the phosphorylation and dephosphorylation of neighboring GPCR kinase sites in the sst2A receptor are subject to differential spatial and temporal regulation. Thus, the pattern of receptor phosphorylation is determined by the duration of agonist stimulation and compartment-specific enzymatic activity.
Keywords: G Protein-coupled Receptors (GPCR), PP2A, Protein Phosphatase, Protein Phosphorylation, Receptor Endocytosis, Receptor Modification, Receptor Regulation, Serine/Threonine Protein Kinase, Serine/Threonine Protein Phosphatase, Somatostatin
Introduction
G protein-coupled receptors (GPCRs)2 are the largest class of mammalian cell surface receptors. When stimulated by hormones, neurotransmitters, or other signaling molecules, GPCRs activate heterotrimeric GTP-binding proteins, which then go on to regulate a variety of enzymes and ion channels to produce changes in second messengers such as cyclic AMP and calcium (1, 2). These signaling events are exquisitely coordinated to produce the desired effect on cell function. Overwhelming evidence has shown that within minutes of receptor activation, GPCR signaling is attenuated by receptor phosphorylation. G protein-coupled receptor kinases (GRKs) phosphorylate ligand-activated GPCRs at cytosolic serine and threonine residues (3, 4). Once phosphorylated, the activated receptors recruit arrestins, cytoplasmic proteins that sterically inhibit receptor-G protein coupling thereby desensitizing this signaling pathway. In addition, because arrestins also bind to numerous cytosolic proteins involved in intracellular signaling and receptor trafficking, the recruitment of arrestins by phosphorylated GPCRs leads to the endocytosis of cell surface receptors and to the initiation of G protein-independent signaling events (5–7). After endocytosis, GPCRs can follow different intracellular trafficking routes (8, 9). Some receptors are sorted to lysosomes where they are degraded, whereas others are dephosphorylated in endocytic compartments and then recycled to the cell surface where they are poised for another round of agonist stimulation.
Because the phosphorylation state of GPCRs determines both their signaling capacity and their subcellular distribution, elucidating the mechanisms controlling receptor phosphorylation and dephosphorylation is key to understanding GPCR regulation. We now know that GPCRs are often phosphorylated on multiple intracellular Ser and Thr residues, although specific phosphorylation sites have been identified in relatively few receptors (10). Furthermore, it seems that each GPCR exists in a mixture of phosphorylation states, with different sites phosphorylated to different extents under various conditions (11–13). Interestingly, experiments with mutant receptors missing specific Ser/Thr residues have indicated that phosphorylation of different sites has distinct effects on receptor function (14). Thus, multisite phosphorylation provides a subtle and complex regulatory mechanism for GPCRs, as for many other proteins, and both the phosphorylation and dephosphorylation of individual receptor sites are likely to be tightly controlled. A number of mathematical models have provided general insights into the molecular mechanisms utilized by cells to regulate protein function by multisite phosphorylation (15, 16). At this time, however, the regulation of GPCR function by multisite phosphorylation is too poorly understood to apply such models in a meaningful way. We know little about how GPCR phosphorylation and dephosphorylation at specific receptor residues are regulated, the kinetics and sequence by which these catalytic events occur, or even the cellular compartments where changes in the phosphorylation state of these receptors are brought about.
Somatostatin receptors are members of the class A family of GPCRs. Of the five known somatostatin receptor subtypes, the sst2A receptor is the most widely distributed in normal tissues as well as in human tumors (17–19). These receptors play a critical role in regulating hormone and neurotransmitter release in the endocrine and neuronal systems and, in addition, modulate a number of gastrointestinal and exocrine processes. Because of its inhibitory effect on tumor cell secretion, the sst2A receptor subtype is an important therapeutic target in the treatment of a variety of neuroendocrine cancers. In addition, it is targeted by the radiolabeled somatostatin analogs used for tumor imaging (20, 21). Thus, modulation of sst2A receptor function will have important therapeutic as well as physiological consequences.
We previously showed that sst2A receptors are rapidly internalized and desensitized following SS14 treatment (22). Both these processes depend on agonist-induced receptor phosphorylation, which occurs independently of G-protein coupling, and is catalyzed by GRKs (22–24). In fact, site-directed mutagenesis has shown that different phosphorylation sites are involved in different aspects of sst2A receptor regulation; phosphorylation of Thr residues in the C terminus of the receptor is required for receptor internalization, and Ser phosphorylation mediates receptor desensitization (25). To develop a better understanding of the functional roles of Ser and Thr residues in the sst2A receptor, we recently identified a cluster of phosphorylation sites within the C terminus and showed that GRKs and protein kinase C (PKC) specifically phosphorylate distinct residues in this cluster (24). In particular, agonist binding rapidly increased phosphorylation on a pair of Ser residues, Ser-341 and Ser-343, and on a pair of Thr residues, Thr-353 and Thr-354 (24). However, we do not know how the phosphorylation and dephosphorylation of these sites are coordinated either spatially or temporally. Therefore, we examined the kinetics of phosphorylation and dephosphorylation at specific receptor residues under conditions of normal receptor trafficking and when receptor internalization was blocked. In addition, using selective inhibitors, we investigated the phosphatases involved in sst2A receptor dephosphorylation. Our results demonstrate that the regulation of GPCR phosphorylation is more complex than previously recognized. Phosphorylation and the dephosphorylation of intracellular Ser/Thr residues are individually controlled leading to an exquisitely choreographed and compartment-specific sequence of catalytic events that produce a pattern of receptor phosphorylation that varies in space and time.
EXPERIMENTAL PROCEDURES
Materials
Cell culture reagents and geneticin (G-418) were purchased from Invitrogen. SS14 was purchased from Bachem (Torrance, CA). The sst2 antagonist, Coy-14 (26), was synthesized at the Salk Institute and generously provided by Dr. Jean Rivier. Lactalbumin hydrolysate, leupeptin, phenylmethylsulfonyl fluoride, soybean trypsin inhibitor, bacitracin, Nonidet P-40, and monoclonal anti-actin antibodies were obtained from Sigma. N-Dodecyl β-d-maltoside was purchased from Calbiochem. Bradford and electrophoresis reagents were obtained from Bio-Rad. Agarose-bound wheat germ agglutinin was purchased from Vector Laboratories (Burlingame, CA). λ-Protein phosphatase was from New England Biolabs Inc. (Beverly, MA). Mouse monoclonal antibody against the hemagglutinin antigen (HA) was from Covance (Berkeley, CA). Anti-GRK antibodies against GRK2 (C-15), GRK3 (C-14), GRK5 (C-20), and GRK6 (C-20) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG were from Kirkegaard & Perry Laboratories (Gaithersburg, MD). 3,3′,5,5′-Tetramethylbenzidine substrate kit was purchased from Pierce; 2,2-azino-di(3-ethylbenzthiozolinesulfonate-(6)) powder was obtained from Roche Applied Science. Chemiluminescence Western blotting detection reagent was purchased from GE Healthcare. Okadaic acid was purchased from LC Laboratories, (Woburn, MA), and fostriecin and FK506 were from Enzo Life Sciences International (Plymouth, PA). All other reagents or chemicals were purchased from common suppliers.
Plasmids
The generation of the pcDNA3 plasmid containing the wild type sst2A receptor with three tandem repeats of the HA epitope at the N terminus has been reported (27). The HA-tagged mutant receptors in which all the serine (Ser−) or threonine (Thr−) residues in the third intracellular loop and the C terminus of the receptor were mutated to alanine have been described (25). Both the WT and mutant receptors are well expressed on the cell surface, and the presence of the HA tag does not interfere with receptor function (25, 27).
Cell Culture
The isolation and properties of clonal CHO cell lines expressing similar levels of wild type and mutant sst2A receptors have been reported (25, 27). Cells were cultured at 37 °C and 5% CO2 in Ham's F-12 medium supplemented with 10% fetal bovine serum and 250 μg/ml G-418. Experimental cultures were plated in either 24- or 6-well plates in Ham's F-12 medium without G-418 and used 2–3 days later for experiments.
Generation and Characterization of Phospho-site-specific Antibodies
The synthesis of phosphorylated antigen peptides and the generation of phospho-site-specific antibodies were described previously (24). The specificity of each antibody for phosphorylated and nonphosphorylated forms of the antigen peptide was determined using a competitive peptide ELISA, and the results are summarized in supplemental Table 1 (24, 28). In addition, the specificity of the phospho-site-directed antibodies for phosphorylated sst2A receptor was validated in Western blots and in an in-cell ELISA (see below). Because each antibody showed optimal sensitivity in different assays, the polyclonal antibodies (Ra-454 and Ra-1124) were usually used for immunoblots, and the mouse monoclonal antibodies (1A2 and 2C12) were used to detect phosphorylated receptor with the in-cell ELISA. As demonstrated below, the results from the two assays were in excellent agreement.
In-cell Enzyme-linked Immunoassay for Receptor Phosphorylation
SS14-induced phosphorylation of the sst2A receptor was measured in intact, fixed cells using phospho-site-specific monoclonal antibodies in a protocol similar to that described previously (29). CHO-K1 cells stably expressing the wild type or a mutant sst2A receptor were washed twice with warm F12LH medium (Ham's F-12 medium containing 5 mg/ml lactalbumin hydrolysate and 20 mm HEPES, pH 7.4) and were then stimulated at 37 °C with SS14 for the times specified. Treated cells were then washed with cold phosphate-buffered saline (PBS) and fixed for 10 min with −20 °C methanol/acetone (1:1) on ice. Following fixation, the plates were washed with PBS and incubated in 5% nonfat dried milk in 150 mm NaCl, 50 mm Tris, 1 mm EDTA, 10 mm NaF, 100 nm sodium orthovanadate, 1% Triton X-100, 0.5% sodium deoxycholate, pH 8.0 (RIPS Buffer) for 1 h at room temperature. Blocked cultures were incubated overnight at 4 °C with a phospho-site-specific monoclonal antibody in either blocking buffer (αThr(P)-353/354) or in 1% BSA in PBS (αSer(P)-341/343). The next day the cells were washed sequentially with 0.05% Tween in PBS and PBS without detergent and then incubated with a horseradish peroxidase-coupled goat anti-mouse IgG for 1 h at room temperature. After additional washes, the reaction was developed with 3,3′,5,5′-tetramethylbenzidine substrate and stopped with 10% sulfuric acid. Absorbance was measured at 450 nm with a Precision microplate reader (Molecular Devices, Union City, CA).
To dephosphorylate the receptor prior to ELISA, CHO-sst2A cells preincubated without or with 10 nm SS14 were washed and fixed with methanol/acetone as described above. Cells were then incubated with 300 units of λ-phosphatase at 37 °C for 3 h in 50 mm HEPES, pH 7.5, containing 0.1 mm Na2EDTA, 5 mm dithiothreitol, 0.01% Brij35, and 2 mm MnCl2. Following enzyme treatment, cells were washed with PBS and an in-cell ELISA was carried out as described above.
To measure receptor dephosphorylation in intact cells, CHO-sst2A cells were treated at 37 °C with 10 nm SS14 for the times indicated, washed with warm F12LH medium, and then incubated in fresh F12LH medium for various times. The dephosphorylation incubation was stopped by placing the cells on ice and fixing with −20 °C methanol/acetone (1:1) for 10 min. The level of receptor phosphorylation was then determined with the in-cell ELISA.
In experiments in which cells were treated with inhibitors, we controlled for effects on receptor levels and cell loss by incubating fixed cells with monoclonal anti-HA antibodies instead of the phospho-specific antibodies. None of the inhibitors used in our studies had a significant effect on receptor levels/well.
Receptor Purification and Immunoblotting
CHO-K1 cells expressing wild type or mutant sst2A receptors were cultured to 70–80% confluency in 6-well plates and stimulated with 10 nm SS14 at 37 °C for various times in F12LH. Cells were then chilled, washed with 4 °C HEPES-buffered saline (0.15 m NaCl, 20 mm HEPES, pH 7.4, 5 mm EDTA, 3 mm EGTA, 10 mm sodium pyrophosphate, 10 mm sodium fluoride, 0.1 mm sodium orthovanadate), and scraped into HEPES buffer containing protease and phosphatase inhibitors (30). Cells were solubilized in lysis buffer (HEPES-buffered saline containing 4 mg/ml dodecyl β-maltoside with protease and phosphatase inhibitors), and receptors were purified by lectin affinity chromatography as described previously (24). Eluted protein samples were denatured by the addition of 2× SDS-PAGE sample buffer (62.5 mm Tris-HCl, 2% SDS, 2% β-mercaptoethanol, 6 m urea, and 20% glycerol, pH 6.8) and heating for 15 min at 60 °C. Proteins were resolved in 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes as reported (31). Membranes were blocked for 2 h at room temperature in blocking buffer (10 mm NaH2PO4, 10% nonfat dry milk, 10% glycerol, and 0.2% Tween 20) and then incubated overnight at 4 °C with phospho-site-specific antibodies in the same buffer. Following a 1-h incubation with HRP-conjugated goat anti-rabbit or anti-mouse secondary antibodies, receptor bands were visualized using ECL chemiluminescent reagent. To determine total receptor levels, the PVDF membranes were stripped and reprobed with anti-HA antibody. The relative intensities of receptor bands were determined by scanning densitometry and analysis using Scion Image (version 1.63; Scion Corp., Frederick, MD). Band intensity with phospho-site-specific antibodies was normalized for total receptor levels in the same gel and expressed as a percentage of the stimulation observed with maximal SS14.
For detection of GRKs, each CHO cell line expressing either the wild type or mutant sst2A receptor was grown to 80% confluency in 6-well plates. Cell lysates were prepared in cold HEPES-buffered saline (0.15 m NaCl, 20 mm HEPES, pH 7.4, 5 mm EDTA, 3 mm EGTA) with protease and phosphatase inhibitors containing 4 mg/ml dodecyl β-maltoside. The cell extract was diluted in 2× SDS sample buffer, heated for 15 min at 95 °C, and then analyzed by SDS-PAGE and immunoblotting with anti-GRK antibodies, as described above. As a loading control, membranes were stripped and re-probed with anti-actin antibodies.
Receptor Internalization
The loss of cell surface receptors upon exposure to SS14 was determined as described previously (27). Briefly, CHO cells expressing HA-tagged sst2A receptor were incubated in F12LH with anti-HA antibody for 2 h at room temperature to label cell surface receptors. Unbound anti-HA antibody was then removed by washing, and the cells were equilibrated at 37 °C for 15 min prior to incubation in the absence or presence of SS14 for the times shown. Cells were then chilled to 4 °C, washed with cold PBS, and fixed with 3% paraformaldehyde. After blocking with 1% bovine serum albumin for 30 min at room temperature, cells were incubated with goat anti-mouse IgG horseradish peroxidase conjugate for 1 h. Subsequently, cells were washed with PBS to remove unbound antibody, and the reaction was developed by incubating with 2,2-azino-di(3-ethylbenzthiozolinesulfonate-(6)) for 30 min. Absorbance was measured at 405 nm.
Data Analysis
Figures show mean ± S.E. (n ≥ 3) or mean ± range (n = 2) of replicate samples in individual experiments and are representative of multiple independent experiments, as described. Where error bars are not visible, they fell within symbol size. Regression analysis was carried out using Prism (version 4.0; GraphPad Software, San Diego). Values for the EC50 were calculated by least squares nonlinear regression analysis of dose-response curves fit to a one-component sigmoidal curve with a Hill coefficient of −1. Half-times were calculated by nonlinear regression analysis using a one-phase exponential association for phosphorylation rates or a one-phase exponential decay for rates of dephosphorylation and receptor internalization. Differences between treatment groups were analyzed using either an unpaired t test or two-way analysis of variance, as appropriate. p values < 0.05 were considered statistically significant.
RESULTS
Development of an Immunoassay for Site-specific Receptor Phosphorylation in Intact Cells
We previously identified four residues in the C-tail of the sst2A receptor that were rapidly phosphorylated following stimulation of cells with SS14, namely Ser-341, Ser-343, Thr-353, and Thr-354 (24). Those studies were facilitated by phospho-site-specific antibodies generated to two peptides, one containing phospho-Ser-341 and -343 (Ser(P)-341/343) and the other with phospho-Thr-353 and -354 (Thr(P)-353/354) (supplemental Table 1). Each peptide antigen was used to produce both polyclonal rabbit and monoclonal mouse antibodies with the expectation that their sensitivity would vary among different assays (see below). Two of the antibodies were characterized previously (24), and two new antibodies were generated for this study. To quantitatively evaluate phospho-site-specificity, each antibody was tested with an ELISA in which antibody binding to the phosphorylated peptide antigen was competed with varying concentrations of the antigen itself and with partially phosphorylated and nonphosphorylated homologs. The results are summarized in supplemental Table 1 and demonstrate that all antibodies bound the diphosphorylated antigen peptide with an affinity that was at least 100-fold higher than the affinity for the nonphosphorylated homolog. Furthermore, all the antibodies bound monophosphorylated peptides with lower affinity than the corresponding diphosphorylated antigen. Thus, each antibody was highly specific for the phosphorylated residues present in the immunizing peptide.
We next determined whether the antibodies recognized the intact, phosphorylated sst2A receptor (supplemental Fig. 1). Both untransfected CHO-K1 cells and cells stably transfected to express the wild type sst2A receptor (CHO-R2) were incubated in the absence and presence of 100 nm SS14 for 15 min. Cells were solubilized, and the sst2A receptor was isolated as described under “Experimental Procedures.” Equal concentrations of cell protein were subjected to SDS-PAGE and analyzed by immunoblotting with one of the phospho-site-directed antibodies and, after stripping, with HA antibody to identify the monomer and dimer receptor bands and provide a measure of total receptor concentration (supplemental Fig. 1). None of the phospho-site-specific antibodies showed any reactivity with nontransfected CHO-K1 cells either with or without SS14 treatment. Furthermore, none of the antibodies recognized sst2A receptors prepared from untreated CHO-R2 cells. However all four phospho-site-specific antibodies reacted with sst2A receptors from SS14-stimulated CHO-R2 cells.
Together, these results demonstrate that our antibody panel specifically recognizes the phosphorylated sst2A receptor in immunoblots. However, use of immunoblots for quantitation of site-specific receptor phosphorylation is labor-intensive. Thus, to expedite the measurement of receptor phosphorylation, we established an ELISA that enabled us to quantitate site-specific sst2A receptor phosphorylation in intact cells without the necessity for receptor solubilization and purification (Fig. 1). Untransfected CHO-K1 cells or cells stably expressing sst2A receptors were incubated without or with SS14 for 30 min. The cells were then fixed, permeabilized, and incubated with monoclonal αSer(P)-341/343 (2C12) or αThr(P)-353/354 (1A2) followed by a secondary antibody-linked peroxidase assay as described under “Experimental Procedures.” The results in Fig. 1 show that, as in immunoblots, both αSer(P)-341/343 (panel A) and αThr(P)-353/354 (panel B) produced a strong signal in cells expressing the WT sst2A receptor but only after SS14 stimulation. No signal was detected with either untransfected CHO-K1 cells or with cells expressing the Ser−/Thr− mutant receptor in which all the Ser and Thr residues in the IC3 loop and C terminus of sst2A were mutated to alanine. These results show that in the whole cell ELISA, as in immunoblots, αSer(P)-341/343 and αThr(P)-353/354 specifically recognized only the agonist-stimulated sst2A receptor.
FIGURE 1.

Phosphorylation-dependent reaction of phospho-site-specific antibodies with the sst2A receptor. The reactivity of the sst2A receptor with the αSer(P)-341/343 and αThr(P)-353/354 monoclonal antibodies was determined using the in-cell ELISA described under “Experimental Procedures.” Each panel shows the mean ± S.E. from a single experiment, representative of at least three independent experiments. Panels A and B, nontransfected CHO cells or CHO cells stably expressing either the wild type (WT) sst2A receptor or the Ser−/Thr− mutant receptor were treated in the absence (□) or presence (■) of 100 nm SS14 for 30 min at 37 °C. Cells were then fixed, blocked, and incubated with either αSer(P)-341/343 (panel A) or αThr(P)-353/354 (panel B). Both antibodies reacted only with the WT receptor and only after SS14 stimulation. Panels C and D, cells expressing the wild type sst2A receptor were treated without (□) or with (■) 10 nm SS14 for 30 min at 37 °C. Following fixation, cells were incubated without or with phosphatase as described under “Experimental Procedures.” Antibody reactivity was measured with either αSer(P)-341/343 (panel C) or αThr(P)-353/354 (panel D) using the in cell ELISA. Phosphatase treatment prevents antibody recognition of the WT sst2A receptor. PPase, phosphatase.
To confirm that antibody recognition was dependent on receptor phosphorylation, we treated CHO-sst2A cells with SS14 as above and then incubated the fixed, permeabilized cells without or with λ-phosphatase prior to the addition of phospho-site-specific antibodies. The results in Fig. 1, panels C and D, show that phosphatase treatment eliminated the ELISA signal produced by both Ser(P)-341/343-specific and Thr(P)-353/354-specific antibodies in SS14-treated cells.
Together our results demonstrate that the ELISA measures site-specific sst2A receptor phosphorylation following SS14 stimulation.
Kinetic Analysis of sst2A Receptor Phosphorylation
GPCR phosphorylation can be catalyzed by multiple kinases that phosphorylate sometimes overlapping and sometimes distinct residues, often at different rates (14). Although our previous studies showed that the overall phosphorylation of the sst2A receptor, as measured by 32PO4 incorporation, occurs with a t½ of 2 min following SS14 stimulation (22), the kinetics of sst2A receptor phosphorylation at distinct sites has not been investigated. Therefore, we measured the rates of sst2A receptor phosphorylation at Ser(P)-341/343 and at Thr(P)-353/354 using both the in-cell ELISA (Fig. 2) and immunoblotting of isolated receptors (Fig. 3) to directly compare results from the two assays.
FIGURE 2.

Sequential phosphorylation of the sst2A receptor. CHO-sst2A cells were stimulated with 10 nm SS14 at 37 °C for the times shown. Cells were then fixed, and receptor phosphorylation was measured at Ser-341/343 (panel A) or Thr-353/354 (panel B) using the in-cell ELISA. The half-times for receptor phosphorylation at Ser(P)-341/343 and Thr(P)-353/354 were 0.44 ± 0.13 and 2.11 ± 0.33 min, respectively (mean ± S.E., n = 4), and were significantly different (p < 0.01).
FIGURE 3.

Rate of receptor phosphorylation is reduced by mutation of upstream or downstream phosphorylation sites. CHO cells stably expressing either the WT receptor or the Ser− or Thr− mutant receptors were stimulated with 10 nm SS14 at 37 °C for various times. Cells were then solubilized, and equal protein samples from each treatment group were purified by adsorption to WGA-agarose followed by SDS-PAGE and immunoblotting with the phospho-specific antibodies shown. Subsequently, membranes were stripped and probed with HA antibody to control for receptor loading. The intensity of the receptor bands was quantitated as described under “Experimental Procedures,” and phosphorylated receptor was normalized for total receptor level and expressed as a percent of the signal at 15 min. Panels A and B show a representative experiment in which phosphorylation of Ser-341/343 was measured in either the WT receptor (panel A) or the Thr− receptor (panel B). Panels D and E show a representative experiment in which phosphorylation on Thr-353/354 was determined in either the WT receptor (panel D) or in the Ser− mutant receptor (panel E). Panels C and F shows the mean ± S.E. for site-specific receptor phosphorylation in WT and mutant receptors from three independent experiments. The half-time for Ser(P)-341/343 phosphorylation was 0.23 ± 0.06 min in the WT receptor and 3.34 ± 0.34 min in the Thr− mutants (p < 0.0001). The half-time for Thr(P)-353/354 phosphorylation was 1.19 ± 0.19 min in the WT receptor and 4.80 ± 1.92 min in the Ser− mutants (p < 0.0001). Panels G and H show the level of GRK2 and GRK6, respectively, in the cells expressing WT receptor and the Ser− or Thr− mutant receptors. Actin was used to determine equal loading.
Stimulation of CHO-sst2A cells with 10 nm SS14 increased phosphorylation at Ser-341/343 with a half-times of 0.44 ± 0.13 min by ELISA (Fig. 2, panel A) and 0.23 ± 0.06 min by immunoblotting (Fig. 3, panels A and C). Phosphorylation at Thr-353/354, although still rapid, was about five times slower than Ser-341/343 phosphorylation in both assays (t½ of 2.11 ± 0.33 min, Fig. 2, panel B, and 1.19 ± 0.19 min, Fig. 3, panels D and F). Steady state phosphorylation of all sites was maintained for at least 2 h in the continued presence of agonist. Thus, the two assays are in excellent agreement and demonstrate that sst2A receptor phosphorylation occurs sequentially, with Ser-341/343 being phosphorylated about five times more quickly than Thr-353/355.
Because receptor phosphorylation occurred in an ordered manner following agonist stimulation, we next asked whether phosphorylation of Ser-341/343 was required for Thr-353/354 phosphorylation. Therefore, we compared the rate at which Thr-353/354 was phosphorylated in the WT receptor and in a Ser− mutant in which all Ser in the C terminus and IC3 of the receptor were replaced with Ala. The results in Fig. 3 (panels D–F) show that the half-time for Thr-353/354 phosphorylation was increased 4-fold, from 1.19 ± 0.19 min in the WT receptor to 4.80 ± 1.92 min in the Ser− mutant. However, because Thr phosphorylation still occurred in the Ser− receptor, phosphorylation of Ser-341/343 was not essential for phosphorylation of Thr-353/354.
The observation that mutating upstream Ser residues reduced the rate of Thr-353/354 phosphorylation prompted us to examine the reverse situation, namely whether mutation of downstream Thr sites affected the phosphorylation of Ser-341/343. The results in Fig. 3 (panels A–C) show that the rate of Ser-341/343 phosphorylation was decreased more than 10-fold by mutating Thr phosphorylation sites; the half-time for Ser-341/343 phosphorylation went from 0.23 ± 0.06 min in the WT receptor to 3.34 ± 0.34 min in the Thr− mutant. Thus, even though Ser-341/343 are phosphorylated prior to Thr-353/354 in the WT sst2A receptor, mutation of the Thr residues markedly reduced the rate of Ser-341/343 phosphorylation.
We had previously shown that the WT, Thr−, and Ser− cell lines expressed similar receptor levels (25). To determine whether differences in GRK concentrations could account for the observed differences in the rates of receptor phosphorylation, we compared GRK expression in the three cell lines. All expressed substantial amounts of GRK2 and -6 (Fig. 3, panels G and H), showed very low GRK5 levels, and no detectable GRK3 (data not shown), consistent with a previous report (32). The concentration of GRK2 and -6 was identical in the three cells lines (Fig. 3, panels G and H) as expected because they were derived by transfection of the same parental cells. Thus, neither differences in receptor density nor differences in GRK concentrations can explain the observed differences in the phosphorylation kinetics of the WT receptors and the Thr− and Ser− mutants. Rather, our results indicate that the rate of sst2A receptor phosphorylation is inhibited by mutations both upstream and downstream of substrate Ser/Thr residues and suggest a high degree of structural specificity for kinase interaction with the receptor.
Dependence of sst2A Receptor Phosphorylation on Agonist Activation
To determine whether receptor phosphorylation at different sites showed the same agonist dependence, we measured the dose response for SS14-induced sst2A receptor phosphorylation at Ser-341/343 and Thr-353/354. CHO-sst2A cells were treated with various concentrations of SS14 for 30 min, and site-specific sst2A receptor phosphorylation was quantitated using the in-cell ELISA. Phosphorylation of Ser-341/343 (Fig. 4, panel A) and Thr-353/354 (panel B) was equally sensitive to agonist. SS14 increased Ser-341/343 and Thr-353/354 phosphorylation with EC50 values of 1.27 ± 0.12 and 1.12 ± 0.13 nm, respectively. A saturating concentration of antagonist (Coy-14) had no effect on sst2A receptor phosphorylation by itself but completely blocked the stimulatory effect of SS14 (Fig. 4, panels C and D).
FIGURE 4.

Agonist dependence for SS14-induced receptor phosphorylation. CHO cells stably expressing the sst2A receptor were treated with the indicated concentrations of SS14 for 30 min at 37 °C. Phosphorylation on either Ser-341/343 (●) (panel A) or Thr-353/354 (○) (panel B) was measured using the intact cell ELISA. Each point shows the mean ± S.E. of triplicate samples from a representative experiment. Where not shown, error bars fall within the symbol size. In multiple independent experiments, the EC50 for SS14-induced phosphorylation at Ser-341/343 (1.27 ± 0.12 nm, n = 11) and Thr-353/354 (1.12 ± 0.13 nm, n = 6) was the same. Panels C and D, CHO cells expressing the wild type sst2A receptor were incubated for 30 min at 37 °C without (□) or with (■) 10 nm SS14 either in the absence (Control) or in the presence of antagonist (Coy-14, 1 μm). Cells were then fixed, and receptor phosphorylation was determined with the in-cell ELISA using the phospho-specific antibodies shown. Antagonist did not by itself affect sst2A receptor phosphorylation but blocked the stimulatory effect of SS14 at both Ser-341/343 and Thr-353/354.
Kinetics and Compartmentalization of sst2A Receptor Dephosphorylation
Given the striking difference in the rates of agonist-induced Ser-341/343 and Thr-353/354 phosphorylation, we next examined the kinetics of receptor dephosphorylation. We first determined whether the presence of excess antagonist during agonist washout altered the rate of receptor dephosphorylation. In fact, the rate of receptor dephosphorylation was unaffected by adding antagonist at the time of SS14 removal (supplemental Fig. 2). Both for this reason and because under physiological conditions hormone stimulation is reversed by simple washout, antagonist was not included in subsequent dephosphorylation incubations.
Some GPCRs must be internalized before they can be dephosphorylated (33), whereas others do not (29, 31, 34). For yet a third group, conflicting results have been reported (35–37). Therefore, we examined sst2A receptor dephosphorylation after different times of agonist treatment. Our previous studies showed that the sst2A receptor is internalized with a half-time of 5 min in CHO-sst2A cells (25), and this was confirmed here (see below). Furthermore, in preliminary experiments, we found that receptor endocytosis stops when SS14 is removed by washing the cells (data not shown). Therefore, we stimulated cells with SS14 for either 2 min (Fig. 5, panel A), at which time over 80% of the receptor is still at the cell surface, or for 15 min (Fig. 5, panel B), at which point ∼70% of the receptor has been internalized (25). After hormone treatment, cells were washed and incubated in fresh 37 °C medium. Receptor phosphorylation was measured by ELISA after various periods of agonist removal. Little dephosphorylation occurred at Ser-341/343 following a 2-min SS14 challenge (Fig. 5, panel A), whereas this site was rapidly dephosphorylated (t½ = 6.7 ± 0.6 min) after 15 min of SS14 treatment (Fig. 5, panel B). These results indicate that dephosphorylation of Ser-341/343 occurs only after receptor internalization. In fact, the small amount of Ser-341/343 dephosphorylation observed after the 2 min of SS14 treatment (Fig. 5, panel A) probably represents the dephosphorylation of receptor internalized during the short agonist exposure.
FIGURE 5.

Duration of agonist stimulation determines subsequent receptor dephosphorylation. CHO-sst2A cells were stimulated at 37 °C with 10 nm SS14 for either 2 min (panel A) or 15 min (panel B). Following a rapid wash, cells were incubated in fresh 37 °C medium without agonist for the times shown. Receptor phosphorylation was determined at Ser-341/343 (●) and Thr-353/354 (○) using the in-cell ELISA. Nonlinear regression curve fitting to a single exponential decay in multiple independent experiments gave a half-time for receptor dephosphorylation at Thr-353/354 of 6.5 ± 0.5 min (n = 5) after 2 min of SS14 treatment and 7.5 ± 0.5 min (n = 4) after 15 min of SS14 stimulation. The half-time for Ser-341/343 dephosphorylation could not be calculated after 2 min of SS14 treatment because so little of the receptor was dephosphorylated. The half-time for Ser-341/343 dephosphorylation following a 15-min SS14 challenge was 6.7 ± 0.6 min (n = 4).
In contrast to the observations with Ser-341/343, extensive Thr-353/354 dephosphorylation occurred after both 2 min (Fig. 5, panel A) and 15 min (Fig. 5, panel B) of SS14 stimulation. Moreover, the rate of Thr-353/354 dephosphorylation was unaffected by the duration of the preceding agonist treatment (t½ = 6.5 ± 0.5 min after 2 min and 7.5 ± 0.5 min after 15 min of SS14 stimulation (n = 4)). Thus, in contrast to Ser-341/343, dephosphorylation of Thr-353/354 can occur either at the cell surface or after receptor endocytosis. Thus, these results demonstrate a striking spatial discrimination in sst2A receptor dephosphorylation; although both Ser-341/343 and Thr-353/354 are dephosphorylated at similar rates after the receptor has internalized, only Thr-353/354 can be dephosphorylated when the receptor resides in the plasma membrane.
To further investigate the extent to which dephosphorylation was spatially constrained, we examined sst2A receptor dephosphorylation after 15 min of SS14 stimulation under normal conditions and when receptor internalization was blocked. Pretreatment of cells with hypertonic sucrose is known to disrupt clathrin-coated pits and, as a result, to inhibit clathrin-dependent receptor endocytosis (38). The GTPase dynamin is essential for clathrin-dependent coated vesicle formation, and dynasore, a noncompetitive inhibitor of dynamin's GTPase activity, has been shown to block dynamin-dependent receptor endocytosis in cells (39). The results in Fig. 6 (panel A) demonstrate that both hypertonic sucrose and dynasore prevent SS14-stimulated sst2A receptor endocytosis. In the absence of inhibitors, SS14 elicited rapid receptor internalization (t½ = 4.9 ± 0.8 min) that was completely blocked by sucrose and dynasore.
FIGURE 6.

Inhibition of receptor internalization selectively blocks receptor dephosphorylation at Ser-341/343. CHO-sst2A cells were equilibrated in the absence (●) or presence of either 0.45 m sucrose (○) or 50 μm dynasore (▿) for 15 min at 37 °C. Panel A, cells were then stimulated with 10 nm SS14 for the times shown in the continued absence or presence of either sucrose or dynasore. After washing with cold PBS, cells were fixed, and cell surface receptors were measured by ELISA as described under “Experimental Procedures.” Surface receptors at each time point were expressed as the percentage of the nonstimulated control group (t = 0). Panel A shows that the rate of receptor internalization in untreated cells followed first order kinetics. In three independent experiments, the half-time for sst2A receptor internalization was 4.9 ± 0.8 min for untreated cells, and sucrose and dynasore completely blocked sst2A receptor internalization. Panels B–E, rate of receptor dephosphorylation was measured in untreated CHO-sst2A cells (●) or in the presence of either 0.45 m sucrose (○) or 50 μm dynasore (▿). Cells were preincubated with inhibitors as above and then stimulated with 10 nm SS14 for an additional 15 min to allow phosphorylation to reach a steady state. Following a rapid wash, cells were incubated at 37 °C for the times shown in fresh medium without SS14 in the continued absence (●) or presence of either sucrose (○) (panels B and D) or dynasore (▿) (panels C and E). Following fixation, receptor phosphorylation was measured on Ser-341/343 (panels B and C) and Thr-353/354 (panels D and E) with an in-cell ELISA. Nonlinear regression curve fitting to a single exponential decay in nine independent experiments gave a half-time of 7.9 ± 0.9 min for Ser-341/343 dephosphorylation and 7.6 ± 0.4 min for Thr-353/354 dephosphorylation in untreated cells. In the presence of sucrose or dynasore, Ser-341/343 dephosphorylation was largely blocked. In contrast, dephosphorylation of Thr-353/354 was unaffected by the endocytosis inhibitors (t½ was 9.5 ± 1.4 min (n = 3) with sucrose and 10.9 ± 1.4 (n = 2) with dynasore).
Because neither sucrose nor dynasore altered the kinetics of sst2A receptor phosphorylation (data not shown), we preincubated cells for 15 min without or with 0.45 m sucrose or 50 μm dynasore and then stimulated cells with 10 nm SS14 for 15 min to achieve maximal phosphorylation (Fig. 6). Thereafter, agonist was removed by washing, and sst2A receptor dephosphorylation was measured in the continued absence or presence of inhibitor.
As before, receptor dephosphorylation occurred rapidly in control cells both at Ser-341/343 (t½ = 7.9 min) (Fig. 6, panels B and C) and at Thr-353/354 (t½ = 7.6 min) (Fig. 6, panels D and E). Strikingly, both sucrose (Fig. 6, panel B) and dynamin (Fig. 6, panel C) completely blocked Ser-341/343 dephosphorylation. In contrast, neither inhibitor altered the rate of Thr-353/354 dephosphorylation in the same experiments (Fig. 6, panels D and E). These results confirm the conclusion that dephosphorylation of Ser-341/343 occurs only after receptor endocytosis, whereas dephosphorylation of Thr-353/354 occurs both at the cell surface and in endocytic vesicles.
Dephosphorylation of Ser-341/343 and Thr-353/354 Is Catalyzed by Different Phosphatases
To investigate the phosphatases responsible for dephosphorylating the sst2A receptor at different phospho-sites and in different cellular compartments, we first examined the effect of okadaic acid. This microbial toxin specifically inhibits PPP-type protein phosphatases blocking PP2A, PP4, PP5, and PP6 at nanomolar concentrations and PP1 at micromolar concentrations (40–42).
Because only Thr-353/354 is dephosphorylated at the plasma membrane, we examined the effect of okadaic acid on receptor dephosphorylation at this phospho-site under conditions where receptor internalization was minimal (Fig. 7). Cells were either incubated with SS14 for 2 min, such that most of the receptors remained at the cell surface (Fig. 7, panel A), or incubated with SS14 for 15 min in the presence of 0.45 m sucrose to block receptor endocytosis (Fig. 7, panel B). Agonist stimulation was carried out in the absence or presence of okadaic acid, and then cells were washed to remove SS14 and incubated further in fresh medium without agonist but in the continued absence or presence of okadaic acid and/or sucrose. Okadaic acid blocked Thr-353/354 dephosphorylation under both conditions.
FIGURE 7.

Okadaic acid inhibits dephosphorylation of cell surface receptors at Thr-353/354. Dephosphorylation of cell surface receptors was determined in CHO-sst2A cells in two ways. Panel A, cells were treated with 10 nm SS14 for 2 min. Panel B, cells were treated with 10 nm SS14 for 15 min in the presence of 0.45 m sucrose to block receptor internalization. In both experiments, cells were preincubated for 15 min without (●) or with (○) 10 nm okadaic acid prior to the SS14 stimulation. Following a rapid wash to remove agonist, cells were incubated for the times shown in fresh 37 °C medium without SS14 in the continued absence (●) or presence (○) of the phosphatase inhibitor. Cells were then fixed, and receptor phosphorylation at Thr-353/354 was measured with an in-cell ELISA. In two independent experiments the half-time for Thr-353/354 dephosphorylation was 7.2 ± 0.4 min after 2 min of SS14 stimulation and 7.1 ± 0.5 min following 15 min of SS14 stimulation in the presence of sucrose. Okadaic acid blocked Thr-353/354 dephosphorylation under both conditions.
To examine the effect of okadaic acid on the dephosphorylation of internalized receptors, CHO-sst2A cells were pretreated with 10 nm okadaic acid and then incubated with SS14 for 15 min. Receptor dephosphorylation was then measured after agonist washout as before. Okadaic acid had little effect on Ser-341/343 dephosphorylation (supplemental Fig. 3, panel A), whereas it again completely blocked the dephosphorylation of Thr-353/354 (supplemental Fig. 3, panel B). Hence, different phosphatases must catalyze the dephosphorylation of the internalized sst2A receptor at Ser-341/343 and at Thr-353/354.
PP1 and PP2A show large differences in their sensitivity to inhibition by okadaic acid (40–42). Thus, we next determined the effect of a range of okadaic acid concentrations on receptor dephosphorylation at both Thr-353/354 and Ser-341/343 (Fig. 8). To allow receptor endocytosis, CHO-sst2A cells were incubated with SS14 for 15 min in the absence and presence of various concentrations of okadaic acid, washed to remove agonist, and then incubated without agonist for an additional 15 min to permit receptor dephosphorylation. Consistent with our previous results, okadaic acid potently blocked Thr-353/354 dephosphorylation (IC50 of 2.45 ± 0.4 nm) (Fig. 8, panel A). However Ser-341/343 dephosphorylation was unaffected even at a concentration of 1 μm okadaic acid (Fig. 8, panel A). These results support the conclusion that a PP2A-like phosphatase catalyzes sst2A receptor dephosphorylation at Thr-353/354. Dephosphorylation of Ser-341/343 is catalyzed by a different phosphatase, one that is insensitive to even high okadaic acid concentrations.
FIGURE 8.

Distinct phosphatases catalyze sst2A receptor dephosphorylation at different phospho-sites. Panel A, CHO-sst2A cells were preincubated with the indicated concentrations of okadaic acid for 15 min and then stimulated for 15 min with 10 nm SS14 in the continued absence or presence of the phosphatase inhibitor. The cells were then washed and incubated in fresh medium without agonist but with okadaic acid for an additional 15 min at 37 °C to allow receptor dephosphorylation to occur. Residual receptor phosphorylation at Ser-341/343 (●) and Thr-353/354 (○) was measured with the in-cell ELISA and expressed as the percent inhibition of the dephosphorylation observed in control cells. In multiple independent experiments, okadaic acid blocked Thr-353/354 dephosphorylation with an EC50 of 2.45 ± 0.40 nm (n = 4) but did not affect Ser-341/343 dephosphorylation even at 1 μm. Panel B, to determine whether okadaic acid inhibition of receptor dephosphorylation at Thr-353/354 was affected by the cellular location of the receptor, CHO-sst2A cells were preincubated with various concentrations of okadaic acid and then treated with 10 nm SS14 for either 2 min (●) or 15 min (○) at 37 °C in the continued presence of the phosphatase inhibitor. The cells were then washed and incubated in fresh medium without agonist but with okadaic acid for an additional 15 min at 37 °C to allow receptor dephosphorylation to occur. Residual phosphorylation at Thr-353/354 was measured with the in-cell ELISA and expressed as the percent inhibition of the dephosphorylation observed in control cells. The EC50 for okadaic acid inhibition of Thr-353/354 dephosphorylation after 2 and 15 min of SS14 stimulation was 2.95 ± 0.52 and 2.45 ± 0.40 nm, respectively.
We next used sensitivity to okadaic acid to compare the enzymes responsible for Thr-353/354 dephosphorylation at the cell surface and in endosomal compartments. CHO-sst2A cells were incubated with SS14 either for 2 min or for 15 min in the absence and presence of varying okadaic acid concentrations. After washing to remove agonist, cells were incubated for an additional 15 min without SS14 in the continued presence of okadaic acid. As shown in Fig. 8, panel B, okadaic acid inhibited Thr-353/354 dephosphorylation with the same potency after stimulating cells with SS14 for 2 min (IC50 2.95 ± 0.52 nm) and for 15 min (IC50 2.45 ± 0.40), indicating that Thr-353/354 dephosphorylation is catalyzed by the same phosphatase both at the plasma membrane and in endocytic vesicles.
Although PP2A and PP1 account for the majority of okadaic acid-sensitive phosphatase activity in cells, protein phosphatases 5 and 6 are also inhibited by low concentrations of okadaic acid. Because fostriecin inhibits PP2A and PP4 at concentrations that are 10,000-fold lower than required for PP5 inhibition (42), we also examined the effect of this shellfish toxin on sst2A receptor dephosphorylation. The results in Fig. 9 show that 5 nm fostriecin had the same effect on Thr-353/354 dephosphorylation as okadaic acid; it blocked receptor dephosphorylation both at the cell surface (Fig. 9, panel A) and intracellularly (Fig. 9, panel B). In contrast, fostriecin had no effect on Ser-341/343 dephosphorylation (Fig. 9, panel C). These results show that dephosphorylation of Thr-353/354 is catalyzed by PP2A or PP4 and further support the conclusion that Ser-341/343 dephosphorylation involves a different phosphatase.
FIGURE 9.
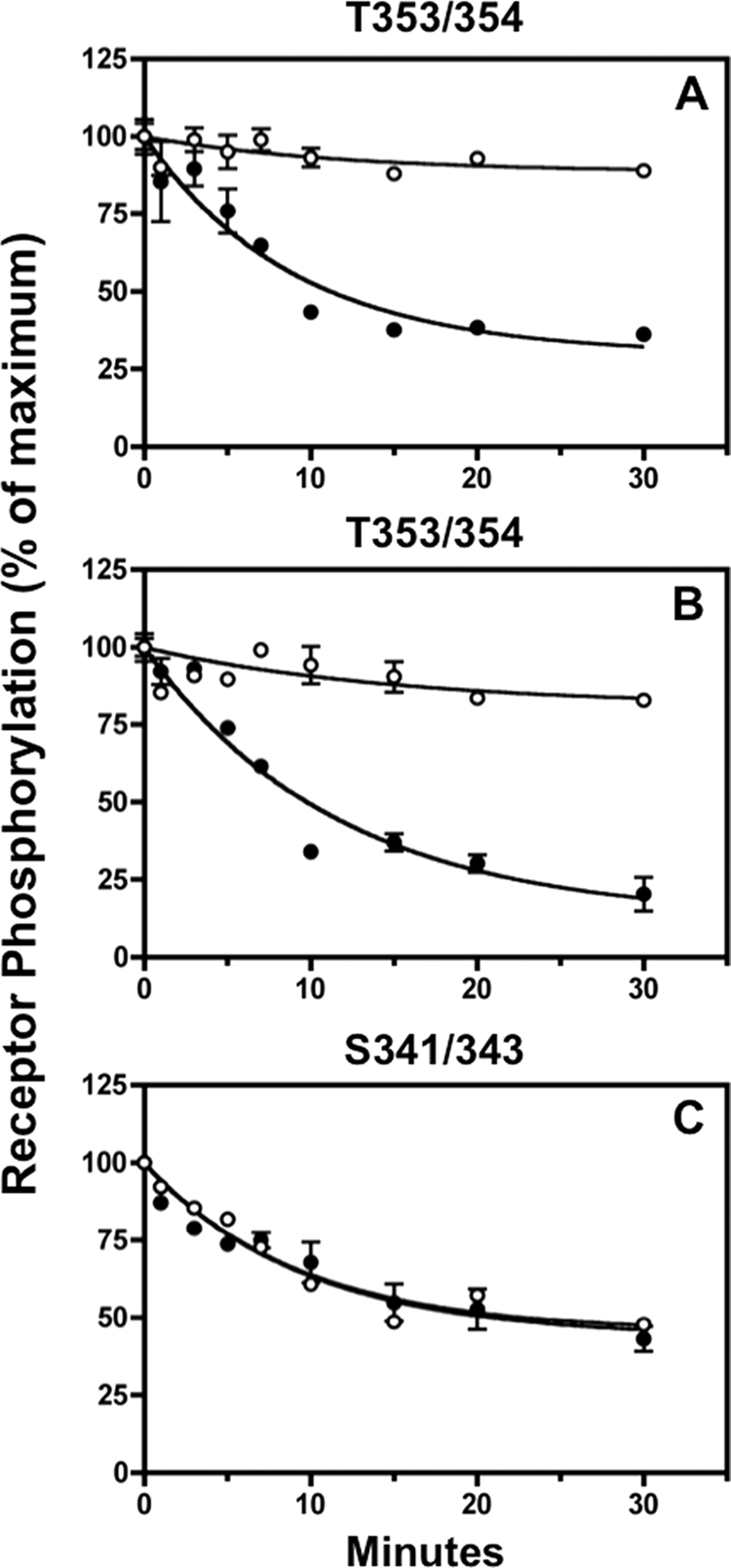
Fostriecin inhibits sst2A receptor dephosphorylation at Thr-353/354 but not at Ser-341/343. Panel A, CHO-sst2A cells were preincubated without (●) or with (○) 5 nm fostriecin for 15 min at 37 °C and then stimulated with 10 nm SS14 for either 2 min (panel A) or 15 min (panels B and C) in the continued absence or presence of the phosphatase inhibitor. Following a rapid wash to remove agonist, cells were subsequently incubated for the times shown in fresh 37 °C medium without SS14 in the continued absence or presence of fostriecin. Cells were then fixed, and the residual receptor phosphorylation was measured with the in-cell ELISA. The graphs show the results of replicate samples in a representative experiment. In multiple independent experiments, the half-time for dephosphorylation of Thr-353/354 in control cells was 6.9 ± 1.1 min (n = 2) after 2 min of agonist exposure (cell surface receptors) and 7.3 ± 0.1 min (n = 2) after 15 min of agonist exposure (internalized receptors). Fostriecin blocked Thr-353/354 dephosphorylation both at the cell surface and intracellularly. In contrast, Ser-341/343 dephosphorylation was unaffected by fostriecin; the half-time for Ser-341/343 dephosphorylation was 8.6 ± 0.7 min (n = 2) min and 7.00 ± 1.0 (n = 2) min in the absence and presence of fostriecin, respectively.
In an effort to identify the phosphatase responsible for sst2A dephosphorylation at Ser-341/343, we determined the effect of FK506, a potent inhibitor of protein phosphatase 2B (calcineurin). However, 1 μm FK506 did not affect sst2A receptor dephosphorylation at Ser-341/343 (supplemental Fig. 4). Furthermore, as expected, FK506 did not inhibit sst2A receptor dephosphorylation at Thr-353/354 either before (data not shown) or after (supplemental Fig. 4) internalization. Thus, the phosphatase responsible for Ser-341/343 dephosphorylation must belong to a phosphatase class, such as PP2C, that is insensitive to known inhibitors.
Table 1 summarizes the effect of inhibitors on sst2A receptor dephosphorylation. Together, the results demonstrate that site-specific sst2A receptor dephosphorylation is catalyzed by at least two different phosphatases.
TABLE 1.
Effect of phosphatase inhibitors on site-specific sst2A receptor dephosphorylation
∅ indicates no inhibition.
| Inhibitor | Target phosphatase | Phospho-site |
||
|---|---|---|---|---|
| Thr(P)-353/354 |
Ser(P)-341/343 intracellular | |||
| Cell surface | Intracellular | |||
| Okadaic acid 10 nm | PP2A, PP4, PP5, PP6 | Block | Block | ∅ |
| Okadaic acid 1 μm | PP1, PP2A, PP4, PP5, PP6 | Block | Block | ∅ |
| Fostriecin 5 nm | PP2A, PP4 | Block | Block | ∅ |
| FK506 1 μm | PP2B | ∅ | ∅ | ∅ |
DISCUSSION
GPCR signaling and trafficking are stringently regulated by receptor phosphorylation, which usually occurs on multiple Ser and Thr residues in the receptor C terminus or third intracellular loop. Studies with individual GPCRs have suggested that phosphorylation at different sites within these highly phosphorylated receptor domains can have distinct consequences for receptor activity, such that the functional state of the receptor is determined by the particular phosphorylated species that can exist (10, 43–46). In fact, this is the case for the sst2A receptor; phosphorylation of Thr residues is necessary for rapid agonist-induced internalization, whereas phosphorylation of Ser residues is responsible for receptor uncoupling (25). Yet because measurement of receptor phosphorylation at individual phospho-sites is experimentally difficult in intact cells, the detailed regulation of GPCR phosphorylation has been investigated for only a few receptors and is not fully understood with any GPCR.
To understand the biochemical mechanisms that control GPCR activity, we characterized the phosphorylation and dephosphorylation of two pairs of GRK sites in the C terminus of the sst2A somatostatin receptor in intact cells. Use of phospho-site-specific antibodies and a sensitive ELISA allowed us to quantitate wild type receptor phosphorylation directly and avoid the uncertainties inherent in mutagenesis-based studies. We found that receptor phosphorylation occurred rapidly and in a sequential manner. Mutation of either the pair of residues that are phosphorylated first (Ser-341/343) or the pair phosphorylated subsequently (Thr-353/354) reduced the rate at which the remaining sites were modified suggesting a stringent structural requirement for GRK catalysis that is independent of the hierarchy of receptor phosphorylation. Unexpectedly, the two pairs of phospho-sites were dephosphorylated by distinct phosphatases and in different cellular compartments. Following removal of agonist, Thr-353/354 was subject to dephosphorylation both at the cell surface and in endocytic vesicles, and these reactions were catalyzed by a PP2A/PP4 phosphatase. In contrast, Ser-341/343 was dephosphorylated only after receptor internalization, and the enzyme involved was insensitive to available inhibitors for different phosphatase classes. Thus, the phosphorylation state of the sst2A receptor is controlled by multiple enzymes acting in specific cellular compartments and, as a result, depends on the duration of receptor activation.
We used two different experimental methods to measure sst2A receptor phosphorylation in intact cells. Immunoblot analysis was used to detect the phosphorylation of purified receptors, as described in detail previously (24). Accurate quantitation of receptor phosphorylation was performed with a rapid in-cell ELISA. The specificities of the antibodies used for immunoblotting and ELISA were very similar (supplemental Table 1), and as shown by multiple control experiments, all antibodies reacted only with the phosphorylated form of the sst2A receptor (Fig. 1 and supplemental Fig. 1). When compared, results from immunoblotting and in cell ELISA agreed closely.
Although the sst2A receptor can be phosphorylated by PKC as well as by GRKs, the phosphorylation reactions investigated here are catalyzed only by GRKs. Early studies, looking at overall phosphorylation by 32PO4 labeling showed that agonist-induced receptor phosphorylation did not involve second messenger-activated kinases (22, 23, 47). More recently, using phospho-site-specific antibodies, we showed that neither forskolin nor phorbol 12-myristate 13-acetate stimulated sst2A receptor phosphorylation at Ser-341/343 or Thr-353/354, whereas SS14 rapidly increased phosphorylation at all these sites (24). Finally, siRNA knockdown of GRK2 inhibited SS14-stimulated receptor phosphorylation at both Ser-341/343 (24) and Thr-353/354.3 Thus, the phosphorylation reactions examined here are catalyzed by GRK2 and possibly other GRKs as well. This conclusion is consistent with our observation that phosphorylation of Ser-341/343 and Thr-353/354 shows the same agonist dose dependence because receptor phosphorylation by second messenger-activated kinases often occurs at much lower agonist concentrations than GRK-catalyzed receptor phosphorylation (48, 49).
Several studies have compared the kinetics of GRKs and a second messenger-activated kinases to phosphorylate different receptor residues in intact cells (46, 48, 49). However, to our knowledge, the rates with which multiple identified GRK sites are phosphorylated has only been determined for rhodopsin (11, 50) and the chemokine receptor CXCR4 (46). Thus, it is interesting to compare the phosphorylation kinetics of the two peptide-activated receptors with the kinetics of rhodopsin phosphorylation. After a flash of light, three Ser residues are phosphorylated in a sequential manner in rhodopsin, with half-times of 0.26 min (Ser-343), 1.4 min (Ser-338), and 13.9 min (Ser-334) (11). Upon exposure to SS14 the sst2A receptor is also phosphorylated sequentially and at remarkably similar rates; the half-times for phosphorylation on Ser-341/343 and Thr-353/354 were ∼0.3 and 1.7 min (summarized in Fig. 10). However, in rhodopsin the sites closest to the C terminus are phosphorylated the fastest, whereas in the sst2A receptor, the Ser residues closest to the plasma membrane are phosphorylated first, and the distal Thr are phosphorylated more slowly. The sequence of GRK-mediated CXCR4 phosphorylation is even more complex; GRK-catalyzed phosphorylation occurs first at Ser-339 followed by Ser-324/325 and then Ser-330 (46). Thus, a general pattern for multisite phosphorylation of GPCRs by GRKs has yet to emerge.
FIGURE 10.

Specificity in the phosphorylation and dephosphorylation of neighboring GRK sites in the sst2A receptor. Upon binding of SS14, the sst2A receptor is activated and becomes rapidly phosphorylated by GRKs first on Ser-341/343 and subsequently on Thr-353/454. In the continued presence of agonist, the phosphorylated receptor is internalized. However, if agonist is removed while the receptor is on the cell surface, Thr(P)-353/354 are rapidly dephosphorylated by an okadaic acid and fostriecin-sensitive phosphatase, shown as PP2A. In contrast, phosphorylated Ser-341/343 are resistant to dephosphorylation while the receptor is on the plasma membrane. If agonist is removed after the receptor has been internalized, the receptor is dephosphorylated at both Ser-341/343 and at Thr-353/354 by two different phosphatases. An okadaic acid/fostriecin-sensitive phosphatase (shown as PP2A) again dephosphorylates Thr-353/354 in internalized receptors. In contrast, Ser-341/343 is dephosphorylated by a phosphatase (PPx), which is resistant to okadaic acid and fostriecin as well as FK506 and thus may belong to the PP2C phosphatase families, for which no inhibitors have been identified. Dephosphorylated sst2A receptors are recycled back to the plasma membrane for another round of signaling.
The reason for a time lag in the GRK-catalyzed phosphorylation of residues only a few amino acids apart in all three receptors is unknown. We tested the hypothesis that Ser-341/343 phosphorylation was required for the subsequent phosphorylation of Thr-353/354 by comparing the rate of Thr-353/354 phosphorylation in WT receptors and in receptors lacking the Ser phosphorylation sites. Indeed, mutating Ser-341/343 to Ala slowed the rate of Thr-353/354 phosphorylation 4-fold, although it did not block it entirely. Unexpectedly, however, mutating Thr-353/354 to Ala also reduced the rate of Ser-341/343 phosphorylation, even though Ser-341/343 is phosphorylated prior to Thr-353/354 in the WT receptor. These results suggest that mutations affect the phosphorylation of distal receptor residues because of the structural changes they produce rather than by interfering with the normal phosphorylation sequence. Although Ser/Thr mutations often disrupt GPCR phosphorylation at distal sites, our results suggest that such an effect does not demonstrate that receptor phosphorylation occurs in a particular order.
An important unanswered question for GPCR regulation is whether dephosphorylation and reactivation of receptors require endocytosis or whether these processes can occur at the plasma membrane. Thus, one of our most striking and unexpected findings was that dephosphorylation of two neighboring GRK sites in the C-tail of the sst2A receptor occurs in different cellular compartments. Because the sst2A receptor internalizes with a half-time of 5 min in the CHO-sst2A cells used in this study (Fig. 6) (25), receptor phosphorylation precedes receptor endocytosis (Fig 10), consistent with mutagenesis studies showing that phosphorylation is required for rapid sst2A receptor internalization (25). Our kinetic measurements show that a short pulse of agonist will lead to the plasma membrane accumulation of receptor initially phosphorylated on Ser-341/343 and then on both Ser-341/343 and Thr-353/354 (Fig 10). Following washout of agonist after such a brief stimulation, Thr-353/354 is rapidly dephosphorylated, whereas dephosphorylation of cell surface receptors at Ser-341/343 does not occur. Thus, a new steady state will be established in which cell surface receptors are phosphorylated on Ser-341/343 but not on Thr-353/354 (Fig 10). This will be the phosphorylation state of sst2A receptors following recovery from a short pulse of agonist, as occurs in neurons. In contrast, longer exposure of cells to agonist, as occurs during endocrine or pharmacological stimulation, will lead to the intracellular accumulation of phosphorylated receptors (25). Removal of agonist after the receptor has been internalized permits receptor dephosphorylation at both Ser-341/343 and Thr-353/354 (Fig 10), and the internalized dephosphorylated receptor is then recycled to the cell surface over the next hour.4
The compartment-specific dephosphorylation of adjacent GRK sites in the sst2A receptor is particularly interesting because previous studies with other GPCRs differ as to whether endocytosis is required for receptor dephosphorylation. For example, internalization of CXCR2 receptors is essential for receptor dephosphorylation following agonist stimulation (33). In contrast, the β2-adrenergic receptor (37, 36) and the thyrotropin-releasing hormone receptor (29) can be dephosphorylated both at the cell membrane and intracellularly. However, to our knowledge, the sst2A receptor provides the first example of a GPCR in which neighboring GRK sites are dephosphorylated in distinct compartments. An important consequence of such site-specific dephosphorylation is that the phosphorylation state of the receptor will be determined by its subcellular distribution and will therefore vary with the time of agonist stimulation (Fig 10). Such differential dephosphorylation may explain why inhibitors of internalization can prevent resensitization of GPCRs in which some dephosphorylation occurs on the cell surface (35, 37).
The phosphatases that catalyze GPCR dephosphorylation have not been identified, and their sensitivity to inhibitors varies greatly. For example, okadaic acid inhibits the dephosphorylation of β2-adrenergic, CCR5, and κ-opioid receptors (48, 51, 52) but does not affect TRH receptor dephosphorylation (29). In intact cells, okadaic acid will inhibit PP2A, PP4, PP5, and PP6 at concentrations of 10–100 nm, although it will also inhibit PP1 at a concentration of 1 μm or greater (41, 42). In our study, okadaic acid did not affect sst2A receptor dephosphorylation at Ser-341/343, even at a concentration of 1 μm. In contrast, nanomolar concentrations of okadaic acid blocked receptor dephosphorylation at Thr-353/354. Interestingly okadaic acid was equipotent at inhibiting Thr-353/354 dephosphorylation at the cell surface and intracellularly, suggesting that the same enzyme is responsible for catalyzing Thr-353/354 dephosphorylation in both compartments. Fostriecin, a potent inhibitor of PP2A and PP4 but not of PP1 and PP5 (42) also had phospho-site-specific effects on sst2A receptor dephosphorylation, inhibiting Thr-353/354 dephosphorylation without affecting dephosphorylation at Ser-341/343. Inhibition of PP2B with FK506 did not block either Ser-341/343 or Thr-353/354 dephosphorylation. Together these results demonstrate that at least two different phosphatases catalyze sst2A receptor dephosphorylation (Table 1 and Fig 10). A PP2A/PP4 phosphatase dephosphorylates the receptor at Thr-353/354, and an inhibitor-resistant phosphatase catalyzes Ser-341/343 dephosphorylation. Unfortunately, inhibitors have yet to be identified for some phosphatase classes (e.g. PP2C) (42), and as a result, the nature of the phosphatase catalyzing sst2A receptor dephosphorylation at Ser-341/343 remains to be characterized. Thus, phosphatases that catalyze sst2A receptor dephosphorylation are determined both by the compartment in which the receptor is localized and the particular phospho-site serving as substrate. How this precise specificity is achieved at a molecular level will be an important area for future investigation.
In conclusion, we show that an unexpectedly complex set of enzymatic mechanisms regulate the phosphorylation state of the sst2A somatostatin receptor. Depending on the duration of agonist exposure and the extent of receptor internalization, this receptor exists in different phosphorylated forms on the cell surface and in intracellular compartments. After agonist stimulation, phosphorylation of plasma membrane receptors occurs rapidly and in a specific order followed by a somewhat slower rate of receptor internalization. Because receptor dephosphorylation is compartment-specific, recovery of a fully dephosphorylated, resensitized receptor at the cell surface cannot occur unless the receptor is internalized prior to agonist washout. After a short burst of agonist stimulation, partially dephosphorylated receptors accumulate on the plasma membrane. Because different phosphorylation sites on the sst2A receptor have specific effects on receptor function (25), the pattern of sst2A receptor phosphorylation is likely to be physiologically important. Neuronal sst2A receptors are subject to short bursts of peptide stimulation and may not have sufficient time to internalize before stimulation halts. In contrast, sst2A receptors in the pituitary undergo longer pulses of agonist exposure. In patients with sst2A receptor-containing endocrine tumors, chronic exposure to somatostatin causes the accumulation of intracellular phosphorylated receptors (53). The extent to which such complex regulatory mechanisms occur with other GPCRs is currently unknown, although it is clear that multiple forms of phosphorylated receptors are produced in cells upon agonist stimulation (12, 13, 46). Because the sequence and kinetics of GPCR phosphorylation and dephosphorylation have been determined at multiple phospho-sites in only a few instances, the existence, the nature, and the functional importance of partially phosphorylated receptor states remain to be characterized. Our studies show that such information will be essential for a full understanding of the activity of differentially phosphorylated receptor states and their significance in GPCR signaling.
Acknowledgments
We thank Dr. Qisheng Liu and Yachu Judy Kao for identifying and characterizing some of the phospho-specific antibodies used in this study.
This work was supported, in whole or in part, by National Institutes of Health Grant DK032234 (to A. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4 and Table 1.
Q. Liu and A. Schonbrunn, unpublished observations.
Y. J. Kao, M. Ghosh, and A. Schonbrunn, submitted for publication.
- GPCR
- G protein-coupled receptor
- GRK
- GPCR kinase
- TMB
- 3,3′,5,5′-tetramethylbenzidine.
REFERENCES
- 1. Park P. S., Lodowski D. T., Palczewski K. (2008) Annu. Rev. Pharmacol. Toxicol. 48, 107–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 3. Krupnick J. G., Benovic J. L. (1998) Annu. Rev. Pharmacol. Toxicol. 38, 289–319 [DOI] [PubMed] [Google Scholar]
- 4. Kohout T. A., Lefkowitz R. J. (2003) Mol. Pharmacol. 63, 9–18 [DOI] [PubMed] [Google Scholar]
- 5. Moore C. A., Milano S. K., Benovic J. L. (2007) Annu. Rev. Physiol. 69, 451–482 [DOI] [PubMed] [Google Scholar]
- 6. Gurevich V. V., Gurevich E. V., Cleghorn W. M. (2008) Handb. Exp. Pharmacol. 186, 15–37 [DOI] [PubMed] [Google Scholar]
- 7. Lefkowitz R. J., Shenoy S. K. (2005) Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 8. Hanyaloglu A. C., von Zastrow M. (2008) Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 9. Seachrist J. L., Ferguson S. S. (2003) Life Sci. 74, 225–235 [DOI] [PubMed] [Google Scholar]
- 10. Tobin A. B. (2008) Br. J. Pharmacol. 153, S167–S176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kennedy M. J., Lee K. A., Niemi G. A., Craven K. B., Garwin G. G., Saari J. C., Hurley J. B. (2001) Neuron 31, 87–101 [DOI] [PubMed] [Google Scholar]
- 12. Trester-Zedlitz M., Burlingame A., Kobilka B., von Zastrow M. (2005) Biochemistry 44, 6133–6143 [DOI] [PubMed] [Google Scholar]
- 13. Vishnivetskiy S. A., Raman D., Wei J., Kennedy M. J., Hurley J. B., Gurevich V. V. (2007) J. Biol. Chem. 282, 32075–32083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tobin A. B., Butcher A. J., Kong K. C. (2008) Trends Pharmacol. Sci. 29, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patwardhan P., Miller W. T. (2007) Cell. Signal. 19, 2218–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salazar C., Höfer T. (2009) FEBS J. 276, 3177–3198 [DOI] [PubMed] [Google Scholar]
- 17. Schonbrunn A. (2004) in Encyclopedia of Biological Chemistry (Lennarz W. J., Lane M. D. eds) pp. 55–60, Elsevier, Oxford [Google Scholar]
- 18. Olias G., Viollet C., Kusserow H., Epelbaum J., Meyerhof W. (2004) J. Neurochem. 89, 1057–1091 [DOI] [PubMed] [Google Scholar]
- 19. Reubi J. C. (2003) Endocr. Rev. 24, 389–427 [DOI] [PubMed] [Google Scholar]
- 20. Weckbecker G., Lewis I., Albert R., Schmid H. A., Hoyer D., Bruns C. (2003) Nat. Rev. Drug Discov. 2, 999–1017 [DOI] [PubMed] [Google Scholar]
- 21. Reubi J. C., Maecke H. R. (2008) J. Nucl. Med. 49, 1735–1738 [DOI] [PubMed] [Google Scholar]
- 22. Hipkin R. W., Friedman J., Clark R. B., Eppler C. M., Schonbrunn A. (1997) J. Biol. Chem. 272, 13869–13876 [DOI] [PubMed] [Google Scholar]
- 23. Hipkin R. W., Wang Y., Schonbrunn A. (2000) J. Biol. Chem. 275, 5591–5599 [DOI] [PubMed] [Google Scholar]
- 24. Liu Q., Bee M. S., Schonbrunn A. (2009) Mol. Pharmacol. 76, 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Q., Dewi D. A., Liu W., Bee M. S., Schonbrunn A. (2008) Mol. Pharmacol. 73, 292–304 [DOI] [PubMed] [Google Scholar]
- 26. Rajeswaran W. G., Hocart S. J., Murphy W. A., Taylor J. E., Coy D. H. (2001) J. Med. Chem. 44, 1305–1311 [DOI] [PubMed] [Google Scholar]
- 27. Liu Q., Cescato R., Dewi D. A., Rivier J., Reubi J. C., Schonbrunn A. (2005) Mol. Pharmacol. 68, 90–101 [DOI] [PubMed] [Google Scholar]
- 28. Gu Y. Z., Schonbrunn A. (1997) Mol. Endocrinol. 11, 527–537 [DOI] [PubMed] [Google Scholar]
- 29. Jones B. W., Song G. J., Greuber E. K., Hinkle P. M. (2007) J. Biol. Chem. 282, 12893–12906 [DOI] [PubMed] [Google Scholar]
- 30. Liu Q., Schonbrunn A. (2001) J. Biol. Chem. 276, 3709–3717 [DOI] [PubMed] [Google Scholar]
- 31. Gardner B., Liu Z. F., Jiang D., Sibley D. R. (2001) Mol. Pharmacol. 59, 310–321 [DOI] [PubMed] [Google Scholar]
- 32. Horie K., Insel P. A. (2000) J. Biol. Chem. 275, 29433–29440 [DOI] [PubMed] [Google Scholar]
- 33. Yang W., Wang D., Richmond A. (1999) J. Biol. Chem. 274, 11328–11333 [DOI] [PubMed] [Google Scholar]
- 34. Brown B. M., Carlson B. L., Zhu X., Lolley R. N., Craft C. M. (2002) Biochemistry 41, 13526–13538 [DOI] [PubMed] [Google Scholar]
- 35. Pippig S., Andexinger S., Lohse M. J. (1995) Mol. Pharmacol. 47, 666–676 [PubMed] [Google Scholar]
- 36. Krueger K. M., Daaka Y., Pitcher J. A., Lefkowitz R. J. (1997) J. Biol. Chem. 272, 5–8 [DOI] [PubMed] [Google Scholar]
- 37. Iyer V., Tran T. M., Foster E., Dai W., Clark R. B., Knoll B. J. (2006) Br. J. Pharmacol. 147, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heuser J. E., Anderson R. G. (1989) J. Cell Biol. 108, 389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kirchhausen T., Macia E., Pelish H. E. (2008) Methods Enzymol. 438, 77–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cohen P., Klumpp S., Schelling D. L. (1989) FEBS Lett. 250, 596–600 [DOI] [PubMed] [Google Scholar]
- 41. Weiser D. C., Shenolikar S. (2003) Curr. Protoc. Mol. Biol. 18, Unit 18.10 [DOI] [PubMed] [Google Scholar]
- 42. Single M., Ni L., Honkanen R. E. (2007) in Protein Phosphatase Protocol (Moorhead G. ed) pp. 23–38, Humana Press Inc., Totowa, NJ [Google Scholar]
- 43. Celver J., Xu M., Jin W., Lowe J., Chavkin C. (2004) Mol. Pharmacol. 65, 528–537 [DOI] [PubMed] [Google Scholar]
- 44. Violin J. D., Ren X. R., Lefkowitz R. J. (2006) J. Biol. Chem. 281, 20577–20588 [DOI] [PubMed] [Google Scholar]
- 45. Zidar D. A., Violin J. D., Whalen E. J., Lefkowitz R. J. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9649–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Busillo J. M., Armando S., Sengupta R., Meucci O., Bouvier M., Benovic J. L. (2010) J. Biol. Chem. 285, 7805–7817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elberg G., Hipkin R. W., Schonbrunn A. (2002) Mol. Endocrinol. 16, 2502–2514 [DOI] [PubMed] [Google Scholar]
- 48. Pollok-Kopp B., Schwarze K., Baradari V. K., Oppermann M. (2003) J. Biol. Chem. 278, 2190–2198 [DOI] [PubMed] [Google Scholar]
- 49. Tran T. M., Friedman J., Qunaibi E., Baameur F., Moore R. H., Clark R. B. (2004) Mol. Pharmacol. 65, 196–206 [DOI] [PubMed] [Google Scholar]
- 50. Ohguro H., Palczewski K., Ericsson L. H., Walsh K. A., Johnson R. S. (1993) Biochemistry 32, 5718–5724 [DOI] [PubMed] [Google Scholar]
- 51. Tran T. M., Friedman J., Baameur F., Knoll B. J., Moore R. H., Clark R. B. (2007) Mol. Pharmacol. 71, 47–60 [DOI] [PubMed] [Google Scholar]
- 52. McLaughlin J. P., Xu M., Mackie K., Chavkin C. (2003) J. Biol. Chem. 278, 34631–34640 [DOI] [PubMed] [Google Scholar]
- 53. Liu Q., Reubi J. C., Wang Y., Knoll B. J., Schonbrunn A. (2003) J. Clin. Endocrinol. Metab. 88, 6073–6079 [DOI] [PubMed] [Google Scholar]