

Abstract
The class I p21-activated kinases (Pak1–3) regulate many essential biological processes, including cytoskeletal rearrangement, cell cycle progression, apoptosis, and cellular transformation. Although many Pak substrates, including elements of MAPK signaling cascades, have been identified, it is likely that additional substrates remain to be discovered. Identification of such substrates, and determination of the consequences of their phosphorylation, is essential for a better understanding of class I Pak activity. To identify novel class I Pak substrates, we used recombinant Pak2 to screen high density protein microarrays. This approach identified the atypical MAPK Erk3 as a potential Pak2 substrate. Solution-based in vitro kinase assays using recombinant Erk3 confirmed the protein microarray results, and phospho-specific antisera identified serine 189, within the Erk3 activation loop, as a site directly phosphorylated by Pak2 in vitro. Erk3 protein is known to shuttle between the cytoplasm and the nucleus, and we showed that selective inhibition of class I Pak kinase activity in cells promoted increased nuclear accumulation of Erk3. Pak inhibition in cells additionally reduced the extent of Ser189 phosphorylation and inhibited the formation of Erk3-Prak complexes. Collectively, our results identify the Erk3 protein as a novel class I Pak substrate and further suggest a role for Pak kinase activity in atypical MAPK signaling.
Keywords: Enzyme Purification, MAPKs, Protein Kinases, Protein-Protein Interactions, Signal Transduction, Atypical MAPK, p21-activated Kinase
Introduction
The class I p21-activated kinases (Pak1–3) are established effectors of the small GTPases Rac1 and Cdc42 (1). Although initially discovered as regulators of the actin cytoskeleton, they have subsequently been implicated in a variety of different signaling pathways, including those that control proliferation, apoptosis, and transformation (2, 3). Not surprisingly, misregulated Pak2 kinase activity is associated with a variety of different pathological conditions (4–6). As Pak kinase activity contributes to several different signaling pathways, a better understanding of the specific role for Paks in any biological response is greatly aided by the identification of the protein(s) they phosphorylate and the consequences of these phosphorylations. Although the current list of Pak kinases substrates is quite large (3, 7), we are particularly interested in how Pak kinases influence MAPK signaling cascades.
MAPK signaling pathways are among the most evolutionarily conserved signal transduction pathways and are critical for many biological responses (8). Various cellular stimuli lead to the enzymatic activation of a family of serine/threonine kinases known as MAP3Ks. Activated MAP3Ks phosphorylate and activate specific MAP2Ks. MAP2Ks are dual specificity kinases that phosphorylate threonine and tyrosine residues within the activation loop of the MAPKs (the conserved TXY motif) to activate them; phosphorylation of both activation loop residues is required for MAPK activation. Activated MAPKs then transphosphorylate a variety of different proteins, including structural proteins, enzymes, and transcription factors that ultimately drive the appropriate cellular response to the initial signal (9). For some signaling pathways, the MAPK substrates are themselves kinases and are defined as MAPK-activated kinases (MAPKAKs or MKs) extending the characteristic three-tiered architecture (10).
Although the central roles of the MAP3K, MAP2K, and MAPK isoforms to signaling pathways are incontrovertible, the intensity and duration of MAPK signaling are additionally regulated by the activity of several noncanonical proteins, including Paks. Pak kinase activity affects Erk1/2 signaling at two distinct points as follows: Pak phosphorylation of the MAP3K c-Raf on serine 338 cooperates with Src-dependent phosphorylation at tyrosine 341 for maximal c-Raf kinase activity (11), and Pak phosphorylation on serine 298 of Mek1 facilitates the interaction of Mek1 with Erk2 (12).
In addition to the classical MAPK families (ERK, p38, and JNK/SAPK), many cells also express atypical MAPK isoforms characterized by the Erk3, Erk4, Nlk (nemo-like kinase), and Erk7/8 proteins (13). Erk3 and -4 are most closely related to Erk1, sharing 45 and 42% amino acid identity, respectively, to the catalytic domain of Erk1. Despite the extent of this similarity, Erk3 cannot phosphorylate validated Erk1/2 substrates in vitro (14). A characteristic that distinguishes the atypical MAPKs from the classical MAPK isoforms is the lack of the characteristic “TXY” motif within their activation loops. In Erk3 and Erk4, the corresponding sequence is SEG, and for Nlk it is TQE (13). Although Erk7 does contain a TXY sequence that is phosphorylated in vivo, this phosphorylation is not catalyzed by any known MAP2K but results from autophosphorylation (15). As such, Erk7 is also generally considered a member of the atypical MAPK family (13).
Unlike the classical MAPKs, dual phosphorylation of the Erk3/Erk4 activation loops is not possible. Mek2, but not Mek1, could poorly phosphorylate Erk3 Ser189 in vitro (16). However, a cell fractionation approach to identify the kinases responsible for Erk3 Ser189 phosphorylation suggested that the cellular Ser189 kinase did not co-fractionate with Erk1/2 kinase activity, excluding Mek1/2 from consideration as cellular Erk3 kinases (17). Similar approaches excluded protein kinase C (PKC) as the cellular Ser189 kinase despite the ability of PKC to phosphorylate Erk3 in vitro (17). The initial observation of Erk3 Ser189 phosphorylation in vivo occurred more than a decade ago (17), but the kinase(s) responsible for this phosphorylation remain unidentified.
Using high density protein microarrays, we identified the Erk3 atypical MAPK as a candidate Pak2 substrate. Pak2 kinase assays using recombinant full-length Erk3 protein in solution confirmed the protein microarray results and suggested that Erk3 is a direct p21-activated kinase substrate in vitro. We further demonstrated that Pak2 targets the Ser189 site, within the activation loop of Erk3, suggesting that p21-activated kinases may contribute to Erk3 activation and furthermore may represent the elusive Erk3 Ser189 kinase (referred to as Kinase X in Ref. 18). A variant of Erk3 lacking this phosphorylation site (S189A) displays an increased nuclear accumulation in fibroblasts, which can be phenocopied with wild type Erk3 by selective inhibition of class I Pak kinase activity. Furthermore, class I kinase inhibition reduced the levels of Erk3 Ser189 phosphorylation in vivo. As Ser189 phosphorylation stabilizes the interaction of Erk3 with its effector Prak, we further demonstrate class I inhibition restricts the formation of Erk3-Prak complexes in cells in a manner dependent on Ser189 phosphorylation. Collectively, our results identified Erk3 as a novel substrate for class I Pak kinase activity and identified Ser189, within the Erk3 activation loop, as a residue phosphorylated by class I kinase activity in vitro and in vivo suggesting a role for class I p21-activated kinases in regulating the subcellular localization and activity of the atypical MAPK Erk3.
EXPERIMENTAL PROCEDURES
Human Protein Microarrays
Kinase substrate identification (KSI) ProtoArrays (version 4.0 arrays containing 8,274 full-length GST fusion proteins provided by Invitrogen) were processed as recommended by the manufacturer with the following modification; the supplied kinase buffer was replaced with 1× phosphobuffer (19). Recombinant His6-Pak2 expressed from Escherichia coli was used as the exogenous kinase. This source of Pak2 is known to be constitutively active3 obviating the need for inclusion of Rac/Cdc42-GTP. “ATP only” and “ATP + Pak2” slides were identically processed in parallel. Radiographic images of the slides were obtained using a Fuji phosphorimager, and spots were identified using GenePix Pro (Molecular Devices). Data analysis was conducted in ProtoArray Prospector version 2.0 software as recommended (Invitrogen). For overlay images, spots were pseudocolored in Adobe Photoshop.
Plasmids and Plasmid Construction
The human Erk3 vector was kindly provided by Dr. Sylvain Meloche, University of Montreal. The Erk3 sequence was cloned into the N-terminal His6 baculovirus transfer vector pFBHTB and used to make high titer baculovirus stocks (P4) as recommended (Invitrogen). For mammalian expression vectors, a human full-length Erk3 ultimate ORF Gateway entry clone was purchased from Invitrogen, and moved to the N-terminal GFP (pcDNA6.2-EmGFP-N) and the N-terminal His6 (pDest26) Gateway destination vectors using the LR Clonase II enzyme mix as recommended (Invitrogen). Site-directed point mutants of Erk3 (S189A, S189D) were created using the Erk3 ultimate ORF plasmid as template and QuickChange II kit (Stratagene) with the following mutagenic oligonucleotides: SA1 (5′-cattattcccataagggtcatcttgctgaaggattggttactaaat-3′); SA2 (5′-atttagtaaccaatccttcagcaagatgacccttatgggaataatg-3′); SD1 (5′-ctcattattcccataagggtcatcttgatgaaggattggttactaaatgg-3′); and SD2 (5′-ccatttagtaaccaatccttcatcaagatgacccttatgggaataatgag-3′). Plasmids encoding only the desired S189A or S189D mutations were identified by DNA sequencing and were then moved to pcDNA6.2-EmGFP-N and pDest26 vectors as described above. GFP-PID (corresponding to amino acids 83–149 of human Pak1) and GFP-PIDL107F constructs were amplified by PCR using human Pak1 and Pak1L107F expression constructs as template and subsequently cloned into pEGFP-C1 (Clontech). mRFP-PID constructs were constructed by Pfusion (New England Biolabs) PCR using the above GFP-PID vectors as templates and the following oligonucleotides, 5′-gcggatccatgcacacaattcatgtcggttttgat-3′ and 5′-gcgaattcctatttatctgtaaagctcatgtattt-3′. The PCR products were digested with BamHI and EcoRI restriction enzymes purified from 1× TBE/agarose gels and ligated into pCS2 + mRFP-N1 (a gift from Dr. Chris Thorpe, Kansas State University) similarly digested. Candidate clones were identified by restriction analysis followed by DNA sequence confirmation.
Purification and Dephosphorylation of Recombinant Erk3
High titer P4 stocks were used to infect suspension cultures of Sf21 cells maintained in SFM-900 III media (Invitrogen). 48 h post-infection, the cultures were pelleted by centrifugation, and the supernatant was discarded, and the pellet was either frozen at −80 °C or resuspended in 30 ml of lysis buffer (15 mm NaHPO4, 300 mm NaCl, 10 mm imidazole, pH 8.0, containing 1 mm PMSF, 1:100 aprotinin (Fisher), and 200 μl of protease inhibitor mixture (Pierce)). The pellet was subjected to 6–8 rounds of a 30-s sonication and 30-s on ice. The lysate was centrifuged for 10 min at 15,000 rpm at 4 °C in an SS-34 rotor. The cleared lysate was transferred to a 50-ml conical tube followed by addition of HisPur beads (Pierce) pre-equilibrated in lysis buffer and rotated for 1 h at 4 °C. The beads were collected by 3000 rpm centrifugation in a swinging bucket rotor Sorvall H1000B (5 min at 4 °C) and washed three times with lysis buffer lacking PMSF and protease inhibitors. The protein was eluted from the beads with lysis buffer containing 200 mm imidazole for 4 h at 4 °C. The 200 mm imidazole eluate containing Erk3 was concentrated using a Viva Spin 15R 30,000 molecular weight cutoff centrifugal concentrator as recommended (Sartorius). The retentate was dialyzed against 10% glycerol/1× Tris-buffered saline (TBS) at 4 °C overnight, then divided into single-use aliquots in 1.5-ml microcentrifuge tubes, and frozen at −80 °C. For dephosphorylation reactions, the Erk3 protein was thawed on ice, reconstituted in 1× PMP buffer (New England Biolabs) supplemented with 1 mm MnCl2, followed by addition of λ phosphatase. Dephosphorylation occurred at 30 °C for 1 h at which time the phosphatase was inactivated by the addition of 10 mm sodium orthovanadate. The protein was then diluted into 1× phosphobuffer, and kinase reactions were conducted as described below omitting [32P]ATP.
Western Analyses
Protein samples were resolved on SDS-polyacrylamide mini-gels and transferred to PVDF membranes as recommended (Bio-Rad). Membranes were blocked in 1× TBST, 5% BSA (Fisher) for greater than 1 h at room temperature. All primary antibodies were incubated at 4 °C overnight in blocking buffer. Primary antibodies used included THETMHis tag mAb (Genscript A00186), total Erk3 (Cell Signaling 4067), Erk3 phospho-Ser189 (Abgent AP3098a), Prak (Santa Cruz Biotechnology sc-46667), and GFP (Cell Signaling 2555). HRP-linked secondary antibodies were from Cell Signaling. Signal detection used the Immobilon chemiluminescent HRP substrate (Millipore).
In Vitro Pak2 Kinase Assays
The Erk3 protein stock was dialyzed into 1× phosphobuffer using a 10,000 molecular weight cutoff mini Slide-a-Lyzer (Pierce). 0, 1, 5, 10, 15, or 20 μl of the Erk3 protein were brought up to a final volume of 30 μl containing 100 μm ATP (final concentration) and 0.25 μCi of [32P]ATP (PerkinElmer Life Sciences). Where indicated, 11 ng of recombinant His6-Pak2 was added. As a positive control for Pak2 kinase activity, myelin basic protein (MBP, Upstate) was included at a final concentration of 0.17 μg/ml. Kinase assays were allowed to proceed for 30 min at 30 °C followed by cooling on ice, and the addition of SDS-PAGE loading buffer to 1× followed by boiling for 5 min. Samples were resolved on 10% acrylamide SDS-polyacrylamide gels, equilibrated into 40% acetic acid, 10% methanol, dried onto Whatman 3MM paper under vacuum, and exposed to a PhosphorImager screen (Cyclone, PerkinElmer Life Sciences).
Cell Culture and Treatments
NIH3T3 and HEK293 cells were obtained from the ATCC. All growth media, serum, and supplements were purchased from Hyclone. NIH3T3 cells were grown in 1× DMEM + 10% calf serum supplemented with penicillin/streptomycin. HEK293 cells were grown in 1× minimal essential medium + 10% fetal bovine serum supplemented with penicillin/streptomycin. Cells were grown at 37 °C, 5% CO2 in a humidified chamber. NIH3T3 cells were transfected in 6-well plates with 2 μg of DNA Arrest-In transfection reagent as recommended (Open Biosystems). 24 h post-transfection, cells were split into 4-well slides (LabTek) and allowed to attach overnight. Cells were fixed in 3.7% formaldehyde (in 1× PBS) for 20 min at room temperature, washed with PBS, pH 7.4, and permeabilized using 0.2% Triton X-100 (in 1× PBS) for 10 min at room temperature. Cells were washed three times in 1× PBS, and DAPI (Roche Applied Science) was added for 5 min prior to addition of ProLong antifade reagent (Invitrogen). Fluorescent images were acquired from a Nikon eclipse E600 microscope equipped with a Qimaging QiCAM camera using Qcapture Pro software. For HEK293 transfections, cells were transfected in 100-mm plates with 5 μg of plasmid DNAs using TransIT-293 reagent (Mirus) or in 6-well plates with the Profection calcium phosphate method (Promega).
Cell Lysis and Immobilized Metal Affinity Chromatography
For His6-Erk3 pulldown assays, HEK293 cells 48 h post-transfection were lysed in RIPA buffer containing 10 mm imidazole, 1 mm sodium orthovanadate, 0.5% sodium deoxycholate, 10 mm β-glycerophosphate, 5 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mm PMSF, and 1:100 aprotinin (Fisher). Lysates were centrifuged at 21,000 × g for 10 min at 4 °C. Cleared lysate protein concentrations were determined by BCA assay (Pierce) and equilibrated with lysis buffer. HisPur beads (Pierce) were added to the cleared lysate and incubated with end/end rotation at 4 °C overnight. The beads were washed twice with lysis buffer, brought to a 100-μl total volume in lysis buffer, followed by addition of 20 μl of 6× SDS-PAGE sample loading buffer.
RESULTS
High Density Protein Microarrays Identify Erk3 as a Potential Pak2 Substrate
Although the list of validated substrates for the p21-activated kinases is quite extensive (2, 20), it is likely that additional substrates remain to be identified. To identify novel Pak kinase substrates, we used constitutively active recombinant Pak2 to screen a high density protein microarray. The protoarrays (KSI slides, Invitrogen) contain 8274 full-length human proteins, including fiduciary kinases that serve as orientation landmarks spotted in duplicate onto microscope slides. Similar arrays have been used to identify novel substrates for a variety of different protein kinases, including Cdk5 (21), and the Arabidopsis thaliana MAPKs Mkp3 and Mkp6 (22). A summary of the proteins identified from the protein microarrays is presented in supplemental Fig. S1. In addition to identifying proteins previously implicated in Rho GTPase signaling (i.e. ARHGEF5 (23) and ARHGAP15 (24)), we also identified the atypical MAPK Erk3 as a potential Pak2 substrate (supplemental Fig. S1). The lack of a signal at the Erk3 coordinates in the ATP only slide suggested that the corresponding signal in the Pak2 kinase slide is not the result of Erk3 autophosphorylation and suggested that Erk3 is a Pak2 phosphoacceptor in vitro.
Pak2 Phosphorylates Full-length Erk3 in Solution
Although KSI slides can rapidly identify potential new kinase substrates, we needed to confirm that Erk3 was a Pak2 phosphoacceptor in solution. This is an important consideration as not every protein identified from protein microarrays recapitulates in solution. KSI experiments using recombinant p38α, CK2, and PKA kinases identified 26 potential substrates, 5 of which (19%) failed confirmation when tested in solution (Invitrogen). As the protein microarrays do not reveal which residues of Erk3 are phosphorylated, we chose to use a baculovirus expression system similar to what has been previously reported for expression of full-length Erk3 (25) and Erk4 (26) (Fig. 2A). Although our Erk3 preparation is not homogeneous, it is highly enriched and cross-reactive with both anti-Erk3 antisera (directed toward residues surrounding leucine 410) and anti-His6 antibodies directed toward the extreme N terminus of the protein (Fig. 2B). Our Erk3 preparations therefore contain presumably full-length (based on electrophoretic mobility) as well as discrete C-terminal truncations that do not extend beyond amino acid 410 (Fig. 2B). We used this source of recombinant Erk3 for Pak2 in vitro kinase assays to confirm the initial protein microarray result. As expected, in the absence of exogenous substrates recombinant Pak2 produced in E. coli undergoes autophosphorylation as monitored by 32P incorporation (Fig. 2C, 1st lane). Inclusion of MBP led to increases in both MBP transphosphorylation and Pak2 autophosphorylation (Fig. 2C, 2nd lane). The increased autophosphorylation of Paks by substrates has been reported previously (27), and we have previously observed that high concentrations of Paktide can lead to Pak1 activation independently of small GTPases (28). Increasing amounts of recombinant Erk3 with a fixed amount of recombinant Pak2 lead to an increase in the extent of Erk3 phosphorylation (Fig. 2C, 3rd to 7th lanes, Erk3 signal is denoted by an asterisk). Omitting Pak2 from samples containing the highest amount of Erk3 (Fig. 2C, 8th lane) indicated that the phospho-Erk3 signal is dependent on Pak2 and argues against Erk3 phosphorylation resulting from autophosphorylation or by an unknown contaminating kinase in our Erk3 preparations. Additionally, although both the His6 and total Erk3 antisera revealed the presence of three major cross-reactive species, the in vitro kinase assays show what appears to be a single phosphorylated species. We do not know whether this results from the inability to specifically resolve the three Erk3 species (each ∼100 kDa) or whether the different C-terminal truncations have differential ability to be phosphorylated by Pak2. This is of specific concern as residues within the extreme C terminus of Erk3 are known to be phosphorylated; Ser684, Ser688, Thr698, and Ser705 are phosphorylated by Cdk1 (29). Together, these results demonstrate that Pak2 kinase is able to selectively phosphorylate GST-Erk3 on protein microarrays (Fig. 1 and supplemental Fig. S1) and His6-Erk3 (Fig. 2C) in vitro.
FIGURE 2.

Erk3 is a Pak2 substrate in solution. A, purification scheme of recombinant Erk3 from baculovirus-infected insect cells. Cell pellets were collected 48 h post-infection with high titer baculovirus stocks and lysed by sonication. Specific fractions were collected and evaluated for purity by electrophoresis on a 10% SDS-polyacrylamide gel followed by Coomassie R-250 staining. Lane 1, total cell lysate; lane 2, cleared lysate; lane 3, HisPur nonbinding fraction; lane 4, HisPur wash 1; lane 5, HisPur wash 2; lane 6, 200 mm imidazole elution fraction; lane 7, Vivaspin 30,000 molecular weight cutoff filtrate; lane 8, Vivaspin 30,000 molecular weight cutoff retentate. The asterisk denotes the slowest migrating and presumably full-length Erk3. B, purified Erk3 protein is recognized by N-terminal hexahistidine and total Erk3 antibodies. Increasing amounts of the 30,000 molecular weight cutoff retentate (A, lane 8) were resolved by electrophoresis on an 8% SDS-polyacrylamide gel and subjected to Western analyses using anti-Erk3 (top) and anti-His6 antibodies (bottom). C, [32P]ATP autoradiogram of Pak2 in vitro kinase assays. For all lanes the amount of Pak2 added remained constant. Lane 1, Pak2 protein alone; lane 2, Pak2 + MBP; lanes 3–7, Pak2 with increasing amounts of Erk3 protein. Lane 8, equivalent amounts of Erk3 as in lane 7 but lacking Pak2. 32P-Erk3 is denoted by asterisk, and 32P-Pak2 and 32P-MBP signals are also identified.
FIGURE 1.

High density protein microarrays identify the atypical MAPK Erk3 as a Pak2 phosphoacceptor. Flow chart for KSI slides (left) were processed as described under “Experimental Procedures” using recombinant His6-Pak2. Statistically significant spots were identified in Protoarray Prospector. A single block of the array from the ATP alone and ATP + Pak2 slides are shown at higher magnification (middle). All substrate proteins are spotted in duplicate; single spots or spots that do not align with the printing grid (gray circles) are excluded from consideration. 33P signals obtained from the phosphorimager were pseudocolored and overlaid (bottom). Yellow signals in the overlay result from autophosphorylation, spotted protein kinases, or by fiduciary kinases (FK) that are arrayed in the upper left and lower right of each block for orientation landmarks. Red signals appearing in the overlay, including Erk3, identify proteins transphosphorylated by recombinant Pak2.
Pak2 Phosphorylates Erk3 on Serine 189
Erk3 is a member of the atypical MAPK family, lacking the characteristic “TXY” motif within the activation loop (in Erk3, the corresponding sequence is Ser189–Glu190–Gly191). Although the Ser189 site is phosphorylated in cells (17, 18), the kinase responsible for this phosphorylation has yet to be identified. As Pak2 can phosphorylate Erk3 in vitro (Figs. 1 and 2C), we asked whether Pak2 could specifically phosphorylate Ser189. Using phospho-specific Ser189 antibodies, we demonstrate that recombinant Erk3 derived from insect cells is pre-phosphorylated at this position (Fig. 3, lane 1), consistent with previous observations for recombinant full-length Erk4 purified from insect cells (26). Addition of ATP alone led to a slight increase in the extent of the phospho-Ser189 signal, as did the inclusion of ATP and Pak2 (Fig. 3, lanes 2 and 3). Although Ser189 cross-reactivity is clearly observed in our Erk3 preparations, we cannot determine the stoichiometry of this phosphorylation by Western blot. To more clearly evaluate whether Pak2 can phosphorylate Ser189, we first dephosphorylated the Erk3 preparations with λ phosphatase followed by inactivation of the phosphatase by addition of sodium orthovanadate. This treatment completely abolished phospho-Ser189 cross-reactivity (Fig. 3, lane 4, top panel) without affecting the total amount of Erk3 (Fig. 3, bottom panel). Addition of ATP alone to the dephosphorylated Erk3 did not promote an increase in phospho-Ser189 cross-reactivity suggesting that the kinases responsible for the slight increase previously observed are sensitive to λ phosphatase. However, addition of Pak2 and ATP to the dephosphorylated Erk3 protein led to an increase in Ser189 cross-reactivity to levels higher than those seen with the original Erk3 preparations (Fig. 3, lane 6 compared with lane 1). Collectively, these results demonstrate that Erk3 is a suitable substrate for Pak2 kinase activity in vitro and that at least one of the residues phosphorylated by Pak2 is serine 189 within the activation loop of Erk3.
FIGURE 3.
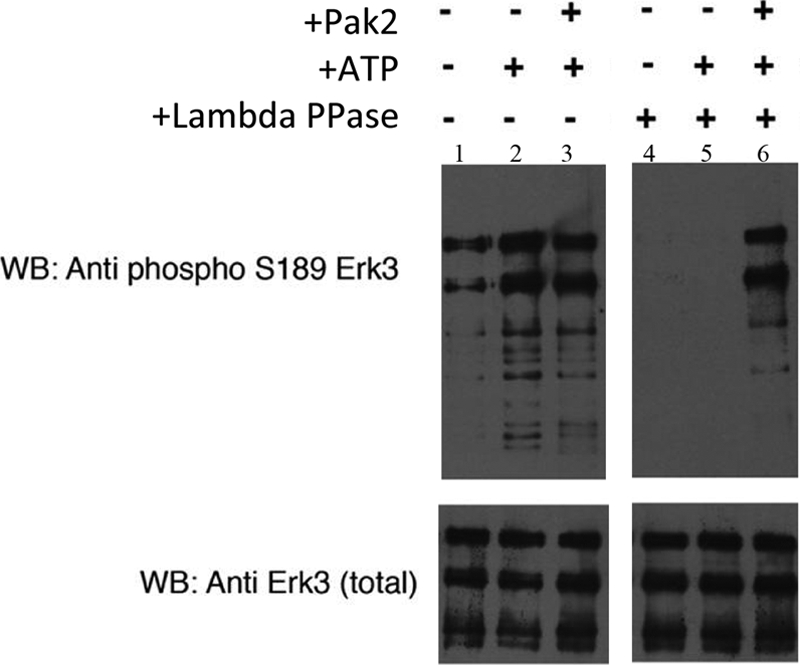
Pak2 phosphorylates serine 189 within the Erk3 activation loop in vitro. The Erk3 protein purified from insect cells is pre-phosphorylated on serine 189. Recombinant Erk3 protein (Fig. 2A, lane 8) was dialyzed into 1× phosphobuffer and subjected to an in vitro kinase assay using recombinant Pak2. Proteins were resolved by electrophoresis on an 8% SDS-polyacrylamide gel and subjected to Western analyses using phospho-Ser189 specific antisera (upper panel) and total Erk3 antibodies (lower panel) to ensure equivalent loading of Erk3. Lane 1, Erk3 protein in 1× phosphobuffer; lane 2, Erk3 protein after 30 min incubation with ATP; lane 3, Erk3 protein after 30 min of incubation with ATP and Pak2. Lanes 4–6 are similar to lanes 1–3 with the exception that the Erk3 source was first dephosphorylated by λ phosphatase (lambda PPase) followed by inactivation of the phosphatase by the addition of sodium orthovanadate. WB, Western blot.
Inhibition of Pak Kinase Activity Reduces the Extent of Erk3 Ser189 Phosphorylation in Cells
Our biochemical results suggested that recombinant Erk3 was phosphorylated by recombinant Pak2 and that at least one of the sites phosphorylated in vitro was within the activation loop of Erk3 prompted us to determine whether this also occurred in cells. As endogenous Erk3 is subjected to degradation by the 26 S proteasome, we relied on co-transfecting HEK293 cells with plasmids encoding GFP-Erk3 and mRFP or mRFP-PID constructs. To evaluate the ability of the PID to inhibit GFP-Erk3, Ser189 phosphorylation requires that cells express both constructs simultaneously. We confirmed this by immunofluorescence of live cells (Fig. 4A); we estimate that the proportion of GFP+/RFP− transfected cells is less than 5% of the total number of GFP-positive cells.
FIGURE 4.

Pak kinase inhibition reduces the extent of Erk3 Ser189 phosphorylation in cells. HEK293 cells were co-transfected with expression constructs for GFP-Erk3 and monomeric RFP, monomeric RFP-PID, or monomeric RFP-PIDL107F. A, 48 h post-transfection, the cells were imaged on an inverted fluorescent microscope to ensure that the majority of cells expressing GFP-Erk3 also expressed an mRFP variant. B, cell lysates were prepared from two independent transfections, including cells imaged in A and separated by electrophoresis prior to Western analyses with variable amounts of lysate loaded to empirically achieve approximately equal levels of total Erk3. These conditions were then used to probe a replicate blot with the phospho-Ser189-specific antibody. Densitometry of the signals acquired from B in ImageJ are presented at right; error bars represent S.D. for three independent transfections.
The co-transfected cells were then lysed and subjected to Western blots using total Erk3 antibodies. Although we generally load equivalent amounts of total protein lysates for Western blots, in this case we had to empirically adjust the amount of lysate loaded to approach equivalent levels of total Erk3 protein as it appears that co-expression of the RFP-PID leads to a decrease in the amount of GFP-Erk3 expressed. Once the appropriate loading conditions were determined, identical membranes were probed with the phospho-Ser189 antibody and total Erk3 antibodies by Western analysis. The relative Ser189 and total Erk3 signals were determined by densitometry. Co-expression of the PID leads to a decrease in the extent of Ser189 cross-reactivity, whereas the mRFP-PIDL107F variant more closely resembled the empty vector control (Fig. 4B). Quantification of three independent transfections suggests that expression of the PID can reduce the Ser189/total Erk3 signal ratio ∼2-fold. These data suggest that selective Pak inhibition in cells leads to a decrease in Erk3 Ser189 phosphorylation and suggests that Erk3 is a class I Pak substrate in cells.
Ser189 Phosphorylation Modulates the Interaction of Erk3 and Prak
Although the kinases responsible for Ser189 phosphorylation had heretofore been uncharacterized, site-directed mutants (S189A) had previously demonstrated that Ser189 phosphorylation was essential for the ability of Erk3 to interact with and activate Prak (18, 26), the only known Erk3 substrate. As Ser189 phosphorylation is required for a productive Erk3/Prak interaction and Pak inhibition leads to decreased Ser189 cross-reactivity in cells (Fig. 4B), we asked whether Pak inhibition also led to a decreased Erk3/Prak interaction. To address this, we co-transfected HEK293 cells with His6-Erk3 expression plasmid and plasmids expressing GFP, GFP-PID, or GFP-PIDL107F. The His6-Erk3 was adsorbed by immobilized metal affinity chromatography, and the extent of endogenous Prak co-purifying with His6-Erk3 was determined by Western analysis. Focusing on the wild type allele (Fig. 5, leftmost panels), expression of the PID, but not GFP alone or the inactive GFP-PIDL107F variant, reduced the amount of Prak co-precipitating with His6-Erk3. This was not due to reduced Prak expression as the levels of endogenous Prak in whole cell lysates was not affected by expression of the PID (Fig. 5). To ensure that the ability of Pak inhibition to reduce the stability of the Erk3-Prak complexes did not result from other residues of Erk3 being phosphorylated by Paks, we repeated the experiments described above using the nonphosphorylatable (S189A) and phospho-mimetic (S189D) variants of His6-Erk3. Consistent with the model proposed for Erk3/Prak interaction (18), mutation of serine 189 to alanine strongly reduced the extent of Prak co-precipitating with His6-Erk3 (Fig. 5, middle panels). Substituting serine 189 with aspartic acid restored the interaction between Erk3 and Prak. Importantly, and unlike the wild type allele, the Erk3S189D/Prak interaction was insensitive to the expression of the PID (Fig. 6, rightmost panels).
FIGURE 5.

Pak kinase inhibition prevents the association of Erk3 with its known effector Prak in an Ser189-dependent manner. HEK293 cells were co-transfected with expression constructs for His6-Erk3, His6-Erk3S189A or His6-Erk3S189D along with expression constructs for GFP, GFP-PID, or GFP-PIDL107F as indicated. 48 h post-transfection, His6 proteins were affinity-adsorbed to HisPur beads. The amount of His6-Erk3 in the His pulldowns was determined by Western analysis using total anti-Erk3 antibodies, and the amount of endogenous Prak was similarly determined using anti-Prak antibodies (upper six panels). To ensure appropriate expression of indicated GFP constructs and that the experimental treatments did not alter the total levels of endogenous Prak, Western analyses of whole cell lysates were conducted using anti-GFP and anti-Prak antibodies (lower six panels). Numbers on the left represent the molecular mass as determined by pre-stained SDS-PAGE loading standards. Results are representative of three independent co-transfections.
FIGURE 6.

Erk3 Ser189 phosphorylation promotes its cytoplasmic redistribution in NIH3T3 cells. A, NIH3T3 cells were transfected with GFP-tagged expression plasmids for wild type Erk3, Erk3S189A, and Erk3S189D. 48 h post-transfection, cells were fixed and counterstained with DAPI to unambiguously identify the nucleus. B, quantification of the subcellular distribution of Erk3. The subcellular localization of the GFP-Erk3 signals was determined for greater than 100 transfected cells. Each GFP-positive cell was scored blind and assigned into one of three categories as follows: uniform GFP signal in the cytoplasm and the nucleus (N = C), predominantly nuclear (N > C), or predominant cytoplasmic staining (N < C). C, inhibition of Pak kinase activity promotes the nuclear retention of GFP-Erk3. NIH3T3 cells were co-transfected with wild type GFP-Erk3 expression plasmids and expression plasmids encoding mRFP, mRFP-PID, or mRFP-PIDL107F. 48 h later, the cells were fixed and analyzed as described above. D, quantification of the subcellular distribution of GFP-Erk3 in cells expressing the indicated mRFP constructs. Bar graphs represent the mean and ± S.E. of at least three different transfections.
Pak Kinase Inhibition Promotes Erk3 Nuclear Accumulation
Erk3 is known to shuttle between the nucleus and the cytoplasm through a CRM1-dependent mechanism (30). To confirm that Ser189 phosphorylation contributes to the localization of Erk3 as was previously reported (18), we assessed subcellular localization of GFP-Erk3, GFP-Erk3S189A, and GFP-Erk3S189D variants in transiently transfected NIH3T3 cells. Wild type and S189D variants are predominantly cytoplasmic, whereas the GFP-Erk3S189A variant displays an increased nuclear localization (Fig. 6, A and B). To determine whether this differential localization was dependent on Pak kinase activity, we co-transfected NIH3T3 cells with wild type GFP-Erk3 and various monomeric red fluorescent protein (mRFP) constructs as follows: mRFP (empty vector control), mRFP-PID (active class I inhibitor), or mRFP-PIDL107F (inactive class I inhibitor). The PID corresponds to residues 83–149 of human Pak1 and acts as a highly selective pan-class I (Pak1–3) inhibitor when expressed in trans by exploiting the activation mechanism of class I Pak (31). Co-expression of mRFP-PID, but not mRFP alone or the inactive mRFP-PIDL107F, significantly increased the extent of nuclear Erk3 (Fig. 6, C and D). These results implicated Pak kinase activity in regulating the subcellular distribution of Erk3.
These results demonstrated that selective class I kinase inhibition in cells modulates the interaction of Erk3 with its only known effector, Prak, in a mechanism that is dependent on Ser189 phosphorylation, and it provides the first evidence that class I kinase activity modulates the activity of Erk3 in cells.
DISCUSSION
Based on their similarity to the classical Erk1 and Erk2 MAPKs, the atypical MAPKs Erk3 and Erk4 were initially identified more than 15 years ago. The pathways regulated by these kinases, however, remain enigmatic with respect to both the cellular upstream-activating signals (functioning analogously to MAP2Ks) and to a slightly lesser extent in regards to downstream effectors. Models for Erk3/Erk4 function have so far been generally restricted to the following properties: (i) the ability of variants of Erk3/Erk4 to bind to Prak; (ii) the ability of Erk3/Erk4 variants to promote Prak Thr-182 phosphorylation and/or activate the kinase activity of Prak, and (iii) the ability of these variants to modulate their own subcellular localization or the subcellular localization of Prak.
Using high density protein microarrays and recombinant Erk3 and Pak2 proteins, we found that Erk3 is a Pak2 substrate in vitro. Phospho-specific antibodies against residues within the Erk3 activation loop further indicated that serine 189 is a site phosphorylated by Pak2 in vitro. Additionally selective class I kinase inhibition led to a decrease in the extent of Ser189 phosphorylation in HEK293 cells (Fig. 4B) suggesting that this kinase substrate relationship between Pak and Erk3 is retained in vivo.
As Erk3 Ser189 phosphorylation is required for the interaction with Prak, we further demonstrated that specific inhibition of class I kinase activity in cells decreases the interaction between His6-Erk3 and endogenous Prak in HEK293 cells (Fig. 5). This effect was specific to the phosphorylation status of Ser189 as Prak interacted poorly with the Erk3S189A variant, whereas Prak interacted strongly with Erk3 phosphomimetic variant (S189D). Importantly, the interaction between Prak and Erk3S189D was insensitive to PID expression indicating that the ability of Pak to stabilize Erk3-Prak complexes derives exclusively from the ability to promote Ser189 phosphorylation.
Specific inhibition of class I kinase activity in fibroblasts led to an increased nuclear retention of GFP-Erk3, suggesting a role for Paks in regulating the subcellular localization of Erk3. This result was rather surprising as Meloche and co-workers (30) had previously reported that nuclear export of Erk3 was independent of both activation loop phosphorylation and kinase activity in NIH3T3 cells, the same cell line used in our studies. The one obvious difference between the two approaches was the way in which Erk3 was detected; Meloche and co-workers (30) detected the localization of Myc-tagged Erk3 by indirect immunofluorescence, whereas we used GFP-Erk3. We do not know whether this accounts for the observed differences, but GFP-tagged variants of Erk3 have been used by other groups that display very similar subcellular localization in HEK293 cells as we observe in NIH3T3 cells (32). We also believe that the similarity in subcellular localization of the GFP-Erk3S189A variant to that seen for GFP-Erk3 in cells co-expressing mRFP-PID (Fig. 6, C and D) supports our conclusion that Ser189 phosphorylation modulates Erk3 subcellular localization. Collectively, our results demonstrate a relationship between the p21-activated kinases and Erk3 and further suggest a role for class I kinase activity in regulating atypical MAPK pathways by promoting Ser189 phosphorylation.
An important question remains. How does Ser189 phosphorylation promote the interaction of Erk3 with Prak? The residues of Erk3 required for Prak binding map to a region distant from the activation loop characterized by the sequence 328FRIEDE333 (33). This interface is distinct from the common docking domain used by classical Erks to bind to both their respective MAP2Ks and their cognate substrates (34). Molecular modeling suggests that Erk4 Ser186 phosphorylation (analogous the Erk3 Ser189) might promote a conformational change that increases the solvent accessibility of isoleucine 330 (within the conserved FRIEDE motif) allowing for efficient interaction with the C terminus of Prak (33).
We believe that the identification of Paks as kinases responsible for Erk3 Ser189 phosphorylation represents an important step forward in our understanding of atypical MAPK signaling cascades. Their identification now affords the ability to modulate atypical MAPK signaling pathways in physiologically relevant settings by specific Pak inhibition. In our opinion, use of the PID affords several important advantages over other experimental approaches, such as pharmacological inhibition or siRNA approaches. These advantages include lack of isoform selectivity, high target specificity, lack of dominant negative effects, and the ability to inhibit endogenous class I activity in trans in tissue culture (31) and in transgenic animals (5). Specific mutants of the PID (L107F) that no longer function as Pak inhibitors further provide an important control to evaluate the specific contribution of class I kinase activity.
Even with the identification of Paks as Ser189 kinases, previous reports of the kinetics of Ser189 phosphorylation raise some important questions. Erk3 Ser189 phosphorylation does not respond to signals that activate the classical MAPKs (18), and Ser189 phosphorylation persists in HEK293 cells rendered quiescent by serum starvation (18). Although the signals that activate class I kinase activity differ from cell type to cell type, serum starvation is sufficient to reduce class I kinase activity in most cells as demonstrated by both Pak activation-specific antisera as well as in-gel kinase assays (19, 31). It is important to note that although we commonly think of Pak kinase activity being “off” in quiescent cells, this activity is still readily detected in serum-starved cells, albeit at much lower levels than in cells stimulated with growth factors. As the relative levels of the inherently unstable Erk3 and class I Paks are unknown, basal class I kinase activity may still be sufficient to promote Ser189 phosphorylation in quiescent cells.
Alternatively, Ser189 could be targeted by kinases other than Pak. Although entirely speculative, other candidate Ser189 kinases include the class II Paks (Pak4–6), which are constitutively active kinases with similar substrate specificity as the class I Paks (28) and/or the related mammalian sterile 20-like kinase (Mst1) as Erk3 was identified in a similar KSI protein microarray screen using recombinant Mst1.4 It is important to note that although these proteins are all members of the Ste20 superfamily, neither the class II Paks nor Mst1 is inhibited by expression of the PID. Irrespective of whether the ability to phosphorylate is unique to class I Paks, our results strongly suggest that class I kinase activity is critical for the formation of the Erk3-Prak complex in cells, which is one of the best metrics for Erk3 signaling, and suggest an important role for class I kinase activity in regulating atypical MAPK signaling.
Acknowledgments
We thank Dr. Sylvain Meloche and co-workers (University of Montreal) for providing the human Erk3 expression construct and for communicating unpublished results.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 CA58836 (to J. C.). This work was also supported by Terry C. Johnson Center for Basic Cancer Research (to A. B.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
A. Beeser and J. Chernoff, unpublished observations.
S. Jalan and J. Chernoff, unpublished observations.
- Pak
- p21-activated kinase
- Prak
- p38-regulated/activated kinase
- KSI
- kinase substrate identification
- mRFP
- monomeric red fluorescent protein
- PID
- protein inhibitory domain
- MBP
- myelin basic protein.
REFERENCES
- 1. Cotteret S., Chernoff J. (2002) Genome Biol. 3, REVIEWS0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kumar R., Gururaj A. E., Barnes C. J. (2006) Nat. Rev. Cancer 6, 459–471 [DOI] [PubMed] [Google Scholar]
- 3. Szczepanowska J. (2009) Acta Biochim. Pol. 56, 225–234 [PubMed] [Google Scholar]
- 4. Allen J. D., Jaffer Z. M., Park S. J., Burgin S., Hofmann C., Sells M. A., Chen S., Derr-Yellin E., Michels E. G., McDaniel A., Bessler W. K., Ingram D. A., Atkinson S. J., Travers J. B., Chernoff J., Clapp D. W. (2009) Blood 113, 2695–2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hayashi M. L., Rao B. S., Seo J. S., Choi H. S., Dolan B. M., Choi S. Y., Chattarji S., Tonegawa S. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 11489–11494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nguyen D. G., Wolff K. C., Yin H., Caldwell J. S., Kuhen K. L. (2006) J. Virol. 80, 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dummler B., Ohshiro K., Kumar R., Field J. (2009) Cancer Metastasis Rev. 28, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krishna M., Narang H. (2008) Cell. Mol. Life Sci. 65, 3525–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Z., Gibson T. B., Robinson F., Silvestro L., Pearson G., Xu B., Wright A., Vanderbilt C., Cobb M. H. (2001) Chem. Rev. 101, 2449–2476 [DOI] [PubMed] [Google Scholar]
- 10. Roux P. P., Blenis J. (2004) Microbiol. Mol. Biol. Rev. 68, 320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mason C. S., Springer C. J., Cooper R. G., Superti-Furga G., Marshall C. J., Marais R. (1999) EMBO J. 18, 2137–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eblen S. T., Slack J. K., Weber M. J., Catling A. D. (2002) Mol. Cell. Biol. 22, 6023–6033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coulombe P., Meloche S. (2007) Biochim. Biophys. Acta 1773, 1376–1387 [DOI] [PubMed] [Google Scholar]
- 14. Cheng M., Boulton T. G., Cobb M. H. (1996) J. Biol. Chem. 271, 8951–8958 [DOI] [PubMed] [Google Scholar]
- 15. Abe M. K., Kuo W. L., Hershenson M. B., Rosner M. R. (1999) Mol. Cell. Biol. 19, 1301–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robinson M. J., Cheng M., Khokhlatchev A., Ebert D., Ahn N., Guan K. L., Stein B., Goldsmith E., Cobb M. H. (1996) J. Biol. Chem. 271, 29734–29739 [DOI] [PubMed] [Google Scholar]
- 17. Cheng M., Zhen E., Robinson M. J., Ebert D., Goldsmith E., Cobb M. H. (1996) J. Biol. Chem. 271, 12057–12062 [DOI] [PubMed] [Google Scholar]
- 18. Déléris P., Rousseau J., Coulombe P., Rodier G., Tanguay P. L., Meloche S. (2008) J. Cell. Physiol. 217, 778–788 [DOI] [PubMed] [Google Scholar]
- 19. Deacon S. W., Beeser A., Fukui J. A., Rennefahrt U. E., Myers C., Chernoff J., Peterson J. R. (2008) Chem. Biol. 15, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hofmann C., Shepelev M., Chernoff J. (2004) J. Cell. Sci. 117, 4343–4354 [DOI] [PubMed] [Google Scholar]
- 21. Schnack C., Hengerer B., Gillardon F. (2008) Proteomics 8, 1980–1986 [DOI] [PubMed] [Google Scholar]
- 22. Feilner T., Hultschig C., Lee J., Meyer S., Immink R. G., Koenig A., Possling A., Seitz H., Beveridge A., Scheel D., Cahill D. J., Lehrach H., Kreutzberger J., Kersten B. (2005) Mol. Cell. Proteomics 4, 1558–1568 [DOI] [PubMed] [Google Scholar]
- 23. Xie X., Chang S. W., Tatsumoto T., Chan A. M., Miki T. (2005) Cell. Signal. 17, 461–471 [DOI] [PubMed] [Google Scholar]
- 24. Seoh M. L., Ng C. H., Yong J., Lim L., Leung T. (2003) FEBS Lett. 539, 131–137 [DOI] [PubMed] [Google Scholar]
- 25. Seternes O. M., Mikalsen T., Johansen B., Michaelsen E., Armstrong C. G., Morrice N. A., Turgeon B., Meloche S., Moens U., Keyse S. M. (2004) EMBO J. 23, 4780–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perander M., Aberg E., Johansen B., Dreyer B., Guldvik I. J., Outzen H., Keyse S. M., Seternes O. M. (2008) Biochem. J. 411, 613–622 [DOI] [PubMed] [Google Scholar]
- 27. Jakobi R., Huang Z., Walter B. N., Tuazon P. T., Traugh J. A. (2000) Eur. J. Biochem. 267, 4414–4421 [DOI] [PubMed] [Google Scholar]
- 28. Rennefahrt U. E., Deacon S. W., Parker S. A., Devarajan K., Beeser A., Chernoff J., Knapp S., Turk B. E., Peterson J. R. (2007) J. Biol. Chem. 282, 15667–15678 [DOI] [PubMed] [Google Scholar]
- 29. Tanguay P. L., Rodier G., Meloche S. (2010) Biochem. J. 428, 103–111 [DOI] [PubMed] [Google Scholar]
- 30. Julien C., Coulombe P., Meloche S. (2003) J. Biol. Chem. 278, 42615–42624 [DOI] [PubMed] [Google Scholar]
- 31. Beeser A., Jaffer Z. M., Hofmann C., Chernoff J. (2005) J. Biol. Chem. 280, 36609–36615 [DOI] [PubMed] [Google Scholar]
- 32. Kant S., Schumacher S., Singh M. K., Kispert A., Kotlyarov A., Gaestel M. (2006) J. Biol. Chem. 281, 35511–35519 [DOI] [PubMed] [Google Scholar]
- 33. Aberg E., Torgersen K. M., Johansen B., Keyse S. M., Perander M., Seternes O. M. (2009) J. Biol. Chem. 284, 19392–19401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bardwell A. J., Frankson E., Bardwell L. (2009) J. Biol. Chem. 284, 13165–13173 [DOI] [PMC free article] [PubMed] [Google Scholar]