

Abstract
Although Arabidopsis (Arabidopsis thaliana) is the best studied plant species, the biological role of one-third of its proteins is still unknown. We developed a probabilistic protein function prediction method that integrates information from sequences, protein-protein interactions, and gene expression. The method was applied to proteins from Arabidopsis. Evaluation of prediction performance showed that our method has improved performance compared with single source-based prediction approaches and two existing integration approaches. An innovative feature of our method is that it enables transfer of functional information between proteins that are not directly associated with each other. We provide novel function predictions for 5,807 proteins. Recent experimental studies confirmed several of the predictions. We highlight these in detail for proteins predicted to be involved in flowering and floral organ development.
Arabidopsis (Arabidopsis thaliana) is the most widely used model organism in plant research. Unraveling the biological processes in this species, therefore, is essential for the understanding of plant biology in general and for the transfer of this knowledge to other species. Fundamental to this goal is the functional annotation of Arabidopsis proteins. While the aim of the National Science Foundation 2010 initiative on Arabidopsis was to reveal the function of each of its proteins by 2010 (Berardini et al., 2004), currently one-third of the proteins still lack a functional annotation in terms of their biological roles. It is unlikely that these missing annotations can be generated solely by large-scale experimental analysis, as these often provide only a general view on the functions of proteins. At the same time, targeted experiments remain time consuming and often require a specific prior hypothesis of function, which for many proteins cannot be formulated. As a complementary approach, therefore, computational methods are needed that can accurately predict protein functions on a large scale or provide leads for hypotheses of function and the design of targeted experiments.
Methods like BLAST (Altschul et al., 1990) and InterProScan (Zdobnov and Apweiler, 2001; Mulder et al., 2005) infer the functions of proteins by identifying their homologs in sequence databases and by the presence of domains that are associated with particular functions, respectively. This homology-based transfer is a powerful approach for functional annotation of novel proteins, but also one that can lead to erroneous inferences because similarity at the sequence level does not necessarily imply that proteins carry out the same function. Also, for lineage-specific or highly divergent proteins, the probability of identifying a functionally characterized homolog is small. Finally, homology transfer cannot deal with subfunctionalization and neofunctionalization of recent paralogs. Besides plain sequences, other types of information need to be integrated to maximize the coverage and accuracy of function prediction (Forslund and Sonnhammer, 2008).
Proteins that participate in the same biological process often interact physically or exhibit correlations in their expression patterns. High-throughput experiments provide genome-wide information on such associations. In addition, protein-protein interactions can be predicted from sequences (Marcotte et al., 1999; Itzhaki et al., 2006; van Dijk et al., 2008). The associations can be viewed as a network with nodes representing proteins and edges representing the interactions between them. Computational methods can employ those networks for function predictions by analyzing the topology of the network to identify sets of proteins with dense interactions between them (Enright et al., 2002) or to analyze their direct relationships (Letovsky and Kasif, 2003; Vazquez et al., 2003) using the guilt-by-association principle. Such methods employ statistical models, the performance of which relies on appropriate selection of the parameters. Recently, we (Kourmpetis et al., 2010) developed a method that accurately estimated these model parameters using a Bayesian approach and that outperformed other related methods.
Integrated approaches for protein function prediction make use of diverse types of data. Peña-Castillo et al. (2008) evaluated such methods using multiple genomic data sets from Mus musculus and concluded that different data sources provide complementary pieces of information on protein function.
For Arabidopsis, various types of genomic data sets are available. The genome sequence is completed (Arabidopsis Genome Initiative, 2000), gene coexpression levels have been calculated using expression values from a wide variety of conditions (Obayashi et al., 2009), and physical protein-protein interactions have been identified through experiments or predicted through homology (Geisler-Lee et al., 2007).
Despite the availability of these data, only a limited number of studies have focused on function prediction of Arabidopsis proteins through the integration of data. Clare et al. (2006) predicted the molecular functions of proteins integrating sequence features with expression experiments. The authors used the decision tree algorithm Q4.5 (Quinlan, 1993) to predict function terms of the controlled vocabularies of Gene Ontology (GO; Ashburner et al., 2000) and the Munich Information Center for Protein Sequences (Frishman et al., 2001). Their algorithm was developed for predictions that required functional classes to be ordered in a hierarchical tree structure. GO has a Directed Acyclic Graph (DAG) structure that is not a tree. Therefore, Clare et al. (2006) restricted their predictions to the GO terms that are related to molecular functions and further to the most general terms that have a tree structure. Lan et al. (2007) predicted the functions of Arabidopsis proteins that are involved in plant response to abiotic stress by combining different gene expression experiments. Horan et al. (2008) grouped Arabidopsis proteins with similar expression patterns by cluster analysis and predicted functions based on overrepresented GO terms in each identified cluster. The agglomerative clustering algorithm assigned each protein to exactly one cluster, while the complex nature of the biological processes led to the expectation that proteins will belong to multiple clusters. Furthermore, the cluster analysis did not provide information on the uncertainty of each prediction. GeneMania (Mostafavi et al., 2008) is a Gaussian Markov Random Fields-based method for protein function prediction that combines multiple networks. In the evaluation experiment of Peña-Castillo et al. (2008), GeneMania was shown to be one of the most accurate methods, and besides predictions for the Mus musculus proteins, it was further applied to several species including Arabidopsis. Bradford et al. (2010) combined sequence data, gene location in the chromosome, phylogenetic profiles, physical protein-protein interactions, and expression levels to predict functions of proteins in Arabidopsis. Using a two-step approach, the authors first constructed ranked lists of proteins that are functionally associated with each query protein. Functions were then inferred by Gene Set Enrichment Analysis (Subramanian et al., 2005) of these lists. Since a large fraction of Arabidopsis proteins lack functional annotations, the ranked lists may contain no or only a few proteins with GO terms assigned to them. The analysis thus has difficulty identifying infrequent GO terms. Lee et al. (2010) derived a composite functional linkage network (Karaoz et al., 2004) for the Arabidopsis proteins by integrating data from sequences, coexpressions, and physical interactions from Arabidopsis and from other species. As in the previous approach, functional inference was only possible when at least one direct neighbor of the query protein had a known function. From the total set of 7,465 Arabidopsis proteins without functional annotation, 2,986 (40%) were not linked to any protein with known function; therefore, function predictions for them was not possible. VirtualPlant (Katari et al., 2010) is visualization software that integrates different sources of data for Arabidopsis, including GO function information on the proteins. VirtualPlant is valuable for bridging the gap between biologists and bioinformaticians by providing an intuitive way to integrate and mine diverse data sources but does not perform de novo function prediction.
In this study, we performed genome-wide function prediction for Arabidopsis proteins by integrating protein sequences, gene expression data, and experimentally derived or predicted protein-protein interactions. We applied Bayesian Markov Random Fields (BMRF; Kourmpetis et al., 2010), a probabilistic method shown to be suitable when the functions of a large number of proteins have to be predicted, such as in the case of Arabidopsis. A powerful feature of BMRF is that it can transfer functional information beyond direct interactions and so can provide function predictions for proteins linked with other proteins of unknown function. In the studies of Bradford et al. (2010) and Lee et al. (2010), such predictions were not possible. We extended the original BMRF to multiple data sources using the framework of Deng et al. (2004), and in a one-step approach, we optimize data source integration for function prediction. Our analysis resulted in 64,721 novel protein function predictions for 5,807 proteins in 867 GO terms that provide detailed functional descriptions. We provide the predictions in the Web site (http://www.ab.wur.nl/bmrf/). After demonstrating the performance of our method using cross-validation as a validation step, we investigated recent experimental evidence for our predictions. As an example of the usefulness of our predictions, we evaluated our predictions on proteins involved in the flowering process in Arabidopsis.
RESULTS
Model Selection
We extended our BMRF function prediction approach (Kourmpetis et al., 2010) to deal with multiple and diverse data sets and applied it to sequence data, protein-protein interaction data, and coexpression data available for Arabidopsis. The first and most crucial step in our study was to identify the best performing function prediction model. We investigated six models of different levels of complexity. Three of those used only one type of data (sequence, protein interaction, or coexpression). The other three used different ways to integrate the various data sources. For benchmarking, we masked the annotation of a set of proteins with known annotations. This set was divided in three strata: network-specific proteins that appear in either (1) the protein-protein interaction network or (2) the coexpression network and (3) those that appear in both. We randomly selected 100 proteins per stratum and predicted their functions. We evaluated the performance by constructing 100 such benchmarking data sets, one for each of 100 GO terms, and used the area under the receiver operating characteristic curve (AUC) as the performance measure.
Table I shows the mean AUC scores for the candidate models in four different evaluation settings (higher value means better performance). Overall, the best performing model is the one that integrates networks and sequence information (BMRF-UNION-DOMAINS). In general, the predictions based on the integrated network outperformed those from single networks (Fig. 1, A and B). The prediction performance improved not only for proteins that appear in both networks but also for network-specific proteins (Fig. 1, A and B; Table I). The latter was unexpected, because the neighborhood of a network-specific protein does not change after the integration of the networks. This performance improvement, therefore, reveals an appealing property of BMRF, namely the propagation of information over long ranges across the network. A more flexible model for network integration that allows the parameters to vary between the networks did not show any improvement compared with the simpler one of constraining the parameters to be equal (Fig. 1C). The model that uses all three data sources clearly had the best performance compared with all other candidate models. We proceeded using this model to make novel predictions for the Arabidopsis proteins.
Table I. Mean AUC scores for the evaluation data sets.
BMRF-PPI (BMRF-COEX) denotes the application of BMRF to the protein-protein interaction (coexpression) network. EN-DOMAINS denotes the application of Elastic Net to the domain information. BMRF-MULTI denotes the integration of the PPI and COEX networks internally by BMRF. BMRF-UNION denotes the application of BMRF to the union of the PPI and COEX networks, whereas BMRF-UNION-DOMAINS also adds the domain information. PPI Only and COEX Only evaluate performance for the masked proteins that appear only in the PPI and COEX networks, “Intersection” for the masked proteins that appear in both networks, and “All” for all masked proteins. The best performing score per category is shown in boldface. NA, Not available.
| Model/Protein Sets | PPI Only | COEX Only | Intersection | All |
| BMRF-PPI | 0.67 | NA | 0.68 | NA |
| BMRF-COEX | NA | 0.66 | 0.67 | NA |
| EN-DOMAINS | 0.61 | 0.63 | 0.61 | 0.62 |
| BMRF-MULTI | 0.71 | 0.70 | 0.74 | 0.70 |
| BMRF-UNION | 0.70 | 0.70 | 0.74 | 0.68 |
| BMRF-UNION-DOMAINS | 0.76 | 0.77 | 0.79 | 0.75 |
Figure 1.

Scatterplots showing the relative performance (AUC score) of different protein function prediction models. The performance was evaluated for 100 GO terms using four sets of masked proteins: those that appear only in the PPI network (light blue) or only in the COEX network (dark blue), proteins that appear in both (red), and the full set of proteins (yellow). A and B, Integrated approach BMRF-UNION against BMRF-PPI (A) and BMRF-COEX (B). For the majority of the cases, the integrated approach performs better not only for proteins that appear in both networks but also for the network-specific proteins (light and dark blue). C, Comparison between the two network integration methods BMRF-UNION and BMRF-MULTI shows little difference in performance. D, Comparison of BMRF-UNION-DOMAINS with the BMRF-UNION and EN-DOMAINS. The performance of the fully integrated approach is significantly better compared with the other methods.
Protein Function Prediction for Arabidopsis
We applied our probabilistic method integrating protein-protein interactions, gene coexpression, and functional domains to predict functions for 8,247 Arabidopsis proteins with unknown biological roles. Our procedure computes a posterior probability for each protein against and for each GO term, which makes the interpretation of the predictions somewhat difficult. To overcome this problem, we constructed a list with positive predictions after obtaining the optimal F-score cutoff on the posterior probability from the set of annotated proteins and applying it to the set of proteins with unknown functions. This resulted in a list of 64,721 predictions for 5,807 proteins against 867 GO terms (the list is available at http://www.ab.wur.nl/bmrf/). For each prediction, we calculated the Precision and Recall at the given probability cutoff in order to facilitate further use (biological interpretation) of the list. Both metrics are high in the list of predictions (Fig. 2). The density of Recall rates shows that an appreciable fraction of proteins received a prediction, while the Precision rates, which are even higher than the Recall rates, show that the list contains a large fraction of correctly predicted proteins.
Figure 2.

Densities for Precision (light blue) and Recall (red) from the list of predictions. The mass for the Recall lies in the region of 0.2 and larger. The precision is high, with its mode at 0.8. [See online article for color version of this figure.]
For validation, we investigated whether there was recent experimental evidence in the literature supporting our predictions and that was not available at the time of our computations. For this purpose, we downloaded the annotation file for Arabidopsis on April 18, 2010, from GO and identified the proteins that were annotated after October 13, 2009 (the date on which we downloaded the annotation file used in our predictions). There were 194 new annotations with GO terms from the Biological Process branch for 103 proteins that were included in our prediction list. In 14 cases, we predicted the exact GO term (Table II) or a more detailed one according to the GO DAG. For 109 new annotations, we predicted one or more GO terms that are more general but related. Hence, in total, we predicted a GO DAG-related function (more general, exact, or more specific) for 123 out of the 194 (63%). This level of performance is highly significant (P < 0.00001) as judged by a permutation test.
Table II. Experimentally verified predictions where BMRF predicted the exact GO term or a more detailed one.
Relation denotes the relation between the GO terms of the new annotation and the BMRF prediction: E, exact prediction; D, the GO term predicted by BMRF is a successor of the annotation according to the GO DAG.
| Protein | Annotation | Reference | BMRF Prediction | Relation |
| AT1G04510 (MAC3A) | Defense response to bacterium | Monaghan et al. (2009) | Defense response to bacterium | E |
| AT2G33340 (MAC3B) | Defense response to bacterium | Monaghan et al. (2009) | Defense response to bacterium | E |
| AT4G28720 (YUC8) | Auxin biosynthetic process | Rawat et al. (2009) | Auxin biosynthetic process | E |
| AT5G48380 (BIR1) | Negative regulation of defense response | Gao et al. (2009) | Negative regulation of defense response | E |
| AT4G15200 (AFH3) | Actin nucleation | Ye et al. (2009) | Actin nucleation | E |
| AT4G23130 (CRK5) | Response to salicylic acid stimulus | Chen et al. (2004) | Response to salicylic acid stimulus | E |
| AT1G18370 (HIK) | Cytokinesis | Oh et al. (2008) | Cytokinesis | E |
| AT3G10570 (CYP77A6) | Flower development | Li-Beisson et al. (2009) | Flower development | E |
| AT3G13220 (WBC27) | Pollen development | Xu et al. (2010) | Pollen development | E |
| AT1G08450 (CRT3) | Defense response to bacterium | Li et al. (2009) | Defense response to bacterium | E |
| AT3G23070 (CFM3A) | Seed development | Asakura et al. (2008) | Embryonic development ending in seed dormancy | D, E |
| AT5G64580 (MUB3.10) | Embryonic development | Mutwil et al. (2010) | Embryonic development ending in seed dormancy | D |
| AT1G77740 (PIP5K2) | Growth | Camacho et al. (2009) | (1) Cell tip growth; (2) developmental cell growth | D |
| AT3G08710 (ATH9) | Cell communication | Meng et al. (2010) | Intracellular signaling cascade | D |
We also compared the prediction performance of BMRF with two recently published integration methods, Aranet (Lee et al., 2010) and GO-AT (Bradford et al., 2010), using the list of new annotations as the validation data set. Each method provides scored predictions from which we calculated Precision and Recall at a series of cutoffs. The Precision of BMRF was higher than the other methods at any given recall rate (Fig. 3).
Figure 3.

Precision versus Recall curves for BMRF (blue), GO-AT (red, dashed), and Aranet (light green) using as validation set the newly annotated proteins (those deposited in the GO database after April 18, 2010). The Precision level at any Recall is higher for BMRF than for the other two methods. [See online article for color version of this figure.]
For the newly annotated proteins, we also make 718 predictions that were not inferred by the newly obtained experimental data. We expect that at least some of our novel predictions will be confirmed in future experiments. Below, we further comment on some of the supported predictions.
Monaghan et al. (2009) performed double mutant analysis on the Arabidopsis proteins MAC3A and MAC3B and showed that they are involved in the defense response against plant pathogens. InterPro searches did not return information related to the function of those proteins, while the BLAST2GO tool predicted the more general term “defense response.” On the other hand, BMRF predicted the GO term “defense response to bacterium” (GO:0042742) for both proteins, which is in complete agreement with the aforementioned experimental study (Fig. 4A). Also, BMRF predicted the involvement of MAC3B in “activation of innate immune response” (GO:0002218), which is also a defense-related process. The same gene was identified to have a ubiquitin-protein ligase molecular function (Wiborg et al., 2008). BMRF predicted that MAC3B is involved in the biological process “protein ubiquitination” (GO:0016567), which is in accordance with that study. Furthermore, Borges et al. (2008) performed a genome-wide transcriptome analysis identifying MAC3B to be involved in “embryonic sac development” (GO:0009553), a function that was also predicted by BMRF. Other examples in which BMRF accurately predicted protein functions include YUC8, which was recently identified by Rawat et al. (2009) to be involved in the “auxin biosynthetic process” (Fig. 4B), and BIR1, which was identified to be involved in “negative regulation of defense response” (Gao et al., 2009; Fig. 4C). BMRF predicted that AT3G8710 is involved in “intracellular signaling cascade” (GO:0023034). On the basis of results described by Meng et al. (2010), this protein is newly assigned to the more general term “cell communication” (GO:0007154).
Figure 4.

Illustrations of three experimentally verified cases where BMRF successfully recovered the exact function of proteins. A, MAC3A and MAC3B were predicted to be involved in “defense response to bacterium.” MAC3A has an experimentally determined interaction with CDC5 as well as one predicted by the interolog from Homo sapiens with PRL1. PRL1 is also predicted to interact with MAC3B by a Saccharomyces cerevisiae interolog. Both CDC5 and PRL1 are involved in defense response to bacterium. B, YUC8 was successfully predicted to be involved in “auxin biosynthetic process.” This protein does not interact with any proteins involved in this process. Still, the prediction was based on the presence of two InterPro domains in its sequence. C, BIR1 is involved in “negative regulation of defense response,” which is correctly predicted by BMRF through its coexpression with four other proteins known to be involved in this process. [See online article for color version of this figure.]
Flowering and Floral Organ Development in Arabidopsis
As a specific example of the usefulness of our method, we here focus on the evaluation of predictions for flowering and floral organ development. Obviously, this is a biological process for which annotation transfer between species is only possible within the plant kingdom. Given the current scarcity of annotation for plants, homology-based methods have limited scope for annotation transfer from other species; therefore, our network-based approach is, in principle, better suited.
GO terms were selected that describe processes related to flowering and floral organ development (Supplemental Table S1). We first discuss a few groups of proteins, including transcription factors (TFs), that are predicted for the selected GO terms and then focus on two particular terms: “floral transition” (GO:0010228) and “corolla development” (GO:0048465).
One important class of TFs with known roles in the regulation of floral transition and in flower development are the MADS domain proteins (Coen and Meyerowitz, 1991; Ng and Yanofsky, 2001; Ferrario et al., 2004). For several members of this family, BMRF predicted additional functions that are consistent with those known functions. For example, for the MADS domain protein Agamous-Like6 (AGL6), the GO term “floral organ development” was predicted, as was the more detailed term for “carpel, gynoecium, and ovule development.” Although in Arabidopsis the function of AGL6 has remained elusive so far (due to the lack of a single loss-of-function mutant exhibiting a clear phenotype), recently it was shown that an AGL6 homolog is involved in petal and anther development in petunia (Petunia hybrida; Rijpkema et al., 2009). Hence, our prediction for floral organ development is supported by independent evidence. A second MADS domain protein predicted for carpel/gynoecium development was AGL15. This protein has a known function in the floral transition process (Adamczyk et al., 2007), but our prediction suggested that it has a broader function in the development of floral organs.
Several other MADS domain proteins with unknown functions to date were predicted to function in flower development, including AGL13, AGL14, AGL71, AGL72, and AGL79. Note that several of these proteins arose through lineage- or species-specific duplications that occurred in the MADS domain protein family. Such duplications render annotation transfer based on orthology inadequate because it cannot deal with subfunctionalization or neofunctionalization, while our network-based method can in principle deal with these cases. Two additional predictions for carpel development, the MADS domain proteins AP3 (AT3G54340) and PI (AT5G20240), seem incorrect in light of existing knowledge that these proteins are only involved in the development of petals and stamens, although it is known that PI is temporarily expressed in the fourth whorl, where carpel formation takes place (Goto and Meyerowitz, 1994).
Regulation of transcription via MADS domain TFs involves histone modification proteins (Hill et al., 2008; Ng et al., 2009). Similarly, chromatin modifications are important in the regulation of the floral transition (for review, see He, 2009). An interesting aspect of our predictions is that several proteins related to chromatin modifications are predicted to be involved in flower development, including histone H3, SPT16 (AT4G10710), and SSRP1 (AT3G28730), which are part of a chromatin-remodeling complex. These predictions do not necessarily imply that those proteins have a very specific function in flower development, as it could well be the case that many different TFs that are involved in various biological processes fulfill their functions via such proteins. Indeed, histone H3 is predicted to be involved in some other developmental processes as well (e.g. leaf morphogenesis).
In addition, two methyltransfersases are predicted to be involved in the floral transition, one of which (PRMT6: AT3G20020) is closely related to a histone-regulating methyltransferase (PRMT10: AT1G04870) with a known function in regulating the floral transition (Niu et al., 2007). Note that for the floral transition in particular, an epigenetic mechanism is biologically meaningful as a way to bridge the temporal separation between the induction of a flowering-competent state by, for example, vernalization and the initiation of flowering in spring (Jung and Müller, 2009).
TFs are important for regulating biological processes in general and flower development in particular. At a lower level, however, target genes with more specific molecular functions are obviously involved in those processes. One particular set of proteins predicted by our method are hydrolases (more specifically, hydrolases that hydrolyze glycosyl compounds). For one of those (AT3G56310, a putative α-galactosidase), which is assigned to the process “positive regulation of flower development,” there is indeed supporting literature evidence (Rojo et al., 2003; van Doorn and Woltering, 2008). Other predicted hydrolases include an α-galactosidase (positive regulation of flower development) and AT3G48700, which is expressed during the petal differentiation and expansion stage, according to The Arabidopsis Information Resource (TAIR). For the GO term 0048573 (photoperiodic control of flowering time), among others, the hydrolases AtXTH17, -18, and -19 are predicted. However, according to Osato et al. (2006), these are preferentially expressed in the roots, and there is evidence for a principal role for the AtXTH18 gene in primary root elongation. Hence, this prediction seems unlikely.
We now briefly discuss our predictions for two particular processes, the floral transition (GO:0010228) and corolla development (GO:0048465). The floral transition refers to the transition from the vegetative to the reproductive phase. The methyltransferase PRMT6, a methyltransferase-related protein (AT5G53920, ribosomal protein L11 methyltransferase-related), and protein AtBAG2 (AT5G62100) were all predicted for floral transition. For PRMT6, the closely related protein PRMT10 is indeed known to be involved in this process (Niu et al., 2007). AtBAG2 is one of the BAG (for Bcl-2-associated athanogene) proteins, which are plant homologs of mammalian regulators of apoptosis. These proteins regulate apoptosis-like processes associated with pathogen attack, abiotic stress, or plant development. For the two BAG family members AtBAG2 and AtBAG6 (AT2G46240), knockouts have been shown to give early flowering (Doukhanina et al., 2006), which provides strong support for our predictions.
Another interesting prediction for floral transition is AT1G10320, which is a U2 snRNP auxiliary factor-related protein (it is predicted as well for photoperiodic control of flowering time). This protein is involved in splicing regulation (Lorković et al., 2000), and evidence is mounting for a role of alternative splicing in the floral transition (Terzi and Simpson, 2008). In particular, some MADS domain proteins have alternative splicing variants with a putative role in this process. One example is FLM (AT1G77080), which via exon-skipping can form two different variants (E.I. Severing, A.D.J. van Dijk, R.G.H. Immink, and R.C.H.J. van Ham, unpublished data).
Corolla development refers to the development of the petals of a flower. Here, several members of the glutaredoxin family were predicted. As there are indeed indications for the involvement of glutaredoxin in petal development (Xing et al., 2005), this prediction seems reasonable.
DISCUSSION
In this study, we apply BMRF, a computational method for protein function prediction, to the proteome of Arabidopsis. By integrating diverse data sources (experimentally identified protein interactions, expression levels, and sequence-derived features), we predict 64,721 novel GO terms for 5,807 Arabidopsis proteins. Performance metrics such as Precision and Recall are estimated for each prediction. We show that our predictions are of high precision and may provide leads for the design of new hypothesis-driven experiments.
It is well known that high-throughput data sets such as those used in our study contain measurement errors. Taking this error into account, by incorporating the edge confidence values in the BMRF model, may lead to further improvement in the prediction performance. The coexpression values that we used in this study capture correlations between expression levels shown in a wide range of biological conditions. However, some proteins may interact only in particular circumstances and therefore have correlated expressions only under those conditions. We plan to work on using such data more efficiently in the BMRF model. Furthermore, the GO annotation files we used do not contain all the available information concerning the functions of Arabidopsis proteins. Integration with additional sources of function information (e.g. by literature mining) may improve the prediction performance of BMRF.
We statistically evaluated the prediction performance of BMRF and compared our predictions with recent new annotations deposited in the GO. From the total of 194 such new annotations, BMRF provided exact or more detailed function predictions for 14 cases and more general but related GO terms for 109 cases. Thus, for 63% of the novel annotations, BMRF was able to predict a relevant function, which is a highly significant result as judged on the basis on a permutation test. BMRF gave better predictions than two recently proposed integrative approaches as judged on the basis of the Precision-Recall curves for the new annotations. We further studied the predictions related to flowering processes and found several cases where our predictions are supported by the literature and therefore may provide information for further experimental validation.
BMRF is a computational method for function prediction that integrates large-scale data sets and transfers functional information between proteins that interact indirectly. These two properties make BMRF a very useful method for protein function prediction in the genomic era, as shown here by the application to the Arabidopsis proteome. Based on the results presented here, we expect that our method will also show its value for other plant and animal species.
MATERIALS AND METHODS
Protein-Protein Interaction Network
Physical interactions between proteins provide valuable information for their functions. Proteins that interact are members of the same complex and involved in the same biological process or pathway. There are around 3,000 experimentally identified interactions between Arabidopsis (Arabidopsis thaliana) proteins. In addition, interactions can be predicted by detecting interacting orthologs. Such predicted interactions are called interologs. Geisler-Lee et al. (2007) used the orthology detection algorithm INPARANOID (Remm et al., 2001) to identify Arabidopsis interologs from several well-studied species, including Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila melanogaster, and Homo sapiens. We downloaded the experimentally derived interactions and the interologs from the TAIR Web site and constructed a protein-protein interaction (PPI) network that contains 7,177 proteins with 72,266 interactions.
Coexpression Network
Besides direct physical interactions, proteins involved in the same biological process present correlations in their gene expression. Genome-wide expression experiments, therefore, provide an important data source for protein function prediction. In the recent study of Lee et al. (2010), gene expression was found to be the most informative data source for protein function prediction. The ATTED-II database (Obayashi et al., 2009) aggregates 58 experiments and 1,388 microarray slides in total. We downloaded the coexpression data from the ATTED-II Web site on July 21, 2009. For each coexpression, a confidence value is provided, which is defined by the mutual rank of the coexpression of two proteins. The authors calculate the Pearson correlation coefficient between all pairs of proteins. Then, for each protein, they rank the Pearson correlation coefficient. As confidence value for an interaction, they calculate the mutual rank (i.e. the square root of the product for the rankings in both directions of the interaction). This coexpression measure is useful for protein function prediction (Obayashi and Kinoshita, 2009). Our coexpression network (referred to as COEX) was constructed by setting the maximum mutual rank to 60. COEX contains 22,133 proteins with 358,540 interactions.
Functional Domains
Sequence signatures are an important source of information concerning the function of a protein. InterPro (Mulder et al., 2005) aggregates the most important tools and databases that are used to identify such sequence patterns and to link them to particular functions, primarily at the molecular level. We used the identified InterPro functional signatures for the proteome of Arabidopsis. This data set was downloaded directly from the TAIR Web site in October 2009.
GO Annotations
We downloaded the annotation file for Arabidopsis from the GO, with version 1.1271 deposited on October 13, 2009. This file contains in total 14,038 Biological Process annotations. Annotations with evidence code “Inferred from Electronic Annotation” were removed from the data set because these are derived from InterPro hits, which we used independently in our study for function prediction. All remaining annotations were up-propagated to their more general terms using the GO DAG structure. In total, there were 2,894 GO terms appearing at least once, but many of them were extremely sparse (i.e. containing less than 10 proteins assigned to them after up-propagation). Our final set contained 1,024 GO terms and 8,247 proteins lacking Biological Process annotations in the Arabidopsis proteome.
Protein Function Prediction
BMRF for a Single Network
We investigated different approaches for protein function prediction. Our starting point was the BMRF method for protein function prediction based on a single network described in an earlier study (Kourmpetis et al., 2010). In particular, given a network that contains N proteins and S edges (indicating interactions between proteins), a particular function of interest (i.e. a GO term) is represented by a N-dimensional binary vector X with element xi = 1 if protein i is annotated as performing the function and xi = 0 otherwise. The elements of X that correspond to unannotated proteins are unknown. The objective of BMRF is to infer the unknowns given the observed part of X using the edges of the protein network. The log odds of the probability that an unannotated protein xi performs the function of interest, given the annotations for all other proteins, denoted by Xi, depends on the number of its direct neighbors performing the function and the number of them that do not perform the function:
where α denotes the intercept, β1 and β0 are interaction parameters, and Si denotes the set of proteins that interact with protein i, so that Mi1 denotes the number of proteins that interact with protein i and perform the function while Mi0 denotes those that interact with protein i but do not perform the function. Inference for the unannotated part of X can be made using a Markov Chain Monte Carlo approach (Kourmpetis et al., 2010). We refer to BMRF-PPI when this method is applied to the PPI network and to BMRF-COEX when it is applied to the COEX network. In both cases, the predictions are limited to the proteins that appear in the network that BMRF is applied to. For example, it is not possible to make predictions for the proteins appearing only in the COEX network by applying BMRF to the PPI network.
BMRF for Multiple Networks
A natural way to integrate multiple networks through BMRF is by using a set of interaction parameters per network. This approach was originally proposed by Deng et al. (2004):
We refer to this model as BMRF-MULTI. A special case of this model is obtained by constraining the interaction parameters between the networks to be equal (i.e. β1PPI = β1COEX and β0PPI = β0COEX). This approach is equivalent to applying BMRF to the single network that is the union of the PPI and COEX networks. The union network has an edge if an edge appears in at least one of the networks. We refer to the latter model as BMRF-UNION.
Elastic Net for Functional Domains
Given the set of M available InterPro domains, we constructed the N × M binary matrix D, where the element dnm is equal to 1 if protein n contains the InterPro domain m and 0 otherwise. The probability that a protein performs the function of interest depends on the presence/absence profile of domains. We write this relationship as a logistic regression with binary variables:
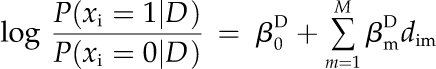 |
The parameter vector βD contains the regression coefficients for the domains and can be estimated using the proteins with known functional annotation. A particular GO term is expected to be related to only a small subset of the domains, and those domains usually act in a concerted way. Therefore, we aim to perform variable selection while keeping highly correlated variables in the model. A suitable method for this purpose is the Elastic Net (EN; Zou and Hastie, 2005) version for logistic regression (Park and Hastie, 2007). EN combines Lasso regression (Tibshirani, 1996) and Ridge regression (Hoerl and Kennard, 1970). In Lasso regression, the sum of the absolute values of the regression coefficients is penalized, while in Ridge regression, their sum of squares is penalized. EN combines both regularization methods using a convex parameter for which on one extreme the model becomes equivalent to Lasso and on the other extreme to Ridge regression. In cases with highly correlated variables, Lasso tends to include only one of those variables in the model. In our application, we aim to obtain a sparse model that includes the set of domains that are related to the function. For this reason, we selected EN as the most appropriate method for this application. EN has two parameters to be set prior to model selection. The first is the convex parameter (taking values between 0 and 1), and the second is the penalty parameter. Usually, those parameters are estimated through cross-validation. We adopted a simple approach by fixing the convex parameter to 0.5 (that gives equal weight to both methods) and by selecting from a series of penalty parameters the one that leads to the largest model containing no more than 10 variables (domains). All computations involving EN were made using the GLMNET R package (Friedman et al., 2010). We refer to this function prediction model as EN-DOMAINS.
Integration of Networks and Functional Domains
Let Pd denote the output (on logit scale) from EN-DOMAINS. We insert Pd into the BMRF model, also adding one more parameter, βd:
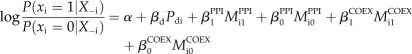 |
Function prediction is further performed by BMRF and updating βd in the same way with the other parameters in the model. We remark that all the quantities in this model are updated during BMRF, while Pdi remains constant.
Performance Evaluation
We estimated the performance of each protein function prediction model by constructing 100 benchmarking data sets, one for each of 100 GO terms randomly selected from different levels of generality. For each GO term, we selected 300 proteins with known function (i.e. known whether it is assigned or not to this particular GO term) to be treated as unknowns. The selection of these “masked” proteins was done using the following procedure. First, the proteins with known functions were classified in three sets: those that appear only in the PPI network, those that appear only in the COEX network, and finally those that appear in both networks. One hundred proteins were randomly selected from each set to be treated as unannotated. Consequently, 200 proteins appear in the PPI network and 200 in the COEX network while 100 appear in both. For the very sparse GO terms (i.e. those with less than 20 proteins assigned to them), we randomly selected exactly half of the proteins that belong to the GO term to be masked. All the protein function prediction models were applied to the 100 benchmarking data sets so obtained. For the evaluation of prediction performance, we used the AUC (Hanley and McNeil, 1982, Fawcett 2006), Precision (Prec), Recall (Rec), and F-score [F-score = 2*Prec*Rec/(Prec + Rec), i.e. the harmonic mean of Recall and Precision]. Recall and Precision are defined as the fraction of proteins correctly predicted of having the function out of the total number of proteins having the function and the fraction of proteins correctly predicted of having the function out of the total number of proteins predicted having the function, respectively. High Recall and high Precision are conflicting aims, and the F-score is a compromise between them that is often used in information retrieval.
All performance metrics were computed using the ROCR R package (Sing et al., 2005).
Construction and Evaluation of a List with Novel Predictions
BMRF computes for each protein the probability of membership in each GO term, except for the most general ones (with more than 3,000 proteins annotated to them). From these membership probabilities, we constructed a list of novel predictions by selecting the cutoff per GO term that maximized the F-score in the set of proteins with known annotations.
We tested this method for cutoff selection on the benchmarking data sets. We first obtained the optimal cutoff using the proteins with known function and then applied this cutoff to the predicted part. Both sets of proteins estimate closely the Recall values (Fig. 5). The Precision when estimated from the set of proteins with known function is a conservative estimation of the one obtained from the set of masked proteins.
Figure 5.

Recall and Precision scores estimated from the held-out set (proteins with “masked” annotation) versus those from the training set (proteins with known annotation; x axis). In supervised learning, performance estimates are based on the held-out set. A, The Recall rates of the training and the held-out set are in accordance. B, Precision estimated from the training set provides a conservative estimate of the true precision estimated by the held-out set.
The novel predictions were compared with the new annotations in the annotation file of April 18, 2010. Because the predictions may be related to the correct annotations by being more general or more specific, we used the GO DAG structure to up-propagate the predicted and the “true” annotations per protein. We define as true positives the set of GO terms that appear in both lists, as false positives those that appear only in the predicted one, and as false negatives those that appear only in the true list. From the measurements, we calculated Precision, Recall, and F-score. The F-score was tested for statistical significance, for which we used a Monte Carlo permutation test, in which the prediction lists of proteins were randomly permuted (shuffled) among proteins that were common to our predictions and the new annotations. After each shuffle, the F-score was calculated. The P value of the test is the rank of the F-score in the data among all the F-scores calculated from the shuffled data divided by the number of permutations. With 100,000 permutations, the lowest obtainable P value is thus 0.00001.
Performance Comparison of BMRF with Other Prediction Methods
We used the list of new annotations as a validation set to compare the performance of BMRF with two state-of-the-art methods that provide function predictions for Arabidopsis proteins, Aranet (Lee et al., 2010) and GO-AT (Bradford et al., 2010). We obtained function prediction lists with confidence scores by querying the Web servers of these two methods. Precision and Recall values were calculated in the full range of scores per method by applying cutoffs and up-propagating the resulting lists. The posterior probabilities from BMRF are uncalibrated in the sense that it is not useful to apply a single cutoff for all the GO terms. For this comparison, we calibrated the probabilities using the function:
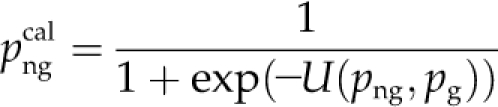 |
with
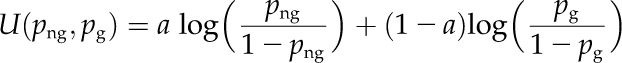 |
and png is the BMRF posterior probability for protein n at GO term g and pg is the prior probability of membership for GO term g (i.e. the proportion of the proteins in our data set that are assigned to this term). After some experimenting using yeast data (Kourmpetis et al., 2010), the parameter a was set to 2, which gives, for sparse GO terms, calibrated probabilities that are the product of png and png/pg. The calibrated probabilities are available from http://www.ab.wur.nl/bmrf/.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Table S1. Selected GO terms for flowering and floral organ development.
Supplementary Material
Acknowledgments
We thank Richard Immink for critically reading the manuscript, Bas te Lintel Hekkert for developing the BMRF Web site, and James Bradford for helping us to obtain the predictions from the GO-AT method. We thank the two reviewers for their valuable comments.
References
- Adamczyk BJ, Lehti-Shiu MD, Fernandez DE. (2007) The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant J 50: 1007–1019 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Asakura Y, Bayraktar OA, Barkan A. (2008) Two CRM protein subfamilies cooperate in the splicing of group IIB introns in chloroplasts. RNA 14: 2319–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardini TZ, Mundodi S, Reiser L, Huala E, Garcia-Hernandez M, Zhang P, Mueller LA, Yoon J, Doyle A, Lander G, et al. (2004) Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol 135: 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges F, Gomes G, Gardner R, Moreno N, McCormick S, Feijó JA, Becker JD. (2008) Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol 148: 1168–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford JR, Needham CJ, Tedder P, Care MA, Bulpitt AJ, Westhead DR. (2010) GO-At: in silico prediction of gene function in Arabidopsis thaliana by combining heterogeneous data. Plant J 61: 713–721 [DOI] [PubMed] [Google Scholar]
- Camacho L, Smertenko AP, Pérez-Gómez J, Hussey PJ, Moore I. (2009) Arabidopsis Rab-E GTPases exhibit a novel interaction with a plasma-membrane phosphatidylinositol-4-phosphate 5-kinase. J Cell Sci 122: 4383–4392 [DOI] [PubMed] [Google Scholar]
- Chen K, Fan B, Du L, Chen Z. (2004) Activation of hypersensitive cell death by pathogen-induced receptor-like protein kinases from Arabidopsis. Plant Mol Biol 56: 271–283 [DOI] [PubMed] [Google Scholar]
- Clare A, Karwath A, Ougham H, King RD. (2006) Functional bioinformatics for Arabidopsis thaliana. Bioinformatics 22: 1130–1136 [DOI] [PubMed] [Google Scholar]
- Coen ES, Meyerowitz EM. (1991) The war of the whorls: genetic interactions controlling flower development. Nature 353: 31–37 [DOI] [PubMed] [Google Scholar]
- Deng M, Chen T, Sun F. (2004) An integrated probabilistic model for functional prediction of proteins. J Comput Biol 11: 463–475 [DOI] [PubMed] [Google Scholar]
- Doukhanina EV, Chen S, van der Zalm E, Godzik A, Reed J, Dickman MB. (2006) Identification and functional characterization of the BAG protein family in Arabidopsis thaliana. J Biol Chem 281: 18793–18801 [DOI] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30: 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. (2006) An introduction to ROC analysis. Pattern Recognit Lett 27: 861–874 [Google Scholar]
- Ferrario S, Immink RG, Angenent GC. (2004) Conservation and diversity in flower land. Curr Opin Plant Biol 7: 84–91 [DOI] [PubMed] [Google Scholar]
- Forslund K, Sonnhammer ELL. (2008) Predicting protein function from domain content. Bioinformatics 24: 1681–1687 [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. (2010) Regularized paths for generalized linear models via coordinate descent. J Stat Software 33: i01. [PMC free article] [PubMed] [Google Scholar]
- Frishman D, Albermann K, Hani J, Heumann K, Metanomski A, Zollner A, Mewes HW. (2001) Functional and structural genomics using PEDANT. Bioinformatics 17: 44–57 [DOI] [PubMed] [Google Scholar]
- Gao M, Wang X, Wang D, Xu F, Ding X, Zhang Z, Bi D, Cheng YT, Chen S, Li X, et al. (2009) Regulation of cell death and innate immunity by two receptor-like kinases in Arabidopsis. Cell Host Microbe 6: 34–44 [DOI] [PubMed] [Google Scholar]
- Geisler-Lee J, O’Toole N, Ammar R, Provart NJ, Millar AH, Geisler M. (2007) A predicted interactome for Arabidopsis. Plant Physiol 145: 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Meyerowitz EM. (1994) Function and regulation of the Arabidopsis floral homeotic gene PISTILLATA. Genes Dev 8: 1548–1560 [DOI] [PubMed] [Google Scholar]
- Hanley JA, McNeil BJ. (1982) The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143: 29–36 [DOI] [PubMed] [Google Scholar]
- He Y. (2009) Control of the transition to flowering by chromatin modifications. Mol Plant 2: 554–564 [DOI] [PubMed] [Google Scholar]
- Hill K, Wang H, Perry SE. (2008) A transcriptional repression motif in the MADS factor AGL15 is involved in recruitment of histone deacetylase complex components. Plant J 53: 172–185 [DOI] [PubMed] [Google Scholar]
- Hoerl AE, Kennard RW. (1970) Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12: 55–57 [Google Scholar]
- Horan K, Jang C, Bailey-Serres J, Mittler R, Shelton C, Harper JF, Zhu JK, Cushman JC, Gollery M, Girke T. (2008) Annotating genes of known and unknown function by large-scale coexpression analysis. Plant Physiol 147: 41–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki Z, Akiva E, Altuvia Y, Margalit H. (2006) Evolutionary conservation of domain-domain interactions. Genome Biol 7: R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CM, Müller AE. (2009) Flowering time control and applications in plant breeding. Trends Plant Sci 14: 563–573 [DOI] [PubMed] [Google Scholar]
- Karaoz U, Murali TM, Letovsky S, Zheng Y, Ding C, Cantor CR, Kasif S. (2004) Whole-genome annotation by using evidence integration in functional-linkage networks. Proc Natl Acad Sci USA 101: 2888–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katari MS, Nowicki SD, Aceituno FF, Nero D, Kelfer J, Thompson LP, Cabello JM, Davidson RS, Goldberg AP, Shasha DE, et al. (2010) VirtualPlant: a software platform to support systems biology research. Plant Physiol 152: 500–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourmpetis YAI, van Dijk ADJ, Bink MCAM, van Ham RCHJ, ter Braak CJF. (2010) Bayesian Markov Random Field analysis for protein function prediction based on network data. PLoS ONE 5: e9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan H, Carson R, Provart NJ, Bonner AJ. (2007) Combining classifiers to predict gene function in Arabidopsis thaliana using large-scale gene expression measurements. BMC Bioinformatics 8: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Ambaru B, Thakkar P, Marcotte EM, Rhee SY. (2010) Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat Biotechnol 28: 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letovsky S, Kasif S. (2003) Predicting protein function from protein/protein interaction data: a probabilistic approach. Bioinformatics (Suppl 1) 19: i197–i204 [DOI] [PubMed] [Google Scholar]
- Li J, Zhao-Hui C, Batoux M, Nekrasov V, Roux M, Chinchilla D, Zipfel C, Jones JDG. (2009) Specific ER quality control components required for biogenesis of the plant innate immune receptor EFR. Proc Natl Acad Sci USA 106: 15973–15978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Beisson Y, Pollard M, Sauveplane V, Pinot F, Ohlrogge J, Beisson F. (2009) Nanoridges that characterize the surface morphology of flowers require the synthesis of cutin polyester. Proc Natl Acad Sci USA 106: 22008–22013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorković ZJ, Wieczorek Kirk DA, Lambermon MHL, Filipowicz W. (2000) Pre-mRNA splicing in higher plants. Trends Plant Sci 5: 160–167 [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. (1999) Detecting protein function and protein-protein interactions from genome sequences. Science 285: 751–753 [DOI] [PubMed] [Google Scholar]
- Meng L, Wong JH, Feldman LJ, Lemaux PG, Buchanan BB. (2010) A membrane-associated thioredoxin required for plant growth moves from cell to cell, suggestive of a role in intercellular communication. Proc Natl Acad Sci USA 107: 3900–3905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan J, Xu F, Gao M, Zhao Q, Palma K, Long C, Chen S, Zhang Y, Li X. (2009) Two Prp19-like U-box proteins in the MOS4-associated complex play redundant roles in plant innate immunity. PLoS Pathog 5: e1000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. (2008) GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol (Suppl 1) 9: S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bradley P, Bork P, Bucher P, Cerutti L, et al. (2005) InterPro, progress and status in 2005. Nucleic Acids Res 33: D201–D205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutwil M, Usadel B, Schütte M, Loraine A, Ebenhöh O, Persson S. (2010) Assembly of an interactive correlation network for the Arabidopsis genome using a novel heuristic clustering algorithm. Plant Physiol 152: 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L, Lu F, Pei Y, Liu C, Cao X. (2007) Regulation of flowering time by the protein arginine methyltransferase AtPRMT10. EMBO Rep 8: 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KH, Yu H, Ito T. (2009) AGAMOUS controls GIANT KILLER, a multifunctional chromatin modifier in reproductive organ patterning and differentiation. PLoS Biol 7: e1000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Yanofsky MF. (2001) Function and evolution of the plant MADS-box gene family. Nat Rev Genet 2: 186–195 [DOI] [PubMed] [Google Scholar]
- Obayashi T, Hayashi S, Saeki M, Ohta H, Kinoshita K. (2009) ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res (Suppl 1) 37:D987–D991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Kinoshita K. (2009) Rank of correlation coefficient as a comparable measure for biological significance of gene coexpression. DNA Res 16: 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SA, Bourdon V, Das 'Pal M, Dickinson H, Twell D. (2008) Arabidopsis kinesins HINKEL and TETRASPORE act redundantly to control cell plate expansion during cytokinesis in the male gametophyte. Mol Plant 1: 794–799 [DOI] [PubMed] [Google Scholar]
- Osato Y, Yokoyama R, Nishitani K. (2006) A principal role for AtXTH18 in Arabidopsis thaliana root growth: a functional analysis using RNAi plants. J Plant Res 119: 153–162 [DOI] [PubMed] [Google Scholar]
- Park MY, Hastie T. (2007) L1-regularization path algorithm for generalized linear models. J R Stat Soc Ser B 69: 659–677 [Google Scholar]
- Peña-Castillo L, Tasan M, Myers CL, Lee H, Joshi T, Zhang C, Guan Y, Leone M, Pagnani A, Kim WK, et al. (2008) A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol (Suppl 1) 9: S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan JR. (1993) C4.5: Programs for Machine Learning. Morgan Kaufmann Publishers, San Francisco [Google Scholar]
- Rawat R, Schwartz J, Jones MA, Sairanen I, Cheng Y, Andersson CR, Zhao Y, Ljung K, Harmer SL. (2009) REVEILLE1, a Myb-like transcription factor, integrates the circadian clock and auxin pathways. Proc Natl Acad Sci USA 106: 16883–16888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M, Storm CEV, Sonnhammer ELL. (2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol 314: 1041–1052 [DOI] [PubMed] [Google Scholar]
- Rijpkema AS, Zethof J, Gerats T, Vandenbussche M. (2009) The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J 60: 1–9 [DOI] [PubMed] [Google Scholar]
- Rojo E, Zouhar J, Carter C, Kovaleva V, Raikhel NV. (2003) A unique mechanism for protein processing and degradation in Arabidopsis thaliana. Proc Natl Acad Sci USA 100: 7389–7394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T. (2005) ROCR: visualizing classifier performance in R. Bioinformatics 21: 3940–3941 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzi LC, Simpson GG. (2008) Regulation of flowering time by RNA processing. Curr Top Microbiol Immunol 326: 201–218 [DOI] [PubMed] [Google Scholar]
- Tibshirani R. (1996) Regression shrinkage and selection via the lasso. J R Stat Soc B 58: 267–288 [Google Scholar]
- van Dijk ADJ, ter Braak CJF, Immink RG, Angenent GC, van Ham RCHJ. (2008) Predicting and understanding transcription factor interactions based on sequence level determinants of combinatorial control. Bioinformatics 24: 26–33 [DOI] [PubMed] [Google Scholar]
- van Doorn WG, Woltering EJ. (2008) Physiology and molecular biology of petal senescence. J Exp Bot 59: 453–480 [DOI] [PubMed] [Google Scholar]
- Vazquez A, Flammini A, Maritan A, Vespignani A. (2003) Global protein function prediction from protein-protein interaction networks. Nat Biotechnol 21: 697–700 [DOI] [PubMed] [Google Scholar]
- Wiborg J, O’Shea C, Skriver K. (2008) Biochemical function of typical and variant Arabidopsis thaliana U-box E3 ubiquitin-protein ligases. Biochem J 413: 447–457 [DOI] [PubMed] [Google Scholar]
- Xing S, Rosso MG, Zachgo S. (2005) ROXY1, a member of the plant glutaredoxin family, is required for petal development in Arabidopsis thaliana. Development 132: 1555–1565 [DOI] [PubMed] [Google Scholar]
- Xu J, Yang C, Yuan Z, Zhang D, Gondwe MY, Ding Z, Liang W, Zhang D, Wilson ZA. (2010) The ABORTED MICROSPORES regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell 22: 91–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Zheng Y, Yan A, Chen N, Wang Z, Huang S, Yang Z. (2009) Arabidopsis formin3 directs the formation of actin cables and polarized growth in pollen tubes. Plant Cell 21: 3868–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. (2001) InterProScan: an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17: 847–848 [DOI] [PubMed] [Google Scholar]
- Zou H, Hastie T. (2005) Regularization and variable selection via the elastic net. J R Stat Soc Ser B 67: 301–320 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.