

Abstract
Accumulating evidence demonstrates that PKCι is an oncogene and prognostic marker that is frequently targeted for genetic alteration in many major forms of human cancer. Functional data demonstrate that PKCι is required for the transformed phenotype of NSCLC, pancreatic, ovarian, prostate, colon and brain cancer cells. Future studies will be required to determine whether PKCι is also an oncogene in the many other cancer types that also overexpress PKCι. Studies of PKCι using genetically defined models of tumorigenesis have revealed a critical role for PKCι in multiple stages of tumorigenesis, including tumor initiation, progression and metastasis. Recent studies in a genetic model of lung adenocarcinoma suggest a role for PKCι in transformation of lung cancer stem cells. These studies have important implications for the therapeutic use of aurothiomalate (ATM), a highly selective PKCι signaling inhibitor currently undergoing clinical evaluation. Significant progress has been made in determining the molecular mechanisms by which PKCι drives the transformed phenotype, particularly the central role played by the oncogenic PKCι-Par6 complex in transformed growth and invasion, and of several PKCι-dependent survival pathways in chemo-resistance. Future studies will be required to determine the composition and dynamics of the PKCι-Par6 complex, and the mechanisms by which oncogenic signaling through this complex is regulated. Likewise, a better understanding of the critical downstream effectors of PKCι in various human tumor types holds promise for identifying novel prognostic and surrogate markers of oncogenic PKCι activity that may be clinically useful in ongoing clinical trials of ATM.
Keywords: tumorigenesis, gene amplification, signal trandsuction, invasion, metastasis, aurothiomalate
Introduction
Protein kinase C (PKC) is a family of structurally related serine/threonine protein kinases whose catalytic activity is regulated by interaction with phospholipid co-factors, inter- and intra-molecular phosphorylation, and specific protein-protein interactions. The PKC enzyme family is divided into three subgroups: the conventional, calcium-dependent cPKCs [alpha (α), beta I (βI), beta II (βII), and gamma (γ)]; the novel, calcium-independent nPKCs [delta (δ), epsilon (ε), eta (η) and theta (θ)]; and the atypical aPKCs [zeta (ζ) and iota (ι) which is also known as lambda (λ) in mice]. This grouping is based on the presence or absence of functional domains that confer specific co-factor and activator requirements. Conventional PKCs are calcium-, diacylglycerol (DAG)- and phosphatidylserine-dependent due to the presence of conserved modular C1 and C2 domains within the regulatory region of the enzyme. Novel PKCs are DAG- and phosphatidylserine-dependent but do not require calcium. Due to the unique structure of their N-terminal regulatory region, atypical PKCs do not require calcium, DAG or phosphatidylserine for activation.
Biochemical and immunologic studies indicate that multiple PKC isozymes are expressed in virtually all cell and tissue types (reviewed in (Fields and Murray, 2008)). The expression of individual PKC isozymes is developmentally regulated and is responsive to the differentiation state of cells and tissues. For these reasons, PKC isozymes are thought to fulfill distinct, non-redundant functions within the cell (reviewed in (Dempsey et al., 2000; Reyland, 2009)). However, the similar activator requirements and substrate specificities of PKC isozymes in vitro have complicated the identification of physiologically relevant, isozyme-specific substrates and cellular functions. The ability to genetically manipulate expression of a specific PKC isozyme and to express mutant forms of individual PKC isozymes with altered kinase activity have proven successful in identifying PKC isozyme-specific functions. Using these genetic techniques, many laboratories, including our own, have demonstrated isozyme- and cell type-specific roles for PKC in cellular proliferation, differentiation, apoptosis and cell polarity (Chalmers et al., 2005; Gokmen-Polar and Fields, 1998; Jansen et al., 2001; Mischak et al., 1993; Murray et al., 1999; Oster and Leitges, 2006). The discovery that PKC is a cellular receptor for the tumor-promoting phorbol esters led to an intense interest in the role of individual PKC isozymes in cancer development (Castagna et al., 1982; Kikkawa et al., 1983). Indeed, the expression level and cellular localization of individual PKC isozymes has been shown to be altered during carcinogenesis in numerous tissue types (reviewed in (Fields and Gustafson, 2003)). However, despite exhaustive investigation, no somatic or germline mutations have been found in the coding regions of any PKCs that are associated with human cancers or other diseases, and to date, only one PKC isozyme, atypical PKCι, has been shown to satisfy the criteria of a human oncogene. This mini-review details the work from our laboratory and others characterizing the role of oncogenic PKCι in rodent carcinogenesis models and human cancer. We also discuss the status of our efforts to therapeutically target this critical oncogene for treatment of cancer patients.
I. PKCι Expression in Primary Human Tumors
PKCι is overexpressed and prognostic in multiple human cancer types
Overexpression of PKCι has been demonstrated in many human cancers (Table 1). PKCι is frequently overexpressed in cancers of the lung (Regala et al., 2005b), pancreas (Scotti et al., 2010), stomach (Takagawa et al., 2010), colon (Murray et al., 2004), esophagus (Yang et al., 2008), liver (Du et al., 2009), bile duct (Li et al., 2008), breast (Kojima et al., 2008), ovary (Eder et al., 2005; Weichert et al., 2003; Zhang et al., 2006), prostate (Ishiguro et al., 2009) and brain (Patel et al., 2008). In addition to these published reports, meta-analysis of publicly available microarray data provides additional support for overexpression of PKCι in many of these tumor types, including lung (Landi et al., 2008) breast (Richardson et al., 2006), pancreas (Segara et al., 2005), prostate (Wallace et al., 2008), ovary (Cancer Genome Atlas) and liver (Wurmbach et al., 2007)(Table 2). Interestingly, increased PKCι in brain cancer was not supported by the available genomic data sets (Lee et al., 2006)(Table 2). Analysis of available microarray datasets revealed that, in addition to lung cancer (Landi et al., 2008), several other squamous-type cancers also express elevated PKCι, including head and neck (Ginos et al., 2004) and tongue cancer (Ye et al., 2008). Significant overexpression of PKCι was also detected in renal cancer (Gumz et al., 2007), bladder cancer (Dyrskjot et al., 2004; Sanchez-Carbayo et al., 2006), melanoma (Talantov et al., 2005) and leukemia (Valk et al., 2004) (Table 2). These studies strongly indicate a role for PKCι in many major forms of human cancer.
TABLE 1.
PKCι Expression in Human Tumor Types
| Tumor type | #Tumor / #Control (p=paired) | Type of Analysis | Result | Reference |
|---|---|---|---|---|
| Lung | 74 / 74 p | blot; qPCR; IHC | Elevated; Amplified; High PKCι predicts poor survival | Regala et al., 2005b |
| PDAC | 28 / 28 p | qPCR; IHC | Elevated; High PKCι predicts poor survival | Scotti et al., 2010 |
| Gastric | 177 | IHC | Elevated; High PKCι predicts disease recurrence | Takagawa et al., 2010 |
| Colon | 5 / 5 p | blot | Elevated | Murray et al., 2004 |
| Esophageal | 108 | IHC; FISH | Elevated; Amplified; gene amplification correlates with tumor size, stage, LN metastasis; High PKCι predicts metastasis | Yang et al., 2008 |
| Hepatocellular carcinoma | 43 / 43 p | RT-PCR; IHC | Elevated; High PKCι correlates with size, metastasis, invasion and stage | Du et al., 2009 |
| Cholangiocarcinoma | 41 / 9 | IHC | Elevated; High PKCι correlates with differentiation, invasion, LN metastasis, stage; Prognostic for poor survival | Li et al., 2008 |
| Breast | 109 | IHC | Elevated | Kojima et al., 2008 |
| Ovarian | 89 | IHC; mRNA; array CGH | Elevated; Amplified; High PKCι correlates with stage | Zhang et al., 2006 |
| Ovarian | 235 | Array CGH; qPCR; IHC | Elevated; Amplified in serous tumors and high copy # predicts poor survival; nonserous tumors: high PKCι predicts poor survival | Eder et al., 2005 |
| Ovarian | 67 / 15 | IHC | Elevated; High PKCι predicts poor survival | Weichert et al., 2003 |
| Prostate | 29 / 29 p | qPCR, blot | Elevated; High PKCι predicts poor recurrence free survival | Ishiguro et al., 2009 |
| Brain | 21 / 12 | blot | Elevated | Patel et al., 2008 |
TABLE 2.
PKCι Expression in Publicly Available Human Tumor Microarray Datasets
| Cancer Type | #Tumor / #Control | Significant in Tumor (P-value) | Reference |
|---|---|---|---|
| Head and Neck | 41 / 13 | Yes (0.04) | Ginos et al., 2004 |
| Tongue | 26 / 12 | Yes (0.009) | Ye et al., 2008 |
| Lung | 58 / 49 | Yes (0.006) | Landi et al., 2008 |
| Superficial Bladder Cancer | 28 / 48 | No (0.248) | SanChez-Carbayo et al., 2006 |
| Infiltrating Bladder Urothelial Carcinoma | 81 / 48 | Yes (5.99E-04) | SanChez-Carbayo et al., 2006 |
| Superficial Bladder Cancer | 28 / 9 | Yes (3.94E-04) | Dyrskjot et al., 2004 |
| Infiltrating Bladder Urothelial Carcinoma | 13 / 9 | Yes (2.57E-05) | Dyrskjot et al., 2004 |
| Brain | 22 / 76 | No (0.068) | Lee et al., 2006 |
| Pancreas | 11 / 6 | Yes (5.37E-04) | Segara et al., 2005 |
| Melanoma | 18 / 7 | Yes (1.74E-04) | Talantov et al., 2005 |
| Ovarian | 38 / 10 | Yes (2.75E-07) | TCGA Ovarian Not Published |
| Liver (HCC) | 35 / 10 | Yes (6.67E-04) | Wurmbach et al., 2007 |
| Breast | 40 / 7 | Yes (0.02) | Richardson et al., 2006 |
| Prostate | 69 / 20 | Yes (0.003) | Wallace et al., 2008 |
| Acute Myeloid Leukemia | 285 / 8 | Yes (0.03) | Valk et al., 2004 |
| Renal | 10 / 10 | Yes (0.003) | Gumz et al., 2007 |
In those tumor types where it has been examined, PKCι expression has also been shown to be of prognostic significance. High PKCι predicts poor patient survival in lung (Regala et al., 2005b) pancreatic (Scotti et al., 2010) bile duct (Li et al., 2008), ovarian (Eder et al., 2005; Weichert et al., 2003) and prostate (Ishiguro et al., 2009) cancer. Elevated PKCι expression predicts disease recurrence in gastric cancer (Takagawa et al., 2010) and metastasis in esophageal cancer (Yang et al., 2008). In our analysis of PKCι expression in lung cancer, PKCι emerged as a prognostic indicator that was comparable to tumor stage in its prognostic value. Interestingly, PKCι expression did not correlate with tumor stage in non-small cell lung cancer (NSCLC); rather PKCι levels were comparable in early and late stage disease indicating that elevated PKCι expression is a very early event in lung tumor development (Regala et al., 2005b). In contrast, PKCι expression correlates with tumor stage in ovarian, bile duct and liver cancer, suggesting that PKCι may regulate disease progression in these tumor types (Du et al., 2009; Li et al., 2008; Zhang et al., 2006).
Thus PKCι expression is elevated in a variety of tumor types, and in many cases, is predictive of poor clinical outcome. Therefore, PKCι expression profiling may identify patients at elevated risk of relapse or disease progression. Since many patients diagnosed with early stage cancer will eventually relapse, PKCι expression profiling may be useful in identifying high risk patients who would be candidates for more aggressive clinical management, perhaps, as will be discussed below, with PKCι-targeted therapy.
PKCι is a target for frequent tumor-specific gene amplification
DNA amplification is one mechanism by which oncogenes are activated in neoplastic tissue. The PKCι gene PRKCI, resides on chromosome 3q26, a chromosomal region frequently amplified in human cancers, particularly squamous cell carcinomas (Brass et al., 1996; Heselmeyer et al., 1997; Lin et al., 2006; Racz et al., 1999; Singh et al., 2002). Therefore, we examined NSCLC tumors for evidence of changes in PRKCI gene copy number (Regala et al., 2005b). PRKCI was found to be amplified in a tumor-specific fashion in 36% of the NSCLC tumors examined. Furthermore, PRKCI amplification correlates with PKCι mRNA and protein expression, and with poor outcome in NSCLC tumors (Regala et al., 2005b). Interestingly, PRKCI amplification was frequently found in lung squamous cell carcinoma (SCC) (~70%) but rarely in lung adenomcarcinoma (LAC) (Regala et al., 2005b), consistent with the distribution of chromosome 3q26 amplification in these tumor types which is confined to SCC (Balsara et al., 1997; Brass et al., 1997). Similar tumor-specific PRKCI amplification has also been observed in ovarian cancers of the serous sub-type (~70%) (Eder et al., 2005; Zhang et al., 2006) and esophageal squamous cell cancer (53%) (Yang et al., 2008). PKCι expression and PRKCI copy number also correlate with chromosome 3q26 gains in these tumors (Eder et al., 2005; Yang et al., 2008; Zhang et al., 2006), indicating that PKCι is a relevant target for tumor-specific chromosome 3q26 amplification. Since chromosome 3q26 amplification is one of the most common chromosomal changes in human cancers, including SCC of the head and neck (Snaddon et al., 2001) and cervix (Sugita et al., 2000), it is likely that PKCι expression and gene copy number are of prognostic significance in these tumors as well.
PRKCI amplification is not the only mechanism by which PKCι expression is elevated in human tumors. PKCι expression is elevated to the same degree and frequency in lung SCC and LAC tumors despite the fact that PKCι gene amplification is largely confined to SCC tumors (Regala et al., 2005b). Furthermore, PKCι is frequently over-expressed in other tumor types, including colon cancers (Murray et al., 2004), pancreatic cancers (Scotti et al., 2010) and leukemia (Gustafson et al., 2004) that do not harbor frequent chromosome 3q26 amplification. We recently demonstrated that Bcr-Abl transcriptionally activates PKCι through Ras/Mek-dependent activation of a specific Elk1 element within the proximal PKCι promoter in chronic myelogenous leukemia (CML) cells (Gustafson et al., 2004). A similar mechanism is likely at play in LAC and pancreatic ductal adenocarcinoma (PDAC) tumors that harbor oncogenic KRAS mutations. Indeed, we have observed a statistically significant positive correlation between the presence of KRAS mutation and PKCι expression in primary LAC tumors (unpublished observations). Likewise in PDAC, in which >90% of tumors harbor a KRAS mutation (Klimstra and Longnecker, 1994), we detected PKCι overexpressed in 96% of tumors (Scotti et al., 2010).
Another potential mechanism for oncogenic activation of PKCι is somatic mutation. However, sequence analysis of all 18 exons of the PKCι gene in 20 LAC cases and 20 SCC cases failed to detect any mutations, suggesting that somatic mutation of PKCι either does not occur or is extremely rare in NSCLC (unpublished observations). In summary, PKCι is the first, and to date only, PKC isozyme shown to be a bonafide human oncogene (Regala et al., 2005b). Current evidence strongly supports the oncogenic role of PKCι in NSCLC, PDAC, ovarian cancer and glioma. Given the widespread overexpression of PKCι in other major tumor types, it appears likely that PKCι will be shown to be an oncogene in many other tumor types as well.
II. PKCι in Cellular Transformation
PKCι as a survival gene in human cancer
The first demonstration that PKCι was important for the transformed phenotype of human cancer cells came from studies in CML cells (Murray and Fields, 1997). CML cells are highly resistant to the apoptotic effects of numerous chemotherapeutic agents as a result of expression of the chimeric tyrosine kinase oncogene Bcr-Abl, the transforming activity that causes CML (Bedi et al., 1995). We found that Bcr-Abl-positive CML cells express high levels of PKCι and that PKCι is activated in response to apoptotic stimuli such as treatment with the chemotherapeutic agent paclitaxel (Jamieson et al., 1999). Disruption of PKCι expression or activity sensitized CML cells to induction of paclitaxel-induced apoptosis (Murray and Fields, 1997). Subsequent studies have established a similar role for PKCι in the survival and chemoresistance of other tumor cell types including prostate (Win and Acevedo-Duncan, 2008), NSCLC (Jin et al., 2005) and glioblastoma (Baldwin et al., 2006).
PKCι and oncogenic ras mediated transformation of intestinal epithelial cells
Studies in mouse fibroblasts first established a functional link between aPKCs and cellular Ras. Ras can activate aPKCs (Diaz-Meco et al., 1994), and aPKC activity is necessary for Ras-mediated effects on the actin-based cytoskeleton in fibroblasts (Bjorkoy et al., 1997; Coghlan et al., 2000; Kampfer et al., 2001; Uberall et al., 1999). Since oncogenic Ras signaling can drive colon carcinogenesis, and oncogenic Ras mutations are detected in ~30% of colon cancers (Slattery et al., 2001; Takayama et al., 2001), we investigated the role of PKCι in Ras-mediated transformation of rat intestinal epithelial (RIE) cells (Murray et al., 2004). We found that oncogenic Ras activates PKCι when introduced into non-transformed RIE cells and that expression of a kinase-deficient, dominant negative PKCι mutant (kdPKCι) in Ras transformed RIE cells inhibits Ras-mediated invasion and anchorage-independent growth (Murray et al., 2004). These results provided direct evidence that PKCι is involved in the establishment of the transformed phenotype by Ras in epithelial cells.
PKCι is required for maintenance of the transformed phenotype of cancer cells
PKCι not only plays a key role in transformation induced by introduction of oncogenic Ras into non-transformed epithelial cells, but also in maintenance of the transformed phenotype of established human cancer cells harboring oncogenic KRAS mutations (Frederick et al., 2008; Regala et al., 2005a; Scotti et al., 2010). Expression of kdPKCι or knock down of PKCι expression using lentiviral-mediated shRNA blocked transformed (anchorage-independent) growth and invasion of human NSCLC cells (Frederick et al., 2008; Regala et al., 2005a) and human PDAC cells (Scotti et al., 2010). Genetic disruption of PKCι also blocks the proliferative and invasive properties of prostate and glioma cell lines in vitro (Baldwin et al., 2008; Ishiguro et al., 2009; Patel et al., 2008). Disruption of PKCι expression also blocks tumorigenicity of NSCLC and PDAC cell tumors injected either subcutaneously, or orthotopically into the lung and pancreas, respectively (Regala et al., 2005a; Scotti et al., 2010). Analysis of human PDAC cells after orthotopic injection into the mouse pancreas revealed that PKCι-deficient tumor cells yielded significantly smaller tumors and significantly fewer metastases to the kidney, liver, diaphragm and mesentery, providing the first evidence that PKCι is important for tumor metastasis in vivo (Scotti et al., 2010). The role of PKCι in transformed growth is not restricted to cell harboring oncogenic KRAS mutations since NSCLC cell lines expressing wild-type KRAS but harboring PRKCI amplification also require PKCι for their transformed phenotype (Regala et al., 2005a; Regala et al., 2005b).
III. PKCι in Tumorigenesis in vivo
PKCι is necessary for oncogenic Kras- and mutant APC-mediated intestinal tumorigenesis
Several transgenic models of tumorigenesis have revealed that PKCι plays a critical promotive role in tumorigenesis in vivo. We established mice in which either kdPKCι or constitutively active (caPKCι) PKCι is expressed specifically in the intestinal epithelium (Murray et al., 2004). Expression of either PKCι mutant had no demonstrable effect on the basal proliferation or differentiation of the colonic epithelium (Murray and Fields, 1997; Murray et al., 2004). However, expression of caPKCι in the colon increased susceptibility to carcinogen (azoxymethane; AOM)-induced formation of colonic preneoplastic lesions, aberrant crypt foci (ACF), whereas expression of kdPKCι significantly inhibited AOM-induced ACF formation (Murray et al., 2004). In addition, caPKCι mice exhibited an increase in the number of AOM-induced colon tumors, and the majority of the tumors in these mice had progressed from benign adenoma to malignant intramucosal carcinoma (Murray et al., 2004). Similar results were obtained in an in vivo model of oncogenic Kras-mediated colon carcinogenesis, the KrasLA2 mouse (Johnson et al., 2001). Expression of kdPKCι in the colonic epithelium of these mice inhibited oncogenic Kras-mediated ACF formation (Murray et al., 2004). These data demonstrated that PKCι is required for oncogenic Kras-mediated transformation of the intestinal epithelium in vivo and constitute the first evidence for a function role of PKCι in tumorigenesis in vivo.
Interestingly, PKCι is also elevated in intestinal tumors formed in ApcMin/+ mice (Murray et al., 2009; Oster and Leitges, 2006). To determine if PKCι plays a role in tumor development in ApcMin/+ mice, the mouse PKCι gene, Prkci, was inactivated in the intestinal epithelium of triple transgenic ApcMin/+/Prkcif/f/villin-Cre mice by Cre-mediated recombination. ApcMin/+/Prkcif/f/villin-Cre mice exhibited loss of intestinal epithelial PKCι expression and a significant decrease in the number of intestinal tumors compared to ApcMin/+/Prkcif/f mice that harbor intact alleles of the Prkci gene (Murray et al., 2009). Thus, PKCι is important for ApcMin/+-induced intestinal epithelial tumorigenesis, indicating that the role of PKCι in colon tumorigenesis in vivo is not limited to Kras-mediated tumors.
Role of PKCι in initiation of lung tumorigenesis: Is PKCι a cancer stem cell gene?
PKCι is an oncogene required for maintenance of the transformed phenotype of non-small cell lung cancer (NSCLC) cells (Frederick et al., 2008; Regala et al., 2005a; Regala et al., 2005b). To address whether PKCι is involved in lung tumor development, we established a mouse model in which oncogenic KrasG12D is activated by Cre-mediated recombination in the lung with or without simultaneous genetic loss of the mouse PKCι gene, Prkci (Regala et al., 2009). Genetic loss of Prkci dramatically inhibits Kras-initiated hyperplasia and subsequent lung tumor formation in vivo. This effect correlates with a defect in the ability of Prkci-deficient bronchioalveolar stem cells (BASCs) to undergo Kras-mediated expansion and morphological transformation in vitro and in vivo (Regala et al., 2009). BASC exhibit stem-like properties and are thought to be the tumor-initiating cells in this model of Kras-mediated lung tumorigenesis (Jackson et al., 2001). Thus, Prkci is required for oncogene-induced expansion and transformation of tumor-initiating, lung stem-like cells. These studies suggest that PKCι may serve a critical role in cancer stem cell biology. Whether PKCι plays a similar role in the maintenance of the cancer stem cell niche in human cancers remains an important topic for future study.
IV. Oncogenic PKCι Signaling Mechanisms
PKCι-mediated survival signaling
PKCι activates multiple survival pathways that confer resistance to apoptosis induced by many stimuli, including TNF-α, carcinogens and chemotherapeutic agents (Figure 1). PKCι is sufficient to mediate the anti-apoptotic effects of Bcr-Abl via transactivation of NF-κB (Jamieson et al., 1999; Lu et al., 2001). Interestingly, in CML cells Bcr-Abl induces PKCι expression through a Ras/Mek-dependent pathway involving a functional Elk1 transcription factor binding site within the proximal promoter of PKCι (Gustafson et al., 2004). Thus Bcr-Abl appears to regulate PKCι at multiple levels to induce a chemoresistant phenotype; not only does Bcr-Abl induce PKCι expression but PKCι is a key effector of Bcr-Abl-mediated survival signaling. PKCι-mediated survival of TNFα-treated prostate cancer cells is also mediated through a NF-κB-dependent mechanism. In prostate cancer cells, PKCι-mediated phosphorylation of IκK leads to activation of the canonical NF-κB pathway and cell survival (Win and Acevedo-Duncan, 2008). In glioblastoma cells, PKCι-mediated survival appears to result from PKCι-induced attenuation of p38 mitogen-activated protein kinase signaling, which protects these cells from cytotoxicity caused by chemotherapeutic agents (Baldwin et al., 2006). In NSCLC cells, the ability of PKCι to enhance resistance to NNK-induced apoptosis appears to be mediated through Src-dependent activation of PKCι, which phosphorylates the proapoptotic protein BAD (Jin et al., 2005). Therefore, PKCι can activate multiple signaling pathways that promote cell survival in different tumor cell types.
Figure 1. Schematic representation of key oncogenic PKCι signaling pathways.
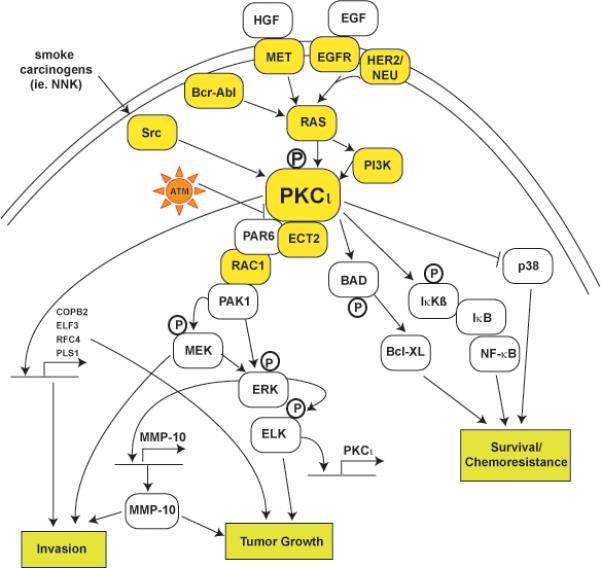
PKCι resides within several major signaling pathways implicated in human cancer. PKCι can be activated by known oncogenes such as Ras, Bcr-Abl, Src and PI3K, cytokines such as TNFα and IL-1, and growth factors such NGF and EGF. PKCι signals to downstream effectors such as Rac1 and NFκB which are important for different aspects of the transformed phenotype. Many components in PKCι-dependent signal pathways are mutated, often by multiple mechanisms (ie. gene amplification and somatic mutation), in human tumors (indicated by yellow boxes). Arrows indicate flow through signaling pathways; touching boxes indicate direct binding of signaling components. Phosphorylation events are indicated by circled Ps.
Rac1 is a critical downstream effector of oncogenic PKCι
The Rho family GTPase Rac1 is activated by oncogenic Ras and is essential for Ras-mediated transformed growth and cellular invasion in fibroblasts (Khosravi-Far et al., 1995; Qiu et al., 1995). PKCι is necessary for Ras-mediated Rac1 activation in RIE cells (Murray et al., 2004); thus, kdPKCι blocks oncogenic Ras-mediated Rac1 activation, and expression of a constitutively active Rac1 allele, RacV12, overcomes dnPKCι-mediated inhibition of cellular invasion (Murray et al., 2004). These studies placed PKCι downstream of oncogenic Kras and upstream of the critical Kras effector Rac1, which activates the Mek-Erk signaling axis to drive transformed growth and also mediates cytoskeletal rearrangement involved in cellular invasion (Murray et al., 2004) (Figure 1). Rac1 is also a critical downstream target of Kras-mediated, PKCι-dependent transformation in NSCLC (Regala et al., 2005a) and PDAC (Scotti et al., 2010). In NSCLC and PDAC cells, KRAS activates a PKCι-Rac1-Pak-Mek-Erk signaling axis that drives transformed growth in vitro and tumorigenicity in vivo (Regala et al., 2005a; Scotti et al., 2010). Interestingly, Rac1 also plays a critical role downstream of oncogenic PKCι in NSCLC cells that do not harbor mutant KRAS indicating that PKCι-Rac1 signaling is not specific to mutant KRAS-mediated transformation (Regala et al., 2005a; Regala et al., 2005b). Expression of RacV12 reconstituted cellular invasion and anchorage-independent growth in PKCι-deficient NSCLC and PDAC cells in a Mek-dependent manner (Regala et al., 2005a; Scotti et al., 2010). Thus, Rac1 is a critical downstream effector of oncogenic PKCι in multiple cancer cell types and is not restricted to tumor cells harboring KRAS mutations.
The role of the PB1 domain of PKCι in oncogenic signaling
The N-terminal regulatory domain of aPKCs is unique in that it contains a Phox/Bem1 (PB1) domain that mediates homo- and heterotypic protein-protein interactions critical for activation and intracellular localization (Lamark et al., 2003). Par6 is a PB1 domain-containing protein that binds atypical PKCs via PB1:PB1 domain interactions (Etienne-Manneville and Hall, 2003; Wilson et al., 2003). Par6 links atypical PKC to cell polarity by forming a complex with atypical PKC and a Rho family GTPase, Rac1 or cdc42 (Etienne-Manneville and Hall, 2003; Joberty et al., 2000; Lin et al., 2000; Noda et al., 2001; Qiu et al., 2000; Suzuki et al., 2003; Suzuki et al., 2001). Since Rac1 is a critical downstream effector of oncogenic PKCι in multiple cell types including the colon, lung and pancreas (Murray et al., 2004; Regala et al., 2005a; Scotti et al., 2010), we assessed the role of the PB1 domain of PKCι in Rac1 activation and NSCLC cell transformation (Regala et al., 2005a). Expression of the PB1 domain of PKCι in NSCLC cells uncouples PKCι and Par6 from Rac1 activation and inhibits transformed growth. Likewise, RNAi-mediated knock down of PKCι, Par6 or Rac1 inhibits transformed growth and cellular invasion in NSCLC cancer cells (Frederick et al., 2008). Expression of wild-type PKCι in PKCι knock down cells restores transformation, whereas expression of a PB1 domain mutant of PKCι, PKCι-D63A, that cannot bind Par6, does not (Frederick et al., 2008). Similarly, expression of wild type Par6 in Par6 knock down cells restores transformation whereas expression of Par6 mutants that either cannot bind PKCι (Par6-K19A) or couple to Rac1 (Par6-ΔCRIB) does not (Frederick et al., 2008). Expression of RacV12 in PKCι- or Par6-depleted NSCLC cells restores transformed growth and cellular invasion (Frederick et al., 2008). The PKCι-Par6 complex functions to activate a Rac1-Mek-Erk signaling axis that drives the transformed growth of NSCLC cells (Frederick et al., 2008). These studies defined a novel PKCι-Par6 complex that is required for NSCLC transformation (Figure 1).
Ect2 binds and activates the PKCι-Par6 complex
Having identified Rac1 as a critical downstream effector of the oncogenic PKCι-Par6 complex, a key question became: how does this complex activate Rac1? To address this question, we utilized a proteomics approach to identify proteins that associate with the PKCι-Par6 complex in NSCLC cells (Justilien and Fields, 2009). The Rho family GTPase guanine nucleotide exchange factor (GEF) Ect2 was identified as a prominent component of the PKCι-Par6 complex (Fields and Justilien, 2010; Justilien and Fields, 2009). RNAi-mediated knock down of Ect2 inhibits Rac1 activity and blocks transformed growth, invasion and tumorigenicity of NSCLC cells, whereas expression of RacV12 restores transformation to Ect2-deficient cells (Justilien and Fields, 2009). Interestingly, the role of Ect2 in NSCLC transformation is distinct from its well-established role in cytokinesis. In fact, NSCLC cells appear to have acquired an Ect2-independent cytokinesis mechanism, which resembles that described in fibrosarcoma H1080 cells (Kanada et al., 2008). Rather, in NSCLC cells Ect2 is mislocalized to the cytoplasm where it binds the PKCι-Par6 complex. Knock down of either PKCι or Par6 causes redistribution of Ect2 to the nucleus and loss of transformed growth and invasion. Therefore, Ect2 and PKCι drive tumor cell proliferation through formation of an oncogenic PKCι-Par6-Ect2 complex. Interestingly, the Ect2 gene ECT2 resides on chromosome 3q26 in close proximity to PRKCI. Studies in primary NSCLC tumors demonstrated that PRKCI and ECT2 are co-amplified and overexpressed in NSCLC (Justilien and Fields, 2009). Thus, Ect2 and PKCι are genetically linked through coordinate gene amplification in NSCLC tumors, and biochemically and functionally linked in NSCLC transformation through formation of an oncogenic PKCι-Par6-Ect2 complex that drives NSCLC cell transformation by activating Rac1 (Figure 1) (Justilien and Fields, 2009).
MMP10 is a critical downstream effector of the oncogenic PKCι-Par6-Rac1 signaling axis
We recently carried out a genomic analysis to identify genes whose expression is modulated by RNAi-mediated knock down of PKCι in NSCLC cells. The matrix metalloproteinase 10 (MMP10; stromolysin 2) emerged from this analysis as a genomic target of PKCι (Frederick et al., 2008). Depletion of PKCι, Par6 or Rac1 by RNAi inhibits MMP10 expression in NSCLC cells, and expression of exogenous wild-type Par6 in Par6 knock down cells restored MMP10 expression, whereas expression of Par6 mutants that either cannot bind PKCι or Rac1 did not, indicating the role of the PKCι-Par6-Rac1 complex in MMP10 expression. RNAi-mediated knock down of MMP10 blocks anchorage-independent growth and cell invasion in NSCLC cells, and the loss of transformed growth and invasion in PKCι knock down or Par6 knock down NSCLC cells can be rescued by the addition of catalytically active MMP10 (Frederick et al., 2008). Taken together, these data defined a PKCι-Par6-Rac1-Pak-Mek-Erk signaling axis that drives anchorage-independent growth and invasion of NSCLC cells, at least in part, through induction of MMP10 expression (Frederick et al., 2008). Interestingly, analysis of primary human lung tumor specimens demonstrated a strong correlation between PKCι and MMP10 expression in primary NSCLC tumors, suggesting a role for the PKCι-Par6-Rac1-Pak-Mek-Erk-MMP10 signaling axis in primary human lung cancers (Frederick et al., 2008). The molecular mechanism by which PKCι-mediated overexpression of MMP10 promotes transformation is currently unexplored and merits further investigation.
In a second genomic study, we conducted a meta-analysis of gene expression in primary lung adenocarcinomas (LAC) from three independent public domain datasets (Erdogan et al., 2009). Our analysis identified four genes, COPB2, ELF3, RFC4 and PLS1, whose expression correlates positively with PKCι in primary LAC tumors in all three databases. QPCR analysis of 60 primary LAC samples showed these four genes are highly overexpressed in tumors, and exhibit a strong positive correlation with PKCι expression (Erdogan et al., 2009). RNAi-mediated knock down of PKCι in LAC cell lines demonstrated that PKCι regulates expression of each of these genes. Furthermore, RNAi-mediated knock down of each of these genes led to significant inhibition of anchorage-independent growth and cellular invasion demonstrating that each of them is important for transformation in LAC cells (Erdogan et al., 2009). Finally, meta-analysis revealed that subsets of these PKCι-regulated genes are coordinately overexpressed with PKCι in other major tumor types including lung squamous cell carcinoma, breast, colon, prostate, pancreatic and glioblastoma cancers (Erdogan et al., 2009). This analysis revealed novel signaling mechanisms that participate in PKCι-mediated transformation (Figure 1) and provide potentially useful biomarkers of PKCι-mediated signaling which may serve as targets for the development of novel prognostic markers and/or therapeutic agents.
V. PKCι as a Therapeutic Target for Treatment of Cancer
The PB1-PB1 domain interaction between PKCι and Par6 is highly specific, and is required for the oncogenic PKCι-Par6-Rac1-MMP10 signaling axis that mediates anchorage-independent growth and invasion of human NSCLC cells in vitro and tumorigenicity in vivo (Frederick et al., 2008). Therefore, we reasoned that this interaction is an attractive target for development of novel mechanism-based therapeutics for treatment of NSCLC.
Using a novel fluorescence resonance energy transfer (FRET)-based assay we identified small molecular weight compounds that can disrupt the PB1-PB1 domain interaction between PKCι and Par6 (Stallings-Mann et al., 2006). Among the most potent inhibitors identified were the gold-containing compounds aurothioglucose (ATG) and aurothiomalate (ATM), which are FDA-approved treatments for rheumatoid arthritis (Messori and Marcon, 2004). ATG and ATM exhibit dose-dependent inhibition of PKCι-Par6 binding with IC50s of ~1μM (Stallings-Mann et al., 2006). Treatment of NSCLC cells with these compounds inhibits PKCι-mediated Rac1 activation and blocks anchorage-independent growth of NSCLC cells in vitro and tumorigenicity in vivo (Stallings-Mann et al., 2006). This inhibition can be rescued by expression of Rac1V12, indicating that ATM targets the interaction between PKCι and Par6 that couples PKCι to Rac1 (Stallings-Mann et al., 2006).
The precise mechanism of action of ATG and ATM in RA is still unknown, however a proposed mechanism of action is the formation of gold-cysteine adducts with target cellular proteins (Bratt et al., 2000; Jeon et al., 2000; Pia Rigobello et al., 2004; Yamashita et al., 2003). The PB1 domain of the atypical PKCs contains a unique cysteine residue, (Cys69) within the conserved OPR, PC and AID (OPCA) motif, which in the crystal structure of the PKCι-Par6 complex resides at the binding interface between PKCι and Par6 (Hirano et al., 2004; Lamark et al., 2003). Mutation of Cys69 to isoleucine (C69I) or valine (C69V), amino acids that frequently reside at this position in other PB1 domains, preserves Par6 binding but makes PKCι resistant to the inhibitory effects of ATM on Par6 binding in vitro (Erdogan et al., 2006). Expression of a C69I PKCι mutant in NSCLC cells supports transformed growth, but renders these cells resistant to the inhibitory effects of ATM on transformed growth (Erdogan et al., 2006). Thus, ATM inhibits PKCι-Par6 interactions in vitro and in vivo, and blocks NSCLC cell transformation by targeting Cys69 within the PB1 domain of PKCι.
Given the clinical potential of ATM as a therapeutic agent we assessed the inhibitory efficacy of ATM on the transformed growth of cell lines representing the major subtypes of lung cancer including lung adenocarcinoma (LAC), lung squamous cell carcinoma (LSCC), large cell carcinoma (LCC), and small cell lung carcinoma (SCLC) (Regala et al., 2008). ATM potently inhibited anchorage-independent growth in all lines tested with IC50s ranging from ~300 nM to 100 μM. The lung cancer cell lines clustered into those that are highly sensitive to ATM (IC50<5 μM) and those that are relatively insensitive to ATM (IC50>40μM). Interestingly, ATM sensitivity did not correlate with tumor sub-type, KRAS mutation status or sensitivity to a panel of standard chemotherapeutic agents frequently used to treat lung cancer patients, including cisplatin, placitaxel and gemcitabine (Regala et al., 2008). Rather, elevated PKCι expression was the major molecular characteristic exhibited by lung cancer cells that were responsive to ATM (Regala et al., 2008). Consistent with our in vitro observations, ATM inhibits tumorigenicity of both sensitive and insensitive lung cell tumors in vivo at plasma drug concentrations consistent with the ATM IC50 of the cell lines in vitro. Furthermore, measurements of plasma drug concentrations demonstrated that both sensitive as well as insensitive cell lines exhibit an anti-tumor response to ATM at plasma levels routinely achieved in RA patients undergoing ATM therapy (Regala et al., 2008). Thus, ATM exhibits anti-tumor activity against major lung cancer subtypes, particularly tumor cells that express high levels of PKCι. PKCι expression profiling revealed that a significant subset of primary NSCLC tumors express PKCι at or above the level associated with ATM sensitivity in vitro (Regala et al., 2008). Therefore, PKCι expression profiling in lung tumor samples may be useful in identifying lung cancer patients most likely to respond to ATM therapy. In addition, the fact that PKCι is overexpressed in many other tumor types (see Table 1 and 2) suggest that ATM may be an effective treatment option for these tumor types as well. Several phase I and phase II clinical trials are currently accruing at Mayo Clinic to determine an appropriate dosing regimen for ATM, and to assess anti-tumor activity of ATM alone and in combination with other targeted therapeutic agents in NSCLC, ovarian cancer and pancreatic cancer.
Summary/Future Directions
Accumulating evidence demonstrates that PKCι is an oncogene that is frequently targeted for genetic alteration in many major forms of human cancer (Tables 1 and 2). Functional data indicate that PKCι is required for the transformed phenotype of NSCLC, pancreatic, ovarian, prostate, colon and brain cancer cells. Future studies will be required to determine whether PKCι is also an oncogene in other cancers. Studies of PKCι using genetically defined models of tumorigenesis have revealed a critical role for PKCι in multiple stages of tumorigenesis, including tumor initiation, progression and metastasis. Recent studies in a genetic model of lung adenocarcinoma suggest a role for PKCι in transformation of lung cancer stem cells. These studies have important implications for the therapeutic use of ATM, particularly if future studies validate PKCι as an important gene in the critical cancer stem cell niche. Significant progress has been made in determining the molecular mechanisms by which PKCι drives the transformed phenotype, particularly the central role played by the oncogenic PKCι-Par6 complex in transformed growth and invasion, and of several PKCι-dependent survival pathways in chemo-resistance. Future studies will be required to determine the composition and dynamics of the PKCι-Par6 complex, and the mechanisms by which oncogenic signaling through this complex is regulated. Likewise, a better understanding of the critical downstream effectors of PKCι in various human tumor types holds promise for identification of novel prognostic and surrogate markers of oncogenic PKCι activity that may be clinically useful in ongoing clinical trials of ATM. Such studies also hold promise of revealing novel therapeutic targets. Similarly, a more complete understanding of the signaling mechanisms by which Kras (and possibly other oncogenes) regulate PKCι expression in human tumors may reveal new therapeutic intervention strategies. Ultimately, ongoing and future clinical trials will be required to establish the usefulness of ATM as an anti-tumor agent, and allow validation of potential surrogate markers and predictors of therapeutic response to PKCι-directed therapy identified in pre-clinical models.
Acknowledgments
The authors wish to thank the members of the Fields and Murray laboratories for their key contributions to the data described in this review. The authors apologize to our colleagues whose important contributions to this area were not cited; though we attempted to cite as much relevant literature as possible, space limitations made comprehensive citation impossible. Work from our laboratories discussed in this article was supported by grants to A.P.F. from the National Institutes of Health (CA081436), the American Lung Association/LUNGevity, The V Foundation for Cancer Research, The James and Esther King Biomedical Research Program and the Mayo Foundation; and to N.R.M. from the National Institutes of Health (CA128661) and the Mayo Clinic SPORE in Pancreatic Cancer Career Development award P50 CA102701 (N.R. Murray).
References
- Baldwin RM, Garratt-Lalonde M, Parolin DA, Krzyzanowski PM, Andrade MA, Lorimer IA. Protection of glioblastoma cells from cisplatin cytotoxicity via protein kinase Ciota-mediated attenuation of p38 MAP kinase signaling. Oncogene. 2006;25(20):2909–2919. doi: 10.1038/sj.onc.1209312. [DOI] [PubMed] [Google Scholar]
- Baldwin RM, Parolin DAE, Lorimer IAJ. Regulation of glioblastoma cell invasion by PKC[iota] and RhoB. Oncogene. 2008;27(25):3587–3595. doi: 10.1038/sj.onc.1211027. [DOI] [PubMed] [Google Scholar]
- Balsara BR, Sonoda G, du Manoir S, Siegfried JM, Gabrielson E, Testa JR. Comparative genomic hybridization analysis detects frequent, often high-level, overrepresentation of DNA sequences at 3q, 5p, 7p, and 8q in human non-small cell lung carcinomas. Cancer Res. 1997;57(11):2116–2120. [PubMed] [Google Scholar]
- Bedi A, Barber JP, Bedi GC, el-Deiry WS, Sidransky D, Vala MS, Akhtar AJ, Hilton J, Jones RJ. BCR-ABL-mediated inhibition of apoptosis with delay of G2/M transition after DNA damage: a mechanism of resistance to multiple anticancer agents. Blood. 1995;86(3):1148–1158. [PubMed] [Google Scholar]
- Bjorkoy G, Perander M, Overvatn A, Johansen T. Reversion of Ras- and phosphatidylcholine-hydrolyzing phospholipase C-mediated transformation of NIH 3T3 cells by a dominant interfering mutant of protein kinase C lambda is accompanied by the loss of constitutive nuclear mitogen-activated protein kinase/extracellular signal-regulated kinase activity. J Biol Chem. 1997;272(17):11557–11565. doi: 10.1074/jbc.272.17.11557. [DOI] [PubMed] [Google Scholar]
- Brass N, Racz A, Heckel D, Remberger K, Sybrecht GW, Meese EU. Amplification of the genes BCHE and SLC2A2 in 40% of squamous cell carcinoma of the lung. Cancer Res. 1997;57(11):2290–2294. [PubMed] [Google Scholar]
- Brass N, Ukena I, Remberger K, Mack U, Sybrecht GW, Meese EU. DNA amplification on chromosome 3q26.1-q26.3 in squamous cell carcinoma of the lung detected by reverse chromosome painting. Eur J Cancer. 1996;32A(7):1205–1208. doi: 10.1016/0959-8049(96)00016-0. [DOI] [PubMed] [Google Scholar]
- Bratt J, Belcher J, Vercellotti GM, Palmblad J. Effects of anti-rheumatic gold salts on NF-kappa B mobilization and tumour necrosis factor-alpha (TNF-alpha)-induced neutrophil-dependent cytotoxicity for human endothelial cells. Clin Exp Immunol. 2000;120(1):79–84. doi: 10.1046/j.1365-2249.2000.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257(13):7847–7851. [PubMed] [Google Scholar]
- Chalmers AD, Pambos M, Mason J, Lang S, Wylie C, Papalopulu N. aPKC, Crumbs3 and Lgl2 control apicobasal polarity in early vertebrate development. Development. 2005;132(5):977–986. doi: 10.1242/dev.01645. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Chou MM, Carpenter CL. Atypical protein kinases Clambda and - zeta associate with the GTP-binding protein Cdc42 and mediate stress fiber loss. Mol Cell Biol. 2000;20(8):2880–2889. doi: 10.1128/mcb.20.8.2880-2889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol. 2000;279(3):L429–L438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco MT, Lozano J, Municio MM, Berra E, Frutos S, Sanz L, Moscat J. Evidence for the in vitro and in vivo interaction of Ras with protein kinase C zeta. J Biol Chem. 1994;269(50):31706–31710. [PubMed] [Google Scholar]
- Du GS, Wang JM, Lu JX, Li Q, Ma CQ, Du JT, Zou SQ. Expression of P-aPKC-iota, E-cadherin, and beta-catenin related to invasion and metastasis in hepatocellular carcinoma. Ann Surg Oncol. 2009;16(6):1578–1586. doi: 10.1245/s10434-009-0423-7. [DOI] [PubMed] [Google Scholar]
- Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, Orntoft TF. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64(11):4040–4048. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- Eder AM, Sui X, Rosen DG, Nolden LK, Cheng KW, Lahad JP, Kango-Singh M, Lu KH, Warneke CL, Atkinson EN, Bedrosian I, Keyomarsi K, Kuo W-l, Gray JW, Yin JCP, Liu J, Halder G, Mills GB. Atypical PKC{iota} contributes to poor prognosis through loss of apical-basal polarity and Cyclin E overexpression in ovarian cancer. PNAS. 2005;102(35):12519–12524. doi: 10.1073/pnas.0505641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogan E, Klee EW, Thompson EA, Fields AP. Meta-analysis of oncogenic protein kinase Ciota signaling in lung adenocarcinoma. Clin Cancer Res. 2009;15(5):1527–1533. doi: 10.1158/1078-0432.CCR-08-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogan E, Lamark T, Stallings-Mann M, Lee J, Pellecchia M, Thompson EA, Johansen T, Fields AP. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281(38):28450–28459. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Cell polarity: Par6, aPKC and cytoskeletal crosstalk. Curr Opin Cell Biol. 2003;15(1):67–72. doi: 10.1016/s0955-0674(02)00005-4. [DOI] [PubMed] [Google Scholar]
- Fields AP, Gustafson WC. Protein kinase C in disease: cancer. Methods Mol Biol. 2003;233:519–537. doi: 10.1385/1-59259-397-6:519. [DOI] [PubMed] [Google Scholar]
- Fields AP, Justilien V. The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv Enzyme Regul. 2010;50(1):190–200. doi: 10.1016/j.advenzreg.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields AP, Murray NR. Protein kinase C isozymes as therapeutic targets for treatment of human cancers. Adv Enzyme Regul. 2008;48:166–178. doi: 10.1016/j.advenzreg.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick LA, Matthews JA, Jamieson L, Justilien V, Thompson EA, Radisky DC, Fields AP. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene. 2008;27(35):4841–4853. doi: 10.1038/onc.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64(1):55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- Gokmen-Polar Y, Fields AP. Mapping of a molecular determinant for protein kinase C betaII isozyme function. J Biol Chem. 1998;273(32):20261–20266. doi: 10.1074/jbc.273.32.20261. [DOI] [PubMed] [Google Scholar]
- Gumz ML, Zou H, Kreinest PA, Childs AC, Belmonte LS, LeGrand SN, Wu KJ, Luxon BA, Sinha M, Parker AS, Sun LZ, Ahlquist DA, Wood CG, Copland JA. Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin Cancer Res. 2007;13(16):4740–4749. doi: 10.1158/1078-0432.CCR-07-0143. [DOI] [PubMed] [Google Scholar]
- Gustafson WC, Ray S, Jamieson L, Thompson EA, Brasier AR, Fields AP. Bcr-Abl regulates protein kinase Ciota (PKCiota) transcription via an Elk1 site in the PKCiota promoter. J Biol Chem. 2004;279(10):9400–9408. doi: 10.1074/jbc.M312840200. [DOI] [PubMed] [Google Scholar]
- Heselmeyer K, Macville M, Schrock E, Blegen H, Hellstrom AC, Shah K, Auer G, Ried T. Advanced-stage cervical carcinomas are defined by a recurrent pattern of chromosomal aberrations revealing high genetic instability and a consistent gain of chromosome arm 3q. Genes Chromosomes Cancer. 1997;19(4):233–240. [PubMed] [Google Scholar]
- Hirano Y, Yoshinaga S, Ogura K, Yokochi M, Noda Y, Sumimoto H, Inagaki F. Solution structure of atypical protein kinase C PB1 domain and its mode of interaction with ZIP/p62 and MEK5. J Biol Chem. 2004;279(30):31883–31890. doi: 10.1074/jbc.M403092200. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Akimoto K, Nagashima Y, Kojima Y, Sasaki T, Ishiguro-Imagawa Y, Nakaigawa N, Ohno S, Kubota Y, Uemura H. aPKClambda/iota promotes growth of prostate cancer cells in an autocrine manner through transcriptional activation of interleukin-6. Proc Natl Acad Sci U S A. 2009;106(38):16369–16374. doi: 10.1073/pnas.0907044106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson L, Carpenter L, Biden TJ, Fields AP. Protein kinase Ciota activity is necessary for Bcr-Abl-mediated resistance to drug-induced apoptosis. J Biol Chem. 1999;274:3927–3930. doi: 10.1074/jbc.274.7.3927. [DOI] [PubMed] [Google Scholar]
- Jansen AP, Verwiebe EG, Dreckschmidt NE, Wheeler DL, Oberley TD, Verma AK. Protein kinase C-epsilon transgenic mice: a unique model for metastatic squamous cell carcinoma. Cancer Res. 2001;61(3):808–812. [PubMed] [Google Scholar]
- Jeon KI, Jeong JY, Jue DM. Thiol-reactive metal compounds inhibit NF-kappa B activation by blocking I kappa B kinase. J Immunol. 2000;164(11):5981–5989. doi: 10.4049/jimmunol.164.11.5981. [DOI] [PubMed] [Google Scholar]
- Jin Z, Xin M, Deng X. Survival function of protein kinase C{iota} as a novel nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-activated bad kinase. J Biol Chem. 2005;280(16):16045–16052. doi: 10.1074/jbc.M413488200. [DOI] [PubMed] [Google Scholar]
- Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2(8):531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009 doi: 10.1038/onc.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampfer S, Windegger M, Hochholdinger F, Schwaiger W, Pestell RG, Baier G, Grunicke HH, Uberall F. Protein kinase C isoforms involved in the transcriptional activation of cyclin D1 by transforming Ha-Ras. J Biol Chem. 2001;276(46):42834–42842. doi: 10.1074/jbc.M102047200. [DOI] [PubMed] [Google Scholar]
- Kanada M, Nagasaki A, Uyeda TQ. Novel functions of Ect2 in polar lamellipodia formation and polarity maintenance during “contractile ring-independent” cytokinesis in adherent cells. Mol Biol Cell. 2008;19(1):8–16. doi: 10.1091/mbc.E07-04-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol. 1995;15(11):6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkawa U, Takai Y, Tanaka Y, Miyake R, Nishizuka Y. Protein kinase C as a possible receptor protein of tumor-promoting phorbol esters. J Biol Chem. 1983;258(19):11442–11445. [PubMed] [Google Scholar]
- Klimstra DS, Longnecker DS. K-ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145(6):1547–1550. [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Akimoto K, Nagashima Y, Ishiguro H, Shirai S, Chishima T, Ichikawa Y, Ishikawa T, Sasaki T, Kubota Y, Inayama Y, Aoki I, Ohno S, Shimada H. The overexpression and altered localization of the atypical protein kinase C lambda/iota in breast cancer correlates with the pathologic type of these tumors. Hum Pathol. 2008;39(6):824–831. doi: 10.1016/j.humpath.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, Bjorkoy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278(36):34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Yang P, Pesatori AC, Consonni D, Bertazzi PA, Wacholder S, Shih JH, Caporaso NE, Jen J. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE. 2008;3(2):e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9(5):391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Li Q, Wang JM, Liu C, Xiao BL, Lu JX, Zou SQ. Correlation of aPKC-iota and E-cadherin expression with invasion and prognosis of cholangiocarcinoma. Hepatobiliary Pancreat Dis Int. 2008;7(1):70–75. [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2(8):540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- Lin M, Smith LT, Smiraglia DJ, Kazhiyur-Mannar R, Lang JC, Schuller DE, Kornacker K, Wenger R, Plass C. DNA copy number gains in head and neck squamous cell carcinoma. Oncogene. 2006;25(9):1424–1433. doi: 10.1038/sj.onc.1209166. [DOI] [PubMed] [Google Scholar]
- Lu Y, Jamieson L, Brasier AR, Fields AP. NF-kappaB/RelA transactivation is required for atypical protein kinase C iota-mediated cell survival. Oncogene. 2001;20(35):4777–4792. doi: 10.1038/sj.onc.1204607. [DOI] [PubMed] [Google Scholar]
- Messori L, Marcon G. Gold complexes in the treatment of rheumatoid arthritis. Met Ions Biol Syst. 2004;41:279–304. [PubMed] [Google Scholar]
- Mischak H, Goodnight JA, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, Blumberg PM, Pierce JH, Mushinski JF. Overexpression of protein kinase C-delta and -epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268(9):6090–6096. [PubMed] [Google Scholar]
- Murray NR, Davidson LA, Chapkin RS, Gustafson WC, Schattenberg DG, Fields AP. Overexpression of protein kinase C betaII induces colonic hyperproliferation and increased sensitivity to colon carcinogenesis. J Cell Biol. 1999;145(4):699–711. doi: 10.1083/jcb.145.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Fields AP. Atypical protein kinase C iota protects human leukemia cells against drug-induced apoptosis. J Biol Chem. 1997;272(44):27521–27524. doi: 10.1074/jbc.272.44.27521. [DOI] [PubMed] [Google Scholar]
- Murray NR, Jamieson L, Yu W, Zhang J, Gokmen-Polar Y, Sier D, Anastasiadis P, Gatalica Z, Thompson EA, Fields AP. Protein kinase C{iota} is required for Ras transformation and colon carcinogenesis in vivo. J Cell Biol. 2004;164(6):797–802. doi: 10.1083/jcb.200311011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Weems J, Braun U, Leitges M, Fields AP. Protein kinase C betaII and PKCiota/lambda: collaborating partners in colon cancer promotion and progression. Cancer Res. 2009;69(2):656–662. doi: 10.1158/0008-5472.CAN-08-3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda Y, Takeya R, Ohno S, Naito S, Ito T, Sumimoto H. Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells. 2001;6(2):107–119. doi: 10.1046/j.1365-2443.2001.00404.x. [DOI] [PubMed] [Google Scholar]
- Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006;66(14):6955–6963. doi: 10.1158/0008-5472.CAN-06-0268. [DOI] [PubMed] [Google Scholar]
- Patel R, Win H, Desai S, Patel K, Matthews JA, Acevedo-Duncan M. Involvement of PKC-iota in glioma proliferation. Cell Prolif. 2008;41(1):122–135. doi: 10.1111/j.1365-2184.2007.00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pia Rigobello M, Messori L, Marcon G, Agostina Cinellu M, Bragadin M, Folda A, Scutari G, Bindoli A. Gold complexes inhibit mitochondrial thioredoxin reductase: consequences on mitochondrial functions. J Inorg Biochem. 2004;98(10):1634–1641. doi: 10.1016/j.jinorgbio.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Abo A, Steven Martin G. A human homolog of the C. elegans polarity determinant Par-6 links Rac and Cdc42 to PKCzeta signaling and cell transformation. Curr Biol. 2000;10(12):697–707. doi: 10.1016/s0960-9822(00)00535-2. [DOI] [PubMed] [Google Scholar]
- Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in Ras transformation. Nature. 1995;374(6521):457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- Racz A, Brass N, Heckel D, Pahl S, Remberger K, Meese E. Expression analysis of genes at 3q26-q27 involved in frequent amplification in squamous cell lung carcinoma. Eur J Cancer. 1999;35(4):641–646. doi: 10.1016/s0959-8049(98)00419-5. [DOI] [PubMed] [Google Scholar]
- Regala RP, Davis RK, Kunz A, Khoor A, Leitges M, Fields AP. Atypical protein kinase C{iota} is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer Res. 2009;69(19):7603–7611. doi: 10.1158/0008-5472.CAN-09-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regala RP, Thompson EA, Fields AP. Atypical protein kinase C iota expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008;68(14):5888–5895. doi: 10.1158/0008-5472.CAN-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005a;280(35):31109–31115. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005b;65(19):8905–8911. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- Reyland ME. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front Biosci. 2009;14:2386–2399. doi: 10.2741/3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9(2):121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24(5):778–789. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- Scotti ML, Bamlet W, Smyrk TC, Fields AP, Murray NR. Protein kinase C iota is required for pancreatic cancer cell transformed growth and tumorigenesis. Cancer Res. 2010;70(5):2064–2074. doi: 10.1158/0008-5472.CAN-09-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segara D, Biankin AV, Kench JG, Langusch CC, Dawson AC, Skalicky DA, Gotley DC, Coleman MJ, Sutherland RL, Henshall SM. Expression of HOXB2, a retinoic acid signaling target in pancreatic cancer and pancreatic intraepithelial neoplasia. Clin Cancer Res. 2005;11(9):3587–3596. doi: 10.1158/1078-0432.CCR-04-1813. [DOI] [PubMed] [Google Scholar]
- Singh B, Stoffel A, Gogineni S, Poluri A, Pfister DG, Shaha AR, Pathak A, Bosl G, Cordon-Cardo C, Shah JP, Rao PH. Amplification of the 3q26.3 locus is associated with progression to invasive cancer and is a negative prognostic factor in head and neck squamous cell carcinomas. Am J Pathol. 2002;161(2):365–371. doi: 10.1016/S0002-9440(10)64191-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery ML, Anderson K, Curtin K, Ma K, Schaffer D, Edwards S, Samowitz W. Lifestyle factors and Ki-ras mutations in colon cancer tumors. Mutat Res. 2001;483(1-2):73–81. doi: 10.1016/s0027-5107(01)00228-7. [DOI] [PubMed] [Google Scholar]
- Snaddon J, Parkinson EK, Craft JA, Bartholomew C, Fulton R. Detection of functional PTEN lipid phosphatase protein and enzyme activity in squamous cell carcinomas of the head and neck, despite loss of heterozygosity at this locus. Br J Cancer. 2001;84(12):1630–1634. doi: 10.1054/bjoc.2001.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006;66(3):1767–1774. doi: 10.1158/0008-5472.CAN-05-3405. [DOI] [PubMed] [Google Scholar]
- Sugita M, Tanaka N, Davidson S, Sekiya S, Varella-Garcia M, West J, Drabkin HA, Gemmill RM. Molecular definition of a small amplification domain within 3q26 in tumors of cervix, ovary, and lung. Cancer Genet Cytogenet. 2000;117(1):9–18. doi: 10.1016/s0165-4608(99)00135-1. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Akimoto K, Ohno S. Protein kinase C lambda/iota (PKClambda/iota): a PKC isotype essential for the development of multicellular organisms. J Biochem (Tokyo) 2003;133(1):9–16. doi: 10.1093/jb/mvg018. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T, Ohno S. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol. 2001;152(6):1183–1196. doi: 10.1083/jcb.152.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagawa R, Akimoto K, Ichikawa Y, Akiyama H, Kojima Y, Ishiguro H, Inayama Y, Aoki I, Kunisaki C, Endo I, Nagashima Y, Ohno S. High expression of atypical protein kinase C lambda/iota in gastric cancer as a prognostic factor for recurrence. Ann Surg Oncol. 2010;17(1):81–88. doi: 10.1245/s10434-009-0708-x. [DOI] [PubMed] [Google Scholar]
- Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, Satoh T, Takimoto R, Kato J, Sakamaki S, Niitsu Y. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121(3):599–611. doi: 10.1053/gast.2001.27203. [DOI] [PubMed] [Google Scholar]
- Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11(20):7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- Uberall F, Hellbert K, Kampfer S, Maly K, Villunger A, Spitaler M, Mwanjewe J, Baier-Bitterlich G, Baier G, Grunicke HH. Evidence that atypical protein kinase C-lambda and atypical protein kinase C-zeta participate in Ras-mediated reorganization of the F-actin cytoskeleton. J Cell Biol. 1999;144(3):413–425. doi: 10.1083/jcb.144.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350(16):1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, Stephens RM, Caporaso NE, Loffredo CA, Ambs S. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68(3):927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- Weichert W, Gekeler V, Denkert C, Dietel M, Hauptmann S. Protein kinase C isoform expression in ovarian carcinoma correlates with indicators of poor prognosis. Int J Oncol. 2003;23(3):633–639. [PubMed] [Google Scholar]
- Wilson MI, Gill DJ, Perisic O, Quinn MT, Williams RL. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol Cell. 2003;12(1):39–50. doi: 10.1016/s1097-2765(03)00246-6. [DOI] [PubMed] [Google Scholar]
- Win HY, Acevedo-Duncan M. Atypical protein kinase C phosphorylates IKKalphabeta in transformed non-malignant and malignant prostate cell survival. Cancer Lett. 2008;270(2):302–311. doi: 10.1016/j.canlet.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, Fiel I, Thung S, Mazzaferro V, Bruix J, Bottinger E, Friedman S, Waxman S, Llovet JM. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45(4):938–947. doi: 10.1002/hep.21622. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Ashino S, Oshima Y, Kawamura S, Ohuchi K, Takayanagi M. Inhibition of TPA-induced NF-kappaB nuclear translocation and production of NO and PGE2 by the anti-rheumatic gold compounds. J Pharm Pharmacol. 2003;55(2):245–251. doi: 10.1211/002235702513. [DOI] [PubMed] [Google Scholar]
- Yang YL, Chu JY, Luo ML, Wu YP, Zhang Y, Feng YB, Shi ZZ, Xu X, Han YL, Cai Y, Dong JT, Zhan QM, Wu M, Wang MR. Amplification of PRKCI, located in 3q26, is associated with lymph node metastasis in esophageal squamous cell carcinoma. Genes Chromosomes Cancer. 2008;47(2):127–136. doi: 10.1002/gcc.20514. [DOI] [PubMed] [Google Scholar]
- Ye H, Yu T, Temam S, Ziober BL, Wang J, Schwartz JL, Mao L, Wong DT, Zhou X. Transcriptomic dissection of tongue squamous cell carcinoma. BMC Genomics. 2008;9:69. doi: 10.1186/1471-2164-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Huang J, Yang N, Liang S, Barchetti A, Giannakakis A, Cadungog MG, O'Brien-Jenkins A, Massobrio M, Roby KF, Katsaros D, Gimotty P, Butzow R, Weber BL, Coukos G. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res. 2006;66(9):4627–4635. doi: 10.1158/0008-5472.CAN-05-4527. [DOI] [PubMed] [Google Scholar]