

Abstract
The use of statins for the prevention or treatment of different neurodegenerative diseases has generated considerable interest albeit with some controversy. Mechanisms of statin-induced neuroprotection are not well understood. Recently, we reported that simvastatin stimulated neuronal gene expression and protein levels of the major antiapoptotic protein Bcl-2 in vivo and in vitro; suppression of Bcl-2 in SH-SY5Y cells reduced simvastatin neuroprotection; effects were independent of cholesterol and other products of the 3-hydroxy-3-methylglutaryl-CoA reductase pathway. Endothelin-1 (ET-1) can increase Bcl-2 abundance via the transcription factor nuclear factor of activated thymocytes (NFATc), and simvastatin was reported to increase ET-1 gene expression. We tested the hypothesis that simvastatin stimulation of Bcl-2 involves up-regulation of ET-1 and binding of NFATc to Bcl-2 promoter sites in SH-SY5Y human neuroblastoma cells. Simvastatin increased both intracellular and secreted ET-1 protein levels. Exogenous ET-1 increased Bcl-2 protein abundance, which was inhibited by ET-1 receptor antagonists. Simvastatin increased translocation of NFATc3 to the nucleus while reducing nuclear NFATc1 and having no effect on NFATc4. Endothelin-1 also increased NFATc3 levels in the nucleus, and this increase was inhibited by ET-1 receptor antagonists. Treatment of cells with simvastatin stimulated binding of NFATc3 to the Bcl-2 promoter. We report novel findings showing that up-regulation of Bcl-2 by simvastatin involves ET-1 and the transcription factor NFATc3. Discovering how statins can selectively alter a specific NFATc isoform that leads to an increase in an antiapoptotic protein will provide a new approach to understanding statin-induced neuroprotection and conditions outside the brain in which apoptosis contributes to pathophysiology.
Keywords: Simvastatin, Bcl-2, Endothelin-1, Alzheimer’s disease, Statins, Neuroprotection, Apoptosis, NFAT
It is well-established that 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors (statins) are the most effective treatment for hypercholesterolemia, and these drugs are commonly prescribed for the management of coronary heart disease [1, 2]. A relatively new use of statins has been proposed, albeit with some controversy, in the treatment of neurodegenerative diseases or conditions such as cerebrovascular disease [3], Alzheimer disease [4], Parkinson’s disease [5], multiple sclerosis [6], and most recently, traumatic brain injury [7]. The mechanism(s) underlying statin-induced neuroprotection generally have been thought to be due to reductions in levels of cholesterol or the isoprenoids farenesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP). Farenesyl pyrophosphate is the precursor of both cholesterol and GGPP, and statins reduce cholesterol, and for the first time, it was reported that statins reduce FPP and GGPP levels in the brain [8]. There is evidence, however, demonstrating that the neuroprotective effects of statins may also involve mechanisms outside the HMG-CoA reductase pathway [9–11]. Statins, for example, reduced cytokine-stimulated production of prostaglandins in microglia and reduced glutamate-induced excitotoxicity in primary cortical neurons [9, 10]. In those studies, statin-induced neuroprotection was not reduced by the concomitant administration of mevalonate or isoprenoids. Adding back mevalonate, the product of HMG-CoA reductase activity and the precursor of isoprenoids and cholesterol counteracts the statin-induced inhibition of HMG-CoA reductase [12]. We have recently shown that simvastatin administration in vivo and in vitro up-regulates gene expression and protein levels of the major antiapoptotic protein Bcl-2 in mouse and guinea pig brain tissue [13, 14] and in mouse primary neurons and SH-SY5Y human neuroblastoma cells [11]. Suppression of Bcl-2 protein levels markedly reduced the neuroprotective effects of simvastatin in SH-SY5Y cells [11]. The protective effects and up-regulation of Bcl-2 by simvastatin were not altered by coincubation with mevalonate.
Simvastatin up-regulation of Bcl-2 was not associated with the HMG-CoA reductase pathway. A potential mechanism for the simvastatin-induced increase in Bcl-2 involves endothelin-1 (ET-1) and the transcription factor nuclear factor of activated T-lymphocytes (NFATc). This hypothesis is based on the following lines of evidence. We had observed in a DNA microarray analysis of brain tissue of mice chronically treated with simvastatin that in addition to a significant increase in Bcl-2 gene expression, there was a significant increase in ET-1 gene expression whose protein product is the precursor of the mature ET-1 protein [13]. Endothelin-1 protein increases Bcl-2 protein levels and reduces apoptosis in cardiomyocytes [15, 16]. Those effects on inhibition of apoptosis and stimulation of Bcl-2 abundance are similar to the effects of simvastatin observed in mouse primary neurons and SH-SY5Y cells [11]. The ET-1-induced increase in Bcl-2 protein levels requires the transcription factor NFATc [15, 16]. An increase in ET-1 protein has been previously reported to initiate activation of a calcium phosphatase resulting in dephosphorylation of NFATc and allowing it to translocate to the nucleus and bind to consensus sequences on the Bcl-2 promoter [16]. The NFATc family of transcription factors consists of four members that are calcium/calcineurin dependent. These members include NFATc1 (NFAT2), NFATc2 (NFAT1), NFATc3 (NFAT4), and NFATc4 (NFAT3) [17]. Nuclear factor of activated T-lymphocytes isoforms are expressed in neurons and astrocytes and have been shown, for example, to play a role in promoting synaptic plasticity and protecting neurons under apoptotic conditions [18, 19], but there is also evidence that specific isoforms may have opposite effects during development [20].
We hypothesize that the simvastatin stimulation of Bcl-2 protein abundance involves up-regulation of ET-1 and binding of NFATc to the Bcl-2 promoter site. The results presented here show that simvastatin increased both intracellular and secreted ET-1 protein levels in SH-SY5Y cells. Incubation of cells with ET-1 protein stimulated Bcl-2 protein abundance, which was inhibited by antagonists to the ET-1 receptors. Simvastatin treatment of cells increased NFATc3 in the nucleus. Surprisingly, simvastatin incubation had the opposite effect on NFATc1 and did not alter levels of NFATc4 in the nucleus. Endothelin-1 treatment of cells increased NFATc3 in the nuclear fraction, and this increase was inhibited by the ET-1 antagonists. We further demonstrated that the functional significance of the simvastatin-induced increase of NFATc3 in the nucleus was to stimulate binding of this transcription factor to the Bcl-2 promoter.
Materials and Methods
Materials
SH-SY5Y human neuroblastoma cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Simvastatin was purchased from EMD Biosciences Inc. (San Diego, CA, USA). Endothelin-1 protein was obtained from Bachem (Torrance, CA, USA), and the ET-1 antagonists BQ-123/BQ788 were purchased from Alexis Biochemicals (San Diego, CA, USA). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified.
Cell Culture
Cell culture procedures were used as we have previously reported [11]. Briefly, cells were plated in 75-cm2 tissue culture flasks (BD Falcon, San Jose, CA, USA) and grown in a 1:1 mixture of DMEM and F-12 Nutrient Mixture (GIBCO, Carlsbad, CA, USA) supplemented with a 1% penicillin–streptomycin–neomycin mix (Invitrogen, Carlsbad, CA, USA) and 10% FBS. Cells were maintained under 5% CO2 at 37°C. Simvastatin was dissolved in ethanol, and a final concentration of 100 nM was added to the cells. We have previously reported that this drug concentration increased Bcl-2 levels, and it was neuroprotective in SH-SY5Y cells and mouse primary neurons [11]. In some experiments, ET-1 was incubated with cells at a concentration of 1.0 μM in culture media for 48 h in the presence or absence of ET-1 receptor ETRA/ETRB antagonists BQ-123/BQ788, respectively. The ET-1 concentration was based on data showing that cardiomyocytes incubated with 1.0 μM increased Bcl-2 gene expression and protein levels [16]. The concentration of the ET-1 receptor antagonists was 2.0 μM and is based on data showing a reduction of ET-1 uptake in astrocytes [21].
ET-1, Bcl-2, and NFATc Protein Levels
ET-1 protein levels were determined using the Endothelin-1 TiterZyme® Enzyme Immunometric Assay (Assay Designs, Ann Arbor, MI, USA) according to the manufacturer’s instructions. Bcl-2 protein levels were determined using a Bcl-2 EIA kit (human total, Assay Designs) according to the manufacturer’s instructions.
NFATc protein levels were determined by Western blot analysis. Nuclear fractions were isolated using the Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA). For the Western blot analysis, 50 μg of total protein was used from each sample and separated on 8% polyacrylamide–SDS gels and analyzed by Western blot with primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA) against NFATc1 (cat # sc-13033), NFATc2 (cat # sc-13034), NFATc3 (cat # sc-8321), and NFATc4 (cat # sc 13036). Primary antibodies were used at a final dilution of 1:800 as recommended by the manufacturer. The secondary antibody, goat anti-rabbit IgG (Pierce Thermo Scientific Life Science Research Products, Rockford, IL, USA), was used at a final concentration of 1:60,000. Lamin A was used as a loading control. Proteins were transferred onto a nitrocellulose membrane and blocked with Superblocker T-20 (Pierce Thermo Scientific Life Science Research Products) for 2 h. The membrane was washed 3× with TBS/Tween 20 and then treated with HRP-conjugated secondary goat anti-rabbit IgG antibody (Pierce) for 2 h. The membrane was washed 3× with TBS/Tween 20, and the immunoreactivity was visualized with Super Signal West Pico chemiluminescent substrate (Pierce). The membrane was then placed on Kodak film and developed using Agfa Developer CP1000. Band density was quantified by densitometry using the Stratagene Eagle Eye II system (Stratagene, La Jolla, CA, USA).
Chromatin Immunoprecipitation
Binding of NFATc3 protein to the NFATc consensus sites on the Bcl-2 promoter were examined using chromatin immunoprecipitation (CHIP). This assay was performed using the SimpleChIP Enzymatic Chromatin IP Kit #9002 (Cell Signaling Technology, Danvers, MA, USA) according to the manufacture’s instructions, and all reagents unless otherwise specified were provided in the kit. Briefly, cells were treated with either 100 nM simvastatin or vehicle control for 72 h and then chemically cross-linked with 1% formaldehyde at room temperature. Chromatin was prepared and then digested with micrococcal nuclease. Digestion and fragmentation size was analyzed by electrophoresis on a 1% agarose gel. DNA was confirmed to be at lengths of approximately 150 to 900 bp (1–5 nucleosomes). DNA fragments were incubated with NFATc3 monoclonal antibody (Santa Cruz Biotechnology), positive control antibody histone H3, and negative control normal rabbit IgG overnight at 4°C. Chromatin immunoprecipitation-grade protein G agarose beads were added to the immune complexes, the beads were washed, and the immunoprecipitates were eluted with an elution buffer contained in the kit. Following reversal of protein-DNA cross-links, DNA was purified using spin columns provided in the CHIP kit. End point polymerase chain reaction (PCR) was then used to quantify DNA. Primers for the human Bcl-2 promoter were designed using National Center for Biotechnology Information’s (NCBI) Primer Basic Local Alignment Search Tool (Primer-BLAST), http://www.ncbi.nlm.nih.gov/tools/primer-blast based on using the accession number EU119400, for the Bcl-2 gene promoter. The location (M region of promoter) that was used to generate possible primers sets was based on the previously described NFATc1 consensus site [15] for the PCR primer template. The primer sequences that were used for the NFATc3 site are as follows: forward strand 5′-AAATAAAACCCTCCCCCACC-3′, reverse strand 5′-GGATAAATGAAGGCAGGACG-3′ (114 bp). Polymerase chain reaction primers for RPL30 control primers were provided in the CHIP kit. Polymerase chain reaction reactions were carried out using the Super Mix II kit (Invitrogen). The PCR products were run on a 2% agarose gel, stained with ethidium bromide, and visualized using the Stratagene Eagle Eye II system (Stratagene). Densitometry was used to measure the relative enrichment of the Bcl-2 DNA sequence by the NFATc3 protein-specific immunoprecipitation in simvastatin and control cells.
Data Analysis
Data were analyzed using a Student t test and the SigmaPlot statistical program (Systat Software Inc., San Jose, CA, USA).
Results
Simvastatin Increases ET-1 Protein Levels
The question initially addressed in the present study is if simvastatin would increase ET-1 protein levels. We had reported that simvastatin administered in vivo increased ET-1 gene expression, but ET-1 protein levels were not examined [13]. Endothelin-1 acts in an autocrine manner, and we determined if simvastatin would increase ET-1 protein abundance in the conditioned media. Figure 1 shows that simvastatin treatment significantly increased (p≤0.001) ET-1 protein levels in the conditioned media. There was a 44% and 50% increase in ET-1 protein abundance when cells were incubated with simvastatin for 72 and 144 h, respectively. The significant increase in secreted ET-1 protein at 72 h is noteworthy because it precedes the simvastatin-induced increase in Bcl-2 protein abundance, which was also observed in SH-SY5Y cells treated with the same drug concentration [11]. Endothelin-1 protein was also significantly increased in the lysate of cells treated with simvastatin (data not shown).
Fig. 1.

Simvastatin increases secreted ET-1 protein levels in SH-SY5Y cells. Cells were incubated with 100 nM simvastatin or vehicle (10 μM ethanol) for 72 and 144 h. Conditioned media were collected and ET-1 protein levels were determined using the Endothelin-1 TiterZyme® Enzyme Immunometric Assay (Assay Designs) (see Materials and Methods). Results are the means ± SEM of three independent experiments performed in triplicate. ***p≤0.001 as compared with control
Exogenous ET-1 Increases Bcl-2 Protein Levels
Simvastatin increased ET-1 protein levels in the conditioned media (Fig. 1). We next examined if exogenous ET-1 would increase Bcl-2 protein levels in cells. Exogenous ET-1 significantly increased (p≤0.05) Bcl-2 protein levels by approximately twofold as compared with control levels as seen in Fig. 2. This finding is consistent with an earlier report in cardiomyocytes [15]. Figure 2 also shows that the stimulatory effect of ET-1 on cellular Bcl-2 protein abundance was significantly inhibited (p≤0.05) when cells were coincubated with antagonists to the ETA and ETB receptors.
Fig. 2.

Endothelin-1 protein increases Bcl-2 protein levels, which is inhibited by ET-1 receptor antagonists in SH-SY5Y cells. Cells were incubated with ET-1 protein (1 μM) alone or in combination with the ET-1 receptor antagonists BQ-123/BQ788 (2 μM) for 48 h (see Materials and Methods). Bcl-2 protein levels were determined in cell lysate using the Bcl-2 EIA kit (human total, Assay Designs). Results are the means ± SEM of three independent experiments performed in triplicate. *p≤0.05 as compared with control cells and *p≤0.05 as compared with ET-1 only
Simvastatin Increases Nuclear NFATc3 Protein Levels
NFATc3 null mice show lower levels of Bcl-2 mRNA and protein as well as increased apoptosis compared with wild-type mice [22]. Endothelin-1 has been reported to increase NFATc3 in the nucleus of human myometrial arteries [23]. Therefore, we first determined if simvastatin treatment would increase NFATc3 protein abundance in the nuclear fraction of treated cells. Data in Fig. 3 demonstrate that simvastatin significantly increased NFATc3 protein levels in the nuclear fraction. The stimulatory effects of simvastatin on NFATc3 protein levels were greatest when cells were incubated with the drug for 72, 96, and 120 h (Fig. 3).
Fig. 3.

Simvastatin increases NFATc3 protein levels in the nuclear fraction of SH-SY5Y cells. Cells were treated with simvastatin (100 nM) for different periods after which time the nuclear fraction was isolated using the Nuclear Extract Kit (Active Motif) (see Materials and Methods). NFATc3 protein levels were determined in the nuclear fraction by Western blot analysis as shown by a representative blot. Band density was quantified by densitometry and expressed relative to lamin A using the Stratagene Eagle Eye II system. Data are percentage change of control and are the means ± SEM of three independent experiments. *p≤0.05 and **p≤0.01 as compared with control cells
The simvastatin-induced increase in NFATc3 could be a specific effect on that isoform or alternatively a drug effect common with the other NFATc isoforms. That possibility was examined and those data are shown in Fig. 4a, b. In the nuclear fraction, NFATc1 was significantly decreased (p≤0.01) by simvastatin treatment (Fig. 4, panel a), while NFATc4 levels were unaffected as compared with control cells (Fig. 4b). NFATc2 was not detectable in SH-SY5Y cells, although as a positive control, it was detected in liver (data not shown).
Fig. 4.

Effects of simvastatin on protein levels of NFATc1 and NFATc4 in the nuclear fraction of SH-SY5Y cells. Cells were incubated with simvastatin for 72 h and levels of NFATc isoforms determined by Western blot analysis as described in the legend to Fig. 3 and in the Materials and Methods section. a NFATc1 as a percentage of control and data are the means ± SEM of three independent experiments. **p≤0.01 as compared with control cells. b NFATc4 as a percentage of control and data are the means ± SEM of three independent experiments
Exogenous ET-1 Increases Nuclear NFATc3
As reported in cardiomyocytes, an increase in Bcl-2 protein is mediated by an ET-1/NFATc calcium/calcineurin-dependent mechanism [15, 16]. We had previously shown that Bcl-2 mRNA and protein levels were increased by simvastatin [11, 13, 14], and data in Figs. 1 to 4 now show that simvastatin stimulates ET-1 protein levels and increases NFATc3 abundance in the nuclear fraction of cells. It was next determined if incubation of cells with ET-1 protein would increase NFATc3 in the nuclear fraction. Data in Fig. 5 show that ET-1 significantly increased (p≤0.001) NFATc3 greater than twofold in the nuclear fraction as compared with control cells. This ET-1-induced increase in nuclear NFATc3 protein was significantly inhibited by the ET-1 receptor antagonists BQ-123/788 as compared with ET-1 treatment alone (Fig. 5).
Fig. 5.

Endothelin-1 protein increases NFATc3 in the nuclear fraction, which is inhibited by ET-1 receptor antagonists in SH-SY5Y cells. Cells were treated with ET-1 (1 μM) for 3 h alone or in combination with the ET-1 receptor antagonists BQ123 and 788 (2 μM), and the nuclear fraction was isolated (see Materials and Methods). NFATc3 protein levels were determined in the nuclear fraction by Western blot analysis. Data are expressed as percentage change of control and are the means ± SEM of three independent experiments. ***p≤0.001 as compared with control; **p≤0.01 as compared with ET-1 only
Simvastatin Increases NFATc3 Binding to the Bcl-2 Promoter
The data in Figs. 3 and 5 indicate that simvastatin and ET-1, respectively, increased levels of the transcription factor NFATc3 in the nuclear fraction of cells. We are proposing that the simvastatin-induced increase in NFATc3 in the nucleus enhances binding of that transcription factor to the Bcl-2 promoter. This hypothesis was tested using a CHIP assay in cells treated with vehicle or 100 nM simvastatin for 72 h. Figure 6 shows that in the absence of simvastatin, NFATc3 was associated with the Bcl-2 promoter. Simvastatin significantly increased (p≤0.01) the binding of NFATc3 to the Bcl-2 promoter as compared with control cells (Fig. 6a, b). There was approximately a 60% increase in DNA binding in the simvastatin-treated cells. The positive control histone H3 binding to the RPL30 promoter was similar in the vehicle and simvastatin-treated cells as was input DNA that had been extracted before the CHIP assay (Fig. 6a). Bands as expected were not detected using the IgG control antibody (Fig. 6a).
Fig. 6.
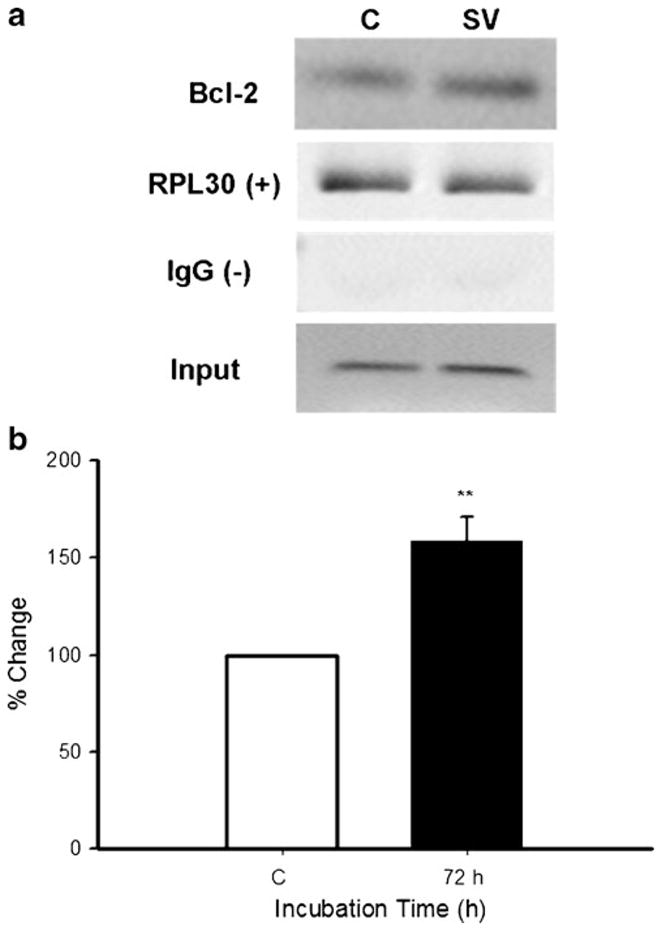
Simvastatin increases NFATc3 binding to the Bcl-2 promoter. Cells were incubated with simvastatin (100 nM) for 72 h. Chromatin immunoprecipitation using the SimpleChIP Enzymatic Chromatin IP Kit #9002 (Cell Signaling Technology) and end point PCR were used to determine NFATc3 binding to the Bcl-2 promoter site (see Materials and Methods). a Bcl-2: changes in DNA binding using primers specific for the Bcl-2 promoter region from control (C) and simvastatin (SV)-treated cells; RPL30: housekeeping gene used as a positive control; IgG: negative control; input: DNA extracted but not immunoprecipitated from C and SV samples. b Relative enrichment of Bcl-2 DNA by NFATc3 protein-specific immunoprecipitation in control and cells incubated with simvastatin for 72 h normalized against input samples. Data are means ± SEM of three independent experiments, **p≤0.01
Discussion
We had previously shown that simvastatin increased Bcl-2 gene expression and protein levels in vivo and in vitro but that effects of the drug on Bcl-2 did not involve the mevalonate/isoprenoid/cholesterol pathway [11, 13, 14]. The present study tested the hypothesis that simvastatin stimulation of Bcl-2 protein abundance involves up-regulation of ET-1 and binding of the transcription factor NFATc to the Bcl-2 promoter. We show that simvastatin increases secreted ET-1 protein levels and that exogenous ET-1 increases Bcl-2 protein levels in SH-SY5Y human neuroblastoma cells. The stimulatory effect of ET-1 on Bcl-2 protein abundance was reduced by ET-1 receptor antagonists. Simvastatin increased NFATc3 levels in the nuclear cell fraction compared with other NFATc isoforms. Binding of NFATc3 to the Bcl-2 promoter was stimulated by simvastatin treatment.
It has been previously reported that simvastatin increased ET-1 gene expression in the cerebral cortex of chronically treated mice [13], but ET-1 protein levels were not determined. The ET-1 gene encodes for the ET-1 precursor protein with eventual formation of mature ET-1 protein involving cleavage by endothelin-converting enzymes [24]. We now demonstrate that simvastatin at a relatively low concentration (100 nM), which previously was shown to be neuroprotective and stimulated Bcl-2 gene expression and protein levels in mouse primary neurons and SH-SY5Y cells [11], significantly increased ET-1 protein abundance (Fig. 1). These results differ with a study in astrocytes in which a high simvastatin concentration (10 μM) down-regulated HIV-Tat-induced ET-1 gene expression and ET-1 protein levels [25]. Statins decreased ET-1 mRNA and ET-1 protein levels in endothelial cells [26, 27]. However, in those studies, statin concentrations (1–10 μM) were several orders of magnitude higher as compared with the present study (100 nM). There is evidence that high concentrations of statins can induce apoptosis and cell death as reviewed in reference [28].
We are proposing that the simvastatin induction of ET-1 protein levels increases Bcl-2, which involves the transcription factor NFATc3 binding to the Bcl-2 promoter. This hypothesis is based on the following lines of evidence. Exogenous ET-1 significantly increased Bcl-2 protein levels in neuroblastoma cells, and this effect was attenuated in the presence of ETA/B receptor antagonists (Fig. 2). These findings are in agreement with an earlier study [15] in cardiomyocytes using the same ET-1 concentration as used in the present study. In the study using cardiomyocytes, it also was found that incubation of cells with ET-1 reduced apoptosis induced by hydrogen peroxide, which could possibly be due to up-regulation of Bcl-2. In ovarian cancer cells, ET-1 was effective in inhibiting apoptosis through the ETA receptor, triggering an antiapoptotic effect that was Bcl-2 dependent and mediated through an AKT pathway [29]. An increase in ET-1 protein has been previously proposed to initiate activation of a calcium phosphatase resulting in dephosphorylation of an NFATc isoform, causing its translocation to the nucleus and binding to the Bcl-2 promoter that has multiple NFATc consensus sequences [16]. We now show that incubation of cells with simvastatin increases NFATc3 abundance in the nucleus. Moreover, simvastatin treatment increased NFATc3 binding to the Bcl-2 promoter (Fig. 6).
A novel observation was that effects of simvastatin on enrichment of NFATc in the nucleus were isoform dependent. Treatment of cells with simvastatin increased NFATc3 abundance in the nucleus but reduced levels of NFATc1 and had no effect on NFATc4 (Fig. 4). Another study found that ET-1 increased NFATc3 in the nucleus of vascular cells [23]. The NFATc family of transcription factors all share the same unique ability among vertebrate transcription factors to translocate into the nucleus in response to an increase in Ca2+/calcineurin activity [30]. Trafficking of NFATc isoforms across the nucleus is dependent on phosphorylation state and various nuclear receptors and kinases. Nuclear factor of activated T-lymphocytes isoforms all contain a conserved PXIXIT and LxVP (also called CnBP-B/CNBR2) sequence motif within the N terminus serving as the main docking site for calcineurin [31]. Binding affinity, however, differs among NFATc isoforms. NFATc1 binds calcineurin in vitro more efficiently than does NFATc2. NFATc3 could have a higher binding affinity for calcineurin compared with other brain NFAT isoforms. The distribution of NFATc protein members between the nucleus and cytosol is dynamically regulated by nuclear kinases [30]. Reducing, for example, GSK-3β activity by lithium, an inhibitor of that kinase, increases the accumulation of NFATc4 and reduces apoptosis in neurons [19]. Effects of lithium on neuroprotection have been suggested to involve a Bcl-2-dependent mechanism [32, 33]. Pretreatment of PC12 cells with lithium increased Bcl-2 abundance and attenuated cell death induced by the Aβ fragment 25 to 35 [34].
NFATc proteins typically cannot bind DNA alone due to their weak binding to DNA and require a cotranscription binding protein, collectively termed NFATn [30]. NFATn proteins are a diverse group of proteins and include, for example, AP-1, GATA, cMAF, and MEF2 family members [35]. The most well-known NFATc cotranscription factor is the AP-1 complex of c-Fos and c-Jun that is associated with NFATc1 [30]. Microarray data from our laboratory [13] showed that mice chronically treated with simvastatin had reduced c-Fos expression, and this is consistent with the finding of less NFATc1 in the nuclear fraction of simvastatin-treated cells (Fig. 4). Given the importance of these cotranscription factors in the function of NFATc family members, identification of NFATc3 cotranscriptional factors would expand understanding of how simvastatin is increasing Bcl-2.
Simvastatin up-regulates gene expression and protein levels of the major antiapoptotic protein Bcl-2 in vivo and in vitro, which was independent of the mevalonate/isoprenoid/cholesterol pathway [11, 13, 14]. Here we provide novel results showing that up-regulation of Bcl-2 by simvastatin involves ET-1 and the transcription factor NFATc3. Notable is that simvastatin had a specific stimulatory effect on NFATc3 translocation to the nucleus as compared with NFATc1 and NFATc4. Discovering how statins can selectively alter a specific NFATc isoform that leads to an increase in Bcl-2 provides a new approach to understanding neuroprotection provided by this class of drugs as well as conditions outside the brain in which apoptosis contributes to cell dysfunction and death.
Acknowledgments
This work was supported by the National Institutes of Health, National Institute on Aging (grants AG23524, AG18357), and the Department of Veterans Affairs.
Contributor Information
Tammy A. Butterick, Department of Pharmacology, Geriatric Research Education and Clinical Center, VA Medical Center, University of Minnesota, Minneapolis, MN, USA
Urule Igbavboa, Department of Pharmacology, Geriatric Research Education and Clinical Center, VA Medical Center, University of Minnesota, Minneapolis, MN, USA.
Gunter P. Eckert, Department of Pharmacology, BiocenterNiederursel, Goethe University, Max-von-Laue-Str. 9, 60438 Frankfurt, Germany
Grace Y. Sun, Department of Biochemistry, Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, Canada
Gary A. Weisman, Department of Biochemistry, Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, Canada
Walter E. Müller, Department of Pharmacology, BiocenterNiederursel, Goethe University, Max-von-Laue-Str. 9, 60438 Frankfurt, Germany
W. Gibson Wood, Email: woodx002@umn.edu, Department of Pharmacology, Geriatric Research Education and Clinical Center, VA Medical Center, University of Minnesota, Minneapolis, MN, USA. Department of Pharmacology, University of Minnesota, 6-120 Jackson Hall, 321 Church Street, SE, Minneapolis, MN 55455, USA.
References
- 1.Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev. 2003;2:517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 2.Law MR, Wald NJ, Rudnicka AR. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta-analysis. Br Med J. 2003;326:1–7. doi: 10.1136/bmj.326.7404.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nassief A, Marsh JD. Statin therapy for stroke prevention. Stroke. 2008;39:1042–1048. doi: 10.1161/STROKEAHA.107.501361. [DOI] [PubMed] [Google Scholar]
- 4.Eckert GP, Wood WG, Müller WE. Statins: drugs for Alzheimer’s disease? J Neural Transm. 2005;112:1057–1071. doi: 10.1007/s00702-004-0273-1. [DOI] [PubMed] [Google Scholar]
- 5.Wahner AD, Bronstein JM, Bordelon YM, Ritz B. Statin use and the risk of Parkinson disease. Neurology. 2008;70:1418–1422. doi: 10.1212/01.wnl.0000286942.14552.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuhaus O, Hartung HP. Evaluation of atorvastatin and simvastatin for treatment of multiple sclerosis. Expert Rev Neurother. 2007;7:547–556. doi: 10.1586/14737175.7.5.547. [DOI] [PubMed] [Google Scholar]
- 7.Tapia-Perez JH, Sanchez-Aguilar M, Torres-Corzo JG, Gordillo-Moscoso AG, Martinez-Perez P, Madeville P, Cruz-Mendoza E, Chalita-Williams J. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758) J Neurotrauma. 2008;25:1011–1017. doi: 10.1089/neu.2008.0554. [DOI] [PubMed] [Google Scholar]
- 8.Eckert GP, Hooff GP, Strandjord DM, Igbavboa U, Volmer DA, Müller WE, Wood WG. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol Dis. 2009;35:251–257. doi: 10.1016/j.nbd.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tringali G, Vairano M, Dello Russo C, Preziosi P, Navarra P. Lovastatin and mevastatin reduce basal and cytokine-stimulated production of prostaglandins from rat microglial cells in vitro: evidence for a mechanism unrelated to the inhibition of hydroxy-methyl-glutaryl CoA reductase. Neurosci Lett. 2004;354:107–110. doi: 10.1016/j.neulet.2003.09.066. [DOI] [PubMed] [Google Scholar]
- 10.Bösel J, Gandor F, Harms C, Synowitz M, Harms U, Djoufack P, Megow D, Dirnagl U, Hörtnagl H, Finks KB, Endres M. Neuroprotective effects of atorvastatin against glutamate-induced excitotoxicity in primary cortical neurones. J Neurochem. 2005;92:1386–1398. doi: 10.1111/j.1471-4159.2004.02980.x. [DOI] [PubMed] [Google Scholar]
- 11.Johnson-Anuna LN, Eckert GP, Franke C, Igbavboa U, Müller WE, Wood WG. Simvastatin protects neurons from cytotoxicity by up-regulating Bcl-2 mRNA and protein. J Neurochem. 2007;101:77–86. doi: 10.1111/j.1471-4159.2006.04375.x. [DOI] [PubMed] [Google Scholar]
- 12.Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- 13.Johnson-Anuna LN, Eckert GP, Keller JH, Igbavboa U, Franke C, Fechner T, Schubert-Zsilavecz M, Karas M, Müller WE, Wood WG. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J Pharmacol Exp Ther. 2005;312:786–793. doi: 10.1124/jpet.104.075028. [DOI] [PubMed] [Google Scholar]
- 14.Franke C, Nöldner M, Abdel-Kader R, Johnson-Anuna LN, Wood WG, Müller WE, Eckert GP. Bcl-2 upregulation and neuroprotection in guinea pig brain following chronic simvastatin treatment. Neurobiol Dis. 2007;25:438–445. doi: 10.1016/j.nbd.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Kakita T, Hasegawa K, Iwai-Kanai E, Adachi S, Morimoto T, Wada H, Kawamura T, Yanazume T, Sasayama S. Calcineurin pathway is required for endothelin-1-mediated protection against oxidant stress-induced apoptosis in cardiac myocytes. Circ Res. 2001;88:1239–1246. doi: 10.1161/hh1201.091794. [DOI] [PubMed] [Google Scholar]
- 16.Kawamura T, Ono K, Morimoto T, Akao M, Iwai-Kanai E, Wada H, Sowa N, Kita T, Hasegawa K. Endothelin-1-dependent nuclear factor of activated T lymphocyte signaling associates with transcriptional coactivator p300 in the activation of the B cell leukemia-2 promoter in cardiac myocytes. Circ Res. 2004;94:1492–1499. doi: 10.1161/01.RES.0000129701.14494.52. [DOI] [PubMed] [Google Scholar]
- 17.Groth RD, Mermelstein PG. NFAT-dependent gene expression in the nervous system: a critical mediator of neurotrophin-induced plasticity. In: Dudek SM, editor. Transcriptional regulation by neuronal activity. Springer; New York: 2008. pp. 187–208. [Google Scholar]
- 18.Schwartz N, Schohl A, Ruthazer ES. Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron. 2009;62:655–669. doi: 10.1016/j.neuron.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, Bonni A. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem. 2005;280:2818–2825. doi: 10.1074/jbc.M408741200. [DOI] [PubMed] [Google Scholar]
- 20.Luoma JI, Zirpel L. Deafferentation-induced activation of NFAT (nuclear factor of activated T-cells) in cochlear nucleus neurons during a developmental critical period: a role for NFATc4-dependent apoptosis in the CNS. J Neurosci. 2008;28:3159–3169. doi: 10.1523/JNEUROSCI.5227-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hasselblatt M, Kamrowski-Kruck H, Jensen N, Schilling L, Kratzin H, Sirén AL, Ehrenreich H. ETA and ETB antagonists synergistically increase extracellular endothelin-1 levels in primary rat astrocyte cultures. Brain Res. 1998;785:253–261. doi: 10.1016/s0006-8993(97)01368-1. [DOI] [PubMed] [Google Scholar]
- 22.Oukka M, Ho IC, de la Brousse FC, Hoey T, Grusby MJ, Glimcher LH. The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity. 1998;9:295–304. doi: 10.1016/s1074-7613(00)80612-3. [DOI] [PubMed] [Google Scholar]
- 23.Nilsson LM, Sun ZW, Nilsson J, Nordström I, Chen YW, Molkentin JD, Wide-Swensson D, Hellstand P, Lydrup ML, Gomez MF. Novel blocker of NFAT activation inhibits IL-6 production in human myometrial arteries and reduces vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C1167–C1178. doi: 10.1152/ajpcell.00590.2005. [DOI] [PubMed] [Google Scholar]
- 24.Haynes WG, Webb DJ. Endothelin as a regulator of cardiovascular function in health and disease. J Hypertens. 1998;16:1081–1098. doi: 10.1097/00004872-199816080-00001. [DOI] [PubMed] [Google Scholar]
- 25.Chauhan A, Hahn S, Gartner S, Pardo CA, Netesan SK, McArthur J, Nath A. Molecular programming of endothelin-1 in HIV-infected brain: role of TAT in up-regulation of ET-1 and its inhibition by statins. FASEB J. 2007;21:777–789. doi: 10.1096/fj.06-7054com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, Sanchez-Pascuala R, Hernandez G, Diaz C, Lamas S. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998;101:2711–2719. doi: 10.1172/JCI1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohkita M, Sugii M, Ka Y, Kitamura A, Mori T, Hayashi T, Takaoka M, Matsumura Y. Differential effects of different statins on endothelin-1 gene expression and endothelial NOS phosphorylation in porcine aortic endothelial cells. Exp Biol Med. 2006;231:772–776. [PubMed] [Google Scholar]
- 28.Wood WG, Eckert GP, Igbavboa U, Müller WE. Statins and neuroprotection: a prescription to move the field forward. Ann NY Acad Sci. 2010 doi: 10.1111/j.1749-6632.2009.05359.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vacca F, Bagnato A, Catt KJ, Tecce R. Transactivation of the epidermal growth factor receptor in endothelin-1-induced mitogenic signaling in human ovarian carcinoma cells. Cancer Res. 2000;60:5310–5317. [PubMed] [Google Scholar]
- 30.Wu H, Peisley A, Graef IA, Crabtree GR. NFAT signaling and the invention of vertebrates. Trends Cell Biol. 2007;17:251–260. doi: 10.1016/j.tcb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 32.Ghribi O, Herman MM, Spaulding NK, Savory J. Lithium inhibits aluminum-induced apoptosis in rabbit hippocampus, by preventing cytochrome c translocation, Bcl-2 decrease, Bax elevation and caspase-3 activation. J Neurochem. 2002;82:137–145. doi: 10.1046/j.1471-4159.2002.00957.x. [DOI] [PubMed] [Google Scholar]
- 33.Youdim MB, Arraf Z. Prevention of MPTP (N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine) dopaminergic neurotoxicity in mice by chronic lithium: involvement of Bcl-2 and Bax. Neuropharmacology. 2004;46:1130–1140. doi: 10.1016/j.neuropharm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Wei H, Leeds PR, Qian Y, Wei W, Chen R, Chuang D. Beta-amyloid peptide-induced death of PC12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur J Pharmacol. 2000;392:117–123. doi: 10.1016/s0014-2999(00)00127-8. [DOI] [PubMed] [Google Scholar]
- 35.Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev. 2001;11:505–512. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]