

Abstract
Formation of the integrin αβ heterodimer is essential for cell surface expression and function. At the core of the αβ interface is a conserved Arg/Lys “finger” from the β-subunit that inserts into a cup-like “cage” formed of two layers of aromatic residues in the α-subunit. We evaluated the role of this residue in heterodimer formation in an αA-lacking and an αA-containing integrin αVβ3 and αMβ2 (CD11b/CD18), respectively. Arg261 of β3 was mutated to Ala or Glu; the corresponding Lys252 of β2 was mutated to Ala, Arg, Glu, Asp, or Phe; and the effects on heterodimer formation in each integrin examined by ELISA and immunoprecipitation in HEK 293 cells cotransfected with plasmids encoding the α- and β-subunits. The Arg261Glu (but not Arg261Ala) substitution significantly impaired cell surface expression and heterodimer formation of αVβ3. Although Lys252Arg, and to a lesser extent Lys252Ala, were well tolerated, each of the remaining substitutions markedly reduced cell surface expression and heterodimer formation of CD11b/CD18. Lys252Arg and Lys252Ala integrin heterodimers displayed a significant increase in binding to the physiologic ligand iC3b. These data demonstrate an important role of the Arg/Lys finger in formation of a stable integrin heterodimer, and suggest that subtle changes at this residue affect the activation state of the integrin.
Integrins are αβ heterodimeric cell surface receptors, which mediate cell-matrix and cell-cell adhesion functions that underlie vital cellular processes such as differentiation, migration, proliferation, and survival (1). Integrins also contribute to many pathologies, including cardiovascular disease, cancer invasion, immune dysfunction, and osteoporosis, thus constituting important therapeutic targets. Normally expressed in an inactive state, integrins switch rapidly to the physiologic ligand-binding (active) state in response to chemical signals generated from within cells (inside-out signaling) (reviewed in Ref. 2). Physiologic ligand binding then transmits conformational signals back to the cytoplasmic tails, thus activating intracellular signal transduction pathways (outside-in signaling). This bidirectional signaling involves conformational changes the nature of which is incompletely understood.
Crystal structure of αVβ3 ectodomain revealed a globular “head” (Fig. 1A) sitting on top of two nearly parallel “legs” (3). The integrin head comprises a seven-bladed β-propeller from the α-subunit and a von Willebrand factor type A (VWFA)4 domain (βA or I-like) from the β-subunit (Fig. 1A). βA is inserted into an Ig-like “hybrid” domain that is inserted in turn into an N-terminal plexin/semaphorin/integrin domain (3, 4). Four epidermal growth factor-like domains followed by a novel β-tail domain form the rest of the β-subunit leg. The α-subunit leg is formed of an Ig-like “thigh” domain followed by two large β-sandwich domains “calf 1” and “calf 2”. The α- and β-subunit legs fold back at a ~135° angle, forming a V-shaped structure, bent at its knees “genu” (between the thigh domain and calf 1 domains of the α-subunit) and EGF-1 and 2 of the β-subunit. The leg from each subunit terminates into a rod-shaped single membrane-spanning segment and a short cytoplasmic tail. The ligand-binding site in integrins is localized on the upper side of the integrin head, formed by a metal-ion-dependent adhesion site (MIDAS) in βA and an adjacent region in the α-subunit’s propeller domain. In some integrins (αA-integrins), the α-subunit contains an extra VWFA domain (αA or I) that acts as a ligand relay, capturing exogenous ligands through its own MIDAS and engaging the βA MIDAS as an endogenous ligand (5).
FIGURE 1.
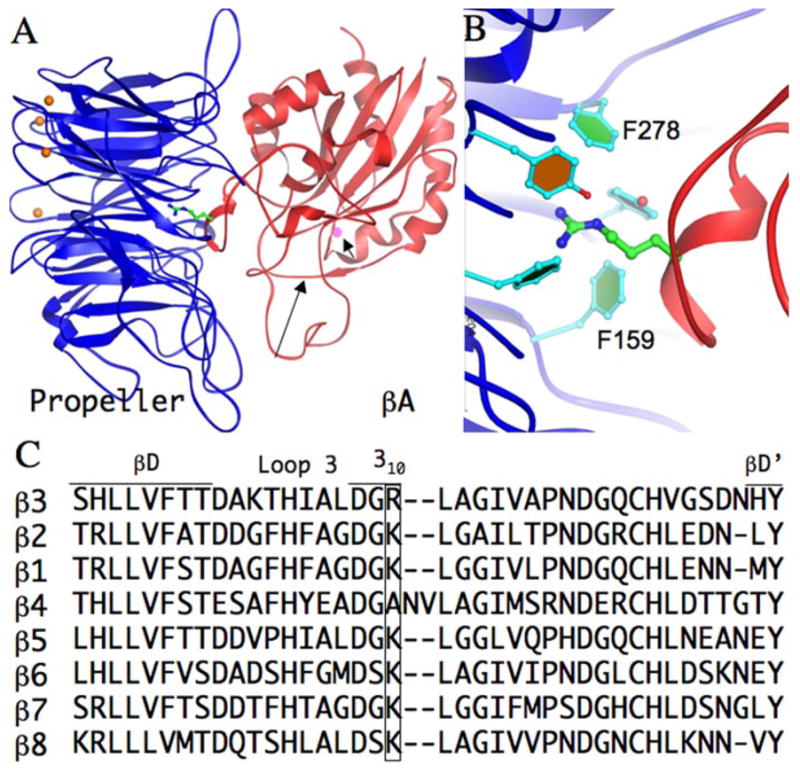
Structure and conservation of the Arg/Lys finger in integrin β-subunits. A, Three-dimensional crystal structure of unliganded integrin αVβ3 head segment (as a ribbon diagram with the αV propeller domain in blue and the βA domain in red) showing the position of the arginine finger (Arg261, carbon atoms in green, amides in blue) projecting from the 310 helix. Loop 3 (long arrow) and the ADMIDAS cation (magenta sphere, short arrow) are indicated. The four metal ions at the base of the propeller are indicated as orange spheres. B, Higher magnification of the Arg/Lys coordination pocket, with a portion of the aromatic side chains forming the upper ring of the cup-like “cage” structure of αV propeller (adapted from Refs. 7 and 33). Orientation is the same as in A. C, Structure alignment of all eight mammalian β-subunits around the Arg/Lys finger (boxed). The amino acid sequence of strands βD and βD′, loop 3 and the 310 helix are indicated in single letter code. The β4-subunit structure model was generated using Modeler (34) with the αVβ3 structure (PDB code: 1jv2) as template.
The largely hydrophobic propeller/βA interface buries a surface area of ~1,600 Å (2), and is responsible for formation of the integrin heterodimer. It bears a striking structural resemblance to the interface between Gβ- (a seven-bladed β-propeller) and Gα (a VWFA domain) subunits of G-proteins (6). At the core of the propeller/βA interface in integrins lies a basic residue (Arg261 in β3, Lys in all other β-subunits except β4) within a 310-helix; its side chain “finger” inserts into a cup-like structure lined by aromatic side chains in the central cavity of the propeller, and stabilized by hydrophobic and cation-π contacts (7) (Fig. 1, A and B). Most naturally occurring mutations that disrupt heterodimer formation in patients with β2- or β3-integrin deficiency (8–11) lie within or in the immediate vicinity of the 310 helix, but do not involve the Arg/Lys finger. A forward genetic screen for mutations in the integrin β subunit (βPS) of Drosophila melanogaster also identified mutations in the vicinity of the 310 helix, all resulting in a strong lethal phenotype (12). These observations suggest an important structural role for the Arg/Lys finger in formation of the integrin heterodimer. In this report, we investigated the role of the Arg/Lys finger in heterodimer formation in the αA-lacking and αA-containing integrins αVβ3 and CD11b/CD18, respectively.
Materials and Methods
Reagents and Abs
Restriction and modification enzymes were obtained from New England Biolabs, Life Technologies, or Fisher Scientific. All cell culture reagents were from Invitrogen Life Technologies. The anti-β3 mAb (mAb) AP3 (IgG1) (13) was from American Type Culture Collection (ATCC); the anti-αV mAb LM142 (IgG1) and the αVβ3 heterodimer-specific mAb LM609 (IgG1) (14), the anti-β3 mAb CBL479 and the anti-αV polyclonal AB1930 were from Millipore. The anti-CD11b mAbs 44a (IgG2a) (15), 903 (IgG1) (16), 107 (IgG1) (17), and a rabbit polyclonal anti-human CD18 Ab have been described previously (18).
DNA constructs
Full-length human wild-type (WT) αV and β3 were provided by Dr. Simon Goodman (Merck-Serono KGaA Darmstadt, Germany) (19). Both were recloned into the pcDNA3 expression vector using standard recombinant DNA protocols. In brief, for generating WT αV in pcDNA3, the unique HindIII site in the multiple cloning region of an empty pcDNA3 vector was removed and WT αV cDNA from the original vector was transferred into this region using EcoRI. For generating WT β3 in pcDNA3, WT β3 cDNA from the original vector was released using XhoI and XbaI restriction enzymes and transferred into the multiple cloning site in an empty pcDNA3 vector. Site-directed mutagenesis in β3- and β2-subunits (CD18) was conducted using standard recombinant DNA protocols (20) in the newly created β3 pcDNA3 and CD18 pcDNA3 expression vectors (21). PCR-amplified DNA was transferred back into the original β3- or CD18-containing expression vector and each mutation was confirmed by DNA sequencing.
Cell culture and transfection
Transient transfection assays were conducted in HEK 293 cells (ATCC) as described (5). In brief, HEK 293 cells were plated on tissue culture treated p-10 plates (BD Clontech) and maintained in complete medium consisting of DMEM (Invitrogen Life Technologies) supplemented with 10% heat-inactivated FBS (Invitrogen Life Technologies) and 50 IU/ml penicillin and streptomycin (Invitrogen Life Technologies) at 37°C. Cells were detached with 0.05% Trypsin-EDTA solution (Invitrogen Life Technologies) and plated in wells of six-well plates at a concentration of 1 million cells per well. Cells at ~70% confluence were transfected with super coiled cDNAs encoding WT or mutant β-subunits together with corresponding α-subunit, using Lipofectamine2000 reagent (Invitrogen Life Technologies), following the manufacturer’s protocol. Transfected cells were grown for 48 h at 37°C in 5% CO2. Cells were then carefully washed and detached with 10 mM EDTA in PBS, counted, and seeded in replicates onto poly-L-lysine (Sigma-Aldrich) coated 48-well plates (BD Clontech) for 2–4 h at 37°C in 5% CO2. Plates were blocked with 1% gelatin in TBS for 2 h at 37°C in 5% CO2. Confluent monolayers in the 48 wells were then used for integrin cell surface expression and ligand-binding assays.
mAb binding studies
Triplicate or quadruplet wells containing confluent monolayers of transfected cells, ~48 h posttransfection, were incubated with mAbs LM142 (10 μg/ml), AP3 (10 μg/ml), LM609 (10 μg/ml), 903 (10 μg/ml), 44a (10 μg/ml), TS1/18 (10 μg/ml), or 107 (10 μg/ml) in TBS buffer containing 0.1% gelatin and 0.02% sodium azide for 1 h at 4°C. Cells were washed and incubated with biotin-labeled rabbit anti-mouse Ab (Vectastain ABC-kit; Vector Laboratories) for 1 h at 4°C, washed three times, and subsequently fixed with 4% paraformaldehyde for 30 min at room temperature. After blocking, wells were developed using streptavidin-alkaline phosphatase (AP, Vectastain ABC-kit; Vector Laboratories) and p-nitrophenyl phosphate (Sigma-Aldrich) to generate a colored product. The enzymatic reaction was quenched with an equal volume of 2N NaOH, and OD (405) was measured. Binding to sham transfected cells conducted in parallel (which did not exceed 5% in all cases) was subtracted.
Immunoprecipitation
Transfected HEK 293 cells in six-well plates were extracted using radio-immunoprecipitation assay buffer (Boston Bioproducts) supplemented with 1 mg/ml DNase and a mixture of protease inhibitors (Complete; Roche Diagnostics). The detergent-soluble fraction was harvested upon centrifugation and immunoprecipitated using the anti-β3 mAb AP3 or the anti-CD18 mAb TS1/18 and Protein G Sepharose-4 (Amersham Pharmacia Biotech). Washed immunoprecipitates or whole cell lysates were separated by SDS-PAGE on a 4–15% gradient Tris-HCl gel (Bio-Rad) under non-reducing (for αVβ3) or reducing (for CD11b/CD18) conditions and electroblotted onto polyvinylidene difluoride membranes (Bio-Rad). After blocking with 5% nonfat milk in 25 mM Tris-HCl (pH 7.4), 137 mM NaCl, 2.7 mM KCl (TBS, Boston Bioproducts), the respective membranes were probed with polyclonal anti-αV, anti-β3 CBL479 mAb, anti-CD11b, or anti-CD18 polyclonal Abs (5). The anti-β3 mAb CBL479 was probed with a polyclonal rabbit anti-mouse Ig Ab. Detection of proteins was performed using HRP linked anti-mouse or anti-rabbit Abs (eBioscience), and SuperSignal chemiluminescent kit (Pierce). The luminescent signal was detected using BioMax x-ray films (Eastman Kodak Company). Band intensity from Western blots was quantitated using the National Institutes of Health ImageJ software (22).
Ligand binding studies
Binding of the activation-dependent physiologic ligand iC3b, and the activation-independent ligand-mimetic mAb 107 to transiently transfected HEK 293 cells expressing CD11b/CD18 was performed in 48-well plates as previously described (5). Sheep erythrocytes coated with iC3b (EiC3b) were prepared following published protocols (5, 21). EiC3b binding was assessed by adding 150 μl of EiC3b (60 × 106/ml) to triplicate confluent wells in a total volume of 300 μl followed by a 15 s spin at 500 rpm. After 35 min of incubation at 37°C, wells were carefully washed and subsequently fixed with 1% glutaraldehyde. Wells were developed using alkaline phosphatase and p-nitrophenyl phosphate as above and binding was quantitated. Specific binding was obtained by subtracting background binding of mock-transfected cells (typically <5% of total binding). Integrin cell surface expression was analyzed simultaneously using anti-CD11b 44a and anti-CD18 TS1/18 mAbs as described above. mAb binding results are reported as histograms representing mean ± SD of triplicate or quadruplet wells. To correct for the effect of differences in surface expression on ligand or ligand-mimic binding, binding of TS1/18 to the WT receptor was considered 100. In the case of EiC3b and 107, relative binding to mutant vs WT integrin was normalized to that obtained in the presence of TS1/18 as previously described (16), according to the following formula: Percent binding to ligand = (percent mutant ligand binding/percent mutant TS1/18 binding)/(percent WT ligand binding/percent WT TS1/18 binding) × 100.
Results
Design of Arg/Lys finger mutations in β3 and CD18 subunits
Sequence and structure alignments show that the Arg/Lys finger is conserved in all mammalian β-chains except β4, where it is replaced with an Ala (Fig. 1C). Conservation of the Arg/Lys finger suggests that this interfacial residue plays an important role in integrin heterodimerization and/or function. Using site-directed mutagenesis, we substituted WT β3 residue Arg261 with Glu (to disrupt the cation-π interaction observed in the αVβ3 crystal structure), and with Ala (replacing the charged residue with a hydrophobic residue). Because the structure of β2 integrin heterodimers is currently not known, we generated a larger number of mutations in the integrin CD11b/CD18 model system by substituting the WT CD18 residue Lys252 (which corresponds to Arg261 in β3) with Arg (a conservative substitution), Asp and Glu (to disrupt the cation-π interaction), Ala (replacing the charged residue with a hydrophobic residue), and Phe (introducing a large hydrophobic side chain).
Effect of β3 Arg261 mutations on cell surface expression and heterodimer formation in αVβ3
We assessed expression of the WT and mutant αVβ3 on the surface of HEK 293 cells, which lack endogenous αVβ3, but endogenously express the related αVβ1 (23). A pcDNA3 expression vector containing a cDNA encoding full-length WT αV was co-transfected with plasmids encoding WT- or mutant β3 into HEK 293 cells using Lipofectamine 2000. Mock transfectants were prepared using the empty pcDNA3 vector. Cell surface expression was measured in an ELISA-based assay, using the anti-αV mAb LM142, the anti-β3 mAb AP3 and the αVβ3 heterodimer-specific mAb LM609. The results, presented in Fig. 2A, show that the expression level of Arg261Ala mutant was comparable to WT, but the surface expression levels of the Arg261Glu mutant integrins was slightly but significantly reduced (to ~75% of WT).
FIGURE 2.
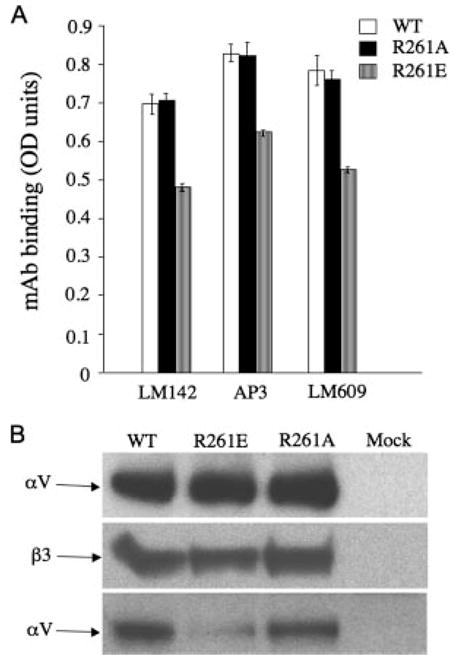
Effect of R261A and R261E on surface expression and αVβ3 heterodimer formation. In this and subsequent figures, amino acids are indicated in the single letter code. A, Histograms showing expression of recombinant WT and mutant αVβ3 on the cell surface of transfected HEK293 cells, probed with the anti-αV mAb LM142, the anti-β3 mAb AP3 or the heterodimer specific mAb LM609. Each histogram represents mean ± SD of triplicate determinations from three independent experiments. B, Western blots, following 4–15% gradient SDS-PAGE, developed with polyclonal anti-αV Ab (upper and lower,) and anti-β3 CBL479 mAb (middle). Equivalent amounts of βV- (upper) and WT or mutant β3-subunits (middle) are detected. Immunoprecipitation with anti-β3 mAb AP3 from HEK 293 cells expressing WT or mutant receptors revealed much reduced levels of the associated αV-subunit in the R261E mutant, compared with WT or R216A receptors (one representative experiment of four conducted).
To determine whether defects in heterodimer formation account for the reduction in cell surface expression, we conducted immunoprecipitation/Western blot studies on detergent-solublized cell lysates from the transiently transfected HEK 293 cells. Probing Western blots of whole lysate from HEK 293 expressing WT or mutant integrins with polyclonal anti-αV or with anti-β3 mAb CBL479 (following immunoprecipitation with anti-β3 AP3 mAbs, due to weak reactivity of this mAb in Western blots) showed that equivalent amounts of normal-sized αV and β3-subunits are present in cells transfected with WT or mutant β3 (Fig. 2B, upper and middle). Thus, the introduced substitutions did not affect the overall structure or expression levels of the respective subunit. However, immunoprecipitation of the β3 subunits from WT- or mutant-expressing cells with mAb AP3 followed by Western blotting with anti-αV polyclonal Ab showed a significant reduction in association of the αV-subunit with Arg261Glu when compared with that associated with WT-or Arg261Ala β3-subunit (Fig. 2B, lower). These findings indicate that the Arg261Glu mutation significantly impairs formation of a stable αVβ3 heterodimer.
Effect of CD18 K252 mutations on cell surface expression and heterodimer formation
To determine whether the above observations are also applicable to the αA-containing integrin subgroup, we examined whether Lys252, the equivalent residue to Arg261 in β2 integrins, is also important in formation of integrin CD11b/CD18. A pcDNA3 expression vector containing a cDNA encoding full-length human WT CD11b was cotransfected with plasmids encoding full-length human WT or mutant CD18 into HEK 293 cells (which do not endogenously express β2 integrins), using Lipofectamine 2000. Mock transfectants were prepared using the empty pcDNA3 vector. Cell surface expression was also measured by ELISA, using the anti-CD11b mAbs 903 and 44a and the anti-CD18 mAb TS1/18. The results, presented in Fig. 3A, show that the expression level of the conserved Lys252Arg substitution in CD11b/CD18 was similar to that of WT, but expression of the other mutants receptors was reduced to various degrees, ranging from ~75% of WT in Lys252Ala mutant to <10% in each of the remaining mutants.
FIGURE 3.
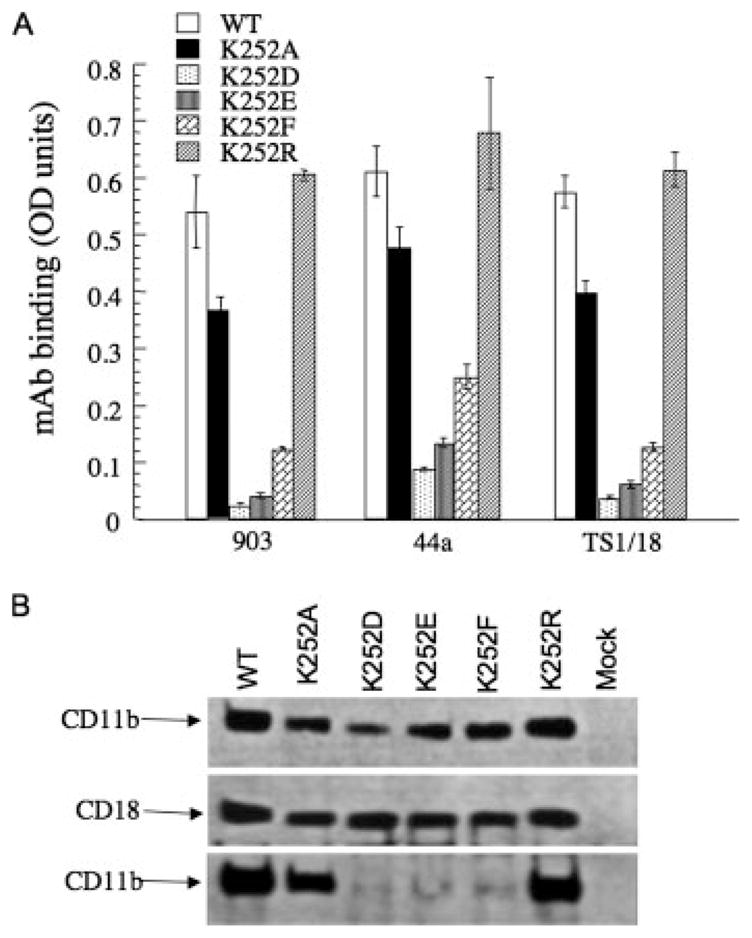
Effect of Arg/Lys mutations on surface expression and formation of integrin CD11b/CD18. A, Surface expression of WT and mutant CD11b/CD18. Histograms (mean ± SD of triplicate determinations from a representative experiment, one of three performed) showing expression levels of WT and mutant CD11b/CD18 receptors in transfected HEK 293 cells. Surface expression was evaluated with the anti-CD11b mAbs 903 and 44a and the anti-CD18 mAbs TS1/18 mAbs. B, Western blots following 4 –15% gradient SDS-PAGE developed with polyclonal anti-CD11b (upper and lower) or anti-CD18 Abs (middle) of whole cell lysate from HEK 293 cells expressing WT or mutant receptors (upper and middle) or anti-CD18 mAb immunoprecipitates probed with polyclonal anti-CD11b Ab (lower) (one representative experiment is shown of three conducted).
Immunoprecipitation/Western blot analyses were then used to evaluate the effect of each mutation on formation of the heterodimer more directly, using detergent-extracted cell lysates from transfected HEK 293 as described (5). Detergent extracts from HEK 293 cells transfected with mutant CD18 (together with WT CD11b) expressed a normal-sized CD11b-subunit, as expected (Fig. 3B, upper), as well as equivalent amounts of normal-sized WT and mutant CD18-subunit (Fig. 3B, middle), indicating that the introduced substitutions did not affect the overall structure or level of mutant CD18. Immunoprecipitation of the CD18 subunit with TS1/18 mAb followed by Western blotting with polyclonal anti-CD11b showed that both Lys252Ala and Lys252Arg mutant CD18 associated with WT CD11b to equivalent degrees as that of WT CD18 (Fig. 3B, lower). However, association of Lys252Asp, Lys252Glu, and Lys252Phe mutant CD18 with WT CD11b was minimal, consistent with the cell surface expression analysis. Thus, charge reversal at the conserved Arg/Lys finger (as in Lys252Asp and Lys252Glu mutants) or introduction of a bulky hydrophobic residue (as in the Lys252Phe mutant) largely abrogates association of the CD11b and the CD18 subunits, whereas the conservative substitution Lys252Arg and the least sterically disruptive substitution Lys252Ala are well tolerated.
Impact of Lys252Ala and Lys252Arg substitutions in CD18 on integrin function
The Arg/Lys finger is just two residues downstream of loop 3 (Fig. 1C), one of the three loops that form the ligand binding MIDAS of the βA domain (1). This structural arrangement is analogous to the activating “switch II” region in G proteins, which is similarly linked to the metal ion binding catalytic site in Gα (1). This structural analogy suggests that the packing of the βA domain against the propeller may also affect integrin activation. We examined the impact of the Lys to Arg or to Ala substitutions on the ability of recombinant CD11b/CD18 heterodimer to bind the physiologic ligand iC3b (16, 24). WT CD11b/CD18 expressed on HEK 293 is partially activated, enabling it to bind the activation-sensitive ligand iC3b in the presence of physiologic divalent cations (Ca2+ plus Mg2+, each at 1 mM) (25). This explains the intrinsic binding of WT CD11b/CD18-expressing HEK 293 to EiC3b (Fig. 4). Lys252Ala and Lys252Arg substitutions in integrin CD11b/CD18 induced a further increase in binding to iC3b by 1.4 and 1.3-fold, respectively, above that of the WT receptor. In contrast, binding of the activation-insensitive ligand-mimetic mAb 107 (17) was similar in both mutant receptors to that of WT (Fig. 4).
FIGURE 4.
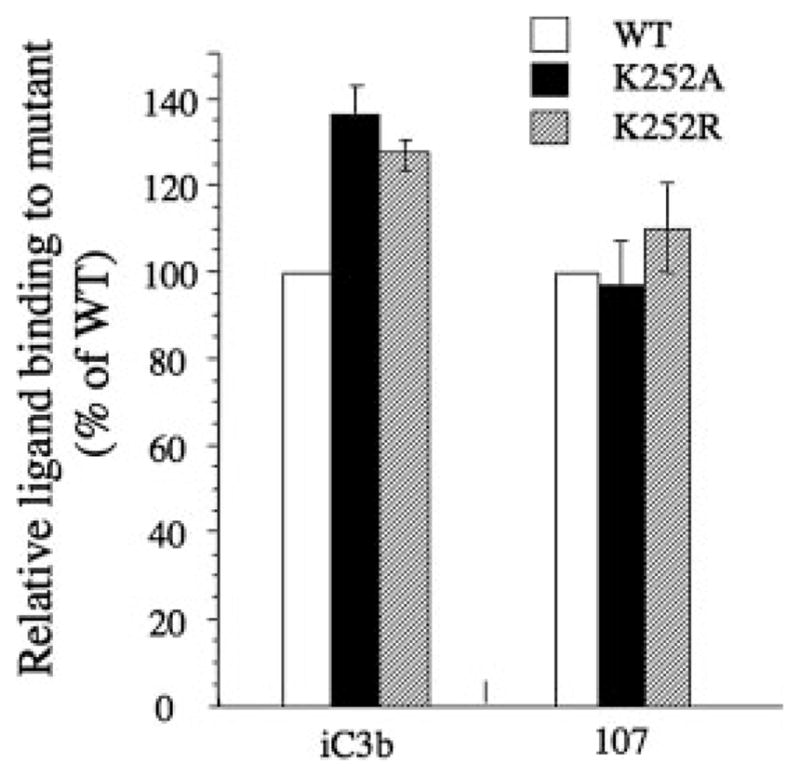
Effect of K252A and K252R on ligand binding. Left, Histograms showing the binding of the physiologic ligand iC3b or the ligand mimic 107 to mutant CD11b/CD18 relative to that of WT (placed at 100), after correcting for receptor expression using mAb TS1/18. Each histogram represents mean ± SD of triplicate (for iC3b) or quadruplet (for 107) determinations from a representative experiment (of three performed). Details of the methods used are described in Materials and Methods.
Discussion
The results presented in this communication validate the model that the Arg/Lys finger plays a important role in formation of a stable integrin heterodimer, and suggest that as in G-proteins, the βA/propeller interface may also affect integrin activation.
Charge reversal of the Arg/Lys finger led to a reduction in surface expression of both the αA-lacking and containing integrins αVβ3 and CD11b/CD18, respectively. Cell surface expression of αVβ3 was less affected by the Arg261Glu substitution in β3 than CD11b/CD18 was by the Lys252Glu in β2 (compare Figs. 2A and 3A), although the detrimental effect of either substitution on stability of the respective heterodimer was comparable in the immunoprecipitation analysis (compare lower in Figs. 2B and 3B). These findings suggest that cell surface expression is a less sensitive measure of integrin heterodimer stability. A similar observation was made in analysis of integrin β3 L262P mutant (11), which was also expressed on the surface of HEK 293 cells, but its effect on heterodimer stability was readily detected under the more stringent immunoprecipitation analysis. These findings may also reflect a generally lower affinity of the αA-containing CD11b subunit to CD18 in the WT integrin than that of αV-subunit for β3. It remains possible that HEK 293 cells, which constitutively express the related αVβ1, may possess the cellular machinery to efficiently package and export the mutant αVβ3 to the cell surface, but not mutant CD11b/CD18 heterodimers that are normally expressed exclusively in a leukocyte context.
The β4-subunit, which pairs only with α6 to form a functional heterodimer, is the only integrin β-subunit lacking the Arg/Lys finger (Fig. 1C). This finger is replaced with an alanine, which is followed by an Asn-Val insertion unique to this subunit. Although there are caveats associated with homology-based modeling, we find that the size and properties of the interface between Ala197AsnVal in β4 and the aromatic cup of α6 is similar to that at the αV/β3 interface, though the main interaction is now mediated by van der Waals’ forces. However, the dipeptide insertion may create a slightly larger interfacial contact with α6, thereby mitigating the reduction in ΔG from the missing cation-π interaction.
Substitution of K252 with Arg or with Ala in the CD11b/CD18 integrin resulted in a significant increase in binding to physiologic ligand by the recombinant integrin (Fig. 4). This increase could result from changes in integrin affinity and/or avidity. Recent evidence suggests that the increase in ligand-binding induced by inside-out signaling in the β2-integrin CD11a is primarily due to affinity modulation rather than avidity changes (26). The proximity of the Arg/Lys finger to the ligand-binding loop 3 of MIDAS, favors an intramolecular (affinity) rather than an intermolecular (clustering/avidity) effect. In contrast to G-proteins, where conformational activation results in complete dissociation of the Gα- and Gβ-subunits, enhanced ligand binding by these two substitutions was elicited with little or no detectable disruption of the integrin heterodimer (Figs. 2B and 3B). These findings suggest that integrin-ligand interactions may be regulated by more subtle allosteric changes at the propeller/βA interface the nature of which remain to be structurally-defined.
The present results also provide new insights into how some naturally-occurring mutations, which occur most frequently in the βA domains of CD18- (27) and β3- (28) integrin subunits, lead to life-threatening bacterial infections and thrombasthenia, respectively. For example, the naturally occurring mutations G251R in CD18 (equivalent to G260 in β3) (29) and L262P in β3 (equivalent to L253 in CD18) (11), known to prevent formation of integrin heterodimers, lie immediately after the critical Arg/Lys finger residue in the 310-helix of the respective integrin. Data presented in this study suggest that these mutations act in part by disrupting the contact of the Arg/Lys finger with the cup-like structure of the propeller. If this interpretation is correct, then mutations in the aromatic cup residues of the propeller domain should likewise impair heterodimer formation. This has recently been borne out: a naturally-occurring Phe171Cys mutation in αIIb (equivalent to Phe159 in αV) (Fig. 1B) found in patients with inherited thrombasthenia, impairs formation of the αIIbβ3 heterodimer and its surface expression in platelets (30). The αVβ3 crystal structure shows that Arg261 is closest to Phe159 of the α-subunit, and contributes the primary cation-π bond; its replacement with cysteine is expected to destabilize the Arg/Lys finger contact with the cup structure. Similarly, a mutation of another aromatic cup residue Phe289 in αIIb (corresponding to the conserved Phe278 in αV) also leads to loss of surface expression and heterodimer formation (31). Other naturally occurring mutations in the cup structure (32) may likewise compromise stable contact with the Arg/Lys finger and hence integrin heterodimer formation.
Acknowledgments
We thank Dora Gaudette for secretarial assistance and Amir H. Qureshi for technical assistance.
Footnotes
This work was supported in part by Grants DK48549, DK50305, and DK068253 from the National Institutes of Health.
Abbreviations used in this paper: VWFA, von Willebrand factor type A; MIDAS, metal-ion-dependent adhesion site; WT, wild type.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol. 2005;21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- 2.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 3.Xiong JP, Stehle T, Goodman SL, Arnaout MA. A novel adaptation of the integrin PSI domain revealed from its crystal structure. J Biol Chem. 2004;279:40252–40254. doi: 10.1074/jbc.C400362200. [DOI] [PubMed] [Google Scholar]
- 4.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alonso JL, Essafi M, Xiong JP, Stehle T, Arnaout MA. Does the integrin αA domain act as a ligand for its βA domain? Curr Biol. 2002;12:R340–R342. doi: 10.1016/s0960-9822(02)00852-7. [DOI] [PubMed] [Google Scholar]
- 6.Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Gi α 1 β 1 γ 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 7.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin α Vβ3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnaout MA, Dana N, Gupta SK, Tenen DG, Fathallah DM. Point mutations impairing cell surface expression of the common β subunit (CD18) in a patient with leukocyte adhesion molecule (Leu-CAM) deficiency. J Clin Invest. 1990;85:977–981. doi: 10.1172/JCI114529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.French DL, Coller BS. Hematologically important mutations: Glanzmann thrombasthenia. Blood Cells Mol Dis. 1997;23:39–51. doi: 10.1006/bcmd.1997.0117. [DOI] [PubMed] [Google Scholar]
- 10.Jackson DE, White MM, Jennings LK, Newman PJ. A Ser162–>Leu mutation within glycoprotein (GP) IIIa (integrin β3) results in an unstable αIIbβ3 complex that retains partial function in a novel form of type II Glanzmann thrombasthenia. Thromb Haemostasis. 1998;80:42–48. [PubMed] [Google Scholar]
- 11.Ward CM, Kestin AS, Newman PJ. A Leu262Pro mutation in the integrin β (3) subunit results in an α(IIb)- β(3) complex that binds fibrin but not fibrinogen. Blood. 2000;96:161–169. [PubMed] [Google Scholar]
- 12.Jannuzi AL, Bunch TA, West RF, Brower DL. Identification of integrin β subunit mutations that alter heterodimer function in situ. Mol Biol Cell. 2004;15:3829–3840. doi: 10.1091/mbc.E04-02-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman PJ, Allen RW, Kahn RA, Kunicki TJ. Quantitation of membrane glycoprotein IIIa on intact human platelets using the monoclonal antibody, AP-3. Blood. 1985;65:227–232. [PubMed] [Google Scholar]
- 14.Cheresh DA, Harper JR. Arg-Gly-Asp recognition by a cell adhesion receptor requires its 130-kDa α subunit. J Biol Chem. 1987;262:1434–1437. [PubMed] [Google Scholar]
- 15.Arnaout MA, Todd RF, III, Dana N, Melamed J, Schlossman SF, Colten HR. Inhibition of phagocytosis of complement C3- or immunoglobulin G-coated particles and of C3bi binding by monoclonal antibodies to a monocyte-granulocyte membrane glycoprotein (Mol) J Clin Invest. 1983;72:171–179. doi: 10.1172/JCI110955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Rieu P, Griffith DL, Scott D, Arnaout MA. Two functional states of the CD11b A-domain: correlations with key features of two Mn2+-complexed crystal structures. J Cell Biol. 1998;143:1523–1534. doi: 10.1083/jcb.143.6.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li R, Haruta I, Rieu P, Sugimori T, Xiong JP, Arnaout MA. Characterization of a conformationally sensitive murine monoclonal antibody directed to the metal ion-dependent adhesion site face of integrin CD11b. J Immunol. 2002;168:1219–1225. doi: 10.4049/jimmunol.168.3.1219. [DOI] [PubMed] [Google Scholar]
- 18.Dana N, Clayton LK, Tennen DG, Pierce MW, Lachmann PJ, Law SA, Arnaout MA. Leukocytes from four patients with complete or partial Leu-CAM deficiency contain the common β-subunit precursor and β-subunit messenger RNA. J Clin Invest. 1987;79:1010–1015. doi: 10.1172/JCI112868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehta RJ, Diefenbach B, Brown A, Cullen E, Jonczyk A, Gussow D, Luckenbach GA, Goodman SL. Transmembrane-truncated αvβ3 integrin retains high affinity for ligand binding: evidence for an ‘inside-out’ suppressor? Biochem J. 1998;330:861–869. doi: 10.1042/bj3300861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- 21.Li R, Arnaout MA. Functional analysis of the β 2 integrins. Methods Mol Biol. 1999;129:105–124. doi: 10.1385/1-59259-249-X:105. [DOI] [PubMed] [Google Scholar]
- 22.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 23.Bodary SC, McLean JW. The integrin β 1 subunit associates with the vitronectin receptor α v subunit to form a novel vitronectin receptor in a human embryonic kidney cell line. J Biol Chem. 1990;265:5938–5941. [PubMed] [Google Scholar]
- 24.Xiong JP, Li R, Essafi M, Stehle T, Arnaout MA. An isoleucine-based allosteric switch controls affinity and shape shifting in integrin CD11b A-domain. J Biol Chem. 2000;275:38762–38767. doi: 10.1074/jbc.C000563200. [DOI] [PubMed] [Google Scholar]
- 25.Shimaoka M, Shifman JM, Jing H, Takagi J, Mayo SL, Springer TA. Computational design of an integrin I domain stabilized in the open high affinity conformation. Nat Struct Biol. 2000;7:674–678. doi: 10.1038/77978. [DOI] [PubMed] [Google Scholar]
- 26.Beals CR, Edwards AC, Gottschalk RJ, Kuijpers TW, Staunton DE. CD18 activation epitopes induced by leukocyte activation. J Immunol. 2001;167:6113–6122. doi: 10.4049/jimmunol.167.11.6113. [DOI] [PubMed] [Google Scholar]
- 27.Arnaout MA, Michishita M. Genetic abnormalities in leukocyte adhesion molecule deficiency. In: Gupta S, Griscelli C, editors. New Concepts in Immunodeficiency Diseases. John Wiley & Sons; Chichester, U.K: 1993. pp. 191–202. [Google Scholar]
- 28.French DL. The molecular genetics of Glanzmann’s thrombasthenia. Platelets. 1998;9:5–20. doi: 10.1080/09537109876951. [DOI] [PubMed] [Google Scholar]
- 29.Hogg N, Stewart MP, Scarth SL, Newton R, Shaw JM, Law SK, Klein N. A novel leukocyte adhesion deficiency caused by expressed but nonfunctional β2 integrins Mac-1 and LFA-1. J Clin Invest. 1999;103:97–106. doi: 10.1172/JCI3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg N, Landau M, Luboshitz J, Rechavi G, Seligsohn U. A novel Phe171Cys mutation in integrin α causes Glanzmann thrombasthenia by abrogating αβ complex formation. J Thromb Haemostasis. 2004;2:1167–1175. doi: 10.1111/j.1538-7836.2004.00758.x. [DOI] [PubMed] [Google Scholar]
- 31.Ambo H, Kamata T, Handa M, Kawai Y, Oda A, Murata M, Takada Y, Ikeda Y. Novel point mutations in the αIIb subunit (Phe289–>Ser, Glu324–>Lys and Gln747–>Pro) causing thrombasthenic phenotypes in four Japanese patients. Br J Haematol. 1998;102:829–840. doi: 10.1046/j.1365-2141.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- 32.Nelson EJ, Li J, Mitchell WB, Chandy M, Srivastava A, Coller BS. Three novel β-propeller mutations causing Glanzmann thrombasthenia result in production of normally stable pro-αIIb, but variably impaired progression of pro-αIIbβ3 from endoplasmic reticulum to Golgi. J Thromb Haemostasis. 2005;3:2773–2783. doi: 10.1111/j.1538-7836.2005.01593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin α Vβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 34.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]