

Abstract
The balance between proliferation and apoptosis is critical for proper development of the nervous system. Yet, little is known about molecules that regulate apoptosis of proliferative neurons. Here we identify a soluble, secreted form of CPG15 expressed in embryonic rat brain regions undergoing rapid proliferation and apoptosis, and show that it protects cultured cortical neurons from apoptosis by preventing activation of caspase 3. Using a lentivirus-delivered small hairpin RNA, we demonstrate that endogenous CPG15 is essential for the survival of undifferentiated cortical progenitors in vitro and in vivo. We further show that CPG15 overexpression in vivo expands the progenitor pool by preventing apoptosis, resulting in an enlarged, indented cortical plate and cellular heterotopias within the ventricular zone, similar to the phenotypes of mutant mice with supernumerary forebrain progenitors. CPG15 expressed during mammalian forebrain morphogenesis may help balance neuronal number by countering apoptosis in specific neuroblasts subpopulations, thus influencing final brain size and shape.
During mammalian evolution, the cerebral cortex has greatly expanded through a tremendous increase in the number of cortical neurons. The surface of the cortical plate has extended and become indented and convoluted as a result of neuron addition in columnar radial units1. At the onset of cortical neurogenesis, the proliferative population of founder cells is confined to the ventricular zone of the embryonic cerebral wall2. Even modest alterations in the size of this progenitor population during its early exponential growth phase can markedly affect final neuronal numbers1,3. Thus, it has been proposed that cellular mechanisms that influence founder cell number may underlie the telencephalic expansion and sculpting that are characteristic of mammalian forebrain development and evolution1. Apoptosis within the founder population is one putative mechanism for influencing eventual brain size and shape4,5.
It has recently been recognized that the role of apoptosis in brain development extends beyond matching of neuronal populations with their appropriate target fields, as specified in the ‘neurotrophic hypothesis’5,6. Caspase 3, a key enzyme in the mammalian apoptotic pathway, is expressed at high levels in the mouse cerebral wall around embryonic day 12 (E12)7, when dying cells are prevalent in proliferative zones of the cerebral cortex8–10. Consistent with these observations, mutant mice deficient in the pro-apoptotic genes Casp3, Casp9 and Apaf1 show gross nervous system malformations resulting from improper expansion of specific neural progenitor populations11–15. The excess neurons in some of these mutants are added as extra radial units, expanding the surface of the cortical plate, rather than influencing its thickness. The cortical plate, with increased size, forms convolutions resembling the gyri and sulci of the primate brain. In addition, later generated cells accumulate below the cortical plate, forming heterotopic cell masses within the ventricles11,12. Despite the essential role of the core apoptotic pathway in brain morphogenesis, little is known about the signals regulating apoptosis of proliferative neurons. Identification of the molecules involved is vital to understanding the complex morphogenetic processes that shape the mammalian brain.
cpg15 (also known as Nrn1) was identified in a screen for activity-regulated genes involved in synaptic plasticity16,17 and encodes a small, highly conserved protein18-19 (also termed neuritin-1). In a membrane-bound form attached by a glycosylphosphatidylinositol (GPI) link, CPG15 has been shown to function non–cell autonomously to coordinately regulate growth of apposing dendritic and axonal arbors, and to promote synaptic maturation19,20. As cpg15 is an activity-regulated gene, late cpg15 expression is contemporaneous with critical periods for activity-dependent plasticity and requires action potential activity. However, cpg15 is also expressed in an activity-independent manner during early brain development before circuit formation and maturation21,22, suggesting that it may have a different role at this stage. We hypothesized that, like the neurotrophic factors, CPG15 has multiple roles during nervous system development. In addition to its previously characterized role as a growth and differentiation factor that affects process outgrowth and synaptic maturation, CPG15 may also function as a survival factor during early brain development. Here we describe the identification of a soluble CPG15 expressed in the embryonic brain that regulates survival of cortical progenitors by preventing caspase-mediated apoptosis.
Results
cpg15 is expressed in embryonic proliferative zones
To examine localization of early, activity-independent cpg15 expression, we performed in situ hybridizations on sections from embryonic rat brains. At the earliest times tested, embryonic days 14 (E14) and 15 (E15), cpg15 mRNA is present in the cortical plate, in the ventricular zone of the dorsal thalamus and in retinal ganglion cells (Fig. 1a–f). At E17–E19, cpg15 is expressed in the telencephalic and dorsal diencephalic subventricular zones (Fig. 1g–l), is expressed in the hippocampal primordia (Fig. 1i–j), and at postnatal day 7 (P7) appears in the external granular layer of the cerebellum (Fig. 1m). In all these regions early cpg15 expression is temporally correlated with expansion of the progenitor pool and apoptotic elimination of superfluous neuroblasts10. cpg15 is not expressed in all proliferative zones and is markedly absent from the olfactory epithelium and ganglionic eminence (Fig. 1a,b,e,f,i,j), suggesting that its function may be cell type specific. cpg15 is also expressed when target-derived trophic support is crucial for protection from apoptosis used to match neuron number with target size. From E19 to P7, cpg15 mRNA is present in the trigeminal ganglia, sensory thalamus and various brainstem nuclei (Fig. 1i,j,m), at times of afferent ingrowth, target selection and synaptogenesis in these structures. From P0, cpg15 expression in the cerebral cortex is downregulated to undetectable levels22, coincident with cessation of apoptosis in this region8,9. At P14, cpg15 mRNA re-appears, not in the ventricular or subventricular zones, but in the differentiated cortical layers (Fig. 1n), where activity-dependent plasticity is thought to occur postnatally. cpg15 mRNA patterns are thus consistent with an early role as a survival factor during brain morphogenesis, and a later role in structural remodeling and synaptic maturation associated with developmental and adult plasticity.
Figure 1.
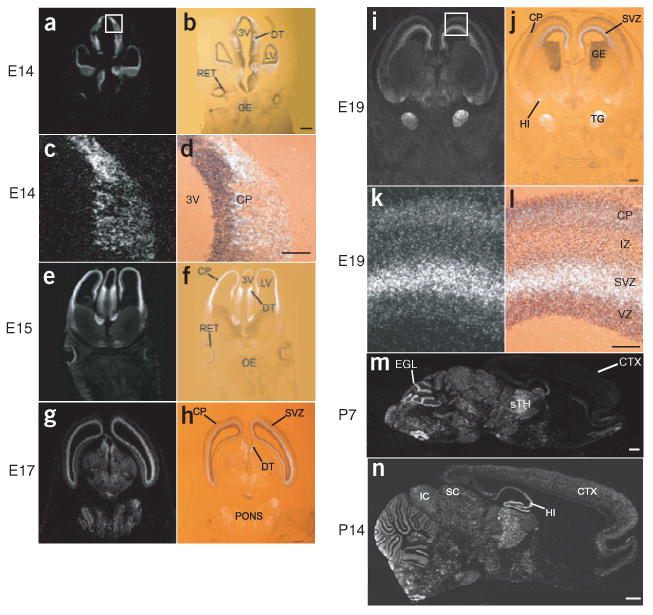
cpg15 mRNA expression is biphasic. (a–n) cpg15 in situ hybridizations on (a–l) coronal sections through the telencephalon during prenatal development, and (m,n) on saggital sections through postnatal brains (ages designated at left). Dark-field photomicrographs of embryonic times are shown in left column; on the right are bright-field photomicrographs of the same sections counterstained with toluidine blue and overlaid with their dark-field views. Marked are the third ventricle (3V), dorsal thalamus (DT), lateral ventricle (LV), retina (RET), olfactory epithelium (OE), cortical plate (CP), subventricular zone (SVZ), ganglionic eminence (GE), trigeminal ganglion (TG), hippocampus (HI), neocortex (CTX), sensory thalamus (sTH), external granular layer (EGL), inferior colliculus (IC) and superior colliculus (SC). Scale bars: a,b,e–j, 0.5 mm; c,d,k,l, 150 μm; m,n, 1 mm.
CPG15 is primarily expressed in a soluble, secreted form
To generate CPG15 for testing in a survival assay, we cloned full-length CPG15 tagged with a Flag epitope into a vector containing an internal ribosomal entry site (IRES) for enhanced green fluorescent protein (EGFP) coexpression (pIRES-EGFP-CPG15-FLAG, Fig. 2a) and expressed it in HEK293T cells. The Flag tag allowed detection and subsequent affinity purification of the CPG15 protein, whereas EGFP marked transfected cells. Consistent with CPG15's GPI link to the cell surface18,19, immunohistochemistry with a monoclonal antibody to Flag (anti-Flag) showed membrane staining of transfected cells (data not shown). Notably, we observed CPG15 staining in untransfected EGFP-negative cells, suggesting intercellular transfer of CPG15 from transfected cells to their untransfected neighbors. To test whether cell-to-cell contact is necessary for intercellular transfer of CPG15, we cocultured transfected and untransfected HEK293T cells on the same coverslip but in distinct locations without physical contact (Fig. 2b). Anti-Flag immunohistochemistry showed CPG15 membrane staining of all untransfected cells (Fig. 2c), suggesting that a soluble form of CPG15 can diffuse between isolated cells. Cells cocultured with cells expressing vector alone (data not shown) or a control Flag-tagged cytoplasmic protein showed no membrane staining (Fig. 2d).
Figure 2.
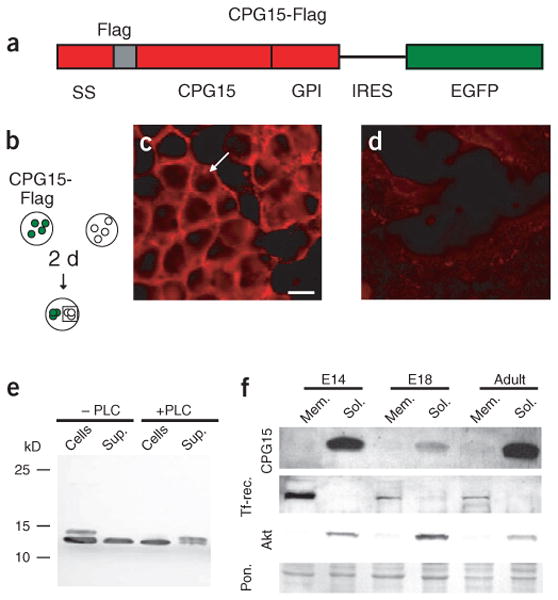
CPG15 is predominantly expressed in a soluble secreted form. (a) Schematic of CPG15-FLAG-IRES-EGFP construct. The Flag peptide sequence (gray) was inserted between the secretion signal (SS) and the CPG15 core domain. Tagged, full-length CPG15 was then cloned upstream of an internal ribosome entry site (IRES) and EGFP. (b) HEK293T cells transfected with CPG15-FLAG-IRES-EGFP (green) were cocultured with untransfected cells (white) for 2 d. (c) Untransfected cells (outlined by square in b) show CPG15 immunoreactivity (white arrow). Scale bar, 10 μm. (d) HEK293T cells cocultured with cells transfected with a Flag-tagged control protein show no membrane staining. (e) Western blots of cell extracts and supernatants from CPG15-FLAG-IRES-EGFP transfected HEK293T cells either untreated or treated with phospholipase C (PLC). (f) Western blots of membrane and soluble fractions from E14 and E18 brains, and adult cortices, probed with antibody against CPG15. Staining for the transferrin receptor (Tf-rec.) and the enzyme Akt serve as membrane- and soluble-fraction controls, respectively. Ponceau staining (Pon.) serves as a loading control.
We verified the presence of soluble CPG15 in supernatants from CPG15-transfected HEK293T cells by western blot analysis (Fig. 2e). Anti-Flag staining showed two CPG15-specific bands of distinct molecular weights in whole-cell extracts. The lower-molecular-weight protein was also present in the supernatant fraction. Treatment of CPG15-transfected cells with phospholipase C (to promote cleavage of GPI anchors and release GPI-anchored proteins from the cell surface23) resulted in a disappearance of the higher molecular weight protein from the cell extracts and its concurrent appearance in the supernatant fraction (Fig. 2e). The lower-molecular-weight protein remained unaffected by phospholipase C treatment. These results suggest that the higher-molecular-weight protein represents membrane-bound GPI-linked CPG15 and that the smaller protein is a soluble form of CPG15.
To determine whether both CPG15 forms are expressed in vivo and at what developmental times, we prepared membrane and soluble protein fractions from brains of E14, E18 and adult rats and examined them by western blot analysis using an antibody against CPG1519. In both embryonic brains and adult cortex, CPG15 was detected predominantly in the soluble protein fractions, with low levels of the membrane-bound protein detected only in the adult (Fig. 2f). As the prevalent form of CPG15 in vivo is soluble, this form is likely the primary mediator of CPG15's early role during embryonic brain development and possibly of its later role as an activity-regulated growth and differentiation factor19,20.
CPG15 rescues cultured cortical neurons from apoptosis
The presence of soluble CPG15 in brain extracts at early developmental times and the localization of its mRNA to specific proliferative populations and to neurons at their target selection phase led us to test whether it may serve as a survival factor that protects against apoptosis. We examined whether soluble CPG15, affinity purified from supernatants of CPG15-Flag–transfected HEK293T cells, was capable of preventing cultured cortical neurons from undergoing apoptosis. Using Hoechst 33324 staining to identify cells with fragmented nuclei, we counted the number of apoptotic neurons in untreated cortical cultures and in cultures after growth factor deprivation (starvation), with or without addition of purified CPG15 (Fig. 3a–c). Growth factor deprivation more than doubled the percentage of apoptotic neurons in the cultures, from approximately 15% to 40%. The increased apoptosis could be completely prevented by addition of soluble CPG15, but not by addition of affinity column elution buffer (Fig. 3d). To confirm that CPG15 was rescuing neurons from apoptotic rather than necrotic cell death, the treated and control cultures were immunostained with an antibody specific to the p17 subunit of activated caspase 3, a key component of the apoptotic pathway in brain development11. All neurons containing pyknotic nuclei visualized by Hoechst staining also stained positive for the p17 cleavage product of activated caspase 3 (Fig. 3e–j). Independent quantification of immunostained neurons expressing cleaved caspase 3 showed that starvation more than doubled their number. CPG15 provision completely prevented the increased caspase 3 activation induced by starvation (Fig. 3k). We conclude that soluble CPG15 protects cortical neurons from apoptosis by preventing activation of caspase pathways induced by growth factor deprivation.
Figure 3.
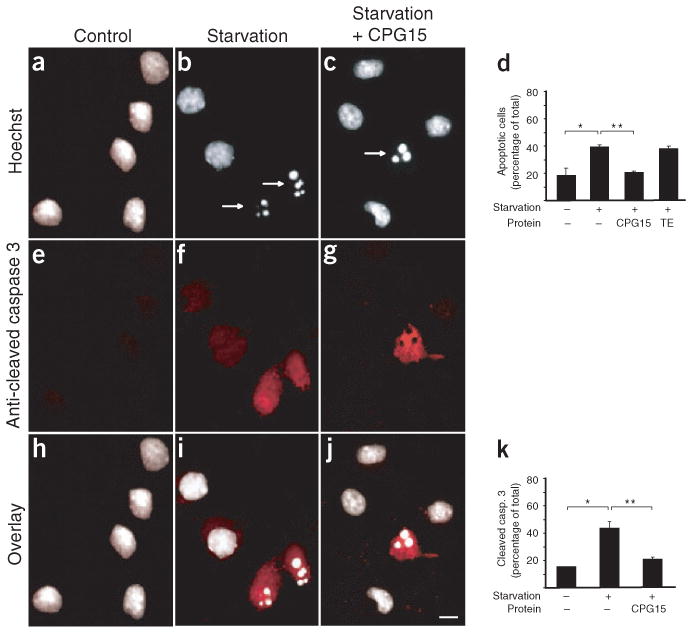
Soluble CPG15 rescues cortical neurons from apoptosis induced by growth factor deprivation (starvation). (a,b) Starvation increased the number of fragmented nuclei seen with Hoechst staining (marked by arrows). (c) CPG15 addition prevented this increase. (d) Quantification of a–c. Starvation significantly increased the percentage of apoptotic neurons (*P < 0.002). CPG15 application prevented this increase (** P < 0.003). (e,f) Starvation induces apoptosis, as seen by increased numbers of neurons immunopositive for cleaved caspase 3. (g) Purified CPG15 prevented the starvation-induced increase in cleaved caspase 3 immunoreactivity. (h–j) Overlay of a and e, b and f, and c and g shows that cells with fragmented nuclei scored by Hoechst staining contain activated caspase 3. (k) Quantification of e–g. Starvation significantly increased the percentage of cleaved caspase 3–immunopositive neurons (* P < 0.001). CPG15 application prevented this increase (**P < 0.002). Scale bar, 10 μm.
Lentivirus-delivered cpg15 shRNA reduces CPG15 levels
To test whether CPG15 also promotes survival of cortical progenitors, we used lentivirus delivery24,25 combined with RNA interference (RNAi)26 to knock down CPG15 expression in vitro and in vivo. We generated lentiviruses expressing CPG15-Flag, a cpg15 shRNA (small hairpin RNA), and a control scrambled cpg15 shRNA with four of the hairpin nucleotides inverted (Fig. 4a). shRNAs are processed to small interfering RNAs (siRNAs) that guide the specific cleavage and elimination of their cognate mRNAs. When primary cortical cultures were coinfected with the cpg15-FLAG and cpg15 shRNA lentiviruses, both the exogenous cpg15-FLAG and the endogenous cpg15 mRNAs were severely reduced as detected by northern blotting (Fig. 4b), but they were unaffected by coinfection with the scrambled cpg15 shRNA control virus. The cpg15 shRNA lentivirus was also effective in knocking down cellular levels of the CPG15 protein as detected by immunocytochemistry (Fig. 4c–f) and by western blotting (Fig. 4g).
Figure 4.
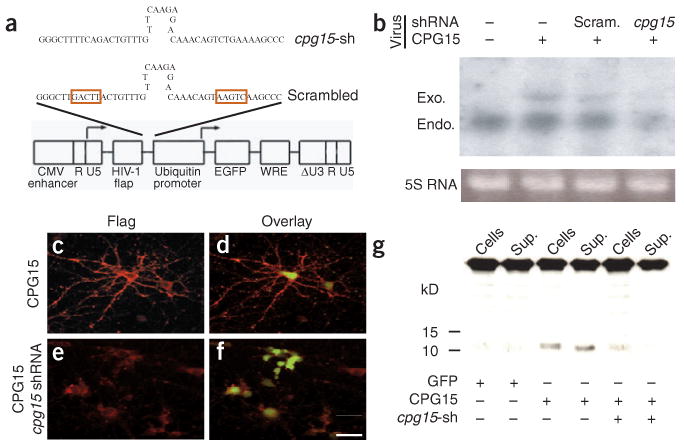
A lentivirus-delivered cpg15 small hairpin RNA (shRNA) knocks down CPG15 expression. (a) Schematic representation of short hairpin sequences and lentivirus vector used for CPG15 knockdown. The shRNA or scrambled shRNA sequences connected to the U6 promoter were inserted upstream of the ubiquitin-C promoter driving EGFP expression (see Methods for details). Red boxes mark the inverted region in the scrambled shRNA. (b–g) In cultured cortical neurons, lentivirus-delivered cpg15 shRNA, but not the scrambled version, markedly reduces cpg15 mRNA assayed by northern blotting (b) and reduces protein expression as assayed by immunocytochemistry (c–f) or by western blotting (g). Neurons were infected with CPG15-FLAG-IRES-EGFP lentivirus, alone (b, lane 2; c,d; and g, lanes 3–4), together with a scrambled cpg15 shRNA control lentivirus (b, lane 3) or together with the cpg15 shRNA lentivirus (b, lane 4; e,f; and g, lanes 5–6). CPG15 immunostaining is red, and EGFP (green) marks infected cells. In g, Sup., supernatant; cpg15-sh, cpg15 shRNA. Scale bar, 25 μm.
CPG15 knockdown increases cortical progenitor apoptosis
To address the role of CPG15 in progenitor cell survival, we first examined the effect of CPG15 knockdown on cultured cortical progenitors that were isolated from E14–E15 embryonic rat cortex and plated in the presence of basic fibroblast growth factor (bFGF). At plating, cells were infected with cpg15 shRNA, scrambled cpg15 shRNA or EGFP lentiviruses. After 4 d in culture the vast majority of cells in uninfected, EGFP-infected or scrambled hairpin–infected control cultures were positive for nestin, a marker of neural progenitors (Fig. 5a,b red staining). In cpg15 shRNA–infected cultures, the number of progenitors was greatly reduced, whereas the number of differentiated neurons (marked by neurofilament-M (Nf-M) staining, blue) was unchanged (Fig. 5c). Quantification of these results shows that the decrease in progenitor numbers in cpg15 shRNA–infected cultures is similar to that seen in cultures deprived of bFGF from plating (Fig. 5d,e). Progenitor loss in the cpg15 shRNA–infected cultures was accompanied by a marked increase in apoptotic cell death identified by Hoechst staining (Fig. 5f). These results demonstrate that depletion of endogenous CPG15 results in increased apoptosis of neuronal progenitors, suggesting that CPG15 is required for their in vitro survival. As acute CPG15 loss has no immediate affect on the number of Nf-M positive neurons, our results further suggest that CPG15 primarily affects progenitor survival.
Figure 5.
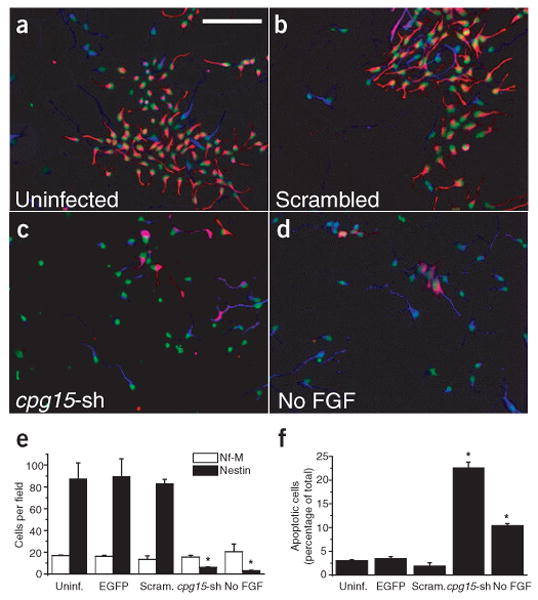
Endogenous CPG15 is required for cortical progenitor survival in vitro. (a–d) Cortical progenitor cultures stained with the progenitor marker nestin (red), the neuronal marker neurofilament-M (Nf-M) (blue) and Hoechst nuclear staining (pseudocolored green). (e) Quantification of a–d and an additional EGFP lentivirus control. Comparison of total number of cells per field staining positive for nestin or Nf-M (*P < 0.001). Infection with the cpg15 shRNA lentivirus (cpg15-sh), but not the scrambled control, leads to a marked decrease in the number of neural progenitors. (f) Quantification of apoptotic cells in a–d and an additional EGFP lentivirus control. Knockdown of CPG15 is accompanied by a significant increase in number of apoptotic cells (*P < 0.001). Scale bar, 100 μm.
Our finding that CPG15 is required for survival of cortical progenitors in culture led us to predict that depletion of endogenous CPG15 at early developmental times would increase apoptosis and reduce survival within the cortical progenitor population in vivo. To test this hypothesis, we delivered cpg15 shRNA lentiviruses or control lentiviruses into E15 embryonic brains by direct ventricular injection. Embryos were harvested at E22 and their brains sectioned. Nissl staining showed moderate shrinkage in the size and ventricular volume of cpg15 shRNA lentivirus–infected brains when compared with uninfected (data not shown), EGFP lentivirus–infected or scrambled shRNA lentivirus–infected brains (Fig. 6a–e), suggesting a decrease in neuronal number. We stained alternate sections by TUNEL and quantified apoptotic neurons in the neocortex, where CPG15 is highly expressed during embryonic development, and in the diencephalon, where CPG15 expression is low in early development (Fig. 1). When compared to uninfected (Fig. 6f), EGFP lentivirus–infected (Fig. 6g) and scrambled shRNA lentivirus–infected brains (Fig. 6h), the cpg15 shRNA brains (Fig. 6i) showed an increase in apoptotic cells in the neocortex. Overlay of TUNEL staining and EGFP staining on the same sections (Fig. 6j–m) demonstrated comparable infection levels by the different lentiviruses. Quantification of TUNEL-stained cells showed that cpg15 shRNA increases apoptosis in the neocortex (Fig. 6n) but not in the diencephalon (Fig. 6o). These results demonstrate that decreasing endogenous CPG15 levels during embryonic development results in increased apoptosis and diminished survival of cortical neurons. Reduced neuronal number is likely to cause shrinkage of the cortical plate and its contraction around the lateral ventricles, as seen in the deformed cpg15 shRNA lentivirus–infected brains (Fig. 6a–e). The specific effect of CPG15 depletion on neocortical neurons suggests that it is not essential for survival in all progenitor populations. Alternatively, the RNAi intervention may be past the critical time when CPG15 is necessary for survival of the diencephalic progenitors. In any case, the lack of effect of the cpg15 shRNA lentivirus on apoptosis in the developing diencephalon demonstrates that the lentivirus-delivered RNAi has no deleterious effect in brain regions surrounding the ventricles.
Figure 6.
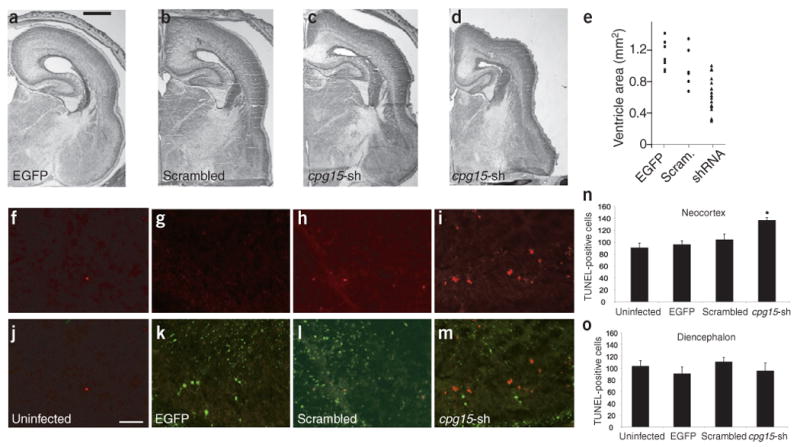
In vivo knockdown of endogenous CPG15 causes shrinkage of the cortical plate and increased apoptosis of cortical neurons. (a–e) In vivo knockdown of endogenous CPG15 results in cortical plate shrinkage. Coronal hemi-sections from (a) brains infected with EGFP lentivirus, (b) scrambled cpg15-shRNA or (c,d) cpg15-shRNA lentivirus. Low to high infection levels shown from left to right. (e) Scatter plot summarizing ventricular area of infected brains. Each symbol represents one hemisphere. (f–i) TUNEL staining of control and cpg15-shRNA lentivirus–infected brains. (j–m) Overlay of TUNEL with EGFP staining shows similar infection levels with control and cpg15-shRNA viruses. (n,o) Quantification of TUNEL-positive cells in the neocortex (n) or diencephalon (o) of control and infected brains (*P < 0.003). Scale bars: a, 1 mm; j, 50 μm.
CPG15 overexpression results in an enlarged cortical plate
In a complementary study, we further examined the role of CPG15 in vivo by overexpressing CPG15-Flag in the developing brain. CPG15-Flag lentivirus was injected into the ventricles of E15 embryonic brains harvested as described above. When compared with control EGFP lentivirus–infected brains, brains overexpressing CPG15 were significantly larger in diameter, with enlarged ventricles but with no alteration in cortical thickness (Fig. 7a–e). Closer examination of the enlarged brains showed sulcus-like indentations (Fig. 7f–k; boxes in a–c are shown at higher magnification in f,h,i, and two additional examples from different CPG15-Flag lentivirus–infected brains are shown in j,k), consistent with a larger surface area resulting from addition of supernumerary neurons in radial units4. Sections from control EGFP lentivirus–infected brains (Fig. 7l) and from brains overexpressing CPG15 (Fig. 7m–p) were double-labeled with nestin (blue) and Nf-M (red), showing heterotopic cell masses within the proliferative ventricular zone. Thus, CPG15 overexpression results in the expansion and involution of the cortical plate, and in heterotopias and discontinuities of the ventricular zone. Malformations of this type are typically seen in mutant mice with an increase in forebrain progenitor numbers11,12,27 and are consistent with an expansion of the progenitor pool.
Figure 7.
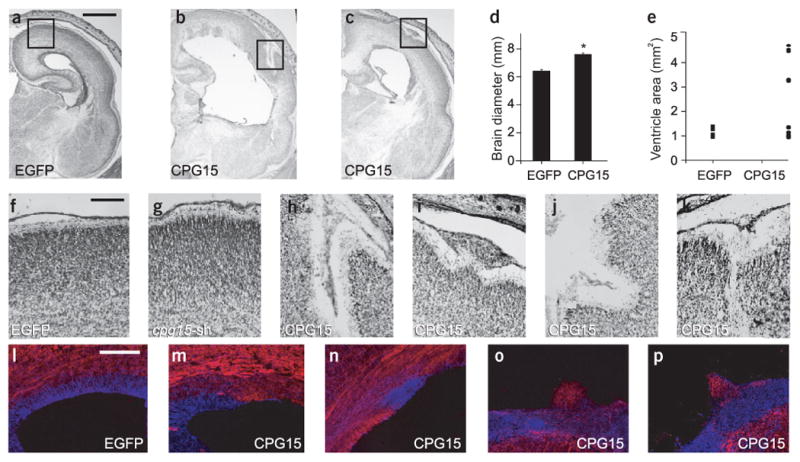
In vivo CPG15 overexpression in the embryonic brain results in an expanded cortical plate and heterotopic cell masses in the ventricular zone. (a–e) CPG15 lentivirus–infected brains are larger in diameter and ventricular area. (a–c) Nissl stained coronal sections from EGFP and CPG15 lentivirus–infected brains. (d) Measurements of brain diameter comparing control and CPG15-overexpressing brains (* P < 0.02). (e) Scatter plot summarizing ventricular area of the same brains. Each symbol represents one hemisphere. (f–k) Nissl-stained coronal sections of brains infected with the indicated viruses. Boxes in (a–c) are shown at higher magnification in f, h and i, respectively. (l–p) Double labeling for nestin in blue and neurofilament in red on EGFP lentivirus–injected (l) and CPG15 lentivirus–injected brains (m–p). Scale bars: a, 1 mm; f, 50 μm; l, 100 μm.
CPG15 expands the progenitor pool by reducing apoptosis
To directly measure an affect of CPG15 on progenitor number, we used BrdU to label proliferating neurons. Pregnant dams were injected with BrdU 3 d after intraventricular injection of the CPG15-Flag lentivirus into embryonic brains. Embryos were harvested 2 or 24 h after BrdU injection (Fig. 8a,b), earlier than for previous experiments to avoid confounding secondary effects of the deformed cortical plate. Low-magnification views showed that 2 h after BrdU injection, the CPG15-Flag lentivirus–infected brains are indistinguishable from uninfected control brains, with normal cellular lamination and cellular distribution within the ventricular and subventricular zones (Fig. 8c,d). However, a higher-magnification view showed a small but significant increase in BrdU-labeled cells within the ventricular zone of CPG15-Flag lentivirus–infected brains, suggesting an increase in progenitor number (Fig. 8e–g).
Figure 8.
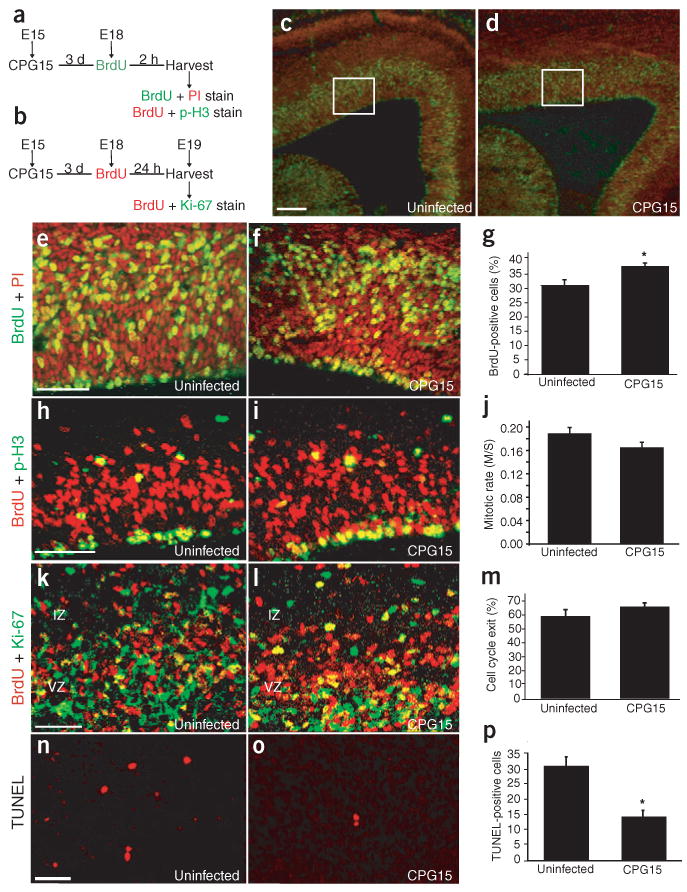
CPG15 overexpression reduces apoptosis in the progenitor pool but does not affect mitotic index or cell cycle exit. (a) Schematic of BrdU labeling experiments shown in c–j. Embryos were injected with virus at E15 and then harvested at E18 2 h after BrdU injection. (b) Schematic of cell cycle exit experiments shown in k–m. Embryos were injected with virus at E15 then harvested at E19 24 h after BrdU injection. (c,d) Low-magnification view of BrdU labeling (green) and propidium iodide nuclear staining (red) in uninfected (c) and CPG15 lentivirus–infected (d) brains. (e,f) Areas outlined in white in c,d, respectively shown at higher magnification. (g) Quantification of BrdU staining shows a significant increase in the percentage of BrdU-labeled cells in CPG15 lentivirus–infected brains (*P = 0.008) as compared to uninfected controls. (h,i) Double labeling for BrdU (red) and the mitosis marker p-H3 (green). (j) Quantification of mitotic rates (ratio of S-phase BrdU-labeled cells to M-phase p-H3–labeled cells) showed no significant difference between CPG15 lentivirus–infected and uninfected brains. (k,l) Double labeling for BrdU (red) and the progenitor cell marker Ki-67 (green). (m) Quantification showed no significant difference in the rate of cell cycle exit between CPG15 lentivirus–infected and control brains. (n,o) TUNEL staining in the ventricular zone of uninfected (n) and CPG15 lentivirus–infected (o) brains. (p) Quantification of TUNEL-positive cells shows a significant decrease in ventricular zone apoptosis in CPG15 lentivirus–infected brains as compared to uninfected controls (*P < 0.001). Scale bars: c, 100 μm; e,h,k,n, 50 μm.
Expansion of the progenitor pool can result from increased mitotic rates, a decrease in cell cycle re-entry, decreased cell death or any combination of these factors. To test for changes in mitotic rates of dividing progenitors, embryos injected with the CPG15-Flag lentivirus and harvested 24 h after BrdU injection at E19 (Fig. 8b) were double stained for BrdU, to mark cells in S phase of the cell cycle, and for phospho-histone-H3 (p-H3), to mark cells in M phase undergoing mitosis (Fig. 8h,i). We found no significant difference between uninfected control and CPG15-Flag lentivirus–infected brains in the ratio of dividing progenitors at M phase to progenitors in S phase (Fig. 8j), indicating that CPG15 does not increase mitotic rate. We next tested whether CPG15 affects the number of progenitors that re-enter the cell cycle rather than progressing towards terminal differentiation. E15 embryos were injected with the CPG15-Flag lentivirus, followed by BrdU injection at E18 (Fig. 8b). Twenty-four hours later, embryos were harvested and double-stained for BrdU, to mark cells that were dividing at the time of injection, and Ki67, to mark progenitors. We identified cells that had exited the cell cycle within the 24 h of BrdU labeling as BrdU+ and Ki67− (red) and divided progenitors in the cell cycle as BrdU+ and Ki67+ (yellow) (Fig. 8k,l). We found that in the CPG15-Flag lentivirus–infected embryos, there was no significant change in the proportion of cortical progenitors exiting the cell cycle as compared to uninfected controls (Fig. 8m).
Finally, to examine whether CPG15 overexpression decreases progenitor apoptosis, we performed TUNEL staining on E18 brains from uninfected control (Fig. 8n) and CPG15-Flag lentivirus–infected (Fig. 8o) embryos 3 d after viral delivery. Counts of TUNEL-stained cells in the cortex of CPG15-Flag lentivirus–infected brains showed a significant decrease in the numbers of apoptotic cells when compared to uninfected control brains (Fig. 8n–p). Together, these studies indicate that the observed increase in progenitor pool size and the expanded cortices seen in CPG15-Flag lentivirus–infected brains are not due to increased progenitor mitotic rates or decreased cell cycle exit, but rather are likely to be a result of decreased apoptosis within the progenitor population.
Discussion
cpg15 expression patterns in the embryonic brain concomitant with periods of rapid cell proliferation and apoptosis, and during circuit formation and target selection, led us to test whether early in development CPG15 might function as a survival factor, similarly to neurotrophins. We found that CPG15 is able to rescue cortical neurons from starvation-induced apoptosis by preventing caspase 3 activation and is also crucial for survival of cortical progenitors. Consequently, manipulating in vivo CPG15 levels in utero had a profound effect on the size and shape of the neocortical plate. Decreasing CPG15 levels caused increased apoptosis of cortical progenitors and shrinkage of the cortical plate, whereas overexpression resulted in decreased progenitor apoptosis and an expanded and convoluted cortical plate.
It is particularly informative to compare the CPG15 overexpression phenotype to developmental mutants with similar presentations. In the case of mutants in the cell-death cascade, expansion of the cortical plate occurs only when the apoptotic pathway is affected in proliferating neurons rather than in postmitotic neurons and is a direct result of an enlarged progenitor pool4,5. Gene-targeting studies have identified Bax, Bcl-XL (Bcl2l1), Apaf1, Casp9 and Casp3 as key elements in neuronal apoptosis occurring during brain development11–15,28,29. By analogy to their counterparts in Caenorhabditis elegans, the proteins are likely to form a linear cell-death cascade, with Apaf1 bound to caspase 9, activating caspase 3, and Bcl-XL acting as an upstream anti-apoptotic regulator of Apaf1 that can be blocked by Bax12,29. Epistatic analysis shows that the upstream components of this pathway, Bax and Bcl-XL, are obligatory only in postmitotic neurons, whereas caspase 3 is unique in its effect on apoptosis of neuronal founder cells29,30. Global formation of the nervous system is unaffected in Bax-deficient embryos31,32, whereas null mutants of Casp3, Casp9 and Apaf1 show severe forebrain malformations that result from hyperplasia and an enlarged neocortical plate11–15. This is consistent with the idea that even modest alterations in the size of the progenitor pool during its exponential growth phase can drastically effect final cortical size and shape1,3. Hyperplasia and an enlarged neocortical plate can also be seen when the cortical progenitor pool is expanded for reasons other than a decrease in cell death: for example, in β-catenin transgenic mice, where cortical progenitors fail to exit the cell cycle after mitosis33. The similar phenotype seen with in vivo CPG15 overexpression suggests that elevated CPG15 expands the progenitor pool, likely by reducing apoptosis. This is supported by results showing that increasing amounts of CPG15 during corticogenesis reduce apoptosis and enlarge the progenitor pool but have no effect on mitotic rates or cell cycle exit. Furthermore, an increased number of apoptotic neurons can be seen in brains with RNAi-mediated knockdown of endogenous CPG15, and in vitro data demonstrates CPG15 function as an anti-apoptotic factor for cortical neurons as well as cortical progenitors.
Other extracellular signaling molecules previously shown to regulate cerebral cortical size through their effect on the progenitor pool are basic fibroblast growth factor34–36, pituitary adenylate cyclase-activating polypeptide37 and, more recently, lysophosphatidic acid38. Basic fibroblast growth factor and pituitary adenylate cyclase–activating polypeptide, respectively, expand or shrink the progenitor pool by acting as mitogenic or anti-mitogenic signals. Lysophosphatidic acid expands the progenitor pool by increasing terminal mitosis and reducing caspase-mediated cell death, and is thus the only factor other than CPG15 that has been shown to decrease apoptosis of cortical progenitors38. Studies of cultured cortical progenitors suggest that BDNF and NT-3 can mediate progenitor cell survival in vitro39. Yet direct intrauterine, intraventricular application of neurotrophins into the embryonic brain does not affect proliferation or apoptosis of cortical progenitors40. Furthermore, classic neurotrophins are absent in most parts of the embryonic brain41, and prenatal CNS defects related to neuronal number have not been seen in neurotrophin single- or double-knockout mice or in animals lacking their receptors42. Thus, CPG15 is one of few molecules shown to be essential for in vivo survival of undifferentiated cortical progenitors. CPG15 expression is specific to a subset of progenitor populations in the developing brain and seems to be required only in these populations. We propose that by countering early apoptosis in specific progenitor subpopulations, CPG15 has a role in regulating brain size and shape during morphogenesis of the mammalian forebrain.
As CPG15 is also expressed in some differentiated neurons during target selection and circuit formation and is able to rescue cortical neurons from starvation-induced apoptosis, it is possible that CPG15 may also function as a target-derived survival factor for differentiated neurons analogous to neurotrophins. It remains to be examined whether CPG15 is required for survival in various populations of differentiated neurons and whether this function of CPG15, as well as its later activity-dependent function as a growth and differentiation factor, are mediated by the soluble form described here. Another possible future direction will be analysis of a cpg15 knockout mouse. As it is technically difficult to intervene by intrauterine delivery of an shRNA earlier than E14–E15, the CPG15 knockdown using RNAi can only affect the very last rounds of cortical progenitor division. In the knockout, cpg15 deletion at an earlier step may result in a more extreme phenotype. However, deleting a gene from the onset of embryogenesis could also lead to compensation and lack of a discernible phenotype, something that can be avoided with an acute knockdown using RNAi at a critical developmental step43. For example, mice with targeted deletion of the doublecortin gene (Dcx) seem to develop a normal neocortex44, whereas electroporation of plasmids encoding shRNA against the doublecortin protein in utero disrupts radial migration in the rat neocortex, resulting in a malformed cerebral cortex45. In this case, acute intervention using RNAi results in a phenotype more similar to the double cortex syndrome seen in humans with mutations in the Dcx gene46 than is seen with the complete Dcx knockout. Thus, RNAi approaches may be complementary to gene knockouts in functional studies of genes important for brain development43.
Methods
All animal work was approved by the Massachusetts Institute of Technology Committee on Animal Care and conforms to US National Institutes of Health guidelines for the use and care of vertebrate animals.
In situ hybridization
In situ hybridizations were performed as previously described22.
CPG15 immunocytochemistry and protein purification
HEK293T cells were grown to 80–90% confluence in 100-mm culture dishes containing 15 ml medium (10% calf serum, 50 U penicillin, 50 μg streptomycin, 4 mM l-glutamate in Dulbecco's Modified Eagles medium (BioWhittaker)) and then transfected with 8 μg of the pIRES-EGFP-CPG15-FLAG plasmid (Fig. 2a) using Lipofectamine 2000 (Invitrogen). For immunocytochemistry, cells were fixed in 4% paraformaldehyde and stained with anti-Flag (Sigma). For protein purification, the medium was harvested 4 d after transfection and debris removed by centrifugation (3,000 rpm, 15 min, 4 °C). The medium was then incubated with 40 μl anti-Flag coupled to agarose (EZview Red ANTI-FLAG M2 Affinity Gel, Sigma) for 12 h at 4 °C on a rotator. The agarose was pelleted by centrifugation for 10 min at 2,000 rpm and washed three times with TBS. The tagged CPG15 protein was eluted from the anti-Flag by incubation for 4 h at 4 °C with 30 μg of 3× Flag peptide (Sigma) diluted in 200 μl TBS; it was recovered by centrifugation for 5 min at 2,000 rpm. Protein concentration in the supernatant was determined by the Bradford assay.
Preparation of membrane and soluble fractions from brain, and western blotting
Membrane and soluble fractions from brains were prepared as detailed in Supplementary Methods online. Western blots were incubated with a polyclonal anti-CPG1519, followed by staining for the transferrin receptor (1:1,000, Zymed) detected by a goat anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibody (1:5,000, Sigma), and by staining for protein Akt (1:1,000, Cell Signaling Technology) detected by a goat anti-rabbit–HRP secondary antibody (1:2,500, Jackson ImmunoResearch).
Growth factor deprivation and apoptosis assays
Primary cortical cultures were done essentially as previously described47. After 6 d in vitro, cortical neurons were washed three times with Neurobasal medium without supplements and then incubated for 12 h in the unsupplemented medium with or without 50 ng ml−1 purified CPG15 protein. After an additional 12 h in feeding medium, cells were fixed in 4% formaldehyde/PBS for 30 min at 4 °C. Hoechst staining and immunocytochemistry were done as described in Supplementary Methods. For quantification, fragmented apoptotic nuclei as well as healthy nuclei were counted blind to experimental treatment using a fluorescence microscope with a UV filter setting for the Hoechst staining (excitation 330–380; emission 420) and rhodamine settings for visualizing the antibody to cleaved caspase 3 (excitation 528–553; emission 600–660). Treatments were repeated in three independent experiments with two coverslips per treatment in each experiment. Each data point represents the mean of 500–600 cells, counted in 40–50 different fields per coverslip. The percentage of apoptotic cells was calculated based on the number of fragmented nuclei divided by the total number of nuclei. Comparisons between groups were analyzed using a Student's t-test.
Immunocytochemistry of neural progenitor cultures
Cortical progenitor cultures were prepared as previously described in detail48. Immunocytochemistry for nestin (1:2,000; Chemicon) and neurofilament M (1:3,000; Chemicon) is described in Supplementary Methods. Quantifications were from three independent experiments (five random images per treatment). Groups were compared using ANOVA post-hoc analysis with the Bonferroni/Dunn method.
Lentivirus generation and infections
Five different cDNA sequences spanning the cpg15 core domain were synthesized, fused to a loop region and then annealed to their antisense sequences and cloned separately into pSilencer1.0-U6 plasmid (Ambion) downstream of the U6 promoter. To test the effectiveness of the cpg15 shRNAs in reducing CPG15 levels, HEK293T cells were separately cotransfected with each one of the pSilencer-cpg15-shRNA plasmids together with the pIRES-EGFP-CPG15-FLAG plasmid at a 40:1 ratio using Lipofectamine 2000 (Invitrogen). CPG15-IRES-EGFP mRNA knockdown was determined by reduced expression of EGFP The most effective small hairpin sequence (5′-GGGCTTTTCAGACTGTTTG-3′) was then amplified with its upstream U6 promoter by PCR and subcloned into the pFUGW lentivirus transfer vector24. To generate the scrambled shRNA construct, the small hairpin sequence was modified to 5′-GGGCTTGACTTACTGTTTG-3′ (the inverted sequence is underlined) and cloned into the pFUGW lentivirus transfer vector as described above. The CPG15-FLAG-IRES-EGFP cDNA was subcloned into pFUGW downstream of the ubiquitin promoter. pFUGW lentivirus was used as the EGFP control. Lentivirus production, concentration and titer determination were done as described24. Typical titers for in vitro experiments were 1 × 106 and for in vivo injections, 5 × 106 to 20 × 106.
For viral infection of primary cortical cultures, culture medium was reduced to 0.5 ml at 3 d in vitro, and viruses were added for >8 h incubation. The medium was then removed and replaced with 0.5 ml saved plating medium and 0.5 ml new feeding medium. Cells were fixed and mounted 4 d later. For infection of progenitor cultures, the different lentiviruses were added to the medium at plating.
In vivo lentivirus injections were performed on timed-pregnant Sprague-Dawley rats (Taconic). Pregnancies were timed with day of plug detection as E1 and birth usually occurred on E23. Neurons and progenitors were infected by injecting 2–2.5 μl of lentivirus into the lateral ventricle of E15 brains. Surgery and injection procedures were as previously described49.
Northern blot hybridization
Northern blot hybridization was performed as previously described50.
Histological and immunohistochemical analysis of embryonic brains
Animals were euthanized at E18, E19 or E22: 3, 4 or 7 d, respectively, after viral injection. Brains were removed and submerged in 4% paraformaldehyde in PBS overnight at 4 °C, transferred to 30% sucrose in PBS at 4 °C until they sank, and then frozen and sectioned at 20 μm using a cryostat. Every sixth section was Nissl stained and used to match sections from different brains. Sections at equivalent levels on the anterior-posterior axis were then processed for TUNEL. Cryosections were blocked in 10% goat serum, 0.3% Triton X-100 for 1 h. Primary antibodies against nestin (1:2,000, Chemicon, mouse monoclonal) and neurofilament (1:1,000, Chemicon, rabbit polyclonal) were diluted in blocking solution. Sections were counterstained with Hoechst 33342 in PBS for 10 min to highlight nuclear DNA. Fluorescent secondary antibodies Alexa-555-conjugated goat anti-mouse (1:200, Molecular Probes) and Alexa-647-conjugated anti-rabbit (1:200, Molecular Probes) were used for visualization as described above.
TUNEL staining on frozen brain sections was performed as described by the manufacturer using the Roche In Situ Cell Death Detection Kit, TMR red. The total number of TUNEL-positive cells present in the neocortex and the diencephalon of uninfected (three each at E18 and E22), EGFP-infected (three at E22) and cpg15 shRNA–infected (four at E22) brains was quantified. Six to twelve sections were analyzed from each brain. Statistical significance was determined by Student's t-test.
To label proliferating cells in the cortex of E18 embryos, BrdU was injected intraperitoneally into the mother at 50 mg kg−1 body weight. BrdU and Ki-67 or p-H3 double labeling was done as previously described for BrdU and Ki6733. Primary antibodies used were BrdU (1:200; Harlan, rat monoclonal), Ki-67 (1:500; Novocastra, rabbit polyclonal), and p-H3 (1:1,000; Upstate, rabbit polyclonal). BrdU-labeled and p-H3–labeled cells were counted from matching sections and percentages were compared by unpaired Student's t-tests. For cell cycle exit studies, a similar approach was used: Ki-67 expression was scored in 50 BrdU-positive cells from each of five randomly chosen fields of view from at least three sections per brain (two brains per each experimental group). Statistical significance was determined by Student's t-test.
Supplementary Material
Acknowledgments
We thank members of the Nedivi laboratory and P. Garrity, J. Hoch, and J. Mintern for helpful comments on the manuscript, J. Cottrell for initiating and help with shRNA cloning and testing, C. Lois for advice on construction and use of lentivirus vectors, C. Walsh and E. Olson for guidance on embryonic injections, and J. Pungor for help with cell counts. This work was sponsored by grants from National Eye Institute and the Ellison Medical Foundation to E. Nedivi. U. Putz was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft, and C. Harwell by a Ford Foundation predoctoral fellowship.
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
Competing Interests Statement: The authors declare that they have no competing financial interests.
References
- 1.Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–388. doi: 10.1016/0166-2236(95)93934-p. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi T, Nowakowski RS, Caviness VSJ. The mathematics of neocortical neuronogenesis. Dev Neurosci. 1997;19:17–22. doi: 10.1159/000111179. [DOI] [PubMed] [Google Scholar]
- 3.Caviness VSJ, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- 4.Haydar TF, Kuan CY, Flavell RA, Rakic P. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex. 1999;9:621–626. doi: 10.1093/cercor/9.6.621. [DOI] [PubMed] [Google Scholar]
- 5.Kuan CH, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- 6.de la Rosa EJ, de Pablo F. Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci. 2000;23:454–458. doi: 10.1016/s0166-2236(00)01628-3. [DOI] [PubMed] [Google Scholar]
- 7.Pompeiano M, Blaschke AJ, Flavell RA, Srinivasan A, Chun J. Decreased apoptosis in proliferative and postmitotic regions of the caspase 3-deficient embryonic central nervous system. J Comp Neurol. 2000;423:1–12. [PubMed] [Google Scholar]
- 8.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 9.Thomaidou D, Mione MC, Cavanagh JFR, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blaschke AJ, Weiner JA, Chun J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol. 1998;396:39–50. doi: 10.1002/(sici)1096-9861(19980622)396:1<39::aid-cne4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 11.Kuida K, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 12.Kuida K, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 13.Hakem R, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 14.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 Homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida H, et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 16.Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature. 1993;363:718–722. doi: 10.1038/363718a0. [DOI] [PubMed] [Google Scholar]
- 17.Hevroni D, et al. Hippocampal plasticity involves extensive gene induction and multiple cellular mechanisms. J Mol Neurosci. 1998;10:75–98. doi: 10.1007/BF02737120. [DOI] [PubMed] [Google Scholar]
- 18.Naeve GS, et al. Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc Natl Acad Sci USA. 1997;94:2648–2653. doi: 10.1073/pnas.94.6.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedivi E, Wu GY, Cline HT. Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science. 1998;281:1863–1866. doi: 10.1126/science.281.5384.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cantallops I, Haas K, Cline HT. Postsynaptic CPG15 promotes synaptic maturation and presynaptic axon arbor elaboration in vivo. Nat Neurosci. 2000;3:1004–1011. doi: 10.1038/79823. [DOI] [PubMed] [Google Scholar]
- 21.Corriveau R, Shatz CJ, Nedivi E. Dynamic regulation of cpg15 during activity-dependent synaptic development in the mammalian visual system. J Neurosci. 1999;19:7999–8008. doi: 10.1523/JNEUROSCI.19-18-07999.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee WCA, Nedivi E. Extended plasticity of visual cortex in dark-reared animals may result from prolonged expression of genes like cpg15. J Neurosci. 2002;22:1807–1815. doi: 10.1523/JNEUROSCI.22-05-01807.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hooper NM. Determination of glycosyl-phosphatidylinositol membrane protein anchorage. Proteomics. 2001;1:748–755. doi: 10.1002/1615-9861(200106)1:6<748::AID-PROT748>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 24.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 25.Rubinson DA, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 26.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 27.Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cyto-architectural distortions in β-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- 28.Motoyama N, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 29.Roth KA, et al. Epistatic and independent functions of Caspase-3 and Bcl-XL in developmental programmed cell death. Proc Natl Acad Sci USA. 2000;97:466–471. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shindler KS, Latham CB, Roth KA. bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J Neurosci. 1997;17:3112–3119. doi: 10.1523/JNEUROSCI.17-09-03112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 32.White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- 34.Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci USA. 1998;95:5672–5677. doi: 10.1073/pnas.95.10.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dono R, Texido G, Dussel R, Ehmke H, Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaccarino FM, et al. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci. 1999;2:246–253. doi: 10.1038/6350. [DOI] [PubMed] [Google Scholar]
- 37.Suh J, Lu N, Nicot A, Tatsuno I, DiCicco-Bloom E. PACAP is an anti-mitogenic signal in developing cerebral cortex. Nat Neurosci. 2001;4:123–124. doi: 10.1038/83936. [DOI] [PubMed] [Google Scholar]
- 38.Kingsbury MA, Rehen SK, Contos JJA, Higgins CMa, Chun J. Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nat Neurosci. 2003;6:1292–1299. doi: 10.1038/nn1157. [DOI] [PubMed] [Google Scholar]
- 39.Barnabé-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunstrom JE, Gray-Swain MR, Osborne PA, Pearlman AL. Neuronal heterotopias in the developing cerebral cortex produced by neurotrophin-4. Neuron. 1997;18:505–517. doi: 10.1016/s0896-6273(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 41.Ernfors P, Merlio JP, Persson H. Cells expressing mRNA for neurotrophins and their receptors during embryonic rat development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- 42.Conover JC, Yancopoulos GD. Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Rev Neurosci. 1997;8:13–27. doi: 10.1515/revneuro.1997.8.1.13. [DOI] [PubMed] [Google Scholar]
- 43.Götz M. Doublecortin finds its place. Nat Neurosci. 2003;6:1245–1247. doi: 10.1038/nn1203-1245. [DOI] [PubMed] [Google Scholar]
- 44.Corbo JC, et al. Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J Neurosci. 2002;22:7548–7557. doi: 10.1523/JNEUROSCI.22-17-07548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai J, et al. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1282. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- 46.Gleeson JG, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 47.Zhou J, Tang XC. Huperzine A attenuates apoptosis and mitochondria-dependent caspase-3 in rat cortical neurons. FEBS Lett. 2002;526:21–25. doi: 10.1016/s0014-5793(02)03107-1. [DOI] [PubMed] [Google Scholar]
- 48.Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 49.Walsh C, Cepko CL. Widespread dispersion of neuronal clones across functional regions of the cerebral cortex. Science. 1992;255:434–440. doi: 10.1126/science.1734520. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.