

Abstract
Fluorescence fluctuation spectroscopy provides information about protein interactions in the intercellular environment from naturally occurring equilibrium fluctuations. We determine the molecular brightness of fluorescent proteins from the fluctuations by analyzing the photon counting histogram (PCH) or its moments and demonstrate the use of molecular brightness in probing the oligomerization state of proteins. We report fluorescence fluctuation measurements of enhanced GFP (EGFP) in cells up to concentrations of 10 μM by using an improved PCH theory. The molecular brightness of EGFP is constant in the concentration range studied. The brightness of a tandem EGFP construct, which carries two fluorophores, increases by a factor of two compared with EGFP alone, demonstrating the sensitivity of molecular brightness as a probe for protein complex formation. Oligomerization of nuclear receptors plays a crucial role in the regulation of gene expression. We probe the oligomerization state of the testicular receptor 4 and the ligand-binding domains of retinoid X receptor and retinoic acid receptor by observing molecular brightness changes as a function of protein concentration. The large concentration range accessible by experiment allows us to perform titration experiments on EGFP fusion proteins. An increase in the molecular brightness with protein concentration indicates the formation of homocomplexes. We observe the formation of homodimers of retinoid X receptor ligand binding domain upon addition of ligand. Resolving protein interactions in a cell is an important step in understanding cellular function on a molecular level. Brightness analysis promises to develop into an important tool for determining protein complex formation in cells.
Every cellular function involves a large number of proteins interacting with one another to fulfill a specific biological task. A characteristic of signaling, metabolic, and other pathways is the reversible association of proteins into complexes as part of a cellular stimulus. The understanding of biological cells requires understanding the intricate and entangled interactions between proteins. Detecting and measuring protein association of cellular proteins is an important first step for ultimately piecing together the inner workings of the cellular machinery. However, we lack spectroscopic tools for directly determining protein interactions in cells. Here, we describe a technique with the capability to detect protein oligomerization in living cells based on fluorescence fluctuation spectroscopy (FFS).
FFS is a very sensitive technique that probes the dynamics and concentration of fluorescent molecules in optical observation volumes of ≈1 fl. Each fluorophore passing through the observation volume is excited by laser light and emits fluorescence, which is registered by a photo detector and produces a small signal fluctuation. Statistical analysis of the signal fluctuations recovers information about fluorophores. Calculation of correlation functions is the most widely used technique and is also known as fluorescence correlation spectroscopy (FCS) (1). Here we will focus on two other ways of analyzing fluctuations, photon counting histogram (PCH) (2) and moment analysis (3). Both techniques determine the molecular brightness and the occupation number in the observation volume. Another brightness technique (fluorescence intensity distribution analysis, FIDA) that is similar to PCH has also been described in the literature (4).
Molecular brightness is defined as the average detected photon count rate for a fluorophore. It depends on instrumental parameters, such as the excitation wavelength and the quantum yield of the detector. If instrumental parameters are kept constant, molecular brightness characterizes a photophysical property of the fluorescent molecule. We use molecular brightness to monitor protein association. If a fluorescently labeled protein diffuses through the observation volume, it will produce a burst of detected photons. The average photon count rate of these bursts determines the molecular brightness of the labeled protein. If such a protein associates to form a homodimer, the new complex will carry two fluorescent labels. The complex will produce, on average, twice as many photons than is the case for the monomeric protein, because two independently fluorescing molecules are participating. Consequently, the molecular brightness of the dimer is twice that of the monomer (5).
Fluorescence fluctuation experiments are very attractive for in vivo applications. The small optical observation volume allows probing any location within a cell with submicron resolution. Fluorescently labeled proteins diffusing through the observation volume give rise to spontaneous signal fluctuations that provide dynamic and static information about the system without the need for external perturbation. The cellular environment is considerably more complex than compared with test tube experiments. Autofluorescence, spatial heterogeneity, and other factors complicate fluorescence fluctuation experiments in living cells. Nevertheless, it has been shown that FCS measurements in living cells are quite feasible (6, 7). In addition, brightness analysis in cells has been successfully demonstrated (8). Cells are essentially picoliter cuvettes with a finite amount of proteins present. Conventional FCS with confocal detection leads to photobleaching of the fluorophore outside of the focal region of the laser beam. The depletion of the reservoir of fluorescent proteins in cells complicates the analysis of fluctuation experiments substantially. Two-photon excitation, originally introduced by Denk et al. (9) for fluorescence imaging, eliminates photobleaching in the out-of-focus regions, because the nonlinear nature of multiphoton absorption restricts the excitation and photobleaching process strictly to the focal region of the laser beam. This reduction in photobleaching is an important advantage of our two-photon fluorescence fluctuation experiments in cells (10).
Fluorescent labeling of proteins in cells is performed by constructing a fusion protein tagged with a GFP. Expression of the fusion protein in the cell produces proteins that are labeled with exactly a single copy of the fluorescent protein. We chose enhanced GFP (EGFP), a variant of GFP, because of its improved folding properties and its superior photostability as compared with the wild type.
We present two-photon fluorescence fluctuation measurements in cells and perform brightness analysis on EGFP. Analysis tools recently developed by us allow quantitative analysis of fluctuation experiments at concentrations as high as 10 μM (11). Previous analysis had been limited to concentrations of ≈250 nM. Thus, we are able to probe the response of proteins over a very wide concentration range. This wide concentration range allows us to perform essentially a titration experiment in a living cell by varying the expressed protein concentration. Our results demonstrate that molecular brightness analysis under in vivo conditions is a robust technique and allows the observation of protein oligomerization. We apply the technique by studying the self-association of nuclear receptors in the presence and absence of ligand. Nuclear receptors form a superfamily of structurally related proteins. These receptors act as transcription factors and provide organisms with a direct control of gene expression in response to developmental and environmental signals. Most of the hormone nuclear receptors form oligomers and changes in oligomerization state are important for initiating consecutive binding of additional cofactors that activate gene expression. Retinoid X receptor (RXR), is special, because it plays a dual role in the nuclear receptor signaling pathway. RXR forms heterodimers with other nuclear receptors, such as retinoid acid receptor (RAR) and vitamin D receptor, but also forms homodimers and is activated by its ligand 9-cis-retinoid acid (12). Here, we probe the oligomeric state of RXR ligand-binding domain (RXR-LBD) and of RAR ligand binding domain (RAR-LBD) in the presence and absence of ligand. In addition, we present data on a full-length orphan nuclear receptor, testicular receptor 4 (TR4).
In vivo experiments examine protein–protein interactions in the presence of all other cellular proteins and probe biological function in their natural environment. Such experiments in living cells will aid in the development of models of cellular function and allow a critical assessment of models based on in vitro experiments by comparison with in vivo results. The concept of brightness analysis is applicable to study protein interactions in general. For example, brightness analysis could detect changes in the aggregation state of proteins upon addition of drugs or signaling molecules and provides a direct measure of drug interactions at specific locations in the intact cell.
Materials and Methods
Experimental Setup. A mode-locked Ti:sapphire laser (Tsunami, Spectra Physics, Mountain View, CA) pumped by an intracavity doubled Nd:YVO4 laser (Millenia, Spectra-Physics) serves as source for two-photon excitation. The laser produces 100-fs pulses with a repetition frequency of 80 MHz (tunable between 700 and 1,000 nm). The experiments were carried out by using a Zeiss Axiovert 200 microscope (Thornwood, NY) with a ×63 Plan Apochromat oil immersion objective (numerical aperture = 1.4). The fluorescence filter turret of the microscope was modified to allow two-photon excitation. In addition to the filter cube used for the fluorescent light path, another filter cube with a dichroic for two-photon excitation (Chroma Technology, Brattleboro, VT) was added, but rotated by 90° with respect to the original cube. This arrangement allows the laser beam to enter the microscope turret from the side, while at the same time preserving the epifluorescence microscope capability of the instrument. We used epifluorescence to locate and position cells and subsequently switched to two-photon microscopy for fluctuation experiments. All measurements were performed with an excitation wavelength of 905 nm, and the power at the sample was 0.6 mW. Photon counts were detected with an avalanche photodiode (APD) (Perkin–Elmer, SPCM-AQ-141). The data acquisition time for individual measurements is ≈50 s. The TTL-output of the APD unit is connected to a PCI data acquisition card (ISS, Champaign, IL), which stores the complete sequence of photon counts by using a sampling frequency of 20 kHz. The recorded photon counts were stored and analyzed with programs written for idl 5.4 (Research Systems, Boulder, CO).
Construction of Expression Vectors, Generation of Stable Cell Lines, and Cell Measurements. A tandem dimeric EGFP (EGFP2) was constructed by cloning EGFP amplified from PCR, and a synthetic linker that encodes 12 aa (GHGTGSTGSGSS) into the pEGFP-C1 plasmid (Clontech) (13). RXR-LBD was amplified from mouse RXRβ (GenBank accession no. X66224) or from human RXRα (GenBank accession no. NM_002957) with a 5′ primer that encodes a XhoI restriction site and a 3′ primer that encodes a EcoRI site. RAR-LBD was amplified from mouse RARα, and TR4 was amplified from mouse (14). RXR-LBD, RAR-LBD, and TR4 cDNAs were subsequently cloned into pEGFP-C1 plasmid. All sequences were checked by automatic sequencing.
COS-1 cells were obtained from American Type Culture Collection and maintained in 10% charcoal/dextran treated FBS (HyClone) and DMEM (without phenol red) media. Transfections were carried out by using polyfect transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. G418-resistant cell clones were selected by culturing cells in media with 250 μg/ml gentamicin sulfate (Mediatech, Herndon, VA) and measured the second week after transfection. The ligands 9-cis- and all-trans-retinoic acid were obtained from Sigma.
Cells were subcultured into eight-well coverglass chamber slides (Nagle Nunc International, Rochester, NY) 24 h before measurements. The growth media was exchanged to Dulbecco's PBS with calcium and magnesium (Biowhittaker, Walkersville, MD) before starting measurements. Hormones were added at 1 μM concentration. FFS measurements were performed 30 min after the addition of ligand. All measurements are carried out in the cell nucleus.
Control Experiments. To avoid problems associated with the presence of endogenous receptors, which would introduce a background of unlabeled receptors into our experiments, we choose COS-1 cells because of their extreme low endogenous concentration of RXR and RAR. We performed Western blot analysis on COS-1 cells to confirm this. Fig. 1 shows the Western blot results of COS-1 cells after transfection with RXR-LBD-EGFP or RXR-EGFP, and nontransfected cells as control. Western blot analysis clearly establishes the presence of RXR-LBD-EGFP and RXR-EGFP, but fails to detect endogenous RXR. Similarly, Western blot analysis failed in detecting endogenous RAR (data not shown). We conclude that the endogenous concentration of both RXR and RAR is too low as to add a background species for the experiments conducted in this study.
Fig. 1.

Western blot analysis of RXR-EGFP and RXR-LBD-EGFP and endogenous RXR from COS-1 cells. COS-1 cells transfected with RXR-EGFP or RXR-LBD-EGFP and nontransfected cells (control) are analyzed. Samples of whole cell lysate (RXR-EGFP, 60 μg; RXR-LBD-EGFP, 60 μg; control, 120 μg) were subjected to SDS/PAGE, transferred to poly(vinylidene difluoride) membrane, and followed by reaction with anti-RXR antibody (Affinity Bioreagents, Golden, CO). The RXR-LBD-EGFP sample displays degraded receptor with lower molecular weight that was not observed in the control.
We also performed control experiments to ensure that photobleaching is absent in our experiments. Photobleaching must be avoided because it produces a nonfluorescent background species that interferes with brightness analysis. Transfected cells in the microscope field are selected by switching the instrument for 1 or 2 s into epifluorescence mode. The bleaching lifetime of our epifluorescence setup was determined to be on the order of 10 min by observing the exponential fluorescence decay of selected cells as a function of illumination time. Thus, photobleaching before the two-photon measurement is negligible.
The fluorescence intensity of two-photon excitation depends quadratically on the laser power as long as saturation and photobleaching effects are negligible. We use a very low excitation power of 0.6 mW at the sample and confirmed that its value is well within the quadratic power dependence. For example, doubling of the excitation power resulted in a 4-fold increase of the fluorescence signal. Controls performed on selected cells verified the quadratic power dependence in all cases (data not shown) and confirm the absence of photobleaching under our experimental conditions.
Theory and Data Analysis. Molecular brightness was determined by analyzing the data using PCH and moment analysis (2, 3). PCH is capable of resolving a binary mixture of molecules that differ in their brightness if the signal statistics is sufficient (5). Laser light of high intensity can lead to photo stress and photobleaching in cells. To avoid such artifacts, the laser light intensity was kept at relatively low levels. No loss in cell viability or increase in autofluorescence was detected under our experimental conditions even after prolonged exposure to laser light. However, the low excitation power reduces the molecular brightness of EGFP to a few thousand counts per second per molecule (cpsm). A direct resolution of a monomer/dimer equilibrium via PCH is not feasible under the given experimental conditions. Consequently, FFS experiments in cells determine an apparent brightness that is a combination of the brightness of all participating fluorescent species. The dependence of the apparent brightness is given by a nonlinear combination of the brightness εi and the occupation number Ni of each species (15),
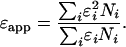 |
[1] |
The apparent brightness of a monomer/dimer mixture will lie between the brightness values of the monomer, ε1, and the dimer, ε2,
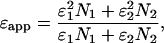 |
[2] |
where N1 and N2 are the average number of molecules of monomer and dimer in the observation volume.
PCH and moment analysis lead to erroneous results at high fluorophore concentrations, because detector deadtime changes the signal statistics. Every detector exhibits a dead time after registering a photon event, during which the system cannot detect further photons. High photon count bursts are more strongly affected by deadtime than low count bursts. This effect leads to an effective lowering of the molecular brightness. To address this problem, we recently developed a new theory for PCH and moment analysis that takes deadtime into account (11). The only additional parameter needed is the deadtime τD of the detector, which we determined to be τD = 50 ns. This theory allowed us to analyze data at concentrations as high as 10 μM. The upper concentration limit corresponds to a photon count rate of ≈2 × 106 cps and reflects the limit of most photon counting experiments.
The concentration of expressed EGFP protein in the cell was determined in the following way. The measured photon count rate 〈k〉 is the product of the molecular brightness and the number of molecules in the observation volume, 〈k〉 = εN (2). If the fluorescence of the protein is not quenched in the fusion protein and upon protein association, then the number of proteins labeled with EGFP is given by N = 〈k〉/ε, where the molecular brightness ε of EGFP has been determined in an independent experiment using the same experimental setup. We calibrate the observation volume by measuring an EGFP solution of known concentration, which we determined by absorption spectroscopy using an extinction coefficient of 53,000 M–1·cm–1 at 489 nm (16). On average, one molecule within the excitation volume corresponds to a concentration of 23 nM. The concentration is therefore calculated by multiplying the occupation number N with 23 nM per molecule. The influence of autofluorescence on brightness analysis is even at the lowest EGFP concentrations measured negligible. This finding is in agreement with a previous study of brightness analysis of EGFP in HeLa cells (8).
Results
Brightness of EGFP as a Function of Concentration. We first characterize the properties of our fluorescent marker, EGFP. COS cells were transiently transfected with EGFP. We identified and aligned transfected cells by using a conventional fluorescence microscope setup and switched to two-photon excitation for fluorescence fluctuation experiments. We determined the molecular brightness by either performing moment or PCH analysis. For PCH analysis, the histogram of the photon counts is calculated from the raw data and subsequently fit by using the PCH model (Fig. 2). The data and the fitted histogram are in good agreement with a reduced χ2 of 1.1. The residuals of the fit are also displayed in Fig. 2. In this experiment, we recovered an EGFP brightness of 4,100 cpsm with a total of 208 molecules inside the excitation volume. The expression level of EGFP varies from cell to cell and is conveniently monitored by the fluorescence intensity, which is proportional to the protein concentration. By picking cells with different expression levels, it is possible to probe the concentration dependence of the molecular brightness. Fig. 3 shows the molecular brightness of EGFP measured on 40 different cells as a function of the photon count rate of the fluorescence intensity. We converted the intensity axis into an EGFP-concentration axis by using the linear relationship between photon count rate and the number of EGFP molecules as outlined in Materials and Methods. We also corrected the relatively minor influence of deadtime on the average fluorescence intensity (11). The molecular brightness of EGFP is constant throughout the measured concentration range as expected, because the photophysics of the fluorophore is independent of concentration. The average brightness of EGFP (solid line in Fig. 3) is 4,000 cpsm with a standard deviation of 300 cpsm. The concentration range covered by the experiments is from ≈50 nM to 5 μM. The upper concentration limit corresponds to ≈1 × 106 cps. The minimum expression level of EGFP measured in the cell population gives the lower concentration limit. We measured EGFP and a fluorescent dye solution under the same brightness conditions as present in the cellular experiments (data not shown). Analysis of these data resulted in the same standard deviation as was found in the intracellular measurements, indicating that the cellular environment does not introduce additional experimental uncertainty as compared with in vitro measurements.
Fig. 2.

Representative PCH of EGFP measured in the nucleus of a COS cell. The power at the sample is 0.6 mW. The solid line represents a fit to a single species PCH model with a detector deadtime of 50 ns. The fit recovers a particle number N of 208 and a brightness ε of 4,100 cpsm. (Lower) The normalized residuals of the fit. The reduced χ2 for this fit is 1.1.
Fig. 3.

Molecular brightness of EGFP and EGFP2 as a function of the average photon count rate and protein concentration. The brightness of EGFP (circles) and EGFP2 (triangles) are concentration independent. Each data point represents the brightness measured in a different cell expressing either EGFP or EGFP2. Note that the brightness of EGFP2 is twice the brightness of EGFP alone. The concentration axis shows the total protein concentration expressed in terms of monomeric EGFP concentration. The average molecular brightness of EGFP (solid line) is 4,000 cpsm with a standard deviation of 300 cpsm. The average molecular brightness of the tandem protein EGFP2 (dashed line) is 7,800 cpsm with a standard deviation of 508 cpsm.
Characterizations of a Tandem Dimer of EGFP. Previously, we compared the molecular brightness of EGFP in vivo and in vitro and found that the molecular brightness of EGFP is identical in the different environments (8), suggesting that brightness might serve as a robust parameter to monitor oligomerization processes. An EGFP tandem dimer was constructed to mimic the conditions of dimeric proteins in cells. COS cells were transiently transfected with the tandem fusion protein EGFP2. Fig. 3 shows the molecular brightness of EGFP2 for 27 cells as a function of protein concentration. The molecular brightness of EGFP2 is concentration independent, as expected. The average molecular brightness of EGFP2 is 7,800 cpsm with a standard deviation of 508 cpsm, whereas the brightness of EGFP is 4,000 cpsm with a standard deviation of 300 cpsm. The figure demonstrates that the tandem protein is clearly distinguishable from monomeric EGFP. In addition, the molecular brightness value of the dimeric tandem protein is within experimental uncertainty twice the value of the molecular brightness of monomeric EGFP. We observe no quenching of the fluorescence in the tandem EGFP construct. This finding suggests that the molecular brightness of EGFP is a robust parameter for detecting protein oligomerization.
Oligomerization of Nuclear Receptors. Dimerization of nuclear receptors is known to play a crucial role in gene regulation (17). RXR plays a prominent role among these receptors because of its potential to form homo- and hetero-complexes (18, 19). We decided to probe its ligand-binding domain RXR-LBD in the nucleus of cells by using brightness analysis, because in vitro data on RXR-LBD are available for comparison (20–24). We performed measurements on COS cells transfected with RXR-LBDα-EGFP that are G-418 resistant and graphed the molecular brightness as a function of protein concentration (Fig. 4A). The brightness of the protein is not constant, but increases as a function of protein concentration. The increase in the apparent molecular brightness indicates a change in the oligomeric composition of the protein solution. At low protein concentrations, the molecular brightness of RXR-LBD-EGFP is the same as the brightness of EGFP measured in a separate experiment (see Fig. 4A), indicating that any protein complex formed with RXR-LBD contains a monomeric unit of RXR-LBD. The increase of the molecular brightness with increasing protein concentration on the other hand requires the formation of homooligomeric protein complexes. If we assume a simple monomer/dimer equilibrium for RXR-LBD, the increase in the brightness signifies an increase in the formation of protein complexes that contain homodimeric RXR-LBD. We expect for the limiting case of purely dimeric RXR-LBD protein complexes an increase of the molecular brightness by a factor of two compared with the brightness of EGFP alone. We conclude that the protein has not reached a purely dimeric composition in the experimentally accessible concentration range. We also performed experiments on RXR-LBDβ-EGFP and recovered the same concentration-dependent brightness response (Fig. 4A).
Fig. 4.

Titration of RXR-LBDα-EGFP and RXR-LBDβ-EGFP in the absence (A) and presence (B) of 1 μM 9-cis-retinoic acid. The receptor concentration is calculated based on the brightness of EGFP (solid line). (A) The apparent brightness εapp increases as a function of protein concentration and indicates the formation of homocomplexes. The response of RXR-LBDα-EGFP and RXR-LBDβ-EGFP are identical. (B) Addition of ligand promotes the formation of homodimers.
The nuclear receptor RXR is activated by its native ligand, 9-cis-retinoic acid. We added the ligand to the cell culture to measure the oligomerization state of the receptor upon ligand activation. Fig. 4B shows the concentration-dependent molecular brightness of RXR-LBDα-EGFP and RXR-LBDβ-EGFP in the presence of ligand. Addition of ligand results in an increase of the molecular brightness of RXR-LBD-EGFP at every measured concentration. In other words, the addition of ligand promotes the formation of RXR-LBD homocomplexes. Both the α and β form of the protein result in identical brightness changes as a function of protein concentration. The apparent molecular brightness increases with protein concentration and reaches a limiting value that is twice the brightness of EGFP. This finding suggests that RXR-LBD forms homodimers and that at high concentrations all RXR-LBD proteins are homodimers. Homooligomers larger than dimers would result in molecular brightness values that exceed twice the brightness of EGFP, which is not observed experimentally. Nevertheless, we cannot rule out the presence of a small fraction of homooligomers larger than dimers. Although the brightness of larger complexes exceeds that of dimers, its contribution to raising the apparent brightness of the mixture at low concentrations will be too small as to be detected experimentally.
Another well characterized nuclear receptor is RAR. In vitro studies of RAR-LBD shows that it does not form homocomplexes (23, 24). We study the behavior of RAR-LBD-EGFP in COS cells. Fig. 5 displays the molecular brightness of the fusion protein as a function of its concentration. The molecular brightness of RAR-LBD is constant over the experimentally studied concentration range. Its average brightness value is identical to the brightness of EGFP, which was determined in an independent experiment. Thus, RAR-LBD is monomeric in any protein complex formed in the cell nucleus. In vitro and in vivo experiments are in agreement with one another. RAR can be activated by either 9-cis- or all-trans-retinoic acid. We tested the response of RAR-LBD to all-trans-retinoic acid. The molecular brightness of RAR-LBD is unchanged and constant over the concentration range studied. Thus, RAR-LBD exists in monomeric form within any protein complex both in the presence and absence of its ligand. We also study an orphan receptor, TR4. Fig. 5 shows the molecular brightness of TR4-EGFP as a function of protein concentration. The brightness of TR4 at the lowest concentration measured is slightly larger than the brightness of EGFP, indicating the presence of homocomplexes of TR4. The molecular brightness of TR4 grows with increasing protein concentration and reaches a value that is close to twice the brightness of EGFP at the highest concentrations measured. This finding suggests that TR4 exists as equilibrium of monomers and homodimers in the cell nucleus. Our result is in accordance with an in vitro study that concludes that the dimeric form of TR4 is the active form of the receptor (14).
Fig. 5.

Titration of RAR-LBD-EGFP and TR4-EGFP nuclear receptors in COS cells. The receptor concentration is calculated based on the brightness of EGFP (solid line). The apparent brightness of RAR-LBD-EGFP in the absence (•) and presence (⊙) of all-trans-retinoic acid is identical to the brightness of EGFP. The brightness of TR4 (□) increases with protein concentration and reaches a brightness that is twice the value of EGFP.
Discussion
Probing cellular functions and mechanisms on a molecular level is crucial for the development of a quantitative understanding of cellular processes. We introduced a spectroscopic tool for observing oligomerization of proteins in the intracellular environment. Molecular brightness analysis of EGFP by FFS provides a useful and robust technique. By using an improved theory that takes detector deadtime into account, we are able to measure at much higher protein concentrations than previously possible. We report quantitative brightness measurements over two orders of magnitude in protein concentration. The highest protein concentration measured is ≈10 μM; the lowest concentration is ≈50 nM, which corresponds to the lowest expression level detected in the transfected cell population. The wide concentration range accessible by the technique allows to titrate protein complexes in cells using molecular brightness as marker for changes in the oligomerization state. The molecular brightness value of EGFP is a remarkably robust parameter, and its value is the same under in vitro and in vivo conditions (8). No quenching of the molecular brightness of EGFP fusion proteins was detected. EGFP2 resulted in twice the molecular brightness compared with EGFP alone. We conclude that brightness is a sensitive marker to observe changes in the oligomerization state of proteins. Here we specifically probed the formation of homocomplexes. Spectroscopic detection of homocomplexes in cells is a difficult problem. Brightness analysis offers a promising and quantitative approach to such studies.
We also determined the diffusion coefficient from the autocorrelation function in addition to PCH analysis. The diffusion coefficient of both RXR-LBD and RAR-LBD decreases upon ligand binding (data not shown), indicating that the protein associates with other protein complexes. However, a quantitative structural interpretation of the diffusion coefficient is complicated, because its value depends not only on the size and shape of the protein complex, but also on its interactions with the cellular environment, which are not well understood. Molecular brightness appears to be a robust parameter and allows the detection of homocomplex formation. The molecular brightness of RAR-LBD corresponds to a monomer, whereas the molecular brightness of RXR-LBD increases with concentrations and reaches the brightness of a homodimer upon addition of ligand. We expect that this technique will detect the formation of larger oligomers, such as tetramers, as well.
A quantitative interpretation of brightness experiments is possible by fitting the brightness curves to a titration model together with Eq. 1. A monomer/dimer equilibrium requires an increase of protein concentration by approximately three orders of magnitude to change the dimer population from 10% to 90% (25). Our experimental brightness curves of TR4 and RXR-LBD in the presence of ligand change from a monomer to a dimer in less than three orders of magnitude of protein concentration, indicating the presence of other biomolecules that interact with the protein.
Brightness analysis rests on the assumption that all proteins of interest are fluorescently labeled. Photobleaching or endogenous proteins introduce a nonfluorescent species that competes with labeled protein in forming binding complexes. The presence of a nonfluorescent species decreases the observed brightness and distorts the measured binding curve. For example, photobleaching of dimeric EGFP2 would establish a dimmer species with one of the two chromophores bleached. In this case, the apparent brightness would fall below the value expected for the dimeric complex. To avoid complications introduced by a nonfluorescent species, we selected experimental conditions where endogenous receptor and photobleaching are absent.
We performed measurements on proteins in the intracellular environment to evaluate the potential of brightness analysis for studying protein oligomerization. Oligomerization processes play an important functional role in signaling pathways and other regulation mechanisms of cells. In this study, we probed the self-association of nuclear receptors by brightness analysis. For RAR-LBD, the in vitro measurements are in agreement with our in vivo results (23, 26). The receptor does not form homocomplexes. However, a very tight tetramer of RXR-LBD with a dissociation coefficient Kd of a few nM has been reported for an in vitro study (27). The tetramer dissociates in vitro into a monomer/dimer mixture upon addition of ligand (23). We could not confirm the existence of the tetramer in the absence of ligand, but observed a monomer/dimer mixture. Upon ligand binding, the monomer/dimer equilibrium shifts to a lower Kd, which means that 9-cis-retinoic acid induces homodimer formation.
Thus, in vitro and in vivo experiment sometimes lead to different results. Such differences are expected, because in vivo experiments probe a protein in a complex environment, where other biomolecules potentially compete or assist with protein binding. Test tube experiments, on the other hand, probe protein interactions in isolation with only one or two protein species present in most experiments. A combination of in vitro and in vivo experiments seems a powerful approach to provide a quantitative characterization of protein interactions, which is necessary for the development of molecular models of cellular function.
The development of quantitative spectroscopic tools for probing biomolecules in living cells is a challenging and timely problem. Here we demonstrate the use of brightness analysis based on fluorescence fluctuation spectroscopy for probing homooligomerization processes. Quantitative brightness analysis in cells covering a wide concentration range seems quite realistic and our results are very encouraging. We demonstrated the technique by using three different proteins. However, the technique itself is applicable to other proteins as well. For example, brightness analysis might open new ways to probe protein complex formation of signaling pathways in cells. Although we concentrated on homocomplex formation, our technique can be extended to study heterocomplex interactions. By using two fluorescent proteins with different emission colors and applying dual color detection, we will be able to detect heterocomplex formation. One goal of proteomics is the characterization of all protein interactions in cells. This is a challenging endeavor and our technique could aid in addressing specific questions about protein interactions. In conclusion, fluorescence fluctuation spectroscopy is a powerful and attractive approach toward quantitative biology in the cellular environment.
Acknowledgments
This work was supported by National Institutes of Health Grant GM64589 and the National Science Foundation Grant MCB-0110831. Y.C. acknowledges support by a postdoctoral fellowship from the National Institutes of Health (5F32GM020853).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FFS, fluorescence fluctuation spectroscopy; cpsm, counts per second per molecule; EGFP, enhanced GFP; EGFP2, tandem EGFP; PCH, photon-counting histogram; RXR, retinoid X receptor; RAR, retinoic acid receptor; LBD, ligand-binding domain; TR4, testicular receptor 4.
References
- 1.Magde, D., Elson, E. & Webb, W. W. (1972) Phys. Rev. Lett. 29, 705–708. [Google Scholar]
- 2.Chen, Y., Müller, J. D., So, P. T. & Gratton, E. (1999) Biophys. J. 77, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian, H. & Elson, E. L. (1990) Proc. Natl. Acad. Sci. USA 87, 5479–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kask, P., Palo, K., Ullmann, D. & Gall, K. (1999) Proc. Natl. Acad. Sci. USA 96, 13756–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Müller, J. D., Chen, Y. & Gratton, E. (2000) Biophys. J. 78, 474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berland, K. M., So, P. T. C. & Gratton, E. (1995) Biophys. J. 68, 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Politz, J. C., Browne, E. S., Wolf, D. E. & Pederson, T. (1998) Proc. Natl. Acad. Sci. USA 95, 6043–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, Y., Müller, J. D., Ruan, Q. & Gratton, E. (2002) Biophys. J. 82, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denk, W., Strickler, J. H. & Webb, W. W. (1990) Science 248, 73–76. [DOI] [PubMed] [Google Scholar]
- 10.Schwille, P., Haupts, U., Maiti, S. & Webb, W. W. (1999) Biophys. J. 77, 2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hillesheim, L. N. & Müller, J. D. (2003) Biophys. J. 85, 1948–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giguere, V. (1999) Endocr. Rev. 20, 689–725. [DOI] [PubMed] [Google Scholar]
- 13.Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A. & Tsien, R. Y. (2002) Proc. Natl. Acad. Sci. USA 99, 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee, C. H., Chinpaisal, C. & Wei, L. N. (1998) J. Biol. Chem. 273, 25209–25215. [DOI] [PubMed] [Google Scholar]
- 15.Chen, Y., Müller, J. D., Tetin, S. Y., Tyner, J. D. & Gratton, E. (2000) Biophys. J. 79, 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller, J. D., Chen, Y. & Gratton, E. (2003) Methods Enzymol. 361, 69–92. [DOI] [PubMed] [Google Scholar]
- 17.Chambon, P. (1996) FASEB J. 10, 940–954. [PubMed] [Google Scholar]
- 18.Yu, V. C., Delsert, C., Andersen, B., Holloway, J. M., Devary, O. V., Naar, A. M., Kim, S. Y., Boutin, J. M., Glass, C. K. & Rosenfeld, M. G. (1991) Cell 67, 1251–1266. [DOI] [PubMed] [Google Scholar]
- 19.Zhang, X. K., Lehmann, J., Hoffmann, B., Dawson, M. I., Cameron, J., Graupner, G., Hermann, T., Tran, P. & Pfahl, M. (1992) Nature 358, 587–591. [DOI] [PubMed] [Google Scholar]
- 20.Chen, H. & Privalsky, M. L. (1995) Proc. Natl. Acad. Sci. USA 92, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kersten, S., Kelleher, D., Chambon, P., Gronemeyer, H. & Noy, N. (1995) Proc. Natl. Acad. Sci. USA 92, 8645–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, B. C., Wong, C. W., Chen, H. W. & Privalsky, M. L. (1997) J. Biol. Chem. 272, 9860–9867. [DOI] [PubMed] [Google Scholar]
- 23.Chen, Z. P., Iyer, J., Bourguet, W., Held, P., Mioskowski, C., Lebeau, L., Noy, N., Chambon, P. & Gronemeyer, H. (1998) J. Mol. Biol. 275, 55–65. [DOI] [PubMed] [Google Scholar]
- 24.Egea, P. F., Rochel, N., Birck, C., Vachette, P., Timmins, P. A. & Moras, D. (2001) J. Mol. Biol. 307, 557–576. [DOI] [PubMed] [Google Scholar]
- 25.Weber, G. (1992) Protein Interactions (Chapman and Hall, New York).
- 26.Chen, Y., Kerimo, A., Khan, S. & Wei, L. N. (2002) Mol. Endocrinol. 16, 2528–2537. [DOI] [PubMed] [Google Scholar]
- 27.Dong, D. & Noy, N. (1998) Biochemistry 37, 10691–10700. [DOI] [PubMed] [Google Scholar]