

Abstract
Objectives
The Pin1 prolyl isomerase acts in concert with proline-directed protein kinases to regulate function of protein substrates through isomerization of peptide bonds that link phosphoserine or phosphothreonine to proline. We sought to determine whether Pin1 interacts with endothelial nitric oxide synthase (eNOS) in endothelial cells in a manner that depends on proline-directed phosphorylation of the eNOS enzyme and whether this interaction influences basal or agonist-stimulated eNOS activity.
Methods and Results
Inhibitors of the ERK 1/2 MAP kinases inhibit proline-directed phosphorylation of eNOS at serine 116 (S116) in bovine aortic endothelial cells (BAECs). Moreover, eNOS and Pin1 can be co-immunoprecipitated from BAECs only when S116 is phosphorylated. In addition, phospho-mimetic S116D eNOS, but not wild-type eNOS, can be co-immunoprecipitated with Pin1 co-expressed in COS-7 cells. Inhibition of Pin1 in BAECs by juglone or by dominant negative Pin1 increases basal and agonist-stimulated NO release from the cells while overexpression of wild-type Pin1 in BAECs suppresses basal and agonist-stimulated NO production. Overexpression of wild-type Pin1 in intact aortae also reduces agonist-induced relaxation of aortic rings.
Conclusions
Our results demonstrate a novel form of eNOS regulation in endothelial cells and blood vessels through S116 phosphorylation-dependent interaction of eNOS with Pin1.
Keywords: eNOS, Pin1, proline-directed phosphorylation, prolyl isomerase, ERK 1/2
Introduction
Endothelial nitric oxide synthase (eNOS), through generation of the vasodilating and vasculoprotective molecule, nitric oxide (NO), plays a key role in blood pressure control and in protection from atherosclerotic lesion formation. eNOS is regulated posttranslationally through the alternative mechanisms of reversible phosphorylation and protein-protein interactions 1, 2. eNOS regulation by phosphorylation is complex and, to date, seven specific sites of regulatory phosphorylation have been identified in bovine eNOS at tyrosine 83 (Y83), serine 116 (S116), threonine 497 (T497), serine 617 (S617), serine 635 (S635), tyrosine 659 (Y659), and serine 1179 (S1179). Equivalent sites are found in human eNOS at Y81, S114, T495, S615, S633, Y657, and S1177. Among these various phosphorylation sites, S1179/S1177 has been particularly well-documented as having an important role in positively modulating eNOS activity2. The eNOS protein-protein interactome is also very complex with many proteins that are known to interact with eNOS either directly or indirectly to influence eNOS activity or subcellular localization. Among these protein-protein interactions, one of the best studied examples is that of caveolin-1. Caveolin-1 binds directly to eNOS and tonically inhibits its catalytic activity1.
Regulation of protein function by reversible phosphorylation is generally thought to occur through direct effects of phosphorylation on the three-dimensional conformation of the phosphorylated protein. However, there is an alternative mechanism by which phosphorylation can affect protein function. This mechanism involves phosphorylation-dependent conformational changes that are induced by the Pin1 prolyl isomerase 3. Pin1 catalyzes the cis to trans isomerization of peptide bonds that link phosphoserine or phosphothreonine to proline. Conformational changes induced by this mechanism are initiated by proline-directed phosphorylation of serines or threonines immediately preceding proline in substrate proteins by one of a large family of proline-directed protein kinases which include the cyclin-dependent kinases (CDKs), the mitogen-activated protein kinases (MAPKs), and glycogen synthase kinase 3 (GSK-3). Subsequent to proline-directed phosphorylation, Pin1 binds to the phosphoserine or phosphothreonine and catalyzes an isomerization reaction at the adjacent peptide bond that can have profound effects on protein conformation and hence on protein function.
Pin1 (Protein Interacting with NIMA (never in mitosis A)) was first discovered and cloned in 19964. Pin1 was initially found to be essential to regulation of mitosis. Subsequently, a large body of literature has been produced implicating Pin1 as also being important in cancer and in Alzheimer’s disease. For example, Pin1 is overexpressed in many human cancers where it functions as a critical enzyme in multiple oncogenic pathways5. In contrast, Pin1 is downregulated in degenerative neurons of Alzheimer’s disease patients, which contributes to age-dependent neurodegeneration 6. A recent report has shown that the inducible nitric oxide synthase (iNOS), which is not expressed in endothelial cells under basal conditions, is negatively regulated by Pin1 in endothelial cells after induction of iNOS by LPS and IFNγ 7. It should be noted that the Pin1 referred to here is distinct from another similarly named protein, PIN, the small protein (89 amino acids) inhibitor of neuronal NOS (nNOS) that inhibits nNOS acitivity by binding to this enzyme and preventing its dimerization 8. In the present study, we have investigated whether eNOS may be regulated in endothelial cells and in blood vessels by the Pin1 prolyl isomerase and whether such regulation occurs in a manner that depends on site-specific, proline-directed phosphorylation of the eNOS enyzme.
Methods
Cell Culture
Primary cultures of bovine aortic endothelial cells (BAECs) were purchased from VEC Technologies Inc. and were used for experiments between passages 2 and 6.
Immunoprecipitation and Immunoblotting
Described in detail in the Online Supplement.
Transfection of COS-7 Cells
COS-7 cells were transfected with various cDNA constructs cloned into the pcDNA3.1/V5-His A,B,C plasmid vector from Invitrogen. DNA-Lipofectamine™2000 complexes (Invitrogen) were added directly to the cells in culture medium according to the manufacturer’s instructions.
DNA Cloning of Pin1
Pin1 cDNA was cloned from human aortic total RNA (United States Biological) by reverse transcriptase-polymerase chain reaction (RT-PCR) using a Phusion RT-PCR Kit from New England Biolabs. Primers used are listed in the Online Supplement.
Site-Directed Mutagenesis
Site-directed mutagenesis of wild-type Pin1 cDNA was performed using a QuickChange Site-Directed Mutagenesis kit (Stratagene) according to the manufacturer’s instructions. Primers used are listed in the Online Supplement. Cloning of wild-type bovine eNOS and mutagenesis of the wild-type sequence to produce S116D eNOS were described previously 9, 10.
Construction, Purification, and Transduction by Adenoviruses
Adenoviruses were generated by the procedure of He et al. 11as described in the Online Supplement.
Measurement of NO Release
NO release was determined by a chemiluminescence assay that measures nitrite levels in conditioned media 12. Media were deproteinized by ethanol precipitation and samples containing nitrite were refluxed in glacial acetic acid containing 65 mmol/L sodium iodide. Under these conditions, nitrite is quantitatively reduced to NO. The resultant NO was purged from the reaction cell with 100% nitrogen and directly quantified after reaction with ozone in an NO-specific chemiluminescence analyzer (Sievers, Model 2801).
Transduction of Mouse Aortic Endothelium with Recombinant Adenoviruses and Determination of Vascular Reactivity of Transduced Vessels
Transduction of mouse aortae with adenoviruses and determination of vascular reactivity of transduced vessels was carried out essentially as reported previously 13and is described in detail in the Online Supplement.
Statistical Analysis
All data are representative of at least 3 separate experiments and are reported as means ± S.E. Overall differences were analyzed by 2-way ANOVA and by repeated measures ANOVA. Differences were considered significant at p<0.05.
Results
Identification of ERK 1/2 as Responsible for eNOS Phosphorylation at S116 in Endothelial Cells
Proline-directed phosphorylation, requiring a proline at the P+1 position, is a prerequisite for Pin1 interaction with (and consequent Pin1-catalyzed isomerization of) Pin1 substrates. Of the five known serine/threonine phosphorylation sites in bovine eNOS, only S116 conforms to the proline at P+1 requirement. Because it has been reported previously that the ERK 1/2 MAP kinase inhibits eNOS activity through phosphorylation of an unidentified site in the eNOS enzyme 14, we considered the possibility that ERK 1/2, which has an absolute requirement for proline in the P+1 position 15, is responsible for S116 phosphorylation of eNOS in endothelial cells under basal conditions. Bovine aortic endothelial cells (BAECs) were either not treated or treated with the selective MEK 1/2 (and hence ERK 1/2 ) inhibitor, PD98059 (50 μM for 1 h) 16. Cells were lysed and lysates were immunoblotted with phospho-S116-specific and nonphospho-specific anti-eNOS antibodies. As shown in Figure 1A, PD98059 blocked S116 phosphorylation of eNOS in BAECs under basal conditions without affecting levels of eNOS protein expression. Densitometric and statistical analysis of blots from repeat experiments showed that S116 phosphorylation was reduced by a statistically significant 87±5% (mean ±S.E., n = 3, P<0.05) by PD98059 treatment. Experiments were also carried out with a structurally distinct selective MEK 1/2 inhibitor, U0126 17. Treatment with this inhibitor (10 μM for 1 h) also significantly reduced basal S116 phosphorylation (70±2%, mean±S.E., n=3, P<0.05) without altering total eNOS expression levels (Figure 1B).
Figure 1.
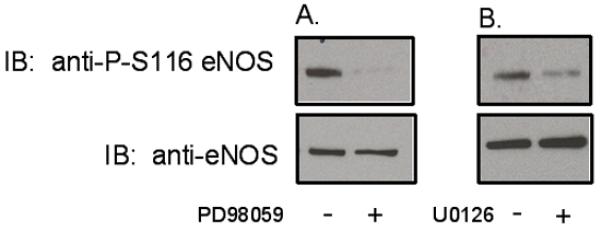
Effects of ERK 1/2 inhibition on basal phosphorylation of eNOS at S116 in BAECs. BAECs were either not treated or treated with: A, PD98059 (50 μM for 1 h) or B, U0126 (10 μM for 1 h). Cells were lysed an lysates were immunoblotted (IB) with phospho-S116-specific and nonphospho-specific anti-eNOS antibodies. Similar results were obtained in at least 3 separate experiments.
eNOS Interacts with Pin1 in Endothelial Cells in a Phosphorylation-Dependent Manner
To determine whether eNOS interacts with Pin1 in endothelial cells, we lysed BAECs and subjected the lysates to immunoprecipitation with anti-eNOS and anti-Pin1 antibodies. Anti-eNOS immunoprecipitates were then immunoblotted with anti-Pin1 antibody and anti-Pin1 immunoprecipitates were immunoblotted with anti-eNOS antibody. As shown in Figure 2A, eNOS (130kDa) was specifically coimmunoprecipitated from endothelial cell lysates by the anti-Pin1 antibody. In addition, Pin1 (18kDa) was specifically coimmunoprecipitated by the anti-eNOS antibody (Figure 2B). Importantly, we also determined whether blockade of S116 phosphorylation of eNOS in BAECs by PD98059 is associated with reduced complex formation of eNOS and Pin1. BAECs were either not pretreated or pretreated with PD98059 (50μM for 1 h) prior to immunoprecipitation from lysates of eNOS with anti-eNOS antibody. Immunoprecipitated proteins were then immunoblotted with anti-eNOS and anti-Pin1 antibodies. As shown in Figure 2C, co-immunoprecipitation of the two proteins was clearly detectable from untreated cells but was significantly reduced from cells in which S116 phosphorylation was blocked by PD98059 treatment. Densitometric and statistical analysis showed an 81±5% decrease (mean±S.E., n=3, P<0.05) in complex formation of eNOS and Pin1 in PD98059-treated cells.
Figure 2.
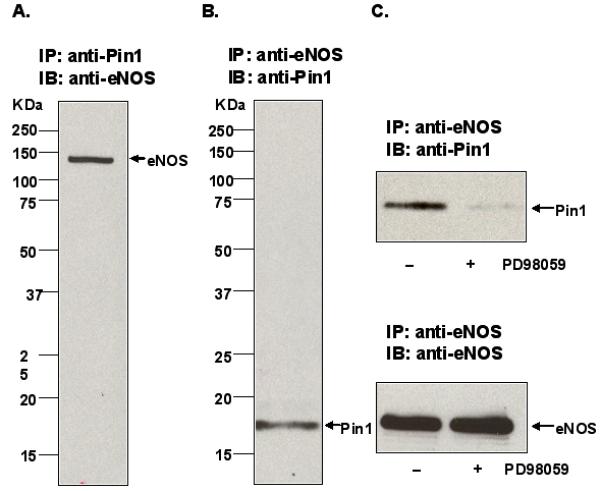
Phosphorylation-dependent association of eNOS and Pin1 in BAECs. A, BAEC lysates were immunoprecipitated (IP) with anti-Pin1 antibody and immunoblotted (IB) with anti-eNOS antibody. B, BAEC lysates were immunoprecipitated (IP) with anti-eNOS antibody and immunoblotted (IB) with anti-Pin1 antibody. C, BAECs were either not pretreated or pretreated with PD98059 (50 μmol/L for 1 h) prior to immunoprecipitation (IP) with anti-eNOS antibody and immunoblotting (IB) with either anti-Pin1 or anti-eNOS antibodies. Results are representative of at least 3 different experiments.
The phosphorylation-dependence of eNOS interactions with Pin1 was further examined using a different experimental model system. COS-7 cells, which ordinarily express no endogenous eNOS, were transfected with either wild-type bovine eNOS (WT eNOS) or a phospho-S116-mimetic form of bovine eNOS (S116D eNOS) in which S116 was changed to an aspartate by site-directed mutagenesis. In addition, full-length human Pin1 cDNA was cloned from human aortic total RNA using reverse transcriptase-polymerase chain reaction (RT-PCR). Pin1 cDNA was then used to co-transfect eNOS-transfected cells. Pin1 was expressed as a Pin1/V5 fusion protein to allow immunoprecipitation of the protein with anti-V5 antibody. Cells were lysed, Pin1/V5 protein was immunoprecipitated with anti-V5 antibody, and immunoprecipitates were immunoblotted with anti-eNOS antibody. Figure 3A shows that, while the phospho-mimetic S116D eNOS was bound by the Pin1/V5 fusion protein in these experiments, WT eNOS was clearly not bound. In addition, we also confirmed that no endogenous S116 phosphorylation of WT eNOS (as occurs in BAECs and promotes Pin1 binding) occurs in WT eNOS-transfected COS-7 cells. Further, we confirmed that equal amounts of Pin1/V5 were immunoprecipitated from each co-transfection condition (Figure 3B). Thus it appears that Pin1 interactions with eNOS are dramatically increased by mimicking phosphorylation of S116, likely due to an increased binding affinity of the phospho-mimetic eNOS protein for Pin1.
Figure 3.
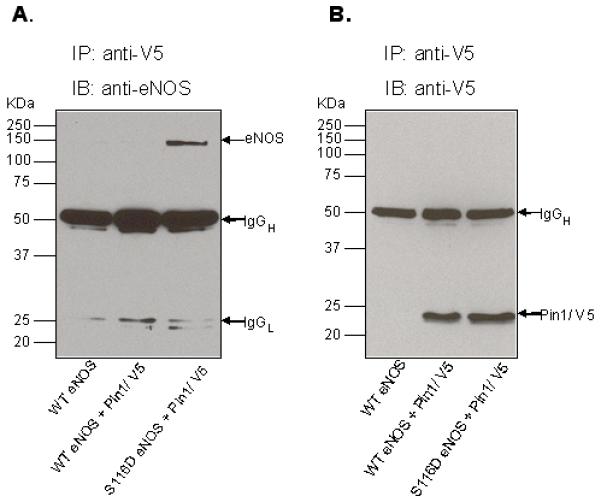
Phosphorylation-dependent association of eNOS and Pin1 following ectopic expression of Pin1 with either wild-type (WT) eNOS or phospho-mimetic (S116D) eNOS. COS-7 cells were transfected with either WT eNOS or S116D with and without cotransfection with V5-tagged Pin1. A, cell lysates were immunoprecipitated (IP) with anti-V5 antibody and immunoblotted (IB) with anti-eNOS antibody. B, cell lysates were immunoprecipitated (IP) with anti-V5 antibody and immunoblotted (IB) with anti-V5 antibody. Similar results were obtained in 3 experiments.
NO Production by Endothelial Cells is Negatively Modulated by Pin1
The effects of eNOS-Pin1 interactions in modulation of eNOS activity in cultured endothelial cells were determined using 3 different approaches. In the first, full-length human Pin1 cDNA was used to prepare and purify a wild-type Pin1 adenovirus for overexpression of the protein in BAECs. Endothelial cells were transduced with either a negative control β-galactosidase (β-gal) adenovirus or with an adenovirus expressing Pin1. After 48 h of virus infection, the amount of basal NO release was measured by quantifying nitrite in conditioned media using an NO-specific chemiluminescence analyzer as described previously 12. As shown in Figure 4A, the amount of NO released from Pin1-overexpressing cells during 48 h of virus infection was significantly reduced (~70%) compared to that from control cells. Media were also changed and cells were treated with 1 μM bradykinin (BK) for 30 min. BK-stimulated NO release was also significantly decreased (~90%) by Pin1 transduction, suggesting that Pin1 effects on eNOS activity are not rapidly reversible by agonist stimulation (Figure 4B). Cells were also lysed and eNOS-Pin1 association was assessed by immunoblotting of anti-eNOS immunoprecipitates with anti-Pin1 antibody. Pin1 binding to eNOS was confirmed to be dramatically increased in Pin1-overexpressing cells (Figure 4C). In an additional set of experiments, Pin1 overexpression produced by adenoviral infection was carried out in the absence and presence of PD98059 (50 μM). As shown in Figure 4D, blocking S116 phosphorylation by PD98059 almost completely prevented the inhibitory effect of Pin1 overexpression to suppress the amount of NO released from the cells, demonstrating clearly the phosphorylation-dependence of the Pin1 effect.
Figure 4.

Effects of Pin1 overexpression on basal and agonist-stimulated NO release from BAECs. A, BAECs were transduced with either an adenovirus expressing β-gal or an adenovirus expressing Pin1. After 48 h, the amount of basal NO release was measured by quantifying nitrite accumulation in conditioned media using the NO analyzer (means ± S.E., n=6, *P<0.05 vs. control). B, 48 h after transduction with either the adenovirus expressing β-gal or the adenovirus expressing Pin1, media was changed and cells were treated with 1 μM bradykinin (BK) for 30 min. Following BK treatment, nitrite in conditioned media was quantified with the NO analyzer (means ± S.E., n=6, *P<0.05 vs. control). C, At the end of the experiment, cells were lysed and lysates were subjected to immunoprecipitation (IP) with anti-eNOS antibody, and immunoblotting (IB) with anti-Pin1 and anti-eNOS antibodies. Results shown are representative of 3 experiments. D, BAECs were transduced with either β-gal or Pin1 adenoviruses in the presence and absence of 50 μM PD98059. After 48 h, basal NO release was measured by quantifying nitrite accumulation in conditioned media using the NO analyzer (means ± S.E., n=6, *P<0.05 vs. control).
The second approach that was used to define the role of Pin1 in regulation of eNOS in cultured endothelial cells was to inhibit endogenous Pin1 activity with a dominant negative form of Pin1. Pin1 is phosphorylated on serine 16 and a S16A Pin1 mutant that is refractory to phosphorylation has been shown to function as a dominant negative mutant 18. In addition, a phospho-mimetic S16E Pin1 mutant has also been shown to exhibit a reduced binding capacity for various Pin1 substrates18. To confirm that phospho-mimetic S16E Pin1 also displays reduced binding to phospho-S116-eNOS, we cotransfected COS-7 cells with either WT Pin1 and S116D eNOS or with S16E Pin1 and S116D eNOS before assessing Pin1-eNOS binding by co-immunoprecipitation. Immunoblotting of lysates showed equivalent levels of expression of S116D eNOS and the two forms of Pin1 in these experiments. Lysates were also immunoprecipitated with anti-Pin1 antibody. Immunoprecipitates were then immunoblotted with anti-eNOS and anti-Pin1 antibodies. As shown in Supplemental Figure I (please see http://atvb.ahajournals.org), phospho-mimetic Pin1 showed a markedly reduced degree of co-immunoprecipitation with eNOS as compared to WT Pin1 when equal amounts of the two forms of Pin1 were immunoprecipitated. Next, we prepared and purified a S16A dominant negative Pin1 adenovirus. BAECs were transduced with either an adenovirus expressing β-gal as a control or an adenovirus expressing dominant negative S16A Pin1. 48 h after virus infection, conditioned medium was analyzed for the amount of NO release using the NO analyzer. As shown in Figure 5A, the amount of NO released from cells infected with dominant negative Pin1 was significantly increased as compared to cells infected with the control virus. Media were also changed and cells were stimulated with BK (1 μM) for 30 min. BK-stimulated NO release was also significantly increased by dominant negative Pin1 infection, again suggesting that Pin1 effects on eNOS activity are not rapidly reversible following agonist stimulation (Figure 5B). Cells were also lysed and eNOS-S16A Pin1 association was assessed using the co-immunoprecipitation protocol. S16A Pin1 association with eNOS was dramatically increased compared to that from control cells (Figure 5C).
Figure 5.
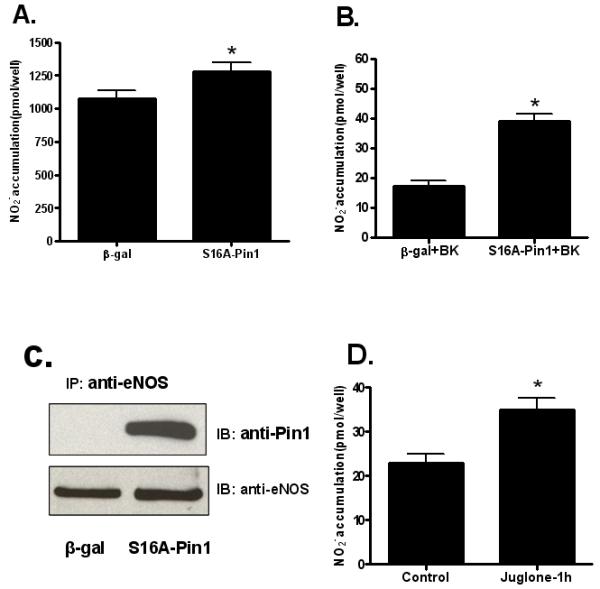
Effects of inhibition of Pin1 on basal and agonist-stimulated NO release from BAECs. A, BAECs were transduced with either an adenovirus expressing β-gal or an adenovirus expressing dominant negative S16A Pin1. After 48 h, basal NO release was measured by quantifying nitrite accumulation in conditioned media using the NO analyzer (means ± S.E., n=6, *P<0.05 vs. control). B, 48 h after transduction with either the adenovirus expressing β-gal or dominant negative S16A Pin1, media were changed and cells were treated with bradykinin (BK, 1 μM for 30 min). Nitrite accumulation following BK treatment was quantified with the NO analyzer (means ± S.E., n=6, *P<0.05 vs. control). C, At the end of the experiment, cells were lysed and lysates were subjected to immunoprecipitation (IP) with anti-eNOS antibody, and immunoblotting (IB) with anti-Pin1 and anti-eNOS antibodies. Results shown are representative of 3 experiments. D, BAECs were incubated without (control) and with 1 μmol/L juglone for 1 h. The amount of NO release during the treatment time was quantified by measuring nitrite levels in the conditioned media using an NO-specific chemiluminescence analyzer (means ± S.E, n=6, *P<0.05 vs. control).
A third approach that was used to determine the effect of Pin1 on eNOS activity was to use the selective pharmacological inhibitor of Pin1 known as juglone (5-hydroxy-1,4-napthoquinone) which covalently binds the thiol groups of cysteines 41 and 69 in the Pin1 protein and thereby irreversibly inhibits the enzyme while leaving other prolyl isomerases such as FK506 binding proteins and cyclophilins unaffected19, 20. BAECs were treated without or with juglone (1μM for 1 h). Juglone was dissolved in DMSO and applied to the cells as a 1000-fold concentrated solution. An equal volume of vehicle solution was applied to control cells. Basal NO release during the treatment time was determined and, as shown in Figure 5D, inhibition of Pin1 activity by juglone, like that of inhibition by the S16A dominant negative Pin1, also resulted in a significant increase in NO production. The requirement of eNOS phosphorylation at S116 for inhibition of eNOS by Pin1 in endothelial cells was also investigated using a nonphosphorylatable dominant negative S116A form of eNOS. Adenoviruses that express wild-type (WT) and S116A forms of eNOS were prepared and BAECs were infected with β-gal (negative control), WT eNOS, and S116A eNOS adenoviruses. Immunoblotting of cell lysates after 48 h of virus infection showed an approximately two-fold increase in total eNOS expression in both the WT eNOS and S116A eNOS adenovirus transduction conditions compared to that of the negative control (Supplemental Figure IIB). After 48 h of infection conditioned media was also analyzed for NO release by measuring nitrite accumulation. As shown in Supplemental Figure IIA, while WT eNOS produced an approximately 2-fold higher level of NO release relative to the control, the S116A dominant negative eNOS produced a statistically significant higher level of NO release (~ 3-fold over control), presumably due to the lack of S116 phosphorylation of S116A eNOS with subsequent loss of phospho-S116-dependent Pin1 binding to eNOS.
Vascular Reactivity of Intact Blood Vessels is Negatively Modulated by Pin1
To determine the role of Pin1 in modulation of vascular reactivity of intact blood vessels, we carried out experiments in which Pin1 was overexpressed in aortae of ICR mice. Mice were anesthetized and exsanguinated followed by perfusion with saline. The thoracic aorta of the mice were then infused with adenoviruses expressing either Pin1 or β-gal as a negative control. The virus-filled vessels were incubated in situ for 2 h and then incubated in vitro overnight. Aortic rings were then prepared for isometric force recording in a multi-myograph apparatus. Rings were preconstricted with 10−5 M serotonin and dose response curves to acetylcholine and sodium nitroprusside (SNP) were constructed. Rings demonstrated similar preconstruction force to serotonin with either Pin1 or β-gal overexpression (2.3±0.2 g vs. 2.2±0.2 g, Pin1vs. β-gal, p=NS). As shown in Figure 6A, rings from aortae of mice infected with the Pin1 adenovirus had significantly reduced relaxant responses to acetylcholine (~30% reduction in maximal relaxation) compared to rings from β-gal-infected control mice, indicating an important role for Pin1 in negatively modulating vascular reactivity. No differences were observed, however, in relaxant responses to SNP demonstrating that the differences observed for acetylcholine-induced relaxation were endothelium-dependent (Figure 6B).
Figure 6.
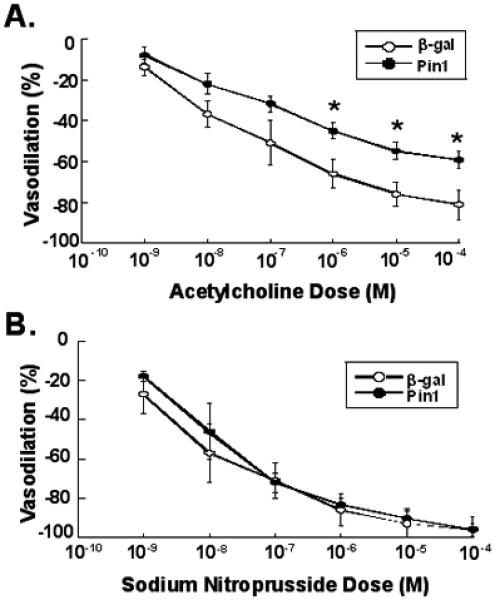
Effects of Pin1 overexpression on vasodilation in serotonin-preconstricted aortic rings of ICR mice. A, aortae were transduced with either β-gal or Pin1 adenoviruses, aortic rings were prepared, and preconstricted with serotonin. Dose response curves of relaxation to acetylcholine were then determined (means ± S.E., n=3, *P<0.05 vs. β-gal control). B, aortae were transduced with either β-gal or Pin1 adenoviruses, aortic rings were prepared, and preconstricted with serotonin. Dose response curves of relaxation to sodium nitroprusside were then determined (means ± S.E., n=3, P=NS vs. β-gal control).
Discussion
The results of the present study identify the ERK 1/2 MAP kinase as being responsible for phosphorylation of eNOS at S116 in endothelial cells under basal conditions. S116 phosphorylation has been shown previously by Kou et al. to be reduced by the protein kinase C (PKC) inhibitor, calphostin C, implicating PKC as a mediator of this specific phosphorylation reaction 21. However, Shaw et al. 22, 23 have recently shown that the AGC kinases (protein kinase A (PKA), protein kinase G (PKG), and PKC) as well as the calmodulin-dependent protein kinases (CAMK kinases) cannot phosphorylate serines or threonines in protein substrates containing a proline at the P+1 position. Proline at P+1 is thus a “veto residue” that precludes phosphorylation by AGC and CAMK kinases. This feature of proline-directed phosphorylation provides very tight control in preventing reciprocal substrate specificity between proline-directed protein kinases and AGC/CAMK kinases. Because S116 in the eNOS amino acid sequence immediately precedes P117, it is reasonable to expect that S116 would be a site of proline-directed phosphorylation by a kinase such as ERK 1/2 rather than a site of PKC or other AGC/CAMK phosphorylation. It is thus likely that the reduced S116 phosphorylation due to PKC inhibition by calphostin C that was reported previously 21 might be explained by an indirect effect of calphostin C such as blockade of PKC phosphorylation of Raf-1. This reaction is known to activate the Raf-1/MEK 1/2/ERK1/2 cascade 24 and its inhibition might contribute to ERK 1/2 inhibition.
The role of ERK 1/2 MAP kinases in eNOS regulation as well as of S116 phosphorylation in eNOS regulation have been subject to alternative explanations in the scientific literature. In various studies, ERK 1/2 inhibition in endothelial cells has been shown to either attenuate 25, not change 26, or enhance 14 eNOS activity. The latter study, whose conclusions are most consistent with our own, showed using co-immunoprecipitation that eNOS exists in a protein-protein complex with ERK 1/2 in endothelial cells and that immunoprecipitated eNOS can be phosphorylated by ERK 2 in the eNOS interactome in vitro resulting in a reduction in eNOS enzyme activity. No specific ERK 1/2 phosphorylation site in eNOS was identified in this study. However, at the time this previous study was carried out, S1179 was the only specific eNOS phosphorylation site yet known. Until recently, some controversy has also existed about whether phosphorylation of eNOS at S116 is stimulatory or inhibitory in nature. For example, phosphorylation of S116 in endothelial cells in response to increased fluid shear stress, a stimulus known to substantially increase eNOS activity, has been demonstrated by mass spectrometry 27. This would tend to suggest that phosphorylation may be stimulatory in nature. An additional study has reported that fluid shear stress, VEGF, and 8-bromo cAMP have no effect on the phosphorylation status of S116 in endothelial cells 28. However, studies by Kou et al. 21 and from our own laboratory 10 have provided convincing evidence that phosphorylation of S116 in eNOS is in fact inhibitory in nature and that phosphorylation of this site significantly reduces eNOS activity. Furthermore, Kou et al. 21 have shown that agonist stimulation of endothelial cells by VEGF induces calcineurin-mediated S116 dephosphorylation of eNOS rather than S116 phosphorylation. In our own studies, we have confirmed this effect of VEGF 10 and have also found that many other eNOS-activating agonists including bradykinin, thapsigargin, ATP, and angiopoietin promote calcineurin-mediated dephosphorylation of eNOS at S116 (unpublished observations).
Our previously published and unpublished results suggest that transient S116 dephosphorylation may contribute to the agonist-stimulated eNOS activation process in the same way that agonist-induced T497 dephosphorylation appears to also contribute to this process29. The results of the current study implicate S116 phosphorylation/dephosphorylation as having a second important role in eNOS regulation that is tonic in nature rather than transient in nature as in the case of agonist activation associated with S116 dephosphorylation. Constitutive S116 phosphorylation in endothelial cells and blood vessels under basal conditions promotes Pin1 interaction with eNOS and suppression of eNOS activity in a manner that is analogous to the tonic suppression of eNOS activity that is produced by eNOS interaction with caveolin-11. This phosphorylation-dependent interaction of Pin1 with eNOS would be expected to induce a conformational change in the eNOS enzyme. Such a conformational change could alter eNOS catalytic activity directly or could affect eNOS activity indirectly by making the enzyme more or less susceptible to phosphorylation/dephosphorylation or to proteolytic degradation. Pin1-induced conformational changes could also affect eNOS activity indirectly by altering its interaction with other protein members of the eNOS interactome. Whether direct or indirect, however, it is clear that S116 phosphorylation-dependent interaction of eNOS with Pin1 serves to suppress eNOS activity under basal and agonist-stimulated conditions in endothelial cells and in intact blood vessels. It is possible that there are additional effects of eNOS phosphorylation at S116 that are not mediated by Pin1. Obtaining definitive evidence for this will likely require future investigations of purified recombinant eNOS before and after phosphorylation at S116, with and without preincubation with purified Pin1. Future studies using various small animal models of cardiovascular disease may also reveal whether altered Pin1 regulation of eNOS has a role in endothelial dysfunction in vascular disease. Deregulation of Pin1 has been clearly shown to contribute to the disease phenotypes of cancer5 and Alzheimer’s disease6. Whether deregulation of Pin1 is similarly associated with a cardiovascular disease phenotype remains to be determined.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by grants from the NIH and the American Heart Association.
Footnotes
Disclosures
None.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–60. doi: 10.1016/j.cardiores.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–9. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- (3).Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–16. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- (4).Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–7. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- (5).Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang D-G. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. American J Pathology. 2004;164:1727–37. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lu KP, Liou Y-C, Vincent I. Proline-directed phosphorylation and isomerization in mitotic regulation and in alzheimer’s disease. BioEssays. 2003;25:174–81. doi: 10.1002/bies.10223. [DOI] [PubMed] [Google Scholar]
- (7).Liu T, Huang Y, Likhotvorik RI, Keshvara L, Hoyt DG. Protein never in Mitosis Gene A Interacting-1 (PIN1) regulates degradation of inducible nitric oxide synthase in endothelial cells. Am J Physiol Renal Physiol. 2008;295:C819–C827. doi: 10.1152/ajpcell.00366.2007. [DOI] [PubMed] [Google Scholar]
- (8).Jaffrey SR, Snyder SH. PIN: An associated protein inhibitor of neuronal nitric oxide synthase. Science. 1996;274:774–7. doi: 10.1126/science.274.5288.774. [DOI] [PubMed] [Google Scholar]
- (9).Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, Nerem RM, Alexander RW, Murphy TJ. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest. 1992;90:2092–6. doi: 10.1172/JCI116092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li C, Ruan L, Sood SG, Papapetropoulos A, Fulton D, Venema RC. Role of eNOS phosphorylation at Ser-116 in regulation of eNOS activity in endothelial cells. Vasc Pharmacol. 2007;47:257–64. doi: 10.1016/j.vph.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).He T-C, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE, Jennings IG, Venema RC. Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. J Biol Chem. 2005;280:35943–52. doi: 10.1074/jbc.M504606200. [DOI] [PubMed] [Google Scholar]
- (13).Fulton D, Ruan L, Sood SG, Li C, Zhang Q, Venema RC. Agonist-stimulated endothelial nitric oxide activation and vascular relaxation: role of eNOS phosphorylation at Tyr-83. Circ Res. 2007;102:497–504. doi: 10.1161/CIRCRESAHA.107.162933. [DOI] [PubMed] [Google Scholar]
- (14).Bernier SG, Haldar S, Michel T. Bradykinin-regulated interactions of the mitogen-activated protein kinase pathway with the endothelial nitric-oxide synthase. J Biol Chem. 2000;275:30707–15. doi: 10.1074/jbc.M005116200. [DOI] [PubMed] [Google Scholar]
- (15).Pearson G, Robinson F, Gibson RB, Xu B-E, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrine Reviews. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- (16).Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- (17).Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–32. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- (18).Lu P-J, Zhou XZ, Liou Y-C, Noel JP, Lu KP. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J Biol Chem. 2002;277:2381–4. doi: 10.1074/jbc.C100228200. [DOI] [PubMed] [Google Scholar]
- (19).Hennig L, Christner C, Kipping M, Schelbert B, Rücknagel KP, Grabley S, Küllertz G, Fischer G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry. 1998;37:5953–60. doi: 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- (20).Siepe D, Jentsch S. Prolyl isomerase Pin1 acts as a switch to control the degree of substrate ubiquitylation. Nat Cell Biol. 2009;11:967–72. doi: 10.1038/ncb1908. [DOI] [PubMed] [Google Scholar]
- (21).Kou R, Greif D, Michel T. Dephosphorylation of endothelial nitric oxide synthase by vascular endothelial growth factor: implications for the vascular responses to cyclosporin A. J Biol Chem. 2002;277:29669–73. doi: 10.1074/jbc.M204519200. [DOI] [PubMed] [Google Scholar]
- (22).Fujii K, Zhu G, Liu Y, Hallam J, Chen L, Herrero J, Shaw S. Kinase peptide specificity: Improved determination and relevance to protein phosphorylation. PNAS. 2004;101:13744–9. doi: 10.1073/pnas.0401881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhu G, Fujii K, Belkina N, Liu Y, James M, Herrero J, Shaw S. Exceptional disfavor for proline at the P+1 position among AGC and CAMK kinases establishes reciprocal specificity between them and the proline-directed kinases. J Biol Chem. 2005;280:10743–8. doi: 10.1074/jbc.M413159200. [DOI] [PubMed] [Google Scholar]
- (24).Kolch W, Heidecker G, Kochs G, Hummel R, Vahidl H, Mischak H, Finkenzeller G, Marmé D, Rapp UR. Protein kinase Cα activates RAF-1 by direct phosphorylation. Nature. 1993;364:249–52. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- (25).Cale JM, Bird IM. Inhibition of MEK/ERK1/2 signalling alters endothelial nitric oxide synthase activity in an agoinist-dependent manner. Biochem J. 2006;398:279–88. doi: 10.1042/BJ20060371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Schmidt K, Givraeil HD, Mayer B. Lack of involvement of extracellular signal-regulated kinase (ERK) in the agonist-induced endothelial nitric oxide synthesis. Biochem Pharmacol. 2002;63:1137–42. doi: 10.1016/s0006-2952(01)00936-4. [DOI] [PubMed] [Google Scholar]
- (27).Corson MA, James NL, Latta SE, Nerem RM, Berk BC, Harrison DG. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ Res. 1996;79:984–91. doi: 10.1161/01.res.79.5.984. [DOI] [PubMed] [Google Scholar]
- (28).Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H. Shear stress stimulates phosphorylation of eNOS at Ser635 by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol. 2002;283:H1819–H1828. doi: 10.1152/ajpheart.00214.2002. [DOI] [PubMed] [Google Scholar]
- (29).Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen Z-P, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J Biol Chem. 2001;276:16587–91. doi: 10.1074/jbc.M100229200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.